

Abstract
The imidazo[1,2-a]pyridine-3-carboxyamides (IAPs) are a unique class of compounds endowed with impressive nanomolar in vitro potency against Mycobacterium tuberculosis (Mtb) as exemplified by clinical candidate Telacebec (Q203). These compounds target mycobacterial respiration through inhibition of the QcrB subunit of cytochrome bc1:aa3 super complex resulting in bacteriostatic efficacy in vivo. Our labs have had a long-standing interest in the design and development of IAPs. However, some of these compounds suffer from short in vivo half-lives, requiring multiple daily dosing or the addition of a cytochrome P450 inhibitor for murine efficacy evaluations. Deuteration has been shown to decrease metabolism as the C-D bond is stronger than the C–H bond. Herein we describe our efforts on design and synthesis of potent deuterated IAPs and the effect that deuteration has upon metabolism through microsomal stability studies.
Keywords: Anti-tuberculosis, Deuterated imidazopyridines, Mycobacterial respiration inhibition, Metabolism
1. Introduction
Tuberculosis (TB) is an ancient but still sinister disease of global importance. It is caused by the air borne pathogenic bacterium Mycobacterium tuberculosis (Mtb). In 2019, about 1/5 of the population was infected with Mtb resulting in 1.4 million deaths [1]. Treatment of drug sensitive Mtb infections require administration of multiple drugs (isoniazid, rifampicin, pyrazinamide and ethambutol) for many months. The COVID-19 pandemic is expected to have a devastating effect with potentially 200,000 to 400,000 of excess TB deaths annually due to lack of TB identification and sustained treatment [1]. In addition, an estimated 1.7 billion people are currently infected with latent TB and drug resistant Mtb remains on the rise further exacerbating the problem with > 480,000 becoming ill with drug resistant TB each year [2]. New treatments are desperately needed to treat latent, drug sensitive and drug resistant Mtb.Fig. 1.
Fig. 1.

Structures of imidazo[1,2-a]pyridine-3-carboxyamides ND-10885 (1) and ND-11176 (2) as compared to Phase-II anti-TB compound Telacebec (Q203).
Our labs were the first to report on the anti-tubercular potency of imidazo[1,2-a]pyridine-3-carboxamides (IAPs) [3]. Through extensive SAR (structure–activity-relationship) studies, we evolved the series to exemplary compounds with nanomolar potency, [4] reported on our scaffold hopping efforts [5] and identified various compounds with in vivo efficacy in the murine model of TB and other life threatening strains of mycobacteria [6] Interestingly, many of the most efficacious IAPs reported violate the medicinal chemistry “rule of five” rubric [7] since they have high molecular weights (>500 amu) and are highly lipophilic (clogP > 5). Despite these guideline infractions, Telacebec (Q203) [8] is an IAP currently undergoing clinical studies as a potential TB treatment [9]. We have found that IAPs can be designed (like ND-10885, 1 and ND-11176, 2) which obey the “rule of five” but often such compounds have short in vivo half-lives resulting in low drug exposure that is not suitable for potential once-a-day oral dosing [10,11]. ND-11176 (2) is very potent compound (MIC = 7–11 nM) and obeys the “rule of five” however when dosed in vivo two major metabolites were observed within rodents and marmosets. One metabolite was a hydroxylation product (presumably upon a CH3) and the other was di-oxidation (transformation of a CH3 to COOH), these metabolites appeared in greater abundance than parent compound within an hour of dosing (data not shown). As such, the rapid metabolism of ND-11176 (2) is a major hindrance to its development potential.
Deuterium, the stable and nonradioactive isotope of hydrogen, has been used to enhance drug exposure in medicinal chemistry for decades [12]. Compared to C–H bonds, C-D bonds are more stable to oxidative processes that may affect enzymatic and metabolic processes in biological systems [13]. The first report on metabolic stabilization of bioactive compounds by deuterium incorporation was in 1961 [14]. Later on, deuterium substitution was also widely used to decrease toxicity, minimize metabolic lability and improve pharmacokinetics [15]. In 2017, the FDA approved the first deuterium drug, deutertrabenazin (3, Fig. 2), to treat Huntington’s disease and tardive dyskinesias [16]. This prompted additional interest in deuterated drugs and as of 2019, there were >20 deuterated compounds undergoing clinical trials with eight in Phase III [17]. Our lab previously designed and synthesized a novel deuterated anti-TB compound D-BTZ043 (4, Fig. 2) [18]. Our studies of D-BTZ043 (4) demonstrated that incorporation of deuterium into the core nitro aromatic warhead of anti-TB agent BTZ043 prolongs the lifetime of the active nitroso oxidation state by the deuterium isotope effect while retaining its low nanomolar anti-TB potency [18]. Herein, we report the design, syntheses and metabolic studies of the first reported deuterated imidazo[1,2-a]pyridine-3-carboxamides with retention of excellent nanomolar anti-TB potency.
Fig. 2.

Structures of Deutertrabenazin (3), BTZ-043 and D-BTZ043 (4).
2. Results and discussion
With over a decade of experience working with the IAP class of compounds and determining structure activity relationship (SAR) trends, we first prepared a small, focused set of analogs and screened them against Mycobacterium tuberculosis H37Rv in two different types of media (7H12 and GAS) in the Alamar blue metabolic readout assay [3,5]. The two different types of media were used to determine if potency could be carbon source dependent as first described by Pethe et. al. [19] Next, selected compounds were evaluated for activity in nonreplicating conditions by the low oxygen recovery assay (LORA), [3,5,20] to assess the potential for killing bacteria that persist due to reduced metabolism, and for toxicity against VERO cells (kidney epithelial cells) [3,5,21].
Common methods to synthesize deuterium compounds include using commercially available deuterium-labeled small building blocks, as well as direct metal catalyzed H-D exchange reactions of advanced intermediates and even final target compounds. Our first effort to synthesize deuterium-labeled imidazopyridines was inspired by the report of Sajiki et.al. [22] and utilized a metal catalyzed H-D exchange reaction to generate the key intermediate deuterated 5-methylpyridin-2-amine 6 (Scheme 1). However, the reported conditions that only used Pd/C as the catalyst required heating in D2O at 160 °C in a sealed tube under a H2 atmosphere. As an alternative, we generated a Pd/C-Pt/C mixed catalyst system that allowed reduction of the reaction temperature to 120 °C while providing the fully deuterated 5-methylpyridin-2-amine (6) in 85 % yield. Subsequent treatment of 6 with commercially available ethyl 2- chloro-3-oxobutanoate (7) at reflux gave the deuterated imidazo[1,2-a] pyridine heterocycle (8). Saponification of 8 with NaOH, followed by acidification gave the versatile deuterated carboxylic acid (9) which was utilized to prepare the panel of desired deuterated analogs (29–47) with variable yields (26–75 %) through classical amide bond formation (HATU or EDC-mediated) with appropriate amines (10 – 28, Scheme 1).
Scheme 1.

Synthesis of deuterated imidazopyridine core and related carboxamides (10-28).
As shown in Table 1, screening revealed that regardless of media carbon source (GAS or 7H12), these deuterated imidazo[1,2-a]pyridine-3-carboxamides are potent anti-TB inhibitors. This observation is consistent with past studies with the non-deuterated imidazo[1,2-a] pyridine-3-carboxamides [3–6]. Also consistent with our previous reports, the deuterated imidazo[1,2-a]pyridine-3-carboxamides are non-toxic against VERO cells (all have IC50 values > 10,000 nM) and at best are weakly active against “latent” TB (MIC range 4,500 to > 10,000 nM). As inhibitors of ATP respiration, specifically the cyt-bc1:aa3 super complex, greater potency against replicating over “latent” (non-replicating) Mtb is expected [21]. SAR trends explored the effect of electron withdrawing groups (4-fluoro, 4-chloro, 4-trifluoromethyl, 4-pentafluorothio, 4-methyl sulfone, and 4-trifluoromethoxy) and electron donating groups (4-tert-butyl, 4-dimethylamine, 4-methoxy, 4-ethoxy and two cyclic ethers). Of the electron withdrawing groups, the 4-trifluoromethoxy (34) and 4-pentafluorothio (32) were the most active compounds with low MIC values of 2.2 nM and 2.9 nM against H37Rv-Mtb in GAS media, respectively (5.5 nM and 5.9 nM in 7H12 media, respectively) which compares well with clinical candidate Q203 (MIC = 0.9 nM and 3.6 nM in GAS and 7H12 media, respectively). The 4-trifluoromethyl (30), 4-chloro (31) and 4-fluoro (29) showed a trend of decreasing in activity with GAS media (MICs of 9.1, 12 and 23.3 nM) but more divergent MICs in the 7H12 media revealing 34 (bearing the 4-CF3) as the most active of these (MIC = 2.2 nM in GAS media). The 4-methyl sulfone (33) was the least active of the compounds containing electron withdrawing groups with MIC values from 174 – 212 nM in both media. Remarkably, analog 35 bearing the electron donating tert-butyl moiety had superior MIC values of 0.5 nM and 2.7 nM in GAS and 7H12 media, respectively. This activity is comparable to that of Q203 (MIC = 0.9 and 3.6 nM in GAS and 7H12 media, respectively). The other analogs with electron donating alkoxy groups, while still very active, had higher MIC values ranging from 9.2 nM (38) to 24.5 nM (37) in GAS media. The two cyclic alkoxy compounds (39 and 40) were the only compounds of the set to show improved activity in 7H12 media with MICs decreasing from 22.5 and 10.1 nM in GAS to 7.2 and 5.1 nM in 7H12, respectively. With the exception of 38, compounds bearing electron donating groups (36–40) had, on average, higher MICs than those bearing electron withdrawing groups. Interestingly, the t-butyl compound (35) was > 8-fold more active in 7H12 media and > 18-fold more active in GAS media than the second most active compound with an electron donating group (38, 4-ethoxy). Looking at SAR trends based on positional considerations of the substituents, we prepared and evaluated the ortho-, meta- and para-chloro isomers (42, 41 and 31, respectively). The 3-chloro (“meta-”, 41) had slightly better activity (MIC of 3.5 and 22.2 nM, GAS and 7H12 media, respectively) than the 4-chloro (“para-”, 31) isomer (MIC of 12.0 and 23.1 nM, GAS and 7H12 media, respectively). The 2-chloro analog (“ortho-”, 42) was the least active of the three with an MIC as high as 112 nM, in 7H12 media (13.4nM in GAS media). To further explore substituent effects at the meta-position we prepared and evaluated three additional analogs (3-trifluoromethoxy, 3-dimethylamine, 3-methoxy). The 3-OCF3 (“meta-”, 43) analog was ~ 2.9-fold less active than the 4-OCF3 (“para-”, 30) in GAS media and > 28-fold less active in 7H12 media. Similarly, the electron donating analog 3-N(CH3)2 (“meta-”, 44) was ~ 3.5-fold less active (in GAS media) and > 5-fold (in 7H12 media) than 3-N(CH3)2 (“para-”, 36). The 3-OCH3 (“meta-”, 45) gave divergent results depending on the media used. In GAS media it was slightly more potent than the 4-OCH3 (“para-”, 37) isomer (20.5 to 24.5 nM, respectively). However, in 7H12 media it was 3-fold less active (105 to 35.4 nM, respectively). This example highlights an advantage of using more than one type of media (or readout) in screening for compound ranking as compounds 32, 34, 35 and Q203, clearly are much more active compounds in both assay conditions. In order to explore the steric and electronic effects of representative di-substituted benzylamides, we synthesized 46 (3,5-difluoro) and 47 (3,5-dichloro). The smaller di- substituted analog 46 (3,5-difluoro) had low MIC values of 7.2 and 12.4 nM, in GAS and 7H12 media, respectively, whereas 47 (3,5-dichloro) was > 2-fold less active (MICs of 18.8 and 40 nM, in GAS and 7H12 media, respectively). The general SAR trend appears to indicate a preference towards size over electronics as both the large 4-pentafluorothio and 4-tert-butyl groups were accommodated and elicited the best activity suggesting a large binding pocket which is consistent with the impressive activity of the large lipophilic amide of Q203. Four non-deuterated analogs of compounds 32, 33, 36 and 46 have been reported in literature and their MICs against H37Rv have been reported [4].a,b,d As anticipated the MIC values of deuterated and non-deuterated compounds were very similar and all (with the exception of 46) were within the 2-fold error of a whole cell screening assay. Non-deuterated analog of 32 had an MIC of 7 nM (compared to 3–6 nM for 32), [4]d analog 33 had an MIC of 400 nM, (compared to 174 – 212 nM for 33), [4]a analog 36 had an MIC of 50 nM (compared to 14–21 nM for 36) [4]a and analog 46 had an MIC of 80 nM (compared to 7–12 nM for 46) [4]b. It should be noted that only the MIC of non-deuterated 32 was determined in the same conditions (MABA assay) [4]d as the deuterated compounds while all the others were determined using an RFU readout [4]b.
Table 1.
Deuterated imidazo[1,2-a]pyridine-3-carboxyamides (29–47) designed and screened against M. tuberculosis H37Rv in two types of media (GAS and 7H12), in low oxygen “latent” (LORA) conditions and for toxicity against Vero cells. Units in nanomolar (nM).
| MIC (nM) | IC50 (nM) | ||||||
|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
||
| Cmpd | MW | ClogP* | Structure | GAS MIC | 7H12 MIC | LORA MIC | Vero |
|
| |||||||
| 29 | 303.36 | 3.74 |

|
23.3 ± 0.5 | 110 ± 11.8 | >10,000 | >10,000 |
| 30 | 353.37 | 4.48 |
|
9.1 ± 5.8 | 3.8 ± 1.3 | >10,000 | >50,000 |
| 31 | 319.82 | 4.31 |

|
12.0 ± 1.5 | 23.1 ± 7.1 | 7,090 | >10,000 |
| 32 | 411.42 | 4.83 |

|
2.9 ± 0.5 | 5.9 ± 0.3 | NT | NT |
| 33 | 363.46 | 1.95 |

|
174 ± 26.9 | 212 ± 28.2 | >10,000 | >10,000 |
| 34 | 369.37 | 4.62 |

|
2.2 ± 0.4 | 5.5 ± 1.8 | >5,000 | >50,000 |
| 35 | 341.48 | 5.42 |

|
0.5 ± 0.2 | 2.7 ± 0.2 | NT | NT |
| 36 | 328.44 | 3.76 |
|
14.4 ± 4.2 | 21.4 ± 11.6 | >10,000 | >10,000 |
| 37 | 315.4 | 3.51 |

|
24.5 ± 0.5 | 35.4 ± 10.0 | >10,000 | >10,000 |
| 38 | 329.43 | 4.04 |

|
9.2 ± 1.3 | 23.6 ± 10.4 | NT | NT |
| 39 | 341.44 | 4.06 |

|
22.5 ± 1.8 | 7.2 ± 0.4 | NT | NT |
| 40 | 343.41 | 3.52 |

|
10.1 ± 1.0 | 5.1 ± 1.6 | NT | NT |
| 41 | 319.82 | 4.31 |

|
3.5 ± 0.3 | 22.2 ± 5.7 | 4,520 | >10,000 |
| 42 | 319.82 | 4.31 |

|
13.4 ± 2.2 | 112 ± 19.5 | 8,490 | >10,000 |
| 43 | 369.37 | 4.62 |

|
26.3 ± 3.6 | 108 ± 7.4 | 4,920 | >10,000 |
| 44 | 328.44 | 3.76 |

|
50.2 ± 6.4 | 111 ± 36.0 | NT | NT |
| 45 | 315.4 | 3.51 |

|
20.5 ± 0.4 | 105 ± 19.1 | NT | NT |
| 46 | 321.35 | 3.88 |

|
7.2 ± 1.6 | 12.4 ± 4.5 | 6,910 | >10,000 |
| 47 | 354.26 | 5.02 |

|
18.8 ± 2.8 | 40.0 ± 13.0 | 8,870 | >10,000 |
| Q203 | 557.01 | 7.62 |

|
0.9 ± 0.4 | 3.6 ± 0.5 | >5,000 | >10,000 |
ClogP was calculated from PerkinElmer Chemdraw® Professional version 16.0.1.4. NT = Not tested.
The impressive activity of the deuterated compounds shown in Table 1, prompted studies to determine if they would also be metabolically more stable. To that end we selected compound 30 as the focal point of our efforts because its non-deuterated analog (ND-11176, 2) had been shown to have in vivo activity in the acute murine model of TB (2 log CFU bacterium reduction at 300 and 500 mg/kg oral dose) but suffered from a short half-life (t1/2 < 2 h) [11]. Previous studies with this parent non-deuterated compound (ND-11176, 2), indicated hydroxylation as the major source of metabolism when it was dosed orally in mice (data not shown). While either the 6-CH3 or 2-CH3 are probable sites of metabolism, the methylene group of the benzyl amine component was also of concern. Therefore, we synthesized a series of deuterium labeled analogs of 2 with incorporation of deuterium at different positions, to further probe whether metabolism could be improved through deuteration (Table 2). Subjection of imidazo[1,2-a] pyridine (49) to the Pd/C-Pt/C catalyst system under a hydrogen atmosphere using different D% of D2O as the solvent yielded different deuterated intermediates. As illustrated in Scheme 2, use of D2O (D, 70 %) gave 50 with deuterium substitution at the 7-H, 8-H and 2-methyl groups but not the 5-H or 6-methyl, while increasing deuterium exposure by use of D2O (D, 99.9 %) provided 53 with additional 2-methyl deuteration. Separate saponification of 50 and 53, gave the corresponding carboxylic acids 51 (2- CD3, 7-D, 8-D) and 54 (2, 6- CD3, 7-D, 8-D) which upon coupling with 4-(trifluoromethyl)benzylamine gave products 52 and 55, respectively. Deuteration of the methylene of 4-CF3-benzylamine (56) was accomplished by reduction of 4-(trifluoromethyl)benzonitrile using the Pd/C-D2 catalyzed system in the presence of DCl (Scheme 3). Thus, inspired by the literature method, [23] D2 was first generated through a Pd/C catalyzed H2-D2 exchange reaction H2-D2O. Addition of 4-(trifluoromethyl)benzonitrile and DCl to the reaction mixture, gave methylene deuterated 4-CF3-benzylamine (56) as a DCl salt in 80 % yield (90 % deuterium incorporation). Standard coupling of 56 with deuterated intermediate 54, produced methylene deuterated product 57 in good yield.
Table 2.
Focused set of four deuterated analogs designed around ND-11176 (2) and screened against M. tuberculosis H37Rv in two types of media (GAS and 7H12), in LORA conditions and for toxicity against Vero cells. Units in nanomolar (nM).
| MIC (nM) | IC50 (nM) | ||||||
|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
||
| Cmpd | MW | ClogP* | Structure | GAS MIC | 7H12 MIC | LORA MIC | Vero |
|
| |||||||
| 30 | 353.37 | 4.48 |

|
9.1 ± 5.8 | 3.8 ± 1.3 | >10,000 | >50,000 |
| 52 | 352.37 | 4.48 |

|
7.4 ± 2.8 | 3.9 ± 1.6 | >10,000 | >50,000 |
| 55 | 355.39 | 4.48 |

|
9.6 ± 2.6 | 4.9 ± 1.3 | >10,000 | >50,000 |
| 57 | 357.40 | 4.48 |

|
5.7 ± 1.2 | 6.7 ± 0.9 | >10,000 | >50,000 |
| 2 | 347.34 | 4.48 |

|
11.3 ± 2.5 | 7.1 ± 2.5 | >10,000 | >50,000 |
| Q203 | 557.01 | 7.62 |

|
0.9 ± 0.4 | 3.6 ± 0.5 | >5,000 | >50,000 |
ClogP was calculated from PerkinElmer Chemdraw® Professional version 16.0.1.4.
Scheme 2.

Synthesis of deuterated methyl substituted imidazopyridine carboxamides (52 and 55).
Scheme 3.

Synthesis of deuterated imidazopyridine with deuterated benzyl carboxamide 57.
These three new deuterated compounds (52, 55, and 57) were tested for anti-TB activity along with 2 (the non-deuterated analog, ND-11176). As anticipated, the anti-TB activity was consistent among all compounds indicating that deuteration did not negatively affect their potency.
To determine whether deuteration improved metabolic stability, we evaluated all the deuterated analogues of 2 (namely, 30, 52, 55 and 57) in mouse, marmoset monkey and human liver microsomes by standard methods and calculated the half-life (t1/2) and clearance (CLint) (Table 3). Gratifyingly, in all cases the deuterated analogs showed better metabolic stability as demonstrated by longer half-lives and lower clearance than non-deuterated 2. In human microsomes, 30 exhibited the best metabolic stability among the analogues (53 min vs 30 min for 30 and 2, respectively). Interestingly, increased deuteration did not always improve metabolism since 55 had a shorter half-life in human microsomes than did 30 and 52. This might be caused by a metabolic switch, in which one metabolic site was blocked while the other site was enhanced [24]. As often seen with metabolism studies, species differences were observed as highly deuterated compound 55 had the best half-life in mouse (t1/2 = 33 min) and monkey (t1/2 = 14 min) but not human (t1/2 = 36 min) compared with other deuterated versions. Furthermore, the monkey microsomes showed the most metabolism when typically, compounds are more rapidly metabolized in rodent species (mouse or rat) [25,26]. Ultimately, it is human microsomal metabolism that is the most important for drug development but having reasonable CLint (<50 mL/min/g protein, an in vitro standard set in our labs for compound advancement to helpful for translation of animal model efficacy to human prediction in vivo assessment) in rodents is very models. To that end, compounds 30 and 2 were evaluated in two additional species (rat and dog) for metabolic stability. Again, deuterium incorporation increased half-life and decreased drug clearance as compound 30 had a half-life of 22 min in dog microsomes (CLint = 64 mL/min/g protein) and 50 min in rat microsomes (CLint = 28 mL/min/ g protein) compared to 18 min in dog microsomes (CLint = 78 mL/min/g protein) and 29 min in rat microsomes (CLint = 48 mL/min/ g protein) for non-deuterated analog 2.
Table 3.
In vitro metabolism determination of four deuterated analogs of ND-11176 (2) in mouse, marmoset and human hepatic microsomes.
| Cmpd | Structure | Mouse | Marmoset | Human | |||
|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
|||
| Half-life (min) |
CLint (ml/min/g protein) | Half-life (min) |
CLint (ml/min/g protein) | Half-life (min) |
CLint (ml/min/g protein) | ||
|
| |||||||
| 30 |

|
20 | 70 | 12 | 116 | 53 | 26 |
| 52 |

|
16 | 87 | 12 | 116 | 46 | 30 |
| 55 |

|
33 | 42 | 14 | 98 | 36 | 38 |
| 57 |

|
18 | 78 | 23 | 60 | 43 | 32 |
| 2 |

|
15 | 92 | 10 | 141 | 30 | 46 |
3. Conclusion
In conclusion, we have developed efficient strategies to deuterate imidazo[1,2-a]pyridines. The in vitro screening confirmed that these deuterated imidazo[1,2-a]pyridine-3-carboxamides have excellent, nanomolar potency against Mtb and are non-toxic to Vero cells. Furthermore, deuteration can improve metabolism by prolonging half-live and decreasing microsomal clearance within multiple animal species particularly in human microsomes. Our future work will include determination in vivo pharmacokinetics and, if warranted, evaluation of efficacy within a murine model of TB infection.
4. Materials and methods
General.
All anhydrous solvents, reagent grade solvents for chromatography and starting materials were purchased from either Aldrich Chemical Co. (Milwaulkee, WI) or Fisher Scientific (Suwanee, GA) unless otherwise noted. Water was distilled and purified through a Milli-Q water system (Millipore Corp., Bedford, MA). General methods of purification of compounds involved the use of silica or silica cartridges purchased from Silicycle (https://www.silicycle.com/) and/or recrystallization. The reactions were monitored by thin-layer chromatography (TLC) on precoated Merck 60 F254 silica gel plates and visualized using UV light (254 nm). All compounds were analyzed for purity by HPLC and characterized by 1H and 13C NMR using a Bruker Ascend Avance III HD Spectrometer (500 MHz). Chemical shifts are re-ported in ppm (δ) relative to the residual solvent peak in the corresponding spectra; chloroform δ 7.27 and δ 77.23, methanol δ 3.31 and δ 49.00 and coupling constants (J) are re-ported in hertz (Hz) (where, s = singlet, bs = broad singlet, d = doublet, dd = double doublet, bd = broad doublet, ddd = double doublet of doublet, t = triplet, tt – triple triplet, q = quartet, m = multiplet) and analyzed using MestreNova NMR data processing. Since each position of each compound is variably susceptible to deuterium exchange, the extent of deuteration at each exchanged position in deuterated compounds obtained was determined by and reported as the 1H NMR integration for the remaining amount of H at each position. For example with compound 8, the peak at 9.09 is labeled as (s, 0.05H) which indicates 95 % exchange of deuterium for hydrogen within the limitations of NMR integration. 19F NMR were run without a standard and are uncorrected. Mass spectra values are reported as m/z. Melting points were measured on a Buchi B-545 melting point instrument and are uncorrected, measured against benzoic acid 118.8–120.2 °C.
LC-MS method “MSU-A”:
Liquid Chromatography-Mass Spectrometry method was performed on an Agilent 1290 infinity coupled to Agilent 6538 Ultra High-Definition Quadrupole Time of Flight (UHD-QTOF) instrument. A separation was achieved by using reverse phase Waters Acquity UPLC HSS T3 1.8 μm (2.1 × 100 mm) column from Waters (Milford, USA). All solvents were purchased from Fisher Scientific LC-MS Optima grade solvents. Water containing 0.1 % formic acid was used as mobile phase A and acetonitrile (CH3CN) containing 0.1 % formic acid was used as mobile phase B. The injection volume was set at 1 μL. Samples were injected in a gradient of 95 % mobile phase A and 5 % mobile phase B in the initial condition to 5 % mobile phase A and 95 % mobile phase B in 9 min. The eluent was held at that composition for an additional 3 min and switched back to the initial condition at 12 min.
The HRMS data acquisition was performed from 50 to 1000 m/z at 1.0 spectra/sec scan rate. The source gas temperature was set at 350 °C with a flow of 8 L/min. The nebulizer gas was set at 55 psig. The capillary voltage was set at 3500 V with fragmentor at 100, skimmer at 45 and octopole RF 500 V. Prior to sample runs, the instrument was calibrated using Agilent low mass calibrant solution.
Data analysis:
The data collected in Agilent LC-MS was analyzed using Agilent Mass Hunter software for HRMS calculation.
LC-MS method “ND-B”:
The liquid chromatography mass spectral (LC-MS) analyses were carried out on a Waters ZQ instrument consisting of Chromatography Module Alliance HT photodiode array detector 2996 and mass spectrometer Micromass ZQ using a 3 X 50 mm Pro C18 YMC reverse phase column and the following mobile phases: 10 mm ammonium acetate in HPLC grade water (A) and HPLC grade acetonitrile (B). A gradient was formed from 5 % to 80 % of B in 10 min with a flow rate of 0.7 mL/min.
4.1. 5-(Methyl-d3)pyridin-3,4,6-d3-2-amine (6)
To a solution of 5-methylpyridin-2-amine (5, 6 mmol, 648 mg) in 10 mL of D2O (DLM-4-PK from Cambridge Isotope, D, 99.9 %) under an argon atmosphere in a sealed tube was added 10 % Pd/C (10 % wt of 5- methylpyridin-2-amine, 65 mg) and 5 % Pt/C (20 % wt of 5-methylpyridin-2-amine, 130 mg). The tube was sealed with a rubber septum and filled with H2 using three vacuum/H2 cycles, then the rubber stopper was replaced with a polytetrafluoroethylene front seal plug. The reaction mixture was heated at 120 °C in an oil bath for 24 h. After cooling to room temperature, the mixture was diluted with EtOAc, purged with argon and then filtered to remove the catalyst. The filtrate was partitioned between EtOAc and water. The aqueous layer was extracted with EtOAc (2 × 15 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo to give 2 as a colorless solid in 85 % yield (5.1 mmol, 581.4 mg). The product was used directly for the next step without any further purification.
4.2. Ethyl 2-methyl-6-(methyl-d3)imidazo[1,2-a]pyridine-3-carboxylate-5,7,8-d3 (8)
To a stirred mixture of 6 (2 mmol, 228 mg), Et3N (2.4 mmol, 326 μL) in 20 mL of CH3CN was added dropwise a solution of ethyl 2-chloroacetoacetate (7, 2.4 mmol, 333 μL) in 5 mL of CH3CN over a period of 10 min. The mixture was stirred overnight at reflux, cooled to room temperature, and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluting with hexanes and EtOAc (3:1) to give 8 as a white solid in 73 % yield (1.46 mmol, 327 mg). 1H NMR (400 MHz, MeOD): δ 1.42 (t, 3H, J = 8.0 Hz), 2.34–2.37 (m, 0.16H), 2.63 (s, 3H), 4.40 (q, 2H, J = 8.0 Hz), 7.37–7.39 (m, 0.07H), 7.45–7.47 (m, 0.05H), 9.09 (s, 0.05H). LC-MS (ESI positive mode) m/z ([M + 1]); Anal. Calcd for C12H9D6N2O2225.15; Found 225.20. HPLCtR = 6.6 min, method ND-B.
4.3. 2-Methyl-6-(methyl-d3)imidazo[1,2-a]pyridine-3-carboxylic-5,7,8-d3 acid (9)
To a solution of 8 (1 mmol, 224 mg) in MeOH was added 2 M NaOH (4 eq, 2 mL). The mixture was stirred at 60 °C for 2 h, and then concentrated under reduced pressure. The residue was dissolved in water and washed with EtOAc. The aqueous layer was adjusted to pH 5.5–6.0 with 2 N HCl at room temperature, and then concentrated under reduced pressure to give 9 as a white solid which was used for the next step without further purification. HRMS (ESI, positive mode) m/z ([M + 1]); Anal. Calcd for C10H5D6N2O2 197.12; Found 197.14. HPLCtR = 2.18 min, method ND-B.
4.4. N-(4-Fluorobenzyl)-2-methyl-6-(methyl-d3)imidazo[1,2-a]pyridine-5,7,8-d3–3-carboxamide (29)
To a solution of intermediate 9 (0.2 mmol, 39 mg) and (4-fluorophenyl)methanamine (0.22 mmol, 0.025 mL) in 6 mL of anhydrous CH3CN was added EDC•HCl (0.4 mmol, 77 mg) and DMAP (0.4 mmol, 49 mg). The mixture was stirred at room temperature for several hours and monitored by LC/MS. When the reaction was completed, the solution was concentrated under reduced pressure and the residue was purified on silica gel chromatography eluting with DCM/MeOH (50:1). Compound 29 was obtained as a white solid in 50 % yield (30 mg, 0.1 mmol). 1H NMR (500 MHz, MeOD) δ 2.30 (s, 0.29H), 2.58 (s, 3H), 4.59 (s, 2H), 7.05–7.08 (m, 2H), 7.26–7.29 (m, 0.1H), 7.41–7.43 (m, 2H), 7.87–7.88 (m, 0.03H), 8.78–8.81 (m, 0.07H). 13C NMR (500 MHz, MeOD) δ 14.11, 42.27, 114.63, 114.93 (d, J = 21.25 Hz), 115.89, 123.23, 125.20, 129.34 (d, J = 7.5 Hz), 130.35, 135.10 (d, J = 3.75 Hz), 144.64, 145.04, 161.32, 162.09, 163.26. HRMS (ESI-TOF, positive mode) m/z ([M + 1]); Anal. Calcd for C17H11D6FN3O 304.1729; Found 304.1727.
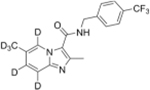
4.5. 2-Methyl-6-(methyl-d3)-N-(4-(trifluoromethyl)benzyl)imidazo [1,2-a]pyridine-5,7,8-d3-3-carboxamide (30)
A mixture of 9 (1 mmol, 196 mg), (4-(trifluoromethyl)phenyl) methanamine (1.2 mmol, 210 mg), HATU (1.5 mmol, 570 mg) in 8 mL of anhydrous DMF was added DIPEA (1.5 mmol, 261 μL). The solution was stirred for 2 h at room temperature, and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluting with DCM and MeOH (50:1) to give 30 (264 mg, 0.75 mmol) as a white solid in 75 % yield. 1H NMR (400 MHz, MeOD): δ 2.33 (s, 0.22H), 2.63 (s, 3H), 4.71 (s, 2H), 7.33 (s, 0.08H), 7.45 (s, 0.07H), 7.59 (d, 2H, J = 8.0 Hz), 7.65 (d, 2H, J = 8.0 Hz), 8.86 (s, 0.06H). 13C NMR (500 MHz, MeOD) δ 14.19, 42.54, 114.61, 115.75, 123.31, 123.48, 125.22 (q, J = 3.75 Hz), 125.63, 127.87, 129.06, 129.31, 130.48, 143.78, 144.70, 145.19, 162.22. HRMS (ESI-TOF, positive mode) m/z ([M + 1]); Anal. Calcd for C18H11D6F3N3O 354.1685; Found 354.1695.
4.6. N-(4-Chlorobenzyl)-2-methyl-6-(methyl-d3)imidazo[1,2-a]pyridine-5,7,8-d3-3-carboxamide (31)
Compound 31 was synthesized according to the method used for 29. Starting with (4-chlorophenyl)methanamine (0.22 mmol, 0.027 mL), compound 31 was obtained as a white solid in 75 % yield (47 mg, 0.15 mmol). 1H NMR (500 MHz, MeOD) δ 2.29 (s, 0.24H), 2.59 (s, 3H), 4.58 (s, 2H), 7.28 (s, 0.07H), 7.31 (d, 2H, J = 10.0 Hz), 7.36 (d, 2H, J = 10.0 Hz), 7.39 (s, 0.11H), 8.80 (s, 0.06H). 13C NMR (500 MHz, MeOD) δ 14.16, 42.30, 114.64, 115.82, 123.23, 125.22, 128.44, 129.05, 130.11, 130.39, 132.77, 137.95, 144.68, 145.12, 162.13. HRMS (ESI-TOF, positive mode) m/z ([M + 1]); Anal. Calcd for C17H11ClD6N3O 320.1428; Found 320.1428.
4.7. 2-Methyl-6-(methyl-d3)-N-(4-(pentafluoro-λ6-sulfaneyl)benzyl) imidazo[1,2-a]pyridine-5,7,8-d3-3-carboxamide (32)
A mixture of 9 (60 mg, 0.31 mmol) and 4-(pentafluorothio)benzylamine (75 mg, 0.31 mmol) were dissolved in dry CH3CN (10 mL) and then EDC•HCl (71 mg, 0.4 mmol) and DMAP (45 mg, 0.4 mmol) were added. The reaction was stirred at RT for 12 h and then it was concentrated to dryness in vacuo. The residue was taken up in CH2Cl2 and washed with 5 % aqueous acetic acid solution (2x), water (1x), saturated aqueous NaHCO3 solution (2x), brine and then dried over sodium sulfate. The drying agent was removed by filtration and organics were concentrated to dryness in vacuo. The resulting residue was purified by silica gel column chromatography eluting with 30 % CH2Cl2: EtOAc to give 32 as a lightly colored oil which became an off white solid upon standing (69 mg, 0.096 mmol, 31 % yield). 1H NMR (400 MHz, CDCl3) δ 7.26 (2H, d, J = 8.6 Hz), 7.49 (2H, d, J = 8.3 Hz), 6.18 (1H, bt, J = 5.4 Hz), 4.76 (2H, d, J = 6.0 Hz), 2.73 (3H, s). 13C NMR (125 MHz, CDCl3) δ 161.7, 153.1 (t, J = 18.1 Hz), 145.5, 145.3, 142.5, 129.9 (t, J = 23.5 Hz), 127.6, 126.5 (q, J = 4.5 Hz), 125.8 (t, J = 29.0 Hz), 122.9, 115.4, (t, J = 26.2 Hz), 114.6, 42.6, 17.0 ppm. 19F NMR (377 MHz, CDCl3) δ 84.4 (quintet, J = 148.6 Hz), 62.9 (d, J = 150.0 Hz). HRMS (ESI-TOF, positive mode) m/z ([M + 1]); Anal. Calcd for C17H11F5N3OSD6 412.1384; Found 412.1414. HPLCtR = 2.7 min, method MSU-A. Mp = 183.9–184.2° C.
4.8. 2-Methyl-6-(methyl-d3)-N-(4-(methylsulfonyl)benzyl)imidazo[1,2-a]pyridine-5,7,8-d3-3-carboxamide (33)
Compound 33 was synthesized according to the method used for compound 29. Starting with (4-(methylsulfonyl)phenyl)methanamine HCl salt (0.22 mmol, 49 mg), Compound 33 was obtained as a white solid in 61 % yield (43 mg, 0.12 mmol). 1H NMR (500 MHz, CDCl3) δ 2.29 (s, 0.4H), 2.68 (s, 3H), 2.99 (s, 3H), 4.74 (d, 2H, J = 5.0 Hz), 6.57 (t, 1H, J = 5.0 Hz), 7.26 (s, 0.04H), 7.43 (s, 0.06H), 7.50 (d, 2H, J = 10.0 Hz), 7.81 (d, 2H, J = 10.0 Hz), 9.15 (s, 0.06H). 13C NMR (500 MHz, CDCl3) δ 17.02, 43.00, 44.75, 114.93, 115.77, 123.11, 126.20, 127.97, 128.31, 130.39, 139.49, 145.38, 145.46, 145.86, 162.07. HRMS (ESI-TOF, positive mode) m/z ([M + 1]); Anal. Calcd for C18H14D6N3O3S 364.1601; Found 364.1596.
4.9. 2-Methyl-6-(methyl-d3)-N-(4-(trifluoromethoxy)benzyl)imidazo [1,2-a]pyridine-5,7,8-d3-3-carboxamide (34)
A mixture of 9 (96 mg, 0.47 mmol) and (4-(trifluoromethoxy) phenyl)methanamine (90 mg, 0.47 mmol) were dissolved in dry CH3CN and then EDC•HCl (134 mg, 0.70 mmol) and DMAP (85 mg, 0.70 mmol) were added. The reaction was stirred at RT for 12 h and was then concentrated to dryness in vacuo. The residue was taken up in CH2Cl2 and washed with 5 % aqueous acetic acid solution (2x), water (1x), saturated aqueous NaHCO3 solution (2x), brine and then dried over sodium sulfate. The drying agent was removed by filtration and organics were concentrated to dryness in vacuo. The resulting residue was purified by silica gel column chromatography eluting with 30 % CH2Cl2: EtOAc to give 34 as a lightly colored oil which became an off white solid upon standing (62 mg, 0.15 mmol, 36 % yield). 34: 1H NMR (400 MHz, CDCl3) δ 7.43 (2H, d, J = 8.5 Hz), 7.23 (2H, d, J = 8.3 Hz), 6.16 – 6.08 (1H, m), 4.71 (2H, d, J = 5.9 Hz), 2.70 (3H, s). 13C NMR (100 MHz, CDCl3) δ 161.7, 148.5, 145.3, 145.2, 137.2, 130.1, 128.9, 126.1, 125.8, 122.8, 120.4 (q, J = 256 Hz), 115.6, 114.8, 42.6, 17.5, 16.9 ppm. 19F NMR (377 MHz, CDCl3) δ –57.89 Hz. HRMS (ESI-TOF, positive mode) m/z ([M + 1]); Anal. Calcd for C18H11F3N3O2D6 370.1644; Found 370.1630. HPLCtR = 2.7 min, method MSU-A. Mp = 189.9–190.6° C.
4.10. N-((4-(tert-Butyl)benzyl)-2-methyl-6-(methyl-d3)imidazo[1,2-a] pyridine-5,7,8-d3-3-carboxamide (35)
A mixture of 9 (60 mg, 0.31 mmol) and 4-(tert-butyl)benzylamine (52 mg, 0.31 mmol) were dissolved in dry CH3CN and then EDC•HCl (71 mg, 0.4 mmol) and DMAP (45 mg, 0.4 mmol) were added. The reaction was stirred at RT for 12 h and then it was concentrated to dryness in vacuo. The residue was taken up in CH2Cl2 and washed with 5 % aqueous acetic acid solution (2x), water (1x), saturated aqueous NaHCO3 solution (2x), brine and then dried over sodium sulfate. The drying agent was removed by filtration and organics were concentrated to dryness in vacuo. The resulting residue was purified by silica gel column chromatography eluting with 30 % CH2Cl2: EtOAc to give 35 as a lightly colored oil which became a white solid upon standing (28 mg, 0.084 mmol, 27 % yield). 35: 1H NMR (400 MHz, CDCl3) δ 7.45 – 7.37 (2H, m), 7.37 – 7.29 (2H, m), 6.06 (1H, bs), 4.68 (2H, d, J = 4.5 Hz), 2.68 (3H, s), 1.33 (9H, s). 13C NMR (100 MHz, CDCl3) δ 161.6, 150.7, 145.1, 145.1, 135.2, 127.4, 125.8, 122.6, 115.6, 115.0, 43.1, 34.5, 31.3, 29.7, 16.8 ppm. HRMS (ESI-TOF, positive mode) m/z ([M + 1]); Anal. Calcd for C21H20N3OD6 342.2477; Found 342.2488. HPLCtR = 2.8 min, method MSU-A. Mp = 137.4–138.4°C.
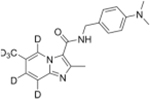
4.11. N-(4-(Dimethylamino))-2-methyl-6-(methyl-d3)imidazo[1,2-a] pyridine-5,7,8-d3-3-carboxamide (36)
Compound 36 was synthesized according to the method used for compound 29. Starting with 4-(aminomethyl)-N,N-dimethylaniline HCl salt (0.22 mmol, 49 mg), 36 was obtained as a white solid in 54 % yield (35 mg, 0.11 mmol). 1H NMR (500 MHz, CDCl3) δ 2.31 (s, 0.22H), 2.62 (s, 3H), 2.94 (s, 6H), 4.57 (d, 2H, J = 5.0 Hz), 6.02 (t, 1H, J = 5.0 Hz), 6.70–6.73 (m, 2H), 7.23 (s, 0.08H), 7.24–7.26 (m, 2H), 7.44 (s, 0.07H), 9.22 (s, 0.05H). 13C NMR (500 MHz, CDCl3) δ 16.83, 40.82, 43.39, 112.99, 115.41, 115.67, 122.81, 125.87, 126.25, 129.10, 130.06, 145.03, 150.39, 161.63. HRMS (ESI-TOF, positive mode) m/z ([M + 1]); Anal. Calcd for C19H17D6N4O 329.2242; Found 329.2243.
4.12. N-(4-Methoxybenzyl)-2-methyl-6-(methyl-d3)imidazo[1,2-a] pyridine-5,7,8-d3-3-carboxamide (37)
Compound 37 was synthesized according to the method used for compound 29. Starting with (4-methoxyphenyl)methanamine (0.22 mmol, 29 mg), compound 37 was obtained as a white solid in 63 % yield (39 mg, 0.13 mmol). 1H NMR (500 MHz, MeOD) δ 2.29 (s, 0.26H), 2.56 (s, 3H), 3.76 (s, 3H), 4.54 (s, 2H), 6.88 (d, 2H, J = 10.0 Hz), 7.27 (s, 0.06H), 7.31 (d, 2H, J = 5.0 Hz), 7.39 (s, 0.05H), 8.78 (s, 0.07H). 13C NMR (500 MHz, CDCl3) δ 14.04, 42.47, 54.48, 113.77, 114.62, 116.01, 123.15, 125.13, 128.81, 130.29, 130.95, 144.61, 144.91, 159.26, 161.99. HRMS (ESI-TOF, positive mode) m/z ([M + 1]); Anal. Calcd for C18H14D6N3O2 316.1918; Found 316.1918.
4.13. N-(4-Ethoxybenzyl)-2-methyl-6-(methyl-d3)imidazo[1,2-a] pyridine-5,7,8-d3-3-carboxamide (38)
A mixture of 9 (100 mg, 0.31 mmol) and 4-(ethoxy)benzylamine (81 mg, 0.51 mmol) were dissolved in dry CH3CN and then EDC•HCl (117 mg, 0.62 mmol) and DMAP (75 mg, 0.61 mmol) were added. The reaction was stirred at RT for 12 h and then concentrated to dryness in vacuo. The residue was taken up in CH2Cl2 and washed with 5 % aqueous acetic acid solution (2x), water (1x), saturated aqueous NaHCO3 solution (2x), brine and then dried over sodium sulfate. The drying agent was removed by filtration and organics were concentrated to dryness in vacuo. The resulting residue was purified by recrystallization from hot CH3CN to give compound 38 as a white solid (39 mg, 0.12 mmol, 23 % yield). 38: 1H NMR (400 MHz, CDCl3) δ 7.31 (2H, d, J = 8.4 Hz), 6.90 (2H, d, J = 8.6 Hz), 6.03 – 5.96 (1H, m), 4.63 (2H, d, J = 5.5 Hz), 4.04 (2H, q, J = 6.9 Hz), 2.65 (3H, s), 1.42 (3H, t, J = 6.9 Hz). 13C NMR (125 MHz, CDCl3) δ 161.5, 158.5, 145.1, 130.1, 129.0, 126.1, 125.8, 125.6, 122.6, 115.6, 115.0, 114.8, 63.5, 43.0, 29.6, 16.8, 14.8 ppm. HRMS (ESI-TOF, positive mode) m/z ([M + 1]); Anal. Calcd for C19H16N3O2D6 330.2083; Found 330.2087. HPLCtR = 2.4 min, method MSU-A. Mp = 162.0–163.0° C.
4.14. N-(Chroman-6-ylmethyl)-2-methyl-6-(methyl-d3)imidazo[1,2-a] pyridine-5,7,8-d3-3-carboxamide (39)
A mixture of 9 (60 mg, 0.31 mmol) and chroman-6-ylmethanamine (53 mg, 0.31 mmol) were dissolved in dry CH3CN and then EDC•HCl (71 mg, 0.4 mmol) and DMAP (45 mg, 0.37 mmol) were added. The reaction was stirred at RT for 12 h and then concentrated to dryness in vacuo. The residue was taken up in CH2Cl2 and washed with 5 % aqueous acetic acid solution (2x), water (1x), saturated aqueous NaHCO3 solution (2x), brine and then dried over sodium sulfate. The drying agent was removed by filtration and organics were concentrated to dryness in vacuo. The resulting residue was purified by recrystallization from hot CH3CN and collected compound 39 as an off white solid (38 mg, 0.11 mmol, 36 % yield). 39: 1H NMR (400 MHz, CDCl3) δ 7.10 (1H, dd, J = 8.3, 1.7 Hz), 7.07 (1H, s), 6.80 (1H, d, J = 8.3 Hz), 6.02 – 5.96 (1H, m), 4.59 (2H, d, J = 5.5 Hz), 4.19 (2H, t, J = 5.0 Hz), 2.77 (2H, t, J = 6.4 Hz), 2.66 (3H, s), 2.01 (2H, dddd, J = 6.4, 6.4, 5.0, 5.0 Hz). 13C NMR (125 MHz, CDCl3) δ 173.6, 161.5, 154.5, 144.9, 129.7, 129.3, 126.8, 122.7, 122.6, 117.1, 115.5, 115.2, 115.0, 66.5, 43.1, 24.9, 22.3, 20.7, 16.7 ppm. HRMS (ESI-TOF, positive mode) m/z ([M + 1]); Anal. Calcd for C20H16N3O2D6 342.2083; Found 342.2083. HPLCtR = 2.4 min, method MSU-A. Mp = 127.6–129.7° C.
4.15. N-((2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)methyl)-2-methyl-6-(methyl-d3)imidazo[1,2-a]pyridine-5,7,8-d3-3-carboxamide (40)
A mixture of 9 (60 mg, 0.31 mmol) and (2,3-dihydrobenzo[b][1,4] dioxin-6-yl)methanamine•HCl (65 mg, 0.31 mmol) were dissolved in dry CH3CN and then EDC•HCl (71 mg, 0.4 mmol) and DMAP (82 mg, 0.67 mmol) were added. The reaction was stirred at RT for 12 h and then concentrated to dryness in vacuo. The residue was taken up in CH2Cl2 and washed with 5 % aqueous acetic acid solution (2x), water (1x), saturated aqueous NaHCO3 solution (2x), brine and then dried over sodium sulfate. The drying agent was removed by filtration and organics were concentrated to dryness in vacuo. The resulting residue was purified by recrystallization from hot CH3CN and gave an off white solid (29 mg, 0.087 mmol, 28 % yield). 40: 1H NMR (400 MHz, CDCl3) δ 6.92–6.89 (1H, m), 6.88–6.85 (2H, m), 6.03–5.97 (1H, m), 4.59 (2H, d, J = 5.6 Hz), 4.29–4.25 (4H, m), 2.67 (3H, s). 13C NMR (125 MHz, CDCl3) δ 161.5, 145.1, 145.0, 143.7, 143.1, 135.5, 122.6, 120.7, 117.6, 116.6, 115.6, 115.0, 64.4, 64.3, 43.0, 29.7, 16.8 ppm. HRMS (ESI-TOF, positive mode) m/z ([M + 1]); Anal. Calcd for C19H14N3O3D6 344.1876; Found 344.1856. HPLCtR = 2.3 min, method MSU-A. Mp = 115.9–116.9° C.
4.16. N-(3-Chlorobenzyl)-2-methyl-6-(methyl-d3)imidazo[1,2-a] pyridine-5,7,8-d3-3-carboxamide (41)
Compound 41 was synthesized according to the method used for 29. Starting with (3-chlorophenyl)methanamine (0.22 mmol, 27 mg), 41 was obtained as a white solid in 57 % yield (36 mg, 0.11 mmol). 1H NMR (500 MHz, MeOD) δ 2.28 (s, 0.16H), 2.59 (s, 3H), 4.59 (s, 2H), 7.23–7.27 (m, 1H), 7.29 (s, 0.1H), 7.30–7.33 (m, 2H), 7.38 (s, 0.09H), 7.41 (s, 1H), 8.79 (s, 0.08H). 13C NMR (500 MHz, MeOD) δ 14.18, 42.43, 114.63, 115.76, 123.24, 125.23, 125.79, 127.09, 127.48, 129.93, 130.40, 141.55, 144.71, 145.16, 162.15. HRMS (ESI-TOF, positive mode) m/z ([M + 1]); Anal. Calcd for C17H11ClD6N3O 320.1433; Found 320.1431.
4.17. N-(2-Chlorobenzyl)-2-methyl-6-(methyl-d3)imidazo[1,2-a] pyridine-5,7,8-d3-3-carboxamide (42)
Compound 42 was synthesized according to the method used for 29. Starting with (2-chlorophenyl)methanamine (0.22 mmol, 27 mg), 42 was obtained as a white solid in 48 % yield (31 mg, 0.1 mmol). 1H NMR (500 MHz, MeOD) δ 2.30 (s, 0.22H), 2.61 (s, 3H), 4.70 (s, 2H), 7.24–7.31 (m, 2H), 7.40–7.42 (m, 1H), 7.46–7.47 (m, 1H), 8.81(s, 0.06H). 13C NMR (500 MHz, MeOD) δ 14.17, 41.06, 114.65, 115.78, 123.26, 125.23, 127.00, 128.72, 129.36, 129.92, 130.13, 130.40, 133.17, 135.93, 144.70, 145.28, 162.16. HRMS (ESI-TOF, positive mode) m/z ([M + 1]); Anal. Calcd for C17H11ClD6N3O 320.1454; Found 320.1431.
4.18. 2-Methyl-6-(methyl-d3)-N-(3-(trifluoromethoxy)benzyl)imidazo [1,2-a]pyridine-5,7,8-d3-3-carboxamide (43)
A mixture of 9 (60 mg, 0.31 mmol) and (3-(trifluoromethoxy) phenyl)methanamine (62 mg, 0.31 mmol) were dissolved in dry CH3CN and then EDC•HCl (71 mg, 0.4 mmol) and DMAP (45 mg, 0.4 mmol) were added. The reaction was stirred at RT for 12 h and then concentrated to dryness in vacuo. The residue was taken up in CH2Cl2 and washed with 5 % aqueous acetic acid solution (2x), water (1x), saturated aqueous NaHCO3 solution (2x), brine and then dried over sodium sulfate. The drying agent was removed by filtration and organics were concentrated to dryness in vacuo. The resulting residue was purified by silica gel column chromatography eluting with 30 % CH2Cl2: EtOAc to give 43 as an off white solid upon standing (29 mg, 0.081 mmol, 26 % yield). 43: 1H NMR (400 MHz, CDCl3) δ 7.41 (1H, dd, J = 7.9, 7.9 Hz), 7.34 (1H, d, J = 7.6 Hz), 7.26 (1H, s), 7.17 (1H, d, J = 7.9 Hz), 6.12 (1H, bt, J = 4.4 Hz), 4.74 (2H, d, J = 5.9 Hz), 2.71 (3H, s). 13C NMR (100 MHz, CDCl3) δ 161.6, 149.6, 145.4, 145.2, 140.8, 130.2, 125.8, 122.9, 120.4, (q, J = 255 Hz), 120.0, 119.9, 119.1, 115.6, 114.7, 42.8, 29.7, 16.9 ppm. 19F NMR (377 MHz, CDCl3) δ – 57.73 ppm. HRMS (ESI-TOF, positive mode) m/z ([M + 1]); Anal. Calcd for C18H11F3N3O2D6 370.1644; Found 370.1655. HPLCtR = 2.6 min, method MSU-A. Mp = 118.3 – 119.2°C.
4.19. N-(3-(Dimethylamino))-2-methyl-6-(methyl-d3)imidazo[1,2-a] pyridine-5,7,8-d3-3-carboxamide (44)
A mixture of 9 (60 mg, 0.31 mmol) and 3-(aminomethyl)-N,N-dimethylaniline (48 mg, 0.31 mmol) were dissolved in dry CH3CN and then EDC•HCl (71 mg, 0.4 mmol) and DMAP (45 mg, 0.4 mmol) were added. The reaction was stirred at RT for 12 h and then concentrated to dryness in vacuo. The residue was taken up in CH2Cl2 and washed with 5 % aqueous acetic acid solution (2x), water (1x), saturated aqueous NaHCO3 solution (2x), brine and then dried over sodium sulfate. The drying agent was removed by filtration and organics were concentrated to dryness in vacuo. The resulting residue was purified by silica gel column chromatography eluting with 30 % CH2Cl2: EtOAc to give 44 as a lightly colored oil which became light tan solid upon standing (39 mg, 0.12 mmol, 39 % yield). 44: 1H NMR (400 MHz, CDCl3) δ 7.25 (1H, dd, J = 8.1, 8.1 Hz), 6.77 – 6.72 (2H, m), 6.69 (1H, dd, J = 8.6, 2.1 Hz), 6.03 (1H, bs), 4.66 (2H, d, J = 5.5 Hz), 2.97 (6H, s), 2.67 (3H, s). 13C NMR (125 MHz, CDCl3) δ 161.5, 151.0, 144.9, 139.0, 129.6, 126.1, 125.8, 125.6, 122.7, 115.6, 115.5, 115.1, 111.8, 111.6, 44.1, 40.5, 29.7, 16.7 ppm. HRMS (ESI-TOF, positive mode) m/z ([M + 1]); Anal. Calcd for C19H17N4OD6 329.2243; Found 329.2222. HPLCtR = 1.9 min, method MSU-A. Mp = 141.9–142.4°C.
4.20. N-(3-Methoxybenzyl)-2-methyl-6-(methyl-d3)imidazo[1,2-a] pyridine-5,7,8-d3-3-carboxamide (45)
Compound 45 was synthesized according to the method used for 29. Starting with (3-methoxyphenyl)methanamine (0.22 mmol, 28 mg), 45 was obtained as a white solid in 64 % yield (41 mg, 0.13 mmol). 1H NMR (500 MHz, CDCl3) δ 2.31 (s, 0.24H), 2.66 (s, 3H), 3.79 (s, 3H), 4.65 (d, 2H, J = 5.0 Hz), 6.15 (brs, 1H), 6.83 (dd, 1H, J1 = 10.0 Hz, J2 = 5.0 Hz), 6.91 (brs, 1H), 6.96 (d, 1H, J = 10.0 Hz), 7.17 (s, 0.08H), 7.27 (t, 1H, J = 10.0 Hz), 7.45 (s, 0.09H), 9.21 (s, 0.07H). 13C NMR (500 MHz, CDCl3) δ 16.94, 43.61, 55.46, 112.58, 113.12, 113.52, 115.21, 115.73, 117.98, 118.89, 120.00, 122.94, 126.27, 130.14, 140.09, 145.29, 160.20, 161.77. HRMS (ESI-TOF, positive mode) m/z ([M + 1]); Anal. Calcd for C18H14D6N3O 316.1920; Found 316.1927.
4.21. N-(3,5-Difluorobenzyl)-2-methyl-6-(methyl-d3)imidazo[1,2-a] pyridine-5,7,8-d3-3-carboxamide (46)
Compound 46 was synthesized according to the method used for 29. Starting with (3,5-difluorophenyl)methanamine (0.22 mmol, 26 mg), 46 was obtained as a white solid in 45 % yield (29 mg, 0.09 mmol). 1H NMR (500 MHz, CDCl3) δ 2.32 (s, 0.16H), 2.70 (s, 3H), 4.66 (d, 2H, J = 5.0 Hz), 6.24 (brs, 1H), 6.71 (t, 1H, J = 5.0 Hz), 6.88 (d, 2H, J = 5.0 Hz), 7.19 (s, 0.03H), 7.46 (s, 0.05H), 9.21 (s, 0.04H). 13C NMR (500 MHz, CDCl3) δ 17.16, 42.81, 102.96 (t, J = 25 Hz), 110.26 (d, J = 5 Hz), 110.41 (d, J = 6.25 Hz), 114.86, 115.85, 123.14, 126.31, 130.10, 130.27 (d, J = 3 Hz), 142.74 (t, J = 7.5 Hz), 145.47, 145.74, 161.91, 162.43 (d, J = 12.5 Hz), 164.41 (d, J = 12.5 Hz). HRMS (ESI-TOF, positive mode) m/z ([M + 1]); Anal. Calcd for C17H10D6F2N3O 322.1642; Found 322.1633.
4.22. N-(3,5-Dichlorobenzyl)-2-methyl-6-(methyl-d3)imidazo[1,2-a] pyridine-5,7,8-d3-3-carboxamide (47)
Compound 47 was synthesized according to the method used for 29. Starting with (3,5-dichlorophenyl)methanamine (0.22 mmol, 39 mg), 47 was obtained as a white solid in 43 % yield (30 mg, 0.086 mmol). 1H NMR (500 MHz, CDCl3) δ 2.30 (s, 0.11H), 2.68 (s, 3H), 4.61 (d, 2H, J = 5.0 Hz), 6.34 (t, 1H, J = 5.0 Hz), 7.13–7.25 (m, 3H), 7.43 (s, 0.04H), 7.95 (s, 0.02H), 9.17 (s, 0.03H). 13C NMR (500 MHz, CDCl3) δ 17.05, 42.58, 114.91, 115.72, 123.16, 125.96, 126.11, 127.31, 127.81, 128.68, 135.48, 135.85, 139.62, 142.29, 145.36, 145.72, 161.86. HRMS (ESI-TOF, positive mode) m/z ([M + 1]); Anal. Calcd for C17H10Cl2D6N3O 354.1036; Found 354.1041.
4.23. Ethyl 6-methyl-2-(methyl-d3)imidazo[1,2-a]pyridine-3-carboxylate-7,8-d2 (50)
To a solution of 49 (1 mmol, 218 mg) in 5 mL of D2O (D, 70 %) under an argon atmosphere in a sealed tube was added 10 % Pd/C (20 % wt of 49, 42 mg) and 5 % Pt/C (20 % wt of 49, 42 mg). The tube was sealed with a rubber septum and filled with H2 using three vacuum/H2 cycles, the rubber septum was then exchanged with a polytetrafluoroethylene front seal plug. The reaction mixture was heated at 130 °C for 24 h. After cooling to room temperature, the tube was purged with argon, the mixture was diluted with EtOAc, and then filtered to remove the catalyst. The filtrate was partitioned between EtOAc and water. The aqueous layer was extracted with EtOAc (2 × 15 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography, eluting with hexanes and EtOAc (4:1, v/v) to give 50 as a white solid in 53 % yield (0.53 mmol, 118 mg). 1H NMR (400 MHz, MeOD): δ 1.43 (t, 3H, J = 8.0 Hz), 2.38–2.40 (m, 2H), 2.65–2.66 (m, 0.56H), 4.42 (q, 2H, J = 8.0 Hz), 7.40–7.42 (m, 0.19H), 7.48–7.50 (m, 0.18H), 9.15 (s, 0.68H). LC-MS (ESI, positive mode) m/z ([M + 1]); Anal. Calcd for C12H10D5N2O2 224.14; Found 224.22. HPLCtR = 6.58 min, method ND-B.
4.24. Ethyl 6-methyl-2-(methyl-d3)imidazo[1,2-a]pyridine-3-carboxylate-7,8-d2 (51)
Compound 26 was synthesized according to the method used for 9. Starting with 50 (1 mmol, 223 mg), 51 was obtained as a white solid which was used directly in the next step without further purification (167 mg, 0.86 mmol, 86 % yield).
4.25. 6-Methyl-2-(methyl-d3)-N-(4-(trifluoromethyl)benzyl)imidazo [1,2-a]pyridine-7,8-d2-3-carboxamide (52)
Compound 52 was prepared according to the method used for 30. Starting from 26 (1 mmol, 195 mg), 52 was obtained as a white solid in 61 % yield (0.61 mmol, 214 mg).). 1H NMR (400 MHz, MeOD): δ 2.34–2.36 (m, 2.19H), 2.59 (s, 0.15H), 4.70 (s, 2H), 7.33 (s, 0.06H), 7.45 (s, 0.08H), 7.58 (d, 2H, J = 8.0 Hz), 7.65 (d, 2H, J = 8.0 Hz), 8.86 (s, 0.66H). HRMS (ESI-TOF, positive mode) m/z ([M + 1]); Anal. Calcd for C18H12D5F3N3O 353.1629; Found 353.1629.
4.26. 2,6-Bis(methyl-d3)imidazo[1,2-a]pyridine-3-carboxylic-7,8-d2 acid (53)
Compound 27 was synthesized according to the method used for 50 by using D2O (D, 99.9 %) as the solvent. Product 53 was obtained as a white solid in 60 % yield (0.6 mmol, 136 mg). 1H NMR (400 MHz, MeOD): δ 1.43 (t, 3H, J = 8.0 Hz), 2.36 (s, 0.06H), 2.59–2.60 (s, 0.06H), 4.41 (q, 2H, J = 8.0 Hz), 7.39 (s, 0.02H), 7.47 (s, 0.02H), 9.11 (s, 0.57H). LC-MS (ESI, positive mode) m/z ([M + 1]); Anal. Calcd for C12H7D8N2O2 227.16; Found, 227.16. HPLCtR = 6.58 min, method ND-B.
4.27. 6-Methyl-2-(methyl-d3)imidazo[1,2-a]pyridine-3-carboxylic-7,8-d2 acid (54)
Compound 54 was prepared according to the method used for 9. Starting from 53, product 54 was obtained as a white solid that was directly used for the next step without further purification (348 mg, 1.76 mmol, 80 % yield).
4.28. 2,6-Bis(methyl-d3)-N-(4-(trifluoromethyl)benzyl)imidazo[1,2-a] pyridine-7,8-d2-3-carboxamide (55)
Compound 55 was prepared according to the method used for 30. Starting from 54 (1 mmol, 198 mg), 55 was obtained as a white solid in 55 % yield (0.55 mmol, 195 mg). 1H NMR (400 MHz, MeOD): δ 2.37 (s, 0.07H), 2.63 (s, 0.06H), 4.71 (s, 2H), 7.62 (d, 2H, J = 8.0 Hz), 7.66 (d, 2H, J = 8.0 Hz), 8.92 (s, 0.47H). 13C NMR (500 MHz, MeOD) δ 42.60, 113.61, 116.02, 121.31, 123.46, 124.23, 125.22, 125.25 (q, J = 3.75 Hz), 125.62, 127.94, 128.24, 128.79 (q, J = 32.5 Hz), 131.03, 143.45, 143.64, 161.63. HRMS (ESI-TOF, positive mode) m/z ([M + 1]); Anal. Calcd for C18H9D8F3N3O 356.1817; Found 356.1820.
4.29. (4-(Trifluoromethyl)phenyl)methan-d2-amine DCl salt (56)
10 % Pd/C (34 mg, 20 % wt of 4-(trifluoromethyl)benzonitrile) and 5 mL of D2O were placed in a 100 mL round-bottom flask. The system was sealed with a rubber septum and filled with H2 using five vacuum/H2 cycles. The mixture was stirred at room temperature for 24 h, then 4-(trifluoromethyl)benzonitrile (1 mmol, 171 mg) was added followed by 1.5 mmol (160 μL) of DCl (D, 99.5 %, DCl 35 % in D2O). The mixture was stirred at room temperature overnight, purged with argon and then filtered to remove the catalyst. The residue was concentrated under reduced pressure to give 56 as a white solid in 80 % yield (171 mg, 0.8 mmol). The product was used for the next step without further purification. 1H NMR (400 MHz, MeOD) δ 4.22 (s, 0.2H), 7.68 (brs, 2H), 7.77 (brs, 2H).
4.30. 2,6-Bis(Methyl-d3)-N-((4-(trifluoromethyl)phenyl)methyl-d2) imidazo[1,2-a]pyridine-7,8-d2-3-carboxamide (57)
To a mixture of 54 (1 mmol, 198 mg), 56 (1 mmol, 214 mg) and HATU (1.5 mmol, 570 mg) in 10 mL of anhydrous DMF was added DIPEA (2.5 mmol, 276 μL). The solution was stirred for 2 h at room temperature, and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluting with DCM and MeOH (50:1, v/v) to give 57 (200 mg, 0.56 mmol) as a white solid in 56 % yield. 1H NMR (400 MHz, DMSO-d6): δ 2.31 (s, 0.11H), 2.59 (s, 0.11H), 4.60 (d, 0.18H, J = 4.0 Hz), 7.60 (d, 2H, J = 8.0 Hz), 7.71 (d, 2H, J = 8.0 Hz), 8.54 (s, 1H), 8.91 (s, 0.62H). 13C NMR (500 MHz, CDCl3) δ 114.12, 115.86, 123.48, 125.22 (q, J = 3.75 Hz), 125.33, 125.63, 127.92, 129.08, 129.34, 130.31, 143.67, 144.63, 144.93, 161.20. HRMS (ESI-TOF, positive mode) m/z ([M + 1]); Anal. Calcd for C18H7D10F3N3O 358.1945; Found 358.1945.
5. Microsomal stability assay
Anti-TB compounds (2 μM) were incubated with human and animal hepatic microsomes (Corning, USA; 0.5 mg/mL) in Tris-HCl buffer (500 mM, pH 7.4, 100 μL) containing MgCl2 (5 mM). The samples were obtained at 0, 10, 20, 30 and 40 min after incubation with NADPH. The samples without NADPH for the 40 min points were set as negative control. Diazepam (2 μM) was set as a reference compound for the dog, rat and mouse liver microsomes incubation systems. Verapamil (2 μM) was used as a reference compound for the human and monkey microsomes incubation systems. All reactions were conducted in duplicate and terminated by adding 100 μL of ice-cold acetonitrile containing the internal standards diazepam and verapamil (1 μM). Compound 30 (2 μM) was set as an internal standard when diazepam was employed as the reference. After centrifugation at 17,000g for 10 min, the upper organic layer was transferred into a polyethylene tube for HPLC analysis.
6. HPLC method
The samples were quantified by HPLC system (Waters, USA) Model 2487 and C18 column (250 m m × 4.6 m m i.d., 5 μm). The wavelength was set as 247 nm. The mobile phase consisted of solvent A (0.1 % formic acid and ammonium formate in water) and solvent B (acetonitrile) with a flow rate of 0.5 mL/min. The gradient was programmed as follows: at 0 min: 20 % B; 0.00–22.00 min: gradient increases to 80 % eluent B; 22.10–30.00 min: 20 % B.
6.1. Data analysis
The half-life and intrinsic hepatic clearance in microsomal stability study was calculated according to equation (1) and (2):
| (1) |
ke is the slope of a linear regression curve in a semilog manner from time-concentration plot.
| (2) |
is protein concentration in the reaction mixture.
Acknowledgements
M. J. M., G. C. M. and R. L. acknowledge funding from the NIH R37AI054193. K. M. was supported by funding to Montana State University from Hsiri Therapeutics.
Footnotes
Competing interests
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: M. J. M. is the CSO of Hsiri Therapeutics. G. C. M. and R. L. are consultants for Hsiri Therapeutics and K. M. was supported by funding to Montana State University from Hsiri Therapeutics.
Declaration of Competing Interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: [Marvin J. Miller reports financial support was provided by National Institutes of Health. Marvin J. Miller reports a relationship with Hsiri Therapeutics that includes: equity or stocks.
M. J. M. is the CSO of Hsiri Therapeutics. G. C. M. and R. L. are consultants for Hsiri Therapeutics and K. M. was supported by funding to Montana State University from Hsiri Therapeutics.].
REFERENCES
- [1].Organization WH Global Tuberculosis Report 2020. https://apps.who.int/iris/bitstream/handle/10665/336069/9789240013131-eng.pdf. [Google Scholar]
- [2].Iacobino A, Fattorini L, Giannoni F, Drug-Resistant Tuberculosis 2020: Where We Stand, Appl. Sci. 10 (2020) 2153, 10.3390/app10062153. [DOI] [Google Scholar]
- [3].Moraski GC, Markley LD, Hipskind PA, Boshoff H, Cho S, Franzblau SG, Miller MJ, Advent of Imidazo[1,2-a]pyridine-3-carboxamides with Potent Multi- and Extended Drug Resistant Antituberculosis Activity, ACS Med. Chem. Lett. 2 (2011) 466–470, 10.1021/ml200036r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].(a) Moraski GC; Markley LD; Cramer J; Hipskind PA; Boshoff H; Bailey M; Alling T; Ollinger J; Parish T; Miller MJ Advancement of Imidazo[1,2- a] pyridines with Improved Pharmacokinetics and Nanomolar Activity Against Mycobacterium tuberculosis ACS Med. Chem. Lett, 2013, 4, 675–679. doi: 10.1021/ml400088y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ollinger J; Bailey MA; Moraski GC; Casey A; Florio S; Alling T; Miller MJ; Parish T A Dual Read-Out Assay to Evaluate the Potency of Compounds Active against Mycobacterium tuberculosis. PLOS ONE 8(4): e60531. 10.1371/journal.pone.0060531. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Moraski GC; Miller PA; Bailey MA; Ollinger J; Parish T; Boshoff HI; Cho S; Anderson JR; Mulugeta S; Franzblau SG; Miller MJ Putting Tuberculosis (TB) To Rest: Transformation of the Sleep Aid, Ambien, and “Anagrams” Generated Potent Antituberculosis Agents ACS Inf. Dis., 2015, 1, 85–90. 10.1021/id500008t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Moraski GC; Bristol R; Seeger N; Boshoff HI; Tsang PS; Miller MJ Preparation and Evaluation of Potent Pentafluorosulfanyl-Substituted Anti-Tuberculosis Compounds, ChemMedChem, 2017, 12, 1108–1115. 10.1002/cmdc.201700170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Moraski GC, Oliver AG, Markley LD, Cho S, Franzblau SG, Miller MJ, Scaffold-switching: An exploration of 5,6-fused bicyclic heteroaromatics systems to afford antituberculosis activity akin to the imidazo[1,2-a]pyridine-3-carboxylates, Bioorg. Med. Chem. Lett. 24 (2014) 3493–3498, 10.1016/j.bmcl.2014.05.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].(a) Cheng Y; Moraski GC; Cramer J; Miller MJ; Schorey JS Bactericidal Activity of an Imidazo[1,2-a]pyridine Using a Mouse M. tuberculosis Infection Model. PLoS One, 2014, 9, e87483. 10.1371/journal.pone.0087483. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Moraski GC; Cheng Y; Cho S; Cramer JW; Godfrey A; Masquelin T; Franzblau SG; Miller MJ; Schorey JS Imidazo[1,2-a]Pyridine-3-Carboxamides Are Active Antimicrobial Agents against Mycobacterium avium Infection In Vivo. Antimicrob Agents Chemother, 2016, 60, 5018–5022. doi: 10.1128/AAC.00618-16. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Scherr N; Bieri R; Thomas SS; Chauffour A; Kalia NP; Schneide P; Ruf MT; Lamelas A; Manimekalai MS; Grüber G; Ishii N; Suzuki K; Tanner M; Moraski GC; Miller MJ; Witschel M; Jarlier V; Pluschke G; Pethe K Nat. Commun, 2018, 9, 1. 10.1038/s41467-018-07804-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].(a) Lipinski CA; Lombardo F; Dominy BW; Feeney PJ Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev, 1997, 23, 3–25. 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]; (b) Benet LZ; Hosey CM; Ursu O; Oprea TI BDDCS, the Rule of 5 and drugability. Adv. Drug Deliv. Rev, 2016, 101, 89–98. doi: 10.1016/j.addr.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lipinski CA Rule of five in 2015 and beyond: Target and ligand structural limitations, ligand chemistry structure and drug discovery project decision Adv. Drug Deliv. Rev, 2016, 101, 34–41. doi: 10.1016/j.addr.2016.04.029. [DOI] [PubMed] [Google Scholar]
- [8].Pethe K; Bifani P; Jang J; Kang S; Park S; Ahn S; Jiricek J; Jung J; Jeon HK; Cechetto J; Christophe T; Lee H; Kempf M; Jackson MM; Lenaerts AJ; Pham H; Jones V; Seo MJ; Kim YM; Seo M; Seo JJ; Park D; Ko Y; Choi I; Kim R; Kim SY; Lim S; Yim S-A; Nam J; Kang H; Kwon H; Oh C-T; Cho Y; Jang Y; Kim J; Chua A; Tan BH; Nanjundappa MB; Rao SPS; Barnes WS; Wintjens R; Walker JR; Alonso S; Lee S; Kim J; Oh S; Oh T; Nehrbass U; Han S-J; No Z; Lee J; Brodin P; Cho S-N; Nam K; Kim J Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med, 2013, 19, 1157–1160 10.1038/nm.3262. [DOI] [PubMed] [Google Scholar]
- [9].de Jager VR, Dawson R, van Niekerk C, Hutchings J, Kim J, Vanker N, van der Merwe L, Choi J, Nam K, Diacon AH, Telacebec (Q203), a New Antituberculosis Agent, N. Engl. J. Med. 382 (2020) 1280–1281, 10.1056/NEJMc1913327. [DOI] [PubMed] [Google Scholar]
- [10].(a) Beites T; O’Brien K; Tiwari D; Engelhart CA; Walters S; Andrews J; Yang HJ; Sutphen ML; Weiner D; Dayao EK; Zimmerman M; Prideaux B; Desai PV; Masquelin T; Via LE; Dartois V; Boshoff HI; Barry CE 3rd; Ehrt S; Schnappinger D Plasticity of the Mycobacterium tuberculosis respiratory chain and its impact on tuberculosis drug development. Nat. Commun., 2019, 10, 4970. 10.1038/s41467-019-12956-2. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Moraski GC; Cheng Y; Cho S; Cramer JW; Godfrey A; Masquelin T; Franzblau SG; Miller MJ Schorey JS Imidazo[1,2-a]Pyridine-3-carboxamides are active antimicrobial agents against Mycobacterium Avium infection in vivo. Antimicrob. Agents Chemother, 2016, 60, 5018–5022. DOI: 10.1128/AAC.00618-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Abrahams KA, Cox JA, Spivey VL, Loman NJ, Pallen MJ, Constantinidou C, Fernandez R, Alemparte C, Remuinan MJ, Barros D, Ballell L, Identification of Novel Imidazo[1,2-a]pyridine Inhibitors Targeting M. tuberculosis QcrB, PloS One. 7 (2012), e52951, 10.1371/journal.pone.0052951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bijani S, Jain V, Padmanabhan D, Pandey B, Shah A, Mixed Pd/C and Pt/C as efficient catalysts for deuteration of Mesalamine, Tetrahedron Lett. 56 (2015) 1211–1214, 10.1016/j.tetlet.2015.01.135. [DOI] [Google Scholar]
- [13].Ito N, Esaki H, Maesawa T, Imamiya E, Maegawa T, Sajiki H, Efficient and Selective Pt/C-Catalyzed H-D Exchange Reaction of Aromatic Rings, Bull. Chem. Soc. Jpn. 81 (2008) 278–286, 10.1246/bcsj.81.278. [DOI] [Google Scholar]
- [14].Liu JF, Harbeson SL, Brummel CL, Tung R, Silverman R, Doller D, Chapter Fourteen - A Decade of Deuteration in Medicinal Chemistry, Ann. Rep. Med. Chem. 50 (2017) 519–542, 10.1016/bs.armc.2017.08.010. [DOI] [Google Scholar]
- [15].Harbeson SL, Tung RD, Deuterium Medicinal Chemistry: A New Approach to Drug Discovery and Development, Medchem. News 2 (2014) 8–22. [Google Scholar]
- [16].Pirali T, Serafini M, Cargnin S, Genazzani AA, Applications of Deuterium in Medicinal Chemistry, J. Med. Chem. 62 (2019) 5276–5297, 10.1021/acs.jmedchem.8b01808. [DOI] [PubMed] [Google Scholar]
- [17].Cargnin S, Serafini M, Piral T, A primer of deuterium in drug design, Future Med. Chem. 11 (2019) 2039–2042, 10.4155/fmc-2019-0183. [DOI] [PubMed] [Google Scholar]
- [18].Liu R, Krchnak V, Brown SN, Miller MJ, Deuteration of BTZ043 Extends the Lifetime of Meisenheimer Intermediates to the Antituberculosis Nitroso Oxidation State, ACS Med Chem Lett. 10 (2019) 1462–1466, 10.1021/acsmedchemlett.9b00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pethe K; Sequeira PC; Agarwalla S; Rhee K; Kuhen K; Phong WY; Patel V; Beer D; Walker JR; Duraiswamy J; Jiricek J; Keller TH; Chatterjee A; Tan MP; Ujjini M; Rao SPS; Camacho L; Bifani P; Mak PA; Ma I; Barnes SW; Chen Z; Plouffe D; Thayalan P; Ng SH; Au M Lee BH; Tan BH; Ravindran S; Nanjundappa M; Lin X; Goh A; Lakshminarayana SB; Shoen C; Cynamon M; Kreiswirth B; Dartois V; Peters EC; Glynne R; Brenner S; Dick T A chemical genetic screen in Mycobacterium tuberculosis identifies carbon-source-dependent growth inhibitors devoid of in vivo efficacy. Nat. Commun, 2010, 1:57 doi: 10.1038/ncomms1060 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cho S, Lee HS, Franzblau S, Microplate Alamar Blue Assay (MABA) and Low Oxygen Recovery Assay (LORA) for Mycobacterium tuberculosis, In Mycobacteria protocols, Humana Press, New York, NY. 281–292 (2015), 10.1007/978-1-4939-2450-9_17. [DOI] [PubMed] [Google Scholar]
- [21].Falzari K, Zhu Z, Pan D, Liu H, Hongmanee P, Franzblau SG, In vitro and in vivo activities of macrolide derivatives against Mycobacterium tuberculosis, Antimicrob. Agents Chemother. 49 (2005) 1447–1454, 10.1128/AAC.49.4.1447-1454.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Esaki H, H.; Ito N; Sakai S; Maegawa T; Monguchi Y; Sajiki H General[25] M Martignoni GM Groothuis R de Kanter, Species differences between mouse, method of obtaining deuterium-labeled heterocyclic compounds using neutral D2O with heterogeneous Pd/C. Tetrahedron, 2006, 62, 10954–10961. 10.1016/j.tet.2006.08.088. [DOI] [Google Scholar]
- [23].Martins A, Lautens M, A Simple, Cost-Effective Method for the Regioselective Deuteration of Anilines, Org. Lett. 10 (2008) 4351–4353, 10.1021/ol801763j [DOI] [PubMed] [Google Scholar]
- [24].Foti RS, Tyndale RF, Garcia KL, Sweet DH, Nagar S, Sharan S, Rock DA, “Target-Site” drug metabolism and transport, Drug Metab. Dispos. 43 (2015) 1156–1168, 10.1124/dmd.115.064576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Martignoni M, Groothuis GM, de Kanter R, Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction, Expert Opin. Drug Metab. Toxicol. 2 (2006) 875–894, 10.1517/17425255.2.6.875. [DOI] [PubMed] [Google Scholar]
- [26].Dixit R, Boelsterli UA, Healthy animals and animal models of human disease(s) in safety assessment of human pharmaceuticals, including therapeutic antibodies, Drug Discov. Today. 12 (2007) 336–342, 10.1016/j.drudis.2007.02.018. [DOI] [PubMed] [Google Scholar]