


Abstract
We report the production, purification and characterization of a DNA ligase encoded by the thermophilic archaeon Methanobacterium thermoautotrophicum. The 561 amino acid Mth ligase catalyzed strand-joining on a singly nicked DNA in the presence of a divalent cation (magnesium, manganese or cobalt) and ATP (Km 1.1 µM). dATP can substitute for ATP, but CTP, GTP, UTP and NAD+ cannot. Mth ligase activity is thermophilic in vitro, with optimal nick-joining at 60°C. Mutational analysis of the conserved active site motif I (KxDG) illuminated essential roles for Lys251 and Asp253 at different steps of the ligation reaction. Mutant K251A is unable to form the covalent ligase–adenylate intermediate (step 1) and hence cannot seal a 3′-OH/5′-PO4 nick. Yet, K251A catalyzes phosphodiester bond formation at a pre-adenylated nick (step 3). Mutant D253A is active in ligase–adenylate formation, but defective in activating the nick via formation of the DNA–adenylate intermediate (step 2). D253A is also impaired in phosphodiester bond formation at a pre-adenylated nick. A profound step 3 arrest, with accumulation of high levels of DNA–adenylate, could be elicited for the wild-type Mth ligase by inclusion of calcium as the divalent cation cofactor. Mth ligase sediments as a monomer in a glycerol gradient. Structure probing by limited proteolysis suggested that Mth ligase is a tightly folded protein punctuated by a surface-accessible loop between nucleotidyl transferase motifs III and IIIa.
INTRODUCTION
DNA ligases catalyze the sealing of 5′-phosphate and 3′-hydroxyl termini at nicks in duplex DNA via three sequential nucleotidyl transfer reactions (1,2). In the first step, attack on the α-phosphorus of ATP or NAD+ by ligase results in release of pyrophosphate or nicotinamide mononucleotide and formation of a covalent intermediate (ligase–adenylate) in which AMP is linked via a phosphoamide bond to the ɛ-amino group of a lysine. In the second step, the AMP is transferred to the 5′-end of the 5′-phosphate-terminated DNA strand to form DNA–adenylate (AppN). In the third step, ligase catalyzes attack by the 3′-OH of the nick on the DNA–adenylate to join the two polynucleotides and release AMP.
DNA ligases are grouped into two families, ATP-dependent ligases and NAD+-dependent ligases, according to the cofactor required for ligase–adenylate formation. The ATP-dependent DNA ligases are found in eubacteria, bacteriophages, archaea, eukarya and eukaryotic viruses, whereas the NAD+-dependent enzymes have been described only in eubacteria. A search of the NCBI database reveals that genes encoding NAD+-dependent DNA ligases have been sequenced from at least 50 eubacterial species, including several thermophiles. NAD+-dependent ligases from thermophilic eubacteria have been characterized biochemically and structurally and they have been used effectively as reagents for detection of DNA mutations (3–9).
The archaea are unicellular organisms that have remarkable biosynthetic capacity and the ability to thrive under extreme environmental conditions. Archaea are thought to be the forerunners of the eukaryotes. Kletzin identified the first DNA ligase gene from an archaeon, Delsulfurolobus ambivalens, and noted amino acid sequence similarity of the Dam ligase polypeptide to eukaryotic viral and cellular ATP-dependent ligases (10). Genes encoding putative DNA ligases have since been sequenced from at least nine other archaeal species. The archaeal ligases are of fairly uniform size (557–619 amino acids) and their primary structures are extensively conserved. Present knowledge of the archaeal ligases is limited to their deduced amino acid sequences.
Here we report the purification and characterization of the DNA ligase encoded by the thermophilic archaeon Methanobacterium thermoautotrophicum ΔH. The 1.75 Mb DNA genome of M.thermoautotrophicum specifies 1855 protein products (11). Smith et al. noted that the primary structures of M.thermoautotrophicum proteins involved in DNA metabolism and gene expression are more similar to eukaryotic proteins than to their eubacterial counterparts (11). The 561 amino acid polypeptide encoded by M.thermoautotrophicum open reading frame 1580 displays strong similarity to ATP-dependent DNA ligases. The putative Mth ligase contains the core catalytic domain common to all ATP-dependent DNA ligases. This catalytic domain is composed of a set of conserved sequence motifs (I, III, IIIa, IV, V and VI) which form the ATP-binding pocket and contribute amino acid functional groups that are essential for catalysis (12–19). Motif I (KxDGxR) includes the lysine nucleophile that becomes covalently linked to AMP during ligase–adenylate formation. In Mth ligase, the presumptive nucleophile is Lys251. The core catalytic domain of Mth ligase is preceded by an ~220 amino acid N-terminal segment related to similarly positioned segments of Saccharomyces cerevisiae DNA ligase Cdc9p, metazoan DNA ligase I and poxvirus DNA ligases.
To elucidate the enzymatic activity of Mth ligase, we produced the polypeptide in Escherichia coli as a His-tagged fusion and purified the recombinant protein to homogeneity. We show that the Mth protein is indeed an ATP-dependent DNA ligase that displays optimal nick-joining activity at 60°C. Mutational analysis of motif I established the essential roles of Lys251 in ligase–adenylate formation (step 1) and Asp253 in DNA–adenylate formation (step 2) and phosphodiester formation (step 3). We found that a selective block to phosphodiester formation (step 3) can be achieved by substituting calcium for magnesium as the divalent cation cofactor. Structure probing by limited proteolysis suggested that the Mth ligase folds similarly to the vaccinia and T7 ligases.
MATERIALS AND METHODS
T7-based vectors for expression of Mth DNA ligase
Oligodeoxynucleotide primers complementary to the 5′- and 3′-ends of the putative Mth ligase gene were used to amplify the open reading frame. Methanobacterium thermoautotrophicum genomic DNA (kindly provided by Dr John Reeve, Ohio State University) was used as the template for PCR. The primers were designed to introduce NdeI and XhoI restriction sites at the 5′- and 3′-ends of the Mth ligase gene. The PCR product was digested with NdeI and XhoI, then cloned into the NdeI and XhoI sites of the T7-based expression plasmid pET16b (Novagen) to yield pET-MthLig. Dideoxy sequencing of the entire insert of pET-MthLig confirmed that no alterations of the genomic DNA sequence were introduced during PCR amplification and cloning of the ligase gene.
Mutations K251A and D253A were introduced into the Mth ligase gene using the two-stage PCR-based overlap extension method (20). pET-MthLig was used as the template for the first-stage PCR reaction. An NdeI–SacII restriction fragment of the mutated second-stage PCR product was inserted into NdeI–SacII-cut pET16-MthLig in lieu of the wild-type restriction fragment. The inserts of the resulting plasmids pET-K251A and pET-D253A were sequenced to confirm the presence of the desired mutations.
Small scale expression and purification of wild-type Mth ligase and mutants K251A and D253A
The pET-MthLig, pET-K251A and pET-D253A plasmids were transformed into E.coli BL21(DE3). Single ampicillin-resistant colonies were inoculated into 100 ml of LB medium containing 0.1 mg/ml ampicillin and grown at 37°C until the A600 reached 0.8. The cultures were placed on ice for 30 min, then adjusted to 0.4 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and subsequently incubated at 17°C for 16 h with continuous shaking. Cells were harvested by centrifugation and the pellets were stored at –80°C. All subsequent procedures were performed at 4°C. Cell lysis was achieved by treatment of thawed, resuspended cells with 0.2 mg/ml lysozyme and 0.1% Triton X-100 in 10 ml of lysis buffer containing 50 mM Tris–HCl (pH 7.5), 0.5 M NaCl and 10% sucrose. Insoluble material was removed by centrifugation at 16 000 r.p.m. for 20 min in a Sorvall SA600 rotor. The supernatants were mixed with 1 ml Ni-NTA–agarose resin (Qiagen) for 30 min. The slurries were poured into columns and the packed resins were washed with lysis buffer. The columns were eluted stepwise with IMAC buffer (50 mM Tris–HCl, pH 7.5, 50 mM NaCl, 10% glycerol) containing 50, 200 and 500 mM imidazole. The polypeptide composition of the column fractions was monitored by SDS–PAGE. The His-tagged Mth ligases were recovered in the 200 mM imidazole fractions. The preparations were dialyzed against buffer containing 50 mM Tris–HCl (pH 7.5), 1 mM DTT, 1 mM EDTA, 10% glycerol, 50 mM NaCl and then stored at –80°C. The protein concentrations of the enzyme preparations were determined using the Bio-Rad dye reagent with bovine serum albumin as the standard.
Large scale purification of Mth ligase
Cultures (12 l) of E.coli BL21(DE3)/pLysS/pET-MthLig cells were grown at 37°C in LB medium containing 0.1 mg/ml ampicillin and 25 µg/ml chloramphenicol until the A600 reached 0.5. The culture was adjusted to 2 mM IPTG and incubation was continued at 37°C for 3 h. Cells were harvested by centrifugation and the pellet (50 g wet wt) was resuspended in 75 ml of buffer L (50 mM Tris–HCl, pH 8.0, 500 mM NaCl, 10% glycerol). All subsequent procedures were performed at 4°C. The cells were lysed by sonication and insoluble material was removed by centrifugation of the lysate for 20 min at 36 000 g. The supernatant fraction was mixed with 5 ml of immobilized nickel resin (ProBound; Invitrogen) for 2 h with gentle shaking. The slurry was then poured into a column and the resin was washed with 25 ml of 10 mM imidazole in buffer L. The bound material was eluted with 10 ml of 500 mM imidazole in buffer L. The eluate was diluted 5-fold with buffer A (20 mM Tris–HCl, pH 7.5, 2 mM DTT, 0.5 mM EDTA, 10% glycerol) and the material was applied to a 5 ml HiTrap-Q column (Pharmacia LKB) that had been equilibrated with 100 mM NaCl in buffer A. The column was washed with 25 ml of buffer A and then developed with a 50 ml linear gradient of 100–600 mM NaCl in buffer A. The elution profile was monitored by SDS–PAGE. Mth ligase eluted at 400 mM NaCl. The peak fractions (comprising 70 mg of apparently homogeneous Mth ligase protein) were pooled and stored at –70°C.
RESULTS
Expression of Mth ligase in bacteria and demonstration of ligase activity
Methanobacterium thermoautotrophicum open reading frame 1580 encoding a ligase-like polypeptide (GenBank accession no. U51624) was PCR amplified from genomic DNA and cloned into a T7 RNA polymerase-based bacterial expression vector so as to fuse the 561 amino acid Mth ligase to a 20 amino acid N-terminal leader peptide containing 10 tandem histidines. The expression plasmid was introduced into E.coli BL21(DE3), a strain that contains the T7 RNA polymerase gene under the control of a lacUV5 promoter. A prominent 68 kDa polypeptide was detectable by SDS–PAGE in whole-cell extracts of IPTG-induced bacteria grown at either 17 or 37°C (not shown). This polypeptide was not present when bacteria containing the pET vector alone were induced with IPTG. After centrifugal separation of the crude lysate, the Mth protein was recovered in the soluble supernatant fraction. The His tag facilitated rapid purification of the recombinant Mth ligase by adsorption to an immobilized nickel resin and subsequent elution with buffer containing imidazole. SDS–PAGE analysis of the imidazole eluate fraction obtained from a small scale purification (from a 100 ml bacterial culture) showed that the preparation was essentially homogenous with respect to the 68 kDa Mth ligase polypeptide (Fig. 1A). The identity of the 68 kDa protein was confirmed by N-terminal sequence analysis (see below).
Figure 1.
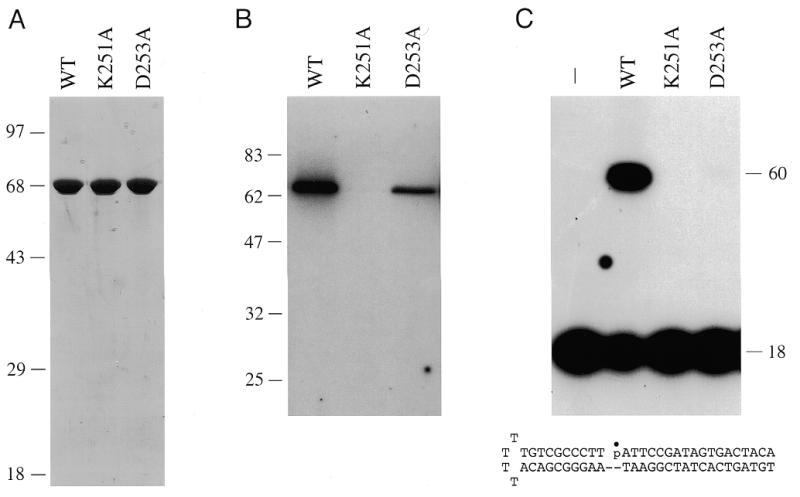
Purification and activity of wild-type Mth ligase and mutants K251A and D253A. (A) Purification. Aliquots (3 µg) of the nickel–agarose fractions were analyzed by SDS–PAGE. Polypeptides were visualized by staining the gel with Coomassie brilliant blue dye. A photograph of the stained gel is shown. The positions and sizes (in kDa) of marker proteins are indicated on the left. (B) Enzyme–adenylate formation. Reaction mixtures (20 µl) containing 50 mM Tris–HCl (pH 7.5), 5 mM DTT, 0.1 mM MgCl2, 40 µM [α-32P]ATP and 4 pmol wild-type (WT) Mth ligase, K251A or D253A were incubated for 15 min at 70°C. Reactions were quenched by adding SDS to 1%. The reaction products were resolved by SDS–PAGE. An autoradiogram of the dried gel is shown. The positions and sizes (in kDa) of marker proteins are indicated on the left. (C) DNA ligation. Reaction mixtures (20 µl) containing 50 mM Tris–HCl (pH 7.5), 5 mM DTT, 10 mM MgCl2, 1 mM ATP, 1 pmol 32P-labeled nicked hairpin DNA and 1 pmol wild-type Mth ligase, K251A or D253A were incubated for 10 min at 60°C. The reactions were quenched with formamide and EDTA. The reaction products were resolved by electrophoresis through a 12% polyacrylamide gel containing 7 M urea in TBE (90 mM Tris–borate, 2.5 mM EDTA). An autoradiogram of the gel is shown. The nicked duplex substrate used in the ligation reactions is illustrated at the bottom.
The initial step in DNA ligation involves formation of a covalent enzyme–adenylate intermediate. In order to assay adenylyl transferase activity of the recombinant Mth protein, we incubated the nickel–agarose protein preparation with [α-32P]ATP and magnesium. This resulted in the formation of an SDS-stable nucleotide–protein adduct that migrated as a single 68 kDa species during SDS–PAGE (Fig. 1B, WT). We conclude that the expressed Mth protein is active in nucleotidyl transfer.
We then assayed the ability of the recombinant Mth protein to seal a hairpin duplex DNA substrate containing a single nick. The structure of the substrate is shown in Figure 1C. ATP and magnesium were included in the assay mixtures. Ligase activity was evinced by conversion of the 5′-32P-labeled 18mer strand to an internally labeled 60mer product (Fig. 1C). These results demonstrate that the Mth protein is indeed a DNA ligase.
Effects of alanine mutations in motif I of Mth ligase
The KxDG sequence in motif I is the signature feature of a superfamily of nucleotidyl transferases that act through a covalent lysyl-NMP intermediate. This family includes the ATP-dependent DNA ligases, ATP-dependent RNA ligases, GTP-dependent mRNA capping enzymes and NAD+-dependent DNA ligases (4,6,12–17,21–25). The contributions of motif I residues Lys251 and Asp253 to the activity of Mth ligase were surmised from the effects of single alanine substitutions. Mutant proteins K251A and D253A were produced in bacteria and purified from soluble lysates by nickel affinity chromatography (Fig. 1A). The K251A mutant was inert in enzyme–adenylate formation (Fig. 1B) and in nick-joining (Fig. 1C). These findings are consistent with Lys251 being the site of covalent adenylation. The D253A protein did react with ATP to form the covalent intermediate (Fig. 1B), but was unable to catalyze the full ligation reaction (Fig. 1C). These results suggest that Asp253 is required for step 2 of the ligase reaction. Similar mutational effects at the motif I lysine and aspartate positions have been noted for the NAD+-dependent Thermus thermophilus and E.coli DNA ligases, the ATP-dependent Chlorella virus DNA ligase and T4 RNA ligase (4,17,21,25). Thus, the archaeal DNA ligase is likely to employ the same constellation of catalytic residues as other polynucleotide ligases.
Sedimentation analysis
Large scale purification of recombinant wild-type Mth ligase from 12 l of IPTG-induced bacteria was achieved by nickel affinity chromatography followed by HiTrap-Q anion exchange chromatography. The HiTrap-Q column preparation (70 mg), which was homogeneous with respect to Mth ligase (not shown), was employed for subsequent characterization of the wild-type enzyme. The native size of the Mth ligase was gauged by zonal velocity sedimentation through a 15–30% glycerol gradient containing 0.2 M NaCl. The gradient fractions were assayed for ligase–AMP complex formation and nick-joining. A single activity peak of enzyme–AMP formation coincided exactly with the ligation activity profile (Fig. 2) and with the sedimentation profile of the Mth ligase polypeptide (not shown). We estimated a sedimentation coefficient of 3.8 S relative to marker proteins bovine serum albumin and cytochrome c that were centrifuged in a parallel gradient (Fig. 2). These findings suggest that the Mth ligase is a monomer.
Figure 2.

Glycerol gradient sedimentation. An aliquot (0.2 ml, 160 µg) of the HiTrap-Q column preparation of Mth ligase was sedimented in a 4.8 ml 15–30% glycerol gradient containing 50 mM Tris–HCl (pH 8.0), 1 mM EDTA, 2 mM DTT, 0.1% Triton X-100 and 200 mM NaCl. The gradient was centrifuged for 22 h at 50 000 r.p.m. in a Beckman SW50 rotor. Fractions (~0.16 ml) were collected from the bottom of the tube. Aliquots of the odd numbered fractions were assayed for DNA nick-joining and ligase adenylation activities. Nick-joining reaction mixtures (20 µl) containing 50 mM Tris–HCl (pH 7.5), 5 mM DTT, 10 mM MgCl2, 1 mM ATP, 1 pmol 32P-labeled nicked DNA and 1/16 µl (4 µl of a 1:63 dilution) of the gradient fractions were incubated for 10 min at 60°C. The reaction products were analyzed by PAGE. The extent of ligation [60mer/(60mer + 18mer)] was determined by scanning the gel with a phosphorimager. Adenylyl transferase reaction mixtures (20 µl) containing 50 mM Tris–HCl (pH 7.5), 5 mM DTT, 10 mM MgCl2, 5 µM [α-32P]ATP and 2 µl of the gradient fractions were incubated for 5 min at 70°C. The reaction products were analyzed by SDS–PAGE. The gel was scanned with a phosphorimager and ligase–adenylate formation was gauged by the signal intensity (PSL) of the radiolabeled ligase polypeptide.
Mth ligase activity is thermophilic in vitro
The extent of nick-joining by Mth ligase was proportional to the amount of input enzyme at all temperatures tested in the range 22–80°C (Fig. 3A). The specific activity was extremely sensitive to reaction temperature. Ligase activity peaked at 60°C (Fig. 3A and B), which is near the optimal in vivo growth temperature for M.thermoautotrophicum (11). Nick-joining activity declined progressively as the temperature was lowered to 22°C (Fig. 3A and B). Apparent activity also declined when the temperature was increased to 70 and 80°C (Fig. 3A and B). It is possible that Mth ligase activity at >60°C was underestimated because the nicked DNA substrate, in which the radiolabeled strand was tethered to the template strand by only 18 bp, inevitably denatures at such elevated reaction temperatures. The temperature optimum of Mth ligase is consistent with the thermophilic behavior of other Mth replication enzymes, including Mth DNA polymerase (optimally active at 60–70°C) and Mth replication factor C (active at 70°C) (26,27).
Figure 3.

Temperature dependence and kinetics of DNA ligation. (A) Reaction mixtures contained 50 mM Tris–HCl (pH 7.5), 5 mM DTT, 10 mM MgCl2, 1 mM ATP, 1 pmol radiolabeled nicked hairpin DNA and the indicated amounts of ligase. Reaction mixtures were incubated at the indicated temperatures for 10 min. The yield of 60mer ligation product at each temperature is plotted as a function of input ligase. (B) Specific activity at each temperature was calculated from the slope of the curve in the linear range of enzyme dependence and then plotted as a function of temperature. (C) Reaction mixtures containing (per 20 µl) 50 mM Tris–HCl (pH 8.0), 5 mM DTT, 10 mM MgCl2, 1 mM ATP, 1 pmol nicked DNA and the indicated amounts of ligase were incubated at 60°C. The reactions were initiated by addition of enzyme. Aliquots (20 µl) were withdrawn at the times indicated and quenched immediately with EDTA and formamide. The time 0 sample was taken before adding enzyme. The extent of ligation is plotted as a function of reaction time.
Kinetics of ligation
The rate of nick-joining at 60°C was proportional to the level of input enzyme. The reaction reached an end point at which ~85% of the labeled donor strand was converted to 60mer (Fig. 3C). This upper limit of ligation probably reflected incomplete annealing of the component strands to form the nicked substrate.
A low level of ligation was detected in the absence of added ATP (Fig. 5A) that was attributed to pre-adenylated Mth ligase in the enzyme preparation. Nick-joining activity was stimulated by inclusion of 1 mM ATP in the reaction mixture (Fig. 4A). GTP, UTP and CTP were inactive at 1 mM concentration, whereas 1 mM dATP was partially active (Fig. 4A). NAD+ (1 mM) was inactive (data not shown). Ligase activity increased with ATP concentration up to 1.2 µM and leveled off at 2–5 µM (Fig. 4B). A Km of 1.1 µM ATP was calculated from a double reciprocal plot of the data (not shown). The apparent Km for dATP was 1.6 µM (not shown).
Figure 5.

Effect of pH and salt on nick-joining activity. (A) Effect of pH. Reaction mixtures containing 5 mM DTT, 10 mM MgCl2, 1 mM ATP, 1 pmol nicked DNA, 100 fmol ligase and either 50 mM Tris–HCl (pH 7.0, 7.5, 8.0, 8.5 or 8.8) or 50 mM sodium cacodylate (pH 6.5) were incubated for 10 min at 60°C. Extent of ligation is plotted as a function of pH of the buffer (determined at 22°C). (B) Reaction mixtures containing 50 mM Tris–HCl (pH 7.5), 5 mM DTT, 10 mM MgCl2, 1 mM ATP, 0.5 pmol nicked DNA, 125 fmol ligase and NaCl as indicated were incubated for 10 min at 60°C. Extent of ligation is plotted as a function of salt concentration.
Figure 4.

Nucleotide cofactor specificity and ATP concentration dependence. (A) Nucleotide specificity. Reaction mixtures (20 µl) containing 50 mM Tris–HCl (pH 7.5), 5 mM DTT, 10 mM MgCl2, 1 pmol nicked DNA, 100 fmol ligase and 1 mM NTP or dNTP as indicated were incubated for 10 min at 60°C. Nucleotide was omitted from a control reaction (none). (B) ATP concentration dependence. Reaction mixtures (20 µl) containing 50 mM Tris–HCl (pH 7.5), 5 mM DTT, 10 mM MgCl2, 1 pmol nicked hairpin DNA, 100 fmol ligase and ATP as indicated were incubated for 10 min at 60°C. The extent of ligation is plotted as a function of ATP concentration.
Effect of pH and salt on ligation
Mth ligase was optimally active between pH 7.5 and 8.8 (Fig. 5A). Higher pH conditions were not tested. Activity declined progressively at pH 7.0 and 6.5. The ligase was extremely sensitive to inhibition by added salt. The standard reaction mixture contained 10 mM NaCl contributed by the DNA substrate solution. Increasing the NaCl concentration to 50 mM reduced activity by a factor of 4, whereas 80 mM NaCl lowered activity by a factor of 10 (Fig. 5B). Activity was reduced 70% in the presence of 50 mM KCl and by 60% in the presence of 10 mM (NH4)2SO4 (not shown). Similar extreme sensitivity to inhibition by monovalent ions was reported for the yeast DNA ligase Cdc9p (28).
Divalent cation dependence and specificity
Nick ligation required an exogenous divalent cation cofactor (Fig. 6A). This requirement could be satisfied by 10 mM magnesium, manganese or cobalt, but not by 10 mM calcium, copper or zinc (Fig. 6A). We noted that reaction of Mth ligase with nicked DNA in the presence of calcium led to an accumulation of the normally undetectable DNA–adenylate intermediate. Trapping of DNA–adenylate was especially pronounced in 2 mM calcium under conditions of ligase excess over input DNA (Fig. 6B). Mth ligase converted >80% of the 5′-32P-labeled 18mer strand into an adenylated species (AppDNA) that migrated ~1 nt slower than the input 18mer during polyacrylamide gel electrophoresis. Only trace levels of ligated product were formed. We conclude that calcium elicits a specific step 3 block in phosphodiester bond formation.
Figure 6.
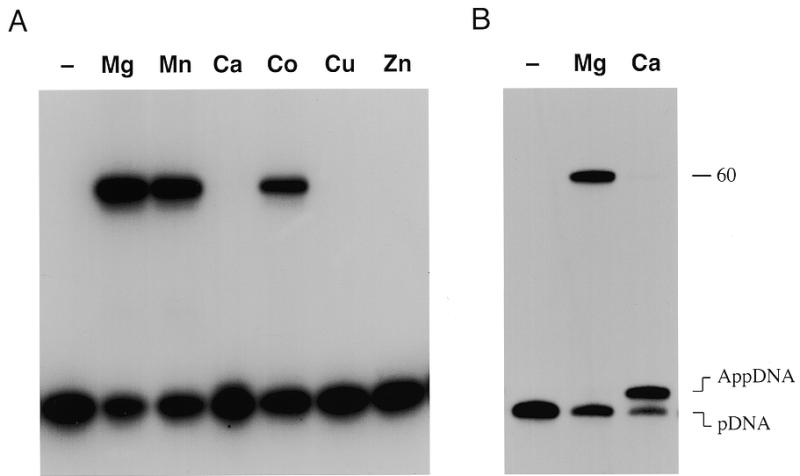
Divalent cation specificity. (A) Reaction mixtures (20 µl) containing 50 mM Tris–HCl (pH 7.5), 1 mM ATP, 1 pmol nicked DNA, 1 pmol ligase and 10 mM indicated divalent cation were incubated for 30 min at 60°C. Magnesium, manganese, calcium and cobalt were added as the chloride salts; copper and zinc were added as sulfates. (B) Reaction mixtures (20 µl) containing 50 mM Tris–HCl (pH 7.5), 1 mM ATP, 1 pmol nicked DNA, 4 pmol ligase and 2 mM MgCl2 or CaCl2 were incubated for 20 min at 60°C. Divalent cation was omitted from control reactions (–). The products were resolved by denaturing PAGE. Autoradiographs of the gels are shown. The positions of the input 5′-monophosphate 18mer strand (pDNA), the adenylated DNA strand (AppDNA) and the 60mer ligation product are indicated on the right.
Phosphodiester bond formation at a pre-adenylated nick
Step 3 of the Mth ligation reaction was assayed by incubating the enzyme with a pre-adenylated nicked DNA substrate (Fig. 7). The adenylated strand used to form this substrate was synthesized as described (17,29) by ligase-mediated AMP transfer to the 5′-32P-labeled strand of a DNA molecule containing a 1 nt gap. The radiolabeled AppDNA strand was purified by denaturing PAGE and then annealed to an unlabeled 42 nt hairpin strand to form the structure shown in Figure 7. This substrate was reacted with excess Mth ligase in the presence of magnesium without added ATP. The wild-type Mth ligase generated a 60mer ligation product (Fig. 7A). The active site mutant K251A also catalyzed the strand-joining step at a pre-adenylated nick. Indeed, more ligated product was formed by K251A than by wild-type ligase (Fig. 7A). We conclude that the lysine nucleophile, which is essential for ligase–adenylate formation, is not essential for phosphodiester bond formation. Similar results were noted for the equivalent motif I lysine mutants of vaccinia and Chlorella virus DNA ligases (17,29).
Figure 7.
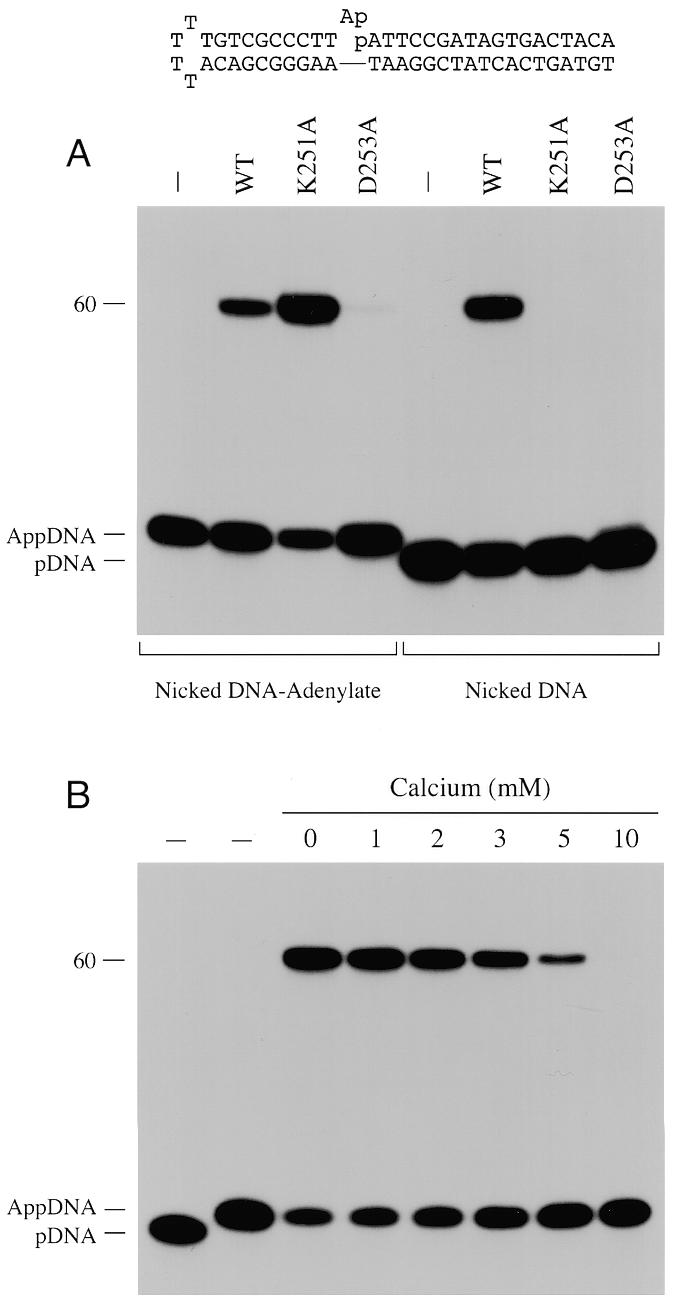
Ligation at a pre-adenylated nick. (A) Reaction mixtures (20 µl) containing 50 mM Tris–HCl (pH 7.5), 5 mM DTT, 10 mM MgCl2, 100 fmol 32P-labeled nicked DNA–adenylate (illustrated at the top of the figure) or 32P-labeled nicked DNA (see Fig. 1) and 2 pmol wild-type Mth ligase (WT), K251A or D253A were incubated for 30 min at 60°C. (B) Reaction mixtures (20 µl) containing 50 mM Tris–HCl (pH 7.5), 200 fmol 32P-labeled nicked DNA–adenylate, 2 pmol K251A ligase, 10 mM MgCl2 and either 0, 1, 2, 3, 5 or 10 mM CaCl2 were incubated for 30 min at 60°C. The reaction products were resolved by denaturing PAGE. Autoradiographs of the gels are shown. Control reaction mixtures lacking ligase are shown in lanes –. The positions of the 5′-32P-labeled 18mer strand (pDNA), the adenylated DNA strand (AppDNA) and the 60mer ligation product are indicated on the left.
Phosphodiester formation on the nicked DNA–adenylate substrate by K251A absolutely required a divalent cation cofactor, which could be magnesium, manganese or cobalt (not shown). Calcium, which elicited an apparent step 3 block in the overall ligation pathway (Fig. 6), failed to support phosphodiester synthesis at the pre-adenylated nick (not shown). The finding that CaCl2 inhibited the ligation reaction of K251A at a pre-adenylated nick in the presence of 10 mM MgCl2 (Fig. 7B) provided evidence that calcium competes with magnesium for an essential metal-binding site on the ligase–(DNA–adenylate) complex. Ligation in 10 mM MgCl2 was reduced by 75% in the presence of 5 mM CaCl2 and by 98% in 10 mM CaCl2 (Fig. 7B). Thus, calcium bound the metal site at least as avidly as magnesium, but did not support step 3 chemistry.
The D253A mutant promoted only trace levels of sealing of the pre-adenylated nicked DNA (Fig. 7A). A parallel reaction of excess D253A with standard nicked DNA resulted in no ligated product, but trace amounts of DNA–adenylate were formed (Fig. 7A). We infer a role for the motif I aspartate side chain in steps 2 and 3 of the Mth ligase reaction.
Structure probing of Mth ligase by limited proteolysis
Recombinant His-tagged Mth ligase was subjected to proteolysis with increasing amounts of trypsin, chymotrypsin and V8 proteases. N-terminal sequencing of the undigested Mth ligase polypeptide by automated Edman chemistry after transfer from an SDS gel to a PVDF membrane confirmed that the N-terminal sequence (GHHHHH) corresponded to that of the recombinant gene product starting from the second residue of the His tag (Fig. 8). Apparently, the ligase suffered removal of the initiating methionine during expression in E.coli. Initial scission of the 68 kDa Mth ligase by trypsin yielded two major products, an ~40 kDa doublet and a 28 kDa species (Fig. 8A). The two components of the 40 kDa doublet had the same N-terminal sequence (HMKELL) arising via trypsin cleavage at an arginine located at the junction between the His tag and the N-terminus of the native Mth protein. The 28 kDa product consisted of a predominant species with N-terminus LTREVP indicative of cleavage at the Arg328/Leu329 peptide bond. A minor component of the 28 kDa band with N-terminal sequence EVPLSL arose via scission at Arg331/Glu332. These sites are denoted by arrows above the polypeptide sequence in Figure 8D. Thus, trypsin cleaved at two tightly clustered sites between motifs III and IIIa, splitting the ligase into a 40 kDa N-terminal fragment and a 28 kDa C-terminal fragment. We surmise that these fragments are themselves tightly folded insofar as they were resistant to digestion by concentrations of trypsin sufficient to cleave all the input ligase.
Figure 8.

Limited proteolysis of Mth DNA ligase. Proteolysis reaction mixtures (20 µl) containing 6 µg of purified Mth ligase and increasing amounts of trypsin (80, 160 or 360 ng) (A), chymotrypsin (100, 200 or 400 ng) (B) or V8 protease (1 or 2 µg) (C) were incubated at 22°C for 15 min. The samples were denatured in SDS and the proteolysis products were resolved by SDS–PAGE. Polypeptides were transferred to a PVDF membrane (Bio-Rad), which was stained with Coomassie brilliant blue and destained as described (31). The membrane was air dried and photographed. The positions and sizes (kDa) of marker proteins are indicated on the left. Slices containing individual proteolytic products were excised. Automated sequencing of the immobilized polypeptide was performed using an Applied Biosystems model 477A microsequencer. The N-terminal sequences are denoted in single letter code. (D) The sites of proteolysis by trypsin and V8 protease are denoted by arrows above the amino acid sequence of Mth ligase. Chymotrypsin cleavage sites are denoted by arrows below the sequence. Conserved motifs I, III, IIIa and IV are highlighted in boxes. Motif I residues Lys251 and Asp253 that were subjected to alanine substitution are denoted by dots.
Treatment of Mth ligase with limiting concentrations of chymotrypsin yielded an ~53 kDa polypeptide, a 28 kDa polypeptide and a ~15 kDa doublet (Fig. 8B). The 53 kDa species, with N-terminus FSQPLT, arose via chymotyptic cleavage between Phe109 and Phe110. The 15 kDa species, which retained the His tag at its N-terminus, presumably constituted the proximal product of the cleavage event at Phe109. The 29 kDa species, with N-terminal sequence DVERLT, was the product of chymotryptic cleavage at Tyr324/Asp325. The 29 kDa fragment became more abundant as chymotrypsin was increased and it remained resistant to digestion by concentrations of chymotrypsin in excess of that sufficient to cleave all the input ligase. In contrast, the 53 kDa species decayed at the higher protease concentrations, giving rise to an ~25 kDa fragment with the same N-terminus at Phe110 (Fig. 8B). We infer that this pattern reflected secondary cleavage of the 53 kDa species at Tyr324/Asp325. Note that the chymotryptic site at Tyr324 is located very close to the tryptic sites at Arg328 and Arg331 (Fig. 8D). Relatively high levels of V8 protease were required to digest Mth ligase (Fig. 8C). The predominant 30 kDa digestion product, with N-terminal sequence GEIIVT, arose via scission of the peptide bond between Glu299 and Gly300. This V8 cleavage site is situated within motif III, just upstream of the clustered sites of trypsin and chymotrypsin cleavage (Fig. 8D). The results of the proteolytic analysis suggest that Mth ligase is a compact protein punctuated by a surface-accessible loop between motifs III and IIIa.
DISCUSSION
We report the biochemical characterization of Mth DNA ligase, the first such study of a DNA ligase from an archaeon. As predicted from sequence comparisons, Mth ligase resembles eukaryotic DNA ligases in its specificity for ATP as the energy cofactor. NAD+, the cofactor for classical eubacterial ligases, did not support strand-joining. The archaeal ligases are smaller than the ATP-dependent ligases of fungi and metazoa (755–1070 amino acids), similar in size to poxvirus ligases (552–564 amino acids) and larger than the virus-encoded ligases of the T bacteriophages (346–487 amino acids) and the ‘minimal’ eukaryotic ligase of Chlorella virus PBCV-1 (298 amino acids). The overall size heterogeneity of the ATP-dependent ligases is principally attributable to the variable N-terminal segments flanking the catalytic core.
Sedimentation analysis of the recombinant Mth ligase suggests that it is a monomeric protein. The native and recombinant versions of E.coli DNA ligase (30), T7 DNA ligase (31), yeast Cdc9p (28), vaccinia DNA ligase (16) and Chlorella virus DNA ligase (32) are also monomeric. Human DNA ligase I undergoes reversible self-association in vitro to form a homotrimer (33).
Limited proteolysis of the Mth ligase suggests that the enzyme is composed of tightly folded protease-resistant segments separated by a protease-sensitive linker region located between conserved motifs III and IIIa. Proteolytic probing of the structures of vaccinia and T7 DNA ligases yielded similar findings, i.e. that the sites of protease accessibility in the vaccinia and T7 ligases were located between motifs III and IIIa (34,35). In the crystal structure of T7 DNA ligase, this polypeptide segment is a disordered loop that extends from the surface of an otherwise tightly folded protein (13). The flexibility and surface exposure would account for the observed protease susceptibility. The observation that the same segment of the Mth DNA ligase is protease sensitive suggests that it is also exposed on the surface.
It is instructive to compare the enzymatic properties of Mth ligase with those of other well-studied DNA ligases. First, the striking feature of Mth ligase is its activity at elevated temperatures. Mth ligase requires high temperatures for nick-joining and the optimal temperature for ligation in vitro agrees well with the favored in vivo growth temperatures for M.thermoautotrophicum. The nick-joining activity of recombinant Mth ligase under multiple turnover conditions at its optimal temperature of 60°C (~0.8 pmol nicks sealed/min/pmol ligase) was low compared to the activities of Chlorella virus ligase (10 min–1) and vaccinia ligase (4 min–1) at 22°C (32,36). Mth ligase specific activity was more in line with that of the thermophilic NAD+-dependent Aquifex aeolicus ligase (2 min–1) measured at 65°C (37).
The spectrum of divalent cations that supported nick-joining by Mth ligase (magnesium, manganese and cobalt) was identical to that reported for Chlorella virus ligase (32). Mth ligase displayed a strong step 3 arrest phenotype in the presence of calcium that resulted in extensive accumulation of the DNA–adenylate intermediate. Calcium caused an accumulation of DNA–adenylate in reactions catalyzed by the thermophilic NAD+-dependent eubacterial ligases from Thermus and Aquifex (5,37).
Mth ligase required a divalent cation to catalyze phosphodiester bond formation at a pre-adenylated nick. This contrasts with the report of Doherty et al. (27) that T7 DNA ligase–adenylate catalyzed steps 2 and 3 in the absence of added divalent cation and in the presence of 10 mM EDTA. Catalysis of step 3 by Mth ligase was promoted by magnesium, manganese or cobalt, but not by calcium. Similarly, vaccinia ligase–adenylate displayed an absolute requirement for a divalent cation for steps 2 plus 3 and this requirement was met by magnesium, manganese or cobalt (36). Of note, the reaction of vaccinia ligase–adenylate with nicked DNA in the presence of calcium resulted in accumulation of DNA–adenylate without phosphodiester formation (S.Shuman, unpublished data). Human DNA ligase I also requires magnesium for phosphodiester formation at a pre-adenylated nick; effects of manganese, cobalt or calcium on the human ligase were not reported (38). The contradictory findings regarding metal dependence for the T7 enzyme versus other ligases force one of the following interpretations: (i) members of the ATP-dependent ligase family have evolved two fundamentally different mechanisms for catalysis of the chemical steps subsequent to ligase–adenylate formation or (ii) the T7 ligase–adenylate complex uniquely contains a tightly bound metal that is resistant to dissociation by EDTA.
The selective effects of mutations in motif I of Mth ligase on individual steps of the ligation pathway confirm and extend earlier mutational analyses of DNA ligases. We find that the motif I lysine is essential for step 1 (ligase–adenylate formation), but not for step 3 (phosphodiester formation) (17,18,29). In contrast, the motif I aspartate is not needed for ligase–adenylate formation, but is needed to facilitate attack of the 5′-phosphate at the nick on ligase–AMP to form DNA–adenylate and subsequently to promote strand sealing (4,17,39). The available crystal structure of T7 DNA ligase with bound ATP does not provide an explanation for the essential catalytic role of the motif I aspartate. (Indeed, it has not yet been shown that the motif I aspartate is required for T7 ligase activity.) Answering outstanding questions concerning catalysis of steps 2 and 3 will require co-crystallization of a ligase bound to nicked DNA and nicked DNA–adenylate. The use of step-arrest ligase mutations such as the motif I Asp→Ala, together with DNA substrate modifications and solution conditions (e.g. calcium) that block specific chemical steps, may facilitate the crystallographic trapping of different functional and conformational states of ligase during its catalytic cycle.
REFERENCES
- 1.Lehman I.R. (1974) Science, 186, 790–797. [DOI] [PubMed]
- 2.Engler M.J. and Richardson,C.C. (1982) Enzymes, 15, 3–29.
- 3.Luo J., Bergstrom,D.E. and Barany,F. (1996) Nucleic Acids Res., 24, 3071–3078. [DOI] [PMC free article] [PubMed]
- 4.Luo J. and Barany,F. (1996) Nucleic Acids Res., 24, 3079–3085. [DOI] [PMC free article] [PubMed]
- 5.Tong J., Cao,W. and Barany,F. (1999) Nucleic Acids Res., 27, 788–794. [DOI] [PMC free article] [PubMed]
- 6.Singleton M.R., Håkansson,K., Timson,D.J. and Wigley,D.B. (1999) Structure, 7, 35–42. [DOI] [PubMed]
- 7.Timson D. and Wigley,D.B. (1999) J. Mol. Biol., 285, 73–83. [DOI] [PubMed]
- 8.Barany F. (1991) Proc. Natl Acad. Sci. USA, 88, 189–193. [DOI] [PMC free article] [PubMed]
- 9.Gerry N.P., Witowski,N.E., Day,J., Hammer,R.P., Barany,G. and Barany,F. (1999) J. Mol. Biol., 292, 251–262. [DOI] [PubMed]
- 10.Kletzin A. (1992) Nucleic Acids Res., 20, 5389–5396. [DOI] [PMC free article] [PubMed]
- 11.Smith D.R., Doucette-Stamm,L.A., Deloughery,C., Lee,H., Dubois,J., Aldredge,T., Bashirzadeh,R., Blakely,D., Cook,R., Gilbert,K. et al. (1997) J. Bacteriol., 179, 7135–7155. [DOI] [PMC free article] [PubMed]
- 12.Shuman S. and Schwer,B. (1995) Mol. Microbiol., 17, 405–410. [DOI] [PubMed]
- 13.Subramanya H.S., Doherty,A.J., Ashford,S.R. and Wigley,D.B. (1996) Cell, 85, 607–615. [DOI] [PubMed]
- 14.Tomkinson A.E., Totty,N.F., Ginsburg,M. and Lindahl,T. (1991) Proc. Natl Acad. Sci. USA, 88, 400–404. [DOI] [PMC free article] [PubMed]
- 15.Kodama,K., Barnes,D.E. and Lindahl,T. (1991) Nucleic Acids Res., 19, 6093–6099. [DOI] [PMC free article] [PubMed]
- 16.Shuman S. and Ru,X. (1995) Virology, 211, 73–83. [DOI] [PubMed]
- 17.Sriskanda V. and Shuman,S. (1998) Nucleic Acids Res., 26, 525–531. [DOI] [PMC free article] [PubMed]
- 18.Sriskanda V. and Shuman,S. (1998) Nucleic Acids Res., 26, 4618–4625. [DOI] [PMC free article] [PubMed]
- 19.Mackey Z.B., Niedergang,C., Menissier-de Murcia,J., Leppard,J., Au,K., Chen,J., de Murcia,G. and Tomkinson,A.E. (1999) J. Biol. Chem., 274, 21679–21687. [DOI] [PubMed]
- 20.Ho S.N., Hunt,H.D., Horton,R.M., Pullen,J.K. and Pease,L.R. (1989) Gene, 77, 51–59. [DOI] [PubMed]
- 21.Heaphy S., Singh,M. and Gait,M.J. (1987) Biochemistry, 26, 1688–1696. [DOI] [PubMed]
- 22.Cong P. and Shuman,S. (1993) J. Biol. Chem., 268, 7256–7260. [PubMed]
- 23.Shuman S., Liu,Y. and Schwer,B. (1994) Proc. Natl Acad. Sci. USA, 91, 12046–12050. [DOI] [PMC free article] [PubMed]
- 24.Håkansson K., Doherty,A.J., Shuman,S. and Wigley,D.B. (1997) Cell, 89, 545–553. [DOI] [PubMed]
- 25.Sriskanda V., Schwer,B., Ho,C.K. and Shuman,S. (1999) Nucleic Acids Res., 27, 3953–3963. [DOI] [PMC free article] [PubMed]
- 26.Kelman Z., Pietrokovsky,S. and Hurwitz,J. (1999) J. Biol. Chem., 274, 28751–28761. [DOI] [PubMed]
- 27.Kelman Z. and Hurwitz,J. (2000) J. Biol. Chem., 275, 7327–7336. [DOI] [PubMed]
- 28.Tomkinson A.E., Tappe,N.J. and Friedberg,E.C. (1992) Biochemistry, 31, 11762–11771. [DOI] [PubMed]
- 29.Sekiguchi J. and Shuman,S. (1997) J. Virol., 71, 9697–9684. [DOI] [PMC free article] [PubMed]
- 30.Modrich P., Ankaru,Y. and Lehman,I.R. (1973) J. Biol. Chem., 248, 7495–7501. [PubMed]
- 31.Doherty A.J., Ashford,S.R., Subramanya,H.S. and Wigley,D.B. (1996) J. Biol. Chem., 271, 11083–11089. [DOI] [PubMed]
- 32.Ho C.K., Van Etten,J.L. and Shuman,S. (1997) J. Virol., 71, 1931–1937. [DOI] [PMC free article] [PubMed]
- 33.Dimitriadis E.K., Prasad,R., Vaske,M.K., Chen,L., Tomkinson,A.E., Lewis,M.S. and Wilson,S.H. (1998) J. Biol. Chem., 273, 20540–20550. [DOI] [PubMed]
- 34.Sekiguchi J. and Shuman,S. (1997) Nucleic Acids Res., 25, 727–734. [DOI] [PMC free article] [PubMed]
- 35.Doherty A.J., Ashford,S.R. and Wigley,D.B. (1996) Nucleic Acids Res., 24, 2282–2287.
- 36.Shuman S. (1995) Biochemistry, 34, 16138–16147. [DOI] [PubMed]
- 37.Tong J., Barany,F. and Cao,W. (2000) Nucleic Acids Res., 28, 1447–1454. [DOI] [PMC free article] [PubMed]
- 38.Yang S.W. and Chan,J.Y.H. (1992) J. Biol. Chem., 267, 8117–8122. [PubMed]
- 39.Odell M. and Shuman,S. (1999) J. Biol. Chem., 274, 14032–14039. [DOI] [PubMed]