


Abstract
A chimeric methylphosphonodiester/phosphodiester 15mer oligodeoxynucleotide of randomly selected sequence was observed to rapidly induce apoptosis in MOLT-4 and Jurkat E6 T lymphocytic leukaemia cells following intracytoplasmic delivery. A series of further methylphosphonate substitutions and mutations and truncations of the oligodeoxynucleotide served to establish that the phosphodiester-linked sequence CGGTA present in the 15mer was responsible for this biological activity. End-protected CpG oligodeoxynucleotide 5mers of sequence type CGNNN exhibited a range of apoptosis-inducing potencies, with CGTTA being the most active. The latter was shown to significantly reduce the rate of RNA synthesis in MOLT-4 cells within 1 h; DNA laddering and redistribution of phosphatidylserine to the outer surface of the plasma membrane were marked by 160 min and mitochondrial transmembrane potential collapsed over roughly the same time scale. Pro-caspase 8 was reduced within 130 min and the proteolytically activated caspase 8 substrate Bid was also down by this time, implicating release of cytochrome c from mitochondria by the active 15 kDa fragment of Bid. Substantial proteolytic activation of pro-caspase 3 was relatively delayed. These findings support a mitochondrial amplification mechanism for apoptosis triggered by CpG 5mers.
INTRODUCTION
Apoptosis or programmed cell death is a process by which a defunct or irreparably damaged cell may be dismantled from within in an orderly fashion and packaged into smaller membrane-bound vesicles to be engulfed and digested by neighbouring cells or macrophages (1). In this way, inflammatory responses which would result from spillage of the cell contents into the extracellular space are avoided. Early events in apoptosis include activation of caspases, cysteine proteases which cleave after aspartate (2), redistribution of phosphatidylserine to the outer surface of the plasma membrane and the appearance of surface blebs (3,4). These may be accompanied sooner or later by DNA laddering, the digestion of the DNA into oligonucleosomal fragments (5–7) and, depending on the apoptotic mechanism engaged, early or late disruption of the mitochondrial transmembrane potential, Δψm (8–10). Signals to undergo apoptosis may be extracellular through interactions at cell surface receptors, such as CD95 (APO-1/Fas) (11), responsible for mediating activation of caspase 8, which in turn proteolytically activates the Bcl-2 family member Bid and initiates a caspase cascade leading to activation of the effector caspase 3 (11–13). Alternatively, they may be intracellular through a variety of interactions which may be dependent on or independent of the tumour suppressor gene product p53 (14–16) and which may or may not require the release of inducers such as ‘apoptosis-inducing factor’ (17) and cytochrome c (18) from the mitochondrial intermembrane space by Bcl-2 family members (19,20). In addition, distinct functional interactions between formally different signalling pathways may predominate depending on the circumstance of the cell. A weak signal generated at CD95 surface receptors may activate insufficient caspase 8 to generate substantial caspase 3 directly, but may suffice for proteolysis of Bid, which in turn promotes release of cytochrome c and entrains a mitochondrial amplification loop mechanism leading to a delayed apoptotic response involving full-blown activation of caspase 8 and caspase 3 (11,21–23).
In the course of evaluating the cellular activity of chimeric, methylphosphonodiester/phosphodiester, antisense oligodeoxynucleotides (24), we observed that a 15mer control of random sequence rapidly induced apoptosis in MOLT-4 and Jurkat E6 T lymphocytic leukaemia cells following intracytoplasmic delivery (25,26). Here we report the results of experiments to establish the basis of this apoptogenic activity which demonstrate that it derives from a phosphodiester-linked five base sequence, CGGTA, present within the oligodeoxynucleotide. We go on to show that isolated ‘CpG oligodeoxynucleotide’ 5mers of sequence type CGNNN (N = A, G, C or T), end-protected against exonucleolytic degradation, also induce apoptosis to varying extents at a given concentration, with CGTTA being more potent than the original 15mer. Apoptosis triggered by this sequence was very rapid and appeared to proceed through a mitochondrial pathway involving the mutually potentiating activation of caspase 8 and Bid.
MATERIALS AND METHODS
Cell lines
The human T cell leukaemia lines MOLT-4 and Jurkat E6, the chronic myeloid leukaemia line K562 and the histiocytic lymphoma line U937 were supplied by the European Collection of Animal Cell Cultures (PHLS Centre for Applied Microbiology and Research, Porton Down, UK). The chronic myeloid leukaemia lines KYO1 and LAMA84 were donated by the LRF Leukaemia Unit, Hammersmith Hospital (London, UK). The myeloma line RPMI 8226 and the Burkitt lymphoma line DAUDI were gifts, respectively, from the Departments of Medicine and Haematology of the University of Liverpool. Cells were maintained in exponential growth in RPMI 1640 medium supplemented with l-glutamine (Gibco Ltd, Paisley, UK) and 10% heat-inactivated fetal bovine serum (Harlan Sera-Lab Ltd, Crawley Down, UK).
Oligodeoxynucleotide synthesis and cell treatment
Chimeric methylphosphonodiester/phosphodiester oligodeoxynucleotides were synthesized as described previously (27) using methylphosphonamidite and phosphoramidite synthons (Glen Research, Sterling, VA; UK supplier Cambio Ltd, Cambridge, UK). 5′-Amino-Modifier C6-TFA (Glen Research) was incorporated in the final cycle of synthesis and the oligodeoxynucleotides were labelled with fluorescein post-synthesis, using Fluos reagent (Roche Diagnostics Ltd, Lewes, UK). Oligodeoxynucleotides terminating at their 3′-ends with phosphodiester internucleoside linkages were synthesized on DMT-C3-succinyl-CPG supports (Peninsula Laboratories Europe Ltd, St Helens, UK) to provide protection against 3′-exonuclease by a 3-hydroxypropyl phosphate group on the 3′-OH. The caspase 3 inhibitor Ac-DEVD-CHO was purchased from Affiniti Research Products Ltd (Mamhead, Exeter, UK). Oligodeoxynucleotides were introduced into the cytoplasm of cells by 10 min reversible permeabilization of the plasma membrane with streptolysin O (Sigma-Aldrich Co. Ltd, Poole, UK) in the presence of external concentrations of 0.2–20 µM oligodeoxynucleotide in serum-free RPMI 1640, as previously described (26; http://www.liv.ac.uk/~giles ). Results presented represent cultures in which >85% of the cells had taken up the oligodeoxynucleotide and excluded propidium iodide as determined by dual parameter flow cytometry on an Ortho Cytoron Absolute (Ortho Diagnostic Systems Ltd, High Wycombe, UK).
Apoptosis assays
Redistribution of plasma membrane phosphatidylserine to the outer cell surface and collapse of the mitochondrial transmembrane potential were measured by flow cytometry on cells loaded with unlabelled oligodeoxynucleotide using FITC-labelled annexin V (Clontech Laboratories UK Ltd, Basingstoke, UK) and the DePsipher kit (R&D Systems Europe Ltd, Abingdon, UK), respectively, according to the manufacturers’ instructions. DNA laddering was monitored by electrophoresis through 0.8% agarose gels containing 1 µg/ml ethidium bromide.
Northern and western blotting
Effects on cellular content of mRNAs and proteins were determined by densitometry of northern and western blots as previously described (28,29). Rabbit antibodies against human caspase 3, pro-caspase 8, pro-caspase 10, Bak and Bax were from Upstate Biotechnology (Waltham, MA; UK supplier TCS Biologicals Ltd, Buckingham, UK) and for human pro-caspase 9 and Bid from R&D Systems Europe Ltd.
Pulse labelling with [5-3H]uridine
Following permeabilisation with streptolysin O and membrane resealing, aliquots of the cultures (1 ml containing 9 × 105 cells) were dispensed into 24-well plates and incubated at 37°C. At various times cells were pulsed (30 min) with 37 kBq [5-3H]uridine (specific activity 999 GBq/mmol; Nycomed Amersham plc, Little Chalfont, UK), then cooled to 4°C and washed twice with 1 ml of ice-cold Tris-buffered saline, pH 7.4, containing 25 mM Tris–HCl, 130 mM NaCl and 5 mM KCl. Uridine mononucleotides were extracted from cell pellets with 60% methanol (1 ml) at –20°C overnight. Pellets were washed with a further 1 ml of 60% methanol and then RNA present in the cell residues was hydrolyzed by addition of 0.5 ml of 1 M NaOH and incubation at 37°C for >4 h. Tubes were cooled in ice and bovine serum albumin solution (10 µl, 200 mg/ml) was added, followed by 0.1 ml of 6 M HCl plus 0.5 ml of 10% trichloroacetic acid to precipitate DNA and protein. Samples of the fractions were combined with PCS (Nycomed Amersham plc) for liquid scintillation counting.
RESULTS
A 15mer, chimeric methylphosphonodiester/phosphodiester oligodeoxynucleotide (24) of randomly selected sequence (Table 1, oligodeoxynucleotide 1, 383) rapidly induced apoptosis in human MOLT-4 T lymphocytic leukaemia cells following intracytoplasmic delivery by reversible plasma membrane permeabilization with streptolysin O (25,26). The oligodeoxynucleotide structure consisted of three non-ionic, nuclease-resistant methylphosphonate (30) internucleoside linkages at each end of the molecule, to protect it against exonuclease degradation in the cells, and a central eight phosphodiester section. This same oligodeoxynucleotide was inactive against MOLT-4 cells when presented exogenously over 24 h and, even when introduced into the cytoplasm, had no significant effect on the growth and survival of human chronic myeloid leukaemia cell lines LAMA84, K562 and KYO1 and a histiocytic lymphoma line U937. However, the human T lymphocytic leukaemia line Jurkat E6 exhibited a similar response to that of MOLT-4 cells. The apoptotic reaction was observed after loading cells with oligodeoxynucleotide from an external concentration range of 1–20 µM, which is the same as that required to achieve substantial RNase H-mediated antisense activity against gene expression with this method of oligodeoxynucleotide delivery (28). Such treatment results in an initial apparent intracellular concentration in MOLT-4 cells of ~0.065–1.3 µM (26), though in reality the oligodeoxynucleotide is rapidly concentrated in the nucleus through binding to nuclear structures (25,31). The bulk of the population of MOLT-4 cells loaded with oligodeoxynucleotide 383 showed overt signs of undergoing apoptosis within 3–4 h, when extensive surface blebbing was apparent (Fig. 1). Redistribution of plasma membrane phosphatidylserine to the outer cell surface (3) was first detectable at ~70 min after delivery of the oligodeoxynucleotide and was widespread by 2.5 h (Fig. 2). The c-myc gene is highly expressed in MOLT-4 cells and whereas the intracellular level of c-myc mRNA, as measured by northern blotting, was lowered by no more than 50% under these treatment conditions, the concentration of Myc protein, determined by western blotting, was reduced to 5–20% of that present in mock treated controls over the course of 4 h (cf. Fig. 7). In contrast, p53 mRNA and protein remained at between 60 and 80% of the levels in untreated control cells at 4 h (data not shown).
Table 1. Oligodeoxynucleotides tested for induction of human apoptosis in human T lymphocytic leukaemia cells.

/, Methylphosphonodiester link; -, phosphodiester link; Fluos, fluorescein label; -ProOH, 3-hydroxypropyl phosphate; Z, 5-methylcytosine; bold type, bases differing from those present in the original 383 sequence (1).
Figure 1.
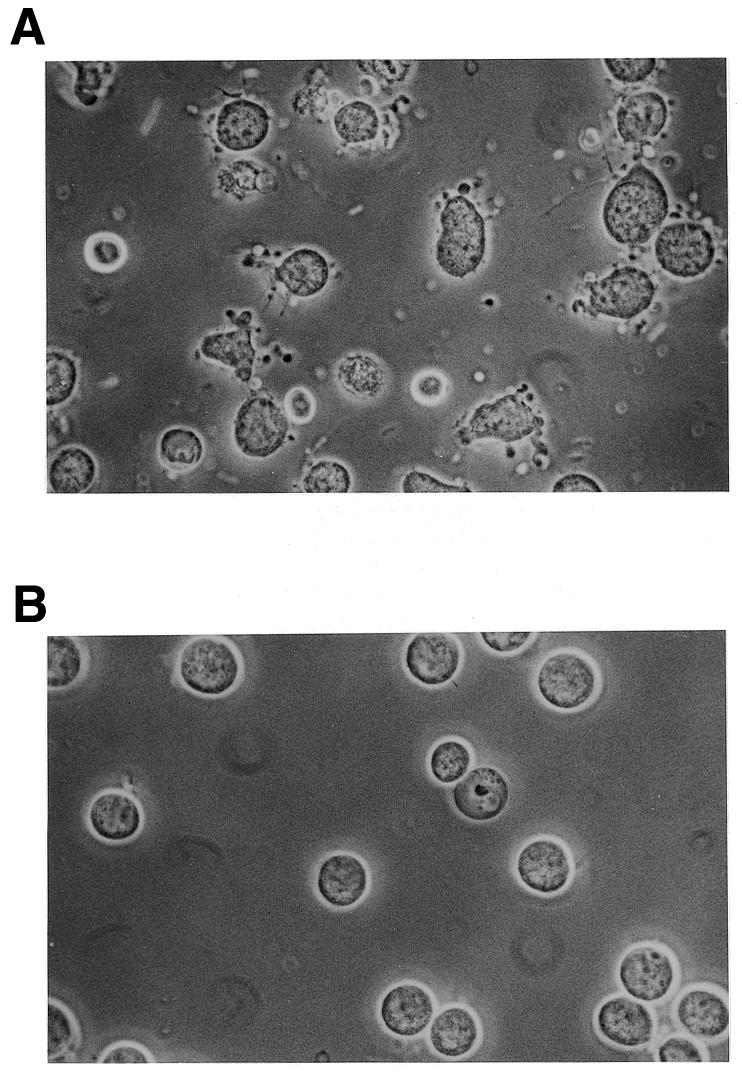
MOLT-4 cells incubated for 3.5 h at 37°C after (A) intracytoplasmic delivery of 383 (Table 1) from a 20 µM external concentration and (B) mock treatment in the absence of oligodeoxynucleotide.
Figure 2.

Redistribution of plasma membrane phosphatidylserine to the outer surface on MOLT-4 cells following intracytoplasmic delivery of oligodeoxynucleotide 383 (Table 1, without fluorescein tag) from an external concentration of 20 µM. Binding of FITC-labelled annexin V to cells excluding propidium iodide counterstain was determined by dual parameter flow cytometry at the indicated times of incubation at 37°C.
Figure 7.

Time course of the effects of intracytoplasmic delivery of CpG oligodeoxynucleotides on intracellular levels of c-myc mRNA (upper) and protein (lower) in MOLT-4 (A and C) and KYO1 (B and D) cells. Cells were loaded with oligodeoxynucleotides from a 20 µM external concentration and were then resuspended in fresh medium at a density of 5 × 105/ml. Band intensities on northern and western blots of isolated RNA and cell lysates, respectively, were determined by densitometry and are normalized to the values for cells treated with streptolysin O alone. The data represent the means ± SD for three replicates.
A range of similar structure, 383-type, chimeric methylphosphonodiester/phosphodiester oligodeoxynucleotides with different base sequences had no effect on the proliferation of MOLT-4 cells. Nor did the active sequence when delivered as a normal, 3′-hydroxyl unprotected, all-phosphodiester oligodeoxynucleotide. This presumably reflected the very short lifetime of all-phosphodiester oligodeoxynucleotides in the cytoplasm of living cells in the absence of the exonuclease protection and reduced endonuclease susceptibility afforded by terminal, nuclease-resistant, methylphosphonate linkages (32). At the same time, these results excluded the possibility that deoxynucleoside monophosphates, released by nuclease-mediated degradation of the oligodeoxynucleotides, were responsible for the observed response. The apoptosis-inducing base sequence in the form of a 545 chimeric methylphosphonodiester/phosphodiester structure (oligodeoxynucleotide 13, Table 1), with five methylphosphonodiester linkages at each end and four central phosphodiesters, had no effect on the proliferation of MOLT-4 cells. Therefore, it was apparent that apoptosis-inducing activity was dependent upon some unmodified internucleoside linkages within the central section of 383.
Further oligodeoxynucleotides were synthesized in order to establish the absolute structural requirements for activity (Table 1). Rearrangement of the base sequence of the central phosphodiester section of 383, while leaving the methylphosphonate sections unchanged, gave oligodeoxynucleotide 383scr1 (oligodeoxynucleotide 3, Table 1), which retained apoptosis-inducing activity of essentially the same potency (Fig. 3A). The only phosphodiester-linked sequence elements surviving this rearrangement were C4G5 and T7A8. However, switching G5 with A8 in 383scr1 to give sequence 383scr2 (oligodeoxynucleotide 5, Table 1) led to complete loss of activity (Fig. 3A). Progressive substitution of phosphodiester internucleoside linkages by methylphosphonates from the 3′-end of 383 did not affect activity (Fig. 3A) in the series 374–347 (oligodeoxynucleotides 6–8 and 12, Table 1), where all oligodeoxynucleotides retained a phosphodiester-linked CGGTA sequence. The apoptosis-inducing effects of oligodeoxynucleotide 347 were in marked contrast to the lack of effect of its isomer 545, which retained a phosphodiester-linked T7A8 but where C4G5 was engulfed by methylphosphonate. Further reduction in the phosphodiester section of 347 by 3′-methylphosphonate substitution gave inactive molecules (oligodeoxynucleotides 14–16, Table 1). These results demonstrated that induction of apoptosis was dependent on phosphodiester-linked C4G5 followed by at least three phosphodiester-linked bases, GTA in 383 and CTA in 383scr1. Taken in conjunction with the lack of apoptosis induction by 383scr2, the absolute requirement for a 5′-CpG dinucleotide was further indicated by mutation of C4 in 356 to a T in 356mut (oligodeoxynucleotide 10, Table 1), which completely abolished activity. Also, it is noteworthy that the isomer of active 356, 653 (oligodeoxynucleotide 11, Table 1), in which methylphosphonate substitution extended through C4G5 but which contained the phosphodiester-linked sequence AACGG, did not induce apoptosis (Fig. 3B), suggesting that both the position and the sequence context of the CpG in the phosphodiester linked 5mer segment might be important.
Figure 3.

Changes in cell number/ml of MOLT-4 cultures over 24 h following intracytoplasmic delivery of oligodeoxynucleotides (Table 1) from 20 µM external concentrations. Cells were resuspended in fresh medium at an initial density of 5 × 105/ml and numbers excluding propidium iodide were measured 24 h later by flow cytometry. The data represent the means ± SD of three replicates. (A) and (B) represent two separate experiments.
Immunostimulation by CpG-containing oligodeoxynucleotides with the optimal consensus sequence R1R2CGY1Y2 is derived through topologically external interactions with B lymphocytes and has been reported to be abolished by methylation of cytosine on the 5 position (33,34). The active sequences of Table 1 induced no response when presented exogenously to MOLT-4 cells over 24 h and, although they would appear to be unrelated to Krieg’s immune stimulatory CpG motifs, it was of interest to establish the effect of cytosine methylation on the induction of apoptosis by these oligodeoxynucleotides. The active oligodeoxynucleotides 383, 383scr1 and 356 were resynthesized with 5-methyl-deoxycytidine replacing phosphodiester-linked deoxycytidine to give Me-383, Me-383scr1 and Me-356 (oligodeoxynucleotides 2, 4 and 9, Table 1). All three C-methylated oligodeoxynucleotides were found to be as effective at inducing apoptosis as the parent molecules (Fig. 3B).
Successive deletions of bases from the termini of the active molecule, 347, (oligodeoxynucleotides 17 and 20–23, Table 1) and mutation within the palindromic 5′-methylphosphonate section (oligodeoxynucleotide 19, Table 1) revealed that the bases within the methylphosphonate-linked portions of the oligodeoxynucleotide were irrelevant and only the phosphodiester linked 5mer portion, CGGTA, was responsible for induction of apoptosis. Since 383scr1 was also effective in induction of apoptosis, it was evident that the active 5mer sequence, CGGTA, could be mutated to CGCTA without loss of activity. However, further mutation to CGCCC, as in 342mut1 (oligodeoxynucleotide 18, Table 1), abolished activity and, therefore, the sequence context of the CpG dinucleotide was clearly important.
A series of all-phosphodiester 5mers was synthesized incorporating successive mutations of bases 3, 4 and 5 of the active sequence CGGTA in order to establish sequence contexts of the CpG dinucleotide which support apoptosis-inducing activity (Table 2). The active sequences CGGTA and CGCTA, present in 383 and 383scr1, respectively, were also able to induce apoptosis in MOLT-4 cells as isolated 5mers following intracytoplasmic delivery. A range of potency was observed for the CpG oligodeoxynucleotides, extending from a substantial reduction in cell numbers over a 24 h period elicited by CGTTA, CGTTG and CGATA to apparent decreases in cell proliferation rate generated with CGGGA (Figs 4 and 5). Exogenous application of the oligomers had no effect on MOLT-4 cells. Oligodeoxynucleotide 5mers synthesized without a 3-hydroxypropyl phosphate protecting group on the 3′-hydroxyl exhibited much reduced potency, consistent with their rapid degradation by 3′-exonuclease within the cells. The 3′-protected oligodeoxynucleotides CGTTA and CGTTG were more effective in inducing apoptosis than the original 383 15mer. Reversal of these sequences 3′→5′ to generate ATTGC and GTTGC controls completely abolished activity, as did reversal of the CpG in CGTTA to give GCTTA. Mutation of the G to give CTTTA and mutation of C to give TGTTA also abrogated induction of apoptosis. Shifting the CpG from positions 1 and 2 of the 5mer to positions 2 and 3 in TCGTT did not eliminate the capacity to induce apoptosis, but reduced potency relative to CGTTG.
Table 2. Apoptogenic activity of CpG 5mer oligodeoxynucleotides tested against MOLT-4 cells.

-, Phosphodiester link; Fluos, fluorescein label; -ProOH, 3-hydroxypropyl phosphate. The activity assignments ++++ to + relate to the proportions of the cell population triggered into apoptosis after loading with oligodeoxynucleotide at a given external concentration.
Figure 4.

Dose–response curves for change in cell number/ml of MOLT-4 cultures over 24 h following intracytoplasmic delivery of CpG 5mers. Cells were loaded with oligodeoxynucleotides from the indicated external concentrations and were then resuspended in fresh medium at an initial density of 5 × 105/ml. Cells excluding propidium iodide were counted 24 h later by flow cytometry. The data are expressed as a percentage of the increase in cell density in cultures mock treated in the absence of oligodeoxynucleotide and represent the means ± SD of three replicates.
Figure 5.

Comparison of the changes in cell number/ml in MOLT-4, KYO1 and Jurkat E6 cultures over 24 h following intracytoplasmic delivery of CpG 5mers. Cells were loaded with oligodeoxynucleotide from a 20 µM external concentration and were then resuspended in fresh medium at an initial density of 5 × 105/ml. Cells excluding propidium iodide were counted 24 h later by flow cytometry. The data represent the means ± SD of three replicates.
The CpG 5mers CGTTA and CGATA were also effective against Jurkat E6 cells (Fig. 5) and the time course of phosphatidylserine externalization was similar to that observed with MOLT-4 cells. Interestingly, although the original 383 CpG oligodeoxynucleotide incorporating the active sequence CGGTA had no significant effect on myeloid leukaemia cell lines, the potent 5mers CGTTA, CGATA (Fig. 5) and CGTTG induced quite significant cell cycle arrest in cultures of the Bcr/Abl-expressing line KYO1; the relative effectiveness of different sequences mirrored their relative apoptotic activity against MOLT-4 cells. Again, the inverted sequences GTTGC and ATTGC were completely inactive. However, KYO1 cells arrested in the cell cycle by CGTTA and CGTTG did not undergo apoptosis, as evidenced by the absence of phosphatidylserine on the outer cell surface over 24 h. In marked contrast, the range of antiproliferative activity of the different CpG 5mers listed in Table 2 against MOLT-4 cells (Figs 4 and 5) was related to differences in the percentage of cells in the treated populations that were triggered into apoptosis at a given concentration, with CGTTA generating the highest and CGGGA the lowest proportion of annexin V staining cells at 160 min. The B cell myeloma line RPMI 8226 was also susceptible to cell cycle arrest by CGTTA and CGATA, while the B lymphoblastoid Burkitt lymphoma cell line DAUDI was completely resistant to these oligodeoxynucleotides. Preliminary work also suggested that the sequence CGTTG may abrogate the in vitro clonogenic potential of primary cells of chronic myelogenous leukaemia from some patients.
Metabolic activity and rates of RNA synthesis were determined by pulse labelling cultures for 30 min with [5-3H]uridine and measuring incorporation of the isotope into soluble mononucleotide and RNA fractions at various times after the initiation of treatment. MOLT-4 cells treated with CGTTA accumulated [5-3H]uridine in the soluble nucleotide pool at the same rate as controls over the first 2 h (Fig. 6A). However, the rate of RNA synthesis fell progressively during this time interval in the CGTTA-treated cells (Fig. 6B). The declining rates of phosphorylation of uridine with time (Fig. 6A) are related to the cells being suspended in fresh medium at 0 h, which initially stimulates the utilisation of exogenous uridine 4- to 5-fold relative to stabilized, exponentially proliferating cultures. The same effect is seen when medium replacement is the only treatment the cells receive. Treatment of MOLT-4 cells with 356, containing the active sequence CGGTA (oligodeoxynucleotide 8, Table 1), or the 5mer CGTTA led to reductions in c-myc mRNA of 40–60% at 2 and 4 h (Fig. 7A) while depletion of c-Myc protein was considerably more dramatic over these intervals (Fig. 7C). Although the effect on c-myc mRNA appeared to be somewhat more transitory in KYO1 cells receiving the same treatments (Fig. 7B), it is noteworthy that the pattern of suppression of c-Myc protein expression in cell cycle-arrested KYO1 cells (Fig. 7D) was similar to that in the apoptotic MOLT-4 cells (Fig. 7C).
Figure 6.

Rates of incorporation of [5-3H]uridine into (A) the mononucleotide pool and (B) RNA following treatment of MOLT-4 cells with the potent apoptosis-inducing CpG 5mer CGTTA (circles), the inverted control ATTGC (diamonds) and streptolysin O alone (triangles). Cells were loaded with oligodeoxynucleotides from a 20 µM external concentration and were pulsed with [5-3H]uridine for 30 min at the indicated times following resuspension in fresh medium. The data correspond to 4.5 × 105 cells and represent the means ± SD of three replicates at each time point. Where error bars are absent, the standard deviation was less than the size of the symbol.
Further experiments were undertaken to characterise the apoptotic process in MOLT-4 cells loaded with CGTTA and CGTTG, using the inverted sequence ATTGC as control. DNA laddering was readily apparent at 160 min (Fig. 8) when the bulk of the cells had redistributed phosphatidylserine to the outer face of the plasma membrane (Fig. 9A). Intracytoplasmic delivery of CpG oligodeoxynucleotides from as low an external concentration as 0.2 µM generated a significant proportion of annexin V-positive cells over background at this time. Collapse of the mitochondrial transmembrane potential, Δψm, began at ~100 min and was widespread in the CGTTA-treated cells by 3 h (Fig. 9B). A similar time course was observed in Jurkat E6 cells. Activation of pro-caspase 3 (Fig. 9C) was somewhat delayed compared to processing of pro-caspase 8 (Fig. 10A). Intracytoplasmic delivery of the caspase 3 inhibitor Ac-DEVD-CHO (from an external concentration of 5–20 µM) together with CGTTA (20 µM) into MOLT-4 cells completely inhibited accumulation of the 17 and 12 kDa active subunits of caspase 3 seen on western blots at 180 min and led to the appearance of the 20 kDa intermediate (11) that was probably produced by the action of another caspase on pro-caspase 3 (data not shown). However, complete inhibition of caspase 3 activity failed to rescue the cells from undergoing apoptosis. Treatment with CGTTA had no effect on the level of pro-caspase 10 in MOLT-4 cells over 280 min (data not shown) and pro-caspase 9 was only minimally affected (Fig. 10C). The Bcl-2 family protein Bax was barely detectable in MOLT-4 cells and Bak expression was minimally reduced during the onset of apoptosis between 100 and 260 min. However, proteolytic processing of the caspase 8 substrate Bid was readily observed (Fig. 10B) over a time course which roughly paralleled that of pro-caspase 8 processing, the collapse of mitochondrial transmembrane potential and externalization of phosphatidylserine.
Figure 8.
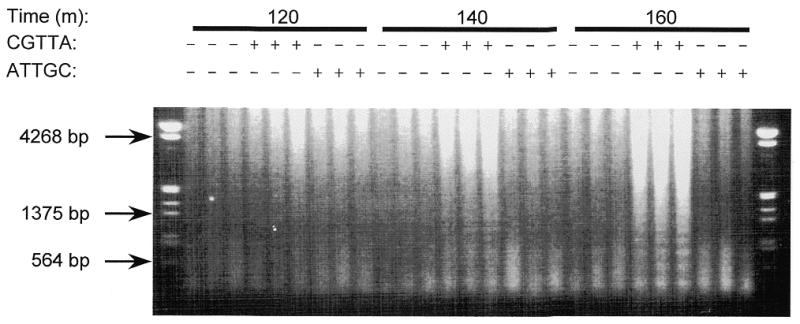
Time course for the appearance of DNA laddering in MOLT-4 cells treated with CGTTA. Intracytoplasmic delivery of oligodeoxynucleotide was from an external concentration of 20 µM and DNA isolated from cell samples taken at the indicated time points was run on a 0.8% agarose–ethidium bromide gel.
Figure 9.

Characteristics of apoptosis in MOLT-4 cells treated with CpG 5mers. Cells were loaded from 20 µM oligodeoxynucleotide (B and C) and the concentrations indicated (A). The dose–response characteristics for the proportion of the cell population exhibiting redistribution of phosphatidylserine at 160 min (A) and the time course of mitochondrial transmembrane potential collapse (B) were determined by flow cytometry. The time course of proteolytic processing of pro-caspase 3 (C) was determined by densitometry of western blots. The data were normalised to the values for cells treated with streptolysin O alone and each point represents the mean ± SD of three replicates.
Figure 10.

The time courses of proteolytic processing of pro-caspase 8 (A), Bid (B) and pro-caspase 9 (C) in MOLT-4 cells following intracytoplasmic delivery of CGTTA. Cells were loaded from an external concentration of 20 µM oligodeoxynucleotide and lysates were prepared at the indicated times after resuspension in fresh medium. Band intensities on western blots were determined by densitometry. The data were normalised to the values for cells treated with streptolysin O alone and each point represents the mean ± SD of three replicates.
DISCUSSION
We have demonstrated that oligodeoxynucleotides as short as five bases in length and containing a CpG dinucleotide at or adjacent to the 5′-end induce apoptosis in MOLT-4 and Jurkat E6 T lymphocytic leukaemia cells and elicit cell cycle arrest in other human leukaemia cell lines. Of the oligodeoxynucleotides tested of sequence type CGNNN, the exonuclease end-protected oligomer CGTTA proved to be the most potent with respect to these activities. It has been suggested that p53 is involved in the induction of apoptosis by X-irradiation in MOLT-4 cells (35,36) and it is not inconceivable that aptameric oligodeoxynucleotides might interact with components of the DNA damage detection/DNA repair machinery of the cell and somehow impinge upon signalling by p53. However, the possible involvement of p53 may be excluded from consideration as to the mechanism of induction of apoptosis by CpG oligodeoxynucleotides, since the p53 system is inoperative in the MOLT-4 cell line used in our experiments. These cells were found to be heterozygous for the p53 gene, with one allele carrying a mutation at the mutation hot-spot, codon 248, where CGG was mutated to CAG, converting an arginine to glutamine in the protein product of the mutant allele (C.Ruddell, PhD thesis in preparation). This mutation has previously been shown to have a dominant negative effect on the activity of the product of the wild-type allele (37).
Overexpression of the oncogene protein c-Myc (38) in otherwise normal cells has been found to lead to apoptosis (16,39). In contrast, c-Myc protein is highly expressed in MOLT-4 cells and its intracellular concentration was reduced to 5–20% of the levels in untreated controls in cells undergoing apoptosis during the 4 h following introduction of a CpG oligodeoxynucleotide. This effect was significantly greater than the more modest reductions in the levels of the corresponding mRNA. Also, it was apparently a direct consequence of the presence of the oligodeoxynucleotide, rather than an indirect result of the onset of apoptosis, since a similar response was observed in KYO1 cells which underwent cell cycle arrest but did not apoptose. Reduction in the intracellular concentration of c-Myc protein might be the result of enhanced rates of degradation following its recruitment to signalling complexes, rather than being a signalling event in its own right, since we have previously observed that reducing c-Myc protein to similar levels or lower with antisense oligodeoxynucleotide analogues did not affect the rate of proliferation of MOLT-4 or KYO1 cells over 24 h (26,40). Decreases in c-Myc have been associated with apoptosis triggered by other factors, but there they have been interpreted as playing a pivotal role (39,41).
Oligodeoxynucleotides incorporating CpG dinucleotides have been reported to inhibit protein tyrosine kinases (42,43). However, our own preliminary work showed no obvious changes in the phosphotyrosine protein profiles of MOLT-4 and KYO1 cells 1 h after loading with CGTTA, as compared to cells treated with ATTGC control oligodeoxynucleotide or mock treated cells (data not shown). The characteristics and kinetics of the apoptotic changes in MOLT-4 cells loaded with CpG oligodeoxynucleotides were very similar to those of type II cells triggered into mitochondria-dependent apoptosis by crosslinking of CD95 (11,21). It would appear that low level activation of caspase 8 leads to cleavage of Bid, the 15 kDa subunit of which promotes release of apoptogenic factors from mitochondria and collapse of the mitochondrial transmembrane potential, accompanied by further activation of caspase 8, Bid cleavage and delayed activation of caspase 3. However, complete inhibition of caspase 3 with Ac-DEVD-CHO failed to protect MOLT-4 cells from CpG oligodeoxynucleotide-induced apoptosis, suggesting that other caspases (2) may also be involved in this process. The upstream target of the oligodeoxynucleotides remains to be identified.
Acknowledgments
ACKNOWLEDGEMENT
This work was supported by the Leukaemia Research Fund.
REFERENCES
- 1.Raff M.C. (1992) Nature, 356, 397–399. [DOI] [PubMed]
- 2.Wolf B.B. and Green,D.R. (1999) J. Biol. Chem., 274, 20049–20052. [DOI] [PubMed]
- 3.Martin S.J., Reutelingsperger,C.P.M., McGahon,A.J., Rader,J.A., van Schie,R.C.A.A., Laface,D.M. and Green,D.R. (1995) J. Exp. Med., 182, 1545–1556. [DOI] [PMC free article] [PubMed]
- 4.Martin S.J., Finucane,D.M., Amarante-Mendes,G.P., O’Brien,G.A. and Green,D.R. (1996) J. Biol. Chem., 271, 28753–28756. [DOI] [PubMed]
- 5.Wyllie A.H. (1980) Nature, 284, 555–556. [DOI] [PubMed]
- 6.Enari M., Sakahira,H., Yokoyama,H., Okawa,K., Iwamatsu,A. and Nagata,S. (1998) Nature, 391, 43–50. [DOI] [PubMed]
- 7.Sakahira H., Enari,M. and Nagata,S. (1998) Nature, 391, 96–99. [DOI] [PubMed]
- 8.Jaattela M., Wissing,D., Kokholm,K., Kallunki,T. and Egeblad,M. (1998) EMBO J., 17, 6124–6134. [DOI] [PMC free article] [PubMed]
- 9.Green D.G. and Reed,J.C. (1998) Science, 281, 1309–1312. [DOI] [PubMed]
- 10.Bortner C.D. and Cidlowski,J.A. (1999) J. Biol. Chem., 274, 21953–21962. [DOI] [PubMed]
- 11.Scaffidi C., Fulda,S., Srinivasan,A., Friesen,C., Li,F., Tomaselli,K.J., Debatin,K.-M., Krammer,P.H. and Peter,M.E. (1998) EMBO J., 17, 1675–1687. [DOI] [PMC free article] [PubMed]
- 12.Tan K.O., Tan,K.M. and Yu,V.C. (1999) J. Biol. Chem., 274, 23687–23690. [DOI] [PubMed]
- 13.Yin X.-M., Wang,K., Gross,A., Zhao,Y., Zinkel,S., Klocke,B., Roth,K.A. and Korsmeyer,S.J. (1999) Nature, 400, 886–891. [DOI] [PubMed]
- 14.Wyllie A. (1997) Nature, 389, 237–238. [DOI] [PubMed]
- 15.Bates S., Phillips,A.C., Clark,P.A., Stott,F., Peters,G., Ludwig,R.L. and Vousden,K.H. (1998) Nature, 395, 124–125. [DOI] [PubMed]
- 16.Evan G. and Littlewood,T. (1998) Science, 281, 1317–1322. [DOI] [PubMed]
- 17.Susin S.A., Lorenzo,H.K., Zamzami,N., Marzo,I., Snow,B.E., Brothers,G.M., Mangion,J., Jacotot,E., Costantini,P., Loeffler,M. et al. (1999) Nature, 397, 441–446. [DOI] [PubMed]
- 18.Zhivotovsky B., Orrenius,S., Brustugun,O.T. and Doskeland,S.O. (1998) Nature, 391–392. [DOI] [PubMed]
- 19.Adams J.M. and Cory,S. (1998) Science, 281, 1322–1326 [DOI] [PubMed]
- 20.Shimizu S., Narita,M. and Tsujimoto,Y. (1999) Nature, 399, 483–487. [DOI] [PubMed]
- 21.Scaffidi C., Schmitz,I., Zha,J., Korsmeyer,S.J., Krammer,P.H. and Peter,M.E. (1999) J. Biol. Chem., 274, 22532–22538. [DOI] [PubMed]
- 22.Watson R.W.G., O’Neill,A., Brannigen,A.E., Coffey,R., Marshall,J.C., Brady,H.R. and Fitzpatrick,J.M. (1999) FEBS Lett., 453, 67–71. [DOI] [PubMed]
- 23.Boesen-de Cock J.G.R., Tepper,A.D., de Vries,E., van Blitterswijk,W.J. and Borst,J. (1999) J. Biol. Chem., 274, 14255–14261. [DOI] [PubMed]
- 24.Tidd D.M. (1996) Perspect. Drug Discov. Design, 4, 51–60.
- 25.Spiller D.G. and Tidd,D.M. (1995) Antisense Res. Dev., 5, 13–21. [DOI] [PubMed]
- 26.Giles R.V., Grzybowski,J., Spiller,D.G. and Tidd,D.M. (1997) Nucl. Nucl., 16, 1155–1163.
- 27.Tidd D.M. (1997) In Gibson,I. (ed.), Antisense and Ribozyme Methodology. Chapman and Hall, Weinheim, Germany, pp. 13–26.
- 28.Giles R.V., Spiller,D.G., Grzybowski,J., Clark,R.E., Nicklin,P. and Tidd,D.M. (1998) Nucleic Acids Res., 26, 1567–1575. [DOI] [PMC free article] [PubMed]
- 29.Spiller D.G., Giles,R.V., Broughton,C.M., Grzybowski,J., Ruddell,C.J., Tidd,D.M. and Clark,R.E. (1998) Antisense Nucleic Acid Drug Dev., 8, 281–293. [DOI] [PubMed]
- 30.Miller P.S. (1998) In Stein,C.A. and Krieg,A.M. (eds), Applied Antisense Oligonucleotide Technology. Wiley-Liss, New York, NY, pp. 3–22.
- 31.Spiller D.G., Giles,R.V., Grzybowski,J., Tidd,D.M. and Clark,R.E. (1998) Blood, 91, 4738–4746. [PubMed]
- 32.Giles R.V., Spiller,D.G., Green,J.A., Clark,R.E. and Tidd,D.M. (1995) Blood, 86, 744–754. [PubMed]
- 33.Krieg A.M., Yi,A.-K., Matson,S., Waldschmidt,T.J., Bishop,G.A., Teasdale,R., Koretzky,G.A. and Klinman,D.M. (1995) Nature, 374, 546–549. [DOI] [PubMed]
- 34.Krieg A.M. (1998) In Stein,C.A. and Krieg,A.M. (eds), Applied Antisense Oligonucleotide Technology. Wiley-Liss, New York, NY, pp. 431–448.
- 35.Zhao Q.L., Kondo,T., Noda,A. and Fujiwara,Y. (1999) Int. J. Radiat. Biol., 75, 493–504. [DOI] [PubMed]
- 36.Nakano H. and Shinohara,K. (1999) Radiat. Res., 151, 686–693. [PubMed]
- 37.O’Connor P.M., Jackman,J., Jondle,D., Bhatia,K., Magrath,I. and Kohn,K.W. (1993) Cancer Res., 53, 4776–4780. [PubMed]
- 38.Ryan K.M. and Birnie,G.D. (1996) Biochem. J., 314, 713–721. [DOI] [PMC free article] [PubMed]
- 39.Thompson E.B. (1998) Annu. Rev. Physiol., 60, 575–600. [DOI] [PubMed]
- 40.Giles R.V., Spiller,D.G., Clark,R.E. and Tidd,D.M. (1999) Antisense Nucleic Acid Drug Dev., 9, 213–220. [DOI] [PubMed]
- 41.Wu M., Bellas,R.E., Shen,J., Yang,W. and Sonenshein,G.E. (1999) J. Immunol., 6530–6535. [PubMed]
- 42.Bergan R.C., Kyle,E., Connell,Y., Neckers,L. (1995) Antisense Res. Dev., 5, 33–38. [DOI] [PubMed]
- 43.Krieg A.M., Matson,S., Cheng,K., Fisher,E., Koretzky,G.A., Koland,J.G. (1997) Antisense Nucleic Acid Drug Dev., 7, 115–123. [DOI] [PubMed]