


Abstract
HAP1, also known as APE/Ref-1, is the major apurinic/apyrimidinic (AP) endonuclease in human cells. Previous structural studies have suggested a possible role for the Asp-210 residue of HAP1 in the enzymatic function of this enzyme. Here, we demonstrate that substitution of Asp-210 by Asn or Ala eliminates the AP endonuclease activity of HAP1, while substitution by Glu reduces specific activity ~500-fold. Nevertheless, these mutant proteins still bind efficiently to oligonucleotides containing either AP sites or the chemically unrelated bulky p-benzoquinone (pBQ) derivatives of dC, dA and dG, all of which are substrates for HAP1. These results indicate that Asp-210 is required for catalysis, but not substrate recognition, consistent with enzyme kinetic data indicating that the HAP1–D210E protein has a 3000-fold reduced Kcat for AP site cleavage, but an unchanged Km. Through analysis of the binding of Asp-210 substitution mutants to oligonucleotides containing either an AP site or a pBQ adduct, we conclude that the absence of Asp-210 allows the formation of a stable HAP1–substrate complex that exists only transiently during the catalytic cycle of wild-type HAP1 protein. We interpret these data in the context of the structure of the HAP1 active site and the recently determined co-crystal structure of HAP1 bound to DNA substrates.
INTRODUCTION
Apurinic/apyrimidinic (AP) sites are one of the major lesions to arise in cellular DNA following exposure to ionising radiation or chemical oxidants (reviewed in 1–5). These sites are also generated during the DNA base excision repair (BER) process in which modified DNA bases are excised by DNA glycosylases. Moreover, AP sites arise at a high frequency ‘spontaneously’ as a result of the inherent lability of the N-glycosyl bond to hydrolytic scission. The biological importance of AP sites is emphasised by the fact that these lesions are not only abundant (~104/cell/day), but also potentially both cytotoxic and mutagenic (1–5). The major enzyme in human cells for repair of AP sites is HAP1 (also known as APE/Ref-1) (6,7; reviewed in 8–10). This enzyme catalyses the hydrolytic cleavage of the phosphodiester bond immediately 5′ to the AP site and hence initiates AP site repair. Although AP sites are probably the primary lesions recognised by the HAP1 protein, this enzyme nevertheless has a broad substrate range. Additional substrates include strand breaks with 3′ phosphoglycolate or 3′ phosphate termini, and the RNA strand of RNA–DNA hybrids (11–17). Moreover, HAP1 displays endonuclease activity towards DNA containing the bulky, p-benzoquinone (pBQ)-derived benzetheno exocyclic adducts pBQ-dC, pBQ-dA and pBQ-dG (18,19). A single catalytic active site is used for all of these activities (19). In addition to functions involved in DNA metabolism, HAP1 displays an apparently unrelated activity through which it regulates the DNA binding ability of a series of transcription factors, including c-Jun, c-Fos and p53 (20). The second function of HAP1 regulates the redox-state of a cysteine residue in the target factor, and is mediated by a domain of HAP1 dispensable for DNA repair function (21,22).
HAP1 is a functional homologue of exonuclease III, the major AP endonuclease in Escherichia coli as evidenced by the fact that expression of the HAP1 cDNA in xth mutants lacking exonuclease III confers some degree of complementation (6,7). Consistent with this, previous X-ray crystallographic studies have shown that HAP1 shares a structural fold with exonuclease III (23,24). Other structural and functional homologues of exonuclease III have been identified in other species, including lower eukaryotes (8–10). More surprisingly, HAP1 and bovine DNase I (a non-specific nuclease) show structural similarity, despite a lack of primary sequence similarity between these proteins (24). A number of amino acid residues are conserved in all members of the exonuclease III family of AP endonucleases. Many of these residues have been shown to play roles in catalysis and/or substrate recognition through analysis of mutant HAP1 derivatives containing single amino acid substitutions (11,12,25–27). These analyses have revealed a key role for His-309, which is proposed to be involved in the hydrolysis reaction (11). This reaction is probably assisted by a number of other residues, including Asp-283 and Asp-308 (11,12,24–27). Moreover, the single divalent cation in the HAP1 active site, which is bound by the carboxylate side chain of Glu-96, is important for catalytic function, and chelation of this co-factor by EDTA drastically reduces the rate of phosphodiester bond cleavage (11,24). This catalytic inhibition by EDTA has been used previously, by ourselves and others, to stabilise (and thus reveal experimentally using band-shift assays) the complex of HAP1 bound to oligonucleotides containing AP sites (25–29). Hence, EDTA acts to dramatically slow the enzyme off-rate by inhibiting phosphodiester bond cleavage, the proposed trigger for enzyme dissociation.
One residue that is conserved in all members of the exonuclease III family, and which is known to be located in the HAP1 active site, is Asp-210 (24). The equivalent residue in E.coli exonuclease III, Asp-151, has been shown to interact with the 5′ phosphate group of a co-crystallised nucleotide (23). Moreover, recent data, derived from an analysis of the co-crystal structure of HAP1 bound to synthetic AP site-containing oligonucleotides (30), showed that the side chain of Asp-210 is aligned appropriately with the scissile P-O3′ bond. Based on these data, it was proposed that the protonation of Asp-210 generates the attacking hydroxyl nucleophile. Hence, in the absence of this residue, it is predicted that HAP1 would be catalytically inactive. To analyse the biochemical function of the Asp-210 residue of HAP1, we have generated mutant HAP1 derivatives containing substitutions at this site. We show that Asp-210 is essential for catalysis of phosphodiester bond cleavage, but is dispensable for substrate recognition by the HAP1 protein.
MATERIALS AND METHODS
Materials
All chemicals used were analytical grade. The [γ-32P]ATP (specific activity 6000 Ci/mmol) was purchased from Amersham (Buckinghamshire, UK). T4 polynucleotide kinase was purchased from USB (OH). OPC cartridges were from Applied Biosystems (CA). The phosphoramidite of 1′,2′-deoxyribose (dSpacer) was purchased from Glen Research (Sterling, VA).
Site-directed mutagenesis
Mutagenesis was performed by the PCR-based method of Landt et al. (31), as described previously. The PCR primers used for each mutant were as follows (altered codons in bold).
HAP1–D210E primer 5′-TGCCACATTGAGCTCTCCACACAGCAC-3′
HAP1–D210N primer 5′-TGCCACATTGAGGTTTCCACACAGCAC-3′
HAP1–D210A primer 5′-TGCCACATTGAGCGCTCCACACAGCAC-3′
The mutated cDNAs were cloned into the XhoI site of pET14b (Invitrogen, Groningen, The Netherlands) as described previously.
Purification of HAP1 and mutant HAP1 proteins
The wild-type and mutant pET-14b–HAP1 constructs were transformed into BL21.pLysS cells, an E.coli strain incorporating a chromosomally integrated T7 polymerase gene under control of the lac promoter. The cells were grown at 30°C in a shaking incubator until the culture reached an OD600 of 0.5. Expression from the T7 promoter of the HAP1 protein, which contained an N-terminal hexahistidine tag, was induced for 2 h by addition of 0.4 mM IPTG (final concentration). The cells were then harvested, lysed by sonication and the cell debris was pelleted by ultracentrifugation (39 000 r.p.m., 4°C for 20 min in a Beckman 45 Ti rotor). The supernatant was then loaded onto a phosphocellulose P11 column and the bound proteins were eluted with a 100–1250 mM NaCl gradient. The fractions containing the HAP1 protein (eluting at ~600 mM NaCl) were pooled and extensively dialysed against binding buffer (5 mM imidazole, 500 mM NaCl, 20 mM Tris–HCl, pH 7.9).
The dialysed protein was loaded onto a Ni2+ charged His-Bind resin column (Novagen, WI) and the column washed with 10 vol binding buffer, followed by 6 vol wash buffer (60 mM imidazole, 500 mM NaCl, 20 mM Tris–HCl, pH 7.9). Bound proteins were eluted with 2 vol elution buffer (1 M imidazole, 500 mM NaCl, 20 mM Tris–HCl, pH 7.9). The purified proteins were stored at –70°C in storage buffer (50% glycerol, 235 mM NaCl, 20 mM Tris–HCl, pH 7.9). All proteins were >95% pure as judged by electrophoresis on 12% SDS–polyacrylamide gels and staining with Coomassie blue.
Oligonucleotide substrates
The double-stranded oligonucleotide containing a single AP site in one strand was identical to that described previously by Rothwell and Hickson (26), and was generated by excision of a uracil residue using uracil DNA glycosylase.
The sequence of 25mer oligonucleotides with a site-specifically inserted pBQ adduct was: 5′-CCGCTAGCGGGTACCGAGCTCGAAT-3′. The pBQ-dC, pBQ-dG and pBQ-dA were placed at positions 8, 7 and 6 from the 5′ terminus, respectively, as shown in bold type. The synthesis of these benzetheno phosphoramidites and their incorporation into oligonucleotides has been described previously by Chenna and Singer (32,33). For competition assays, the following oligonucleotide containing a single tetrahydrofuran residue (X) was also used: 5′-TGCTCCCXCCCTGCT-3′ (34). The oligonucleotides were synthesised using an Applied Biosystems model 392 DNA synthesiser, and purified by HPLC followed by denaturing polyacrylamide gel electrophoresis.
Band-shift assays
For analysis of HAP1 binding to AP site-containing oligonucleotides, the method described by Rothwell and Hickson (26) was employed. For assays utilising pBQ adducts, oligonucleotides were 5′ end-labelled with [γ-32P]ATP and each annealed to its complementary oligonucleotides as described previously (35). An aliquot of 18 fmol of a 32P-labelled pBQ adduct-containing oligonucleotide duplex was then incubated with various concentrations of HAP1 or mutant HAP1 proteins for 5 min on ice in a binding buffer (final volume 25 µl) comprising 25 mM Tris–HCl, pH 7.0, 0.5 mM DTT, 0.5 mM spermidine, 50 mM KCl, 5 mM EDTA, pH 8.0, 0.5 mM MgCl2, 0.2 µg poly(dl-dC)·poly(dl-dC), 200 µg/ml BSA and 10% glycerol. The samples (without dye added) were loaded onto a pre-cooled (4°C) non-denaturing 6% polyacrylamide gel (the ratio of acrylamide:bis-acrylamide was 19:1). The gel was run in cold 0.5× TBE buffer (45 mM Tris-borate, 1 mM EDTA, pH 8.0) at 4°C for ~1 h at 150 V. The gel was dried and autoradiographed. Competition assays were performed under the same conditions except that the molar excess of unlabelled competitor oligonucleotide duplex containing either an unmodified base or pBQ-dC or a synthetic AP site was added to the reaction mixture prior to the addition of 32P-labelled probe.
AP endonuclease assays
The AP endonuclease activities of the native and mutant HAP1 proteins were assayed using a double-stranded oligonucleotide labelled at the 5′-end with 32P and containing a single AP site, as described by Barzilay et al. (11,12). Reactions were performed as described previously (11,12,26) and the reaction products were electophoresed on a 12% denaturing polyacrylamide gel. The gels were dried onto Whatman 3MM paper and exposed to a 32P-sensitive phosphor screen (Molecular Dynamics, CA). The screens were scanned on a PhosphorImager 425 (Molecular Dynamics) and analysed using the ImageQuant software. The amount of cleavage was quantified by reference both to the uncleaved strand (to equalize for gel loading) and to a positive control for AP site cleavage (incubation with 500 mM piperidine, 98°C for 30 min).
Enzyme kinetics
Kinetic analyses were performed using the AP endonuclease assay described above. Initial rates of AP site cleavage were determined over a range of substrate concentrations (5–500 nM). By determining the initial rates (ν) at each substrate concentration [S], a plot of substrate concentration against reaction rate was produced. The data were then fitted to an ascending exponential using the Kaleidograph package, which produced values for Km, Vmax and Kcat.
Redox assays
The redox activity of HAP1 was assayed by monitoring the ability of the protein to reactivate oxidized Jun protein to a form that binds DNA in gel retardation assays, as described previously (20,21).
Structural studies
Molecular modelling of the HAP1 active site was based on the previously published X-ray crystallographic structure of HAP1 (24). Figures were produced using PREPI (courtesy of S. Islam and M. Sternberg, Imperial Cancer Research Fund, London, UK).
RESULTS
Asp-210 is conserved in the exonuclease III family of AP endonucleases and forms part of a hydrogen-bonding network in the active site
Alignment of the primary sequences of members of the exonuclease III family of AP endonucleases from bacteria, lower eukaryotes and mammalian cells, revealed the presence of a conserved motif DXN (one letter amino acid code: X = any residue) (Fig. 1). Previous structural studies on both exonuclease III and HAP1 have shown that these invariant aspartate and asparagine residues (Asp-210 and Asn-212 in HAP1) lie within the catalytic active site in each case (23,24).
Figure 1.

A conserved motif in the exonuclease III family of AP endonucleases. The amino acid sequence of HAP1 has been aligned with those of AP endonucleases from different species: bovine (BAP1), murine (APEX), Arabidopsis thaliana (Arp1), Drosophila melanogaster (Rrp1), Streptococcus pneumoniae (ExoA) and E.coli (exonuclease III). The conserved residues, Asp-210 and Asn-212 in HAP1, are shown in bold type with an asterisk below.
The active site of HAP1 lies in a pocket at the base of an α/β sandwich, and is surrounded by loop regions (Fig. 2). Within the active site, the imidazole ring of His-309 hydrogen bonds with the carboxyl groups of Asp-283 and Asp-210. Asn-68 hydrogen bonds with the main chain amide of Asp-70 and the carboxyl group of Asp-210. The latter also hydrogen bonds to the main chain amide of Asn-212. The hydroxyl group of Tyr-171 hydrogen bonds to the metal binding residue, Glu-96. These bonds, together with those mediated via water molecules, form an extensive hydrogen-bonding network in the active site of HAP1.
Figure 2.
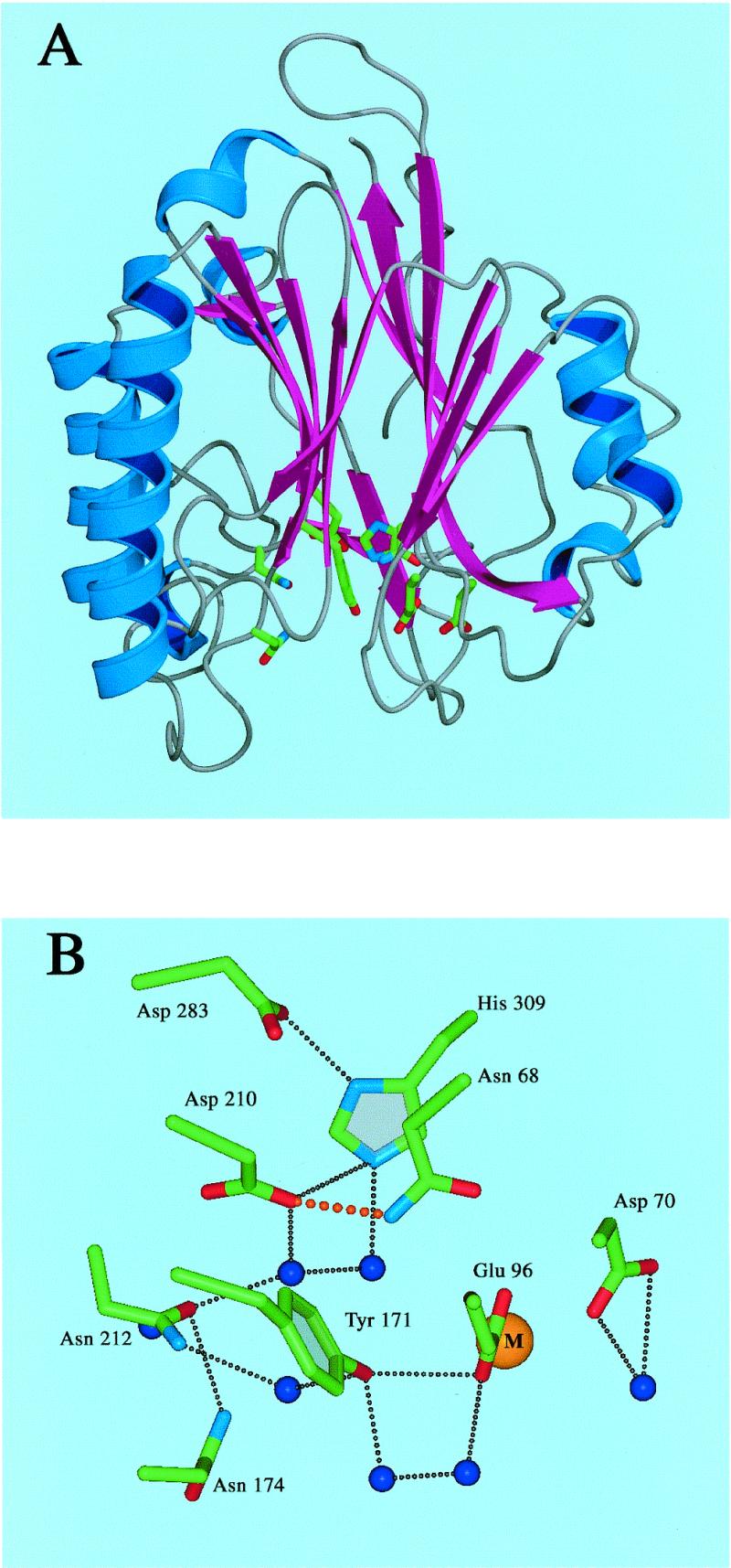
(A) Overall view of HAP1 (NDB/PDB accession no. 1BIX). α helices are shown in blue, and β strands are shown in magenta. Active site residues are represented as atom coloured sticks. (B) Close-up view of active site residues. Side chains are represented as atom coloured sticks, water molecules are shown as blue spheres, the bound metal ion is shown as a gold sphere. Hydrogen bonds are represented by black dots. The hydrogen bond between Asp-210 and Asn-68 is shown in red. For clarity, hydrogen bonds to main chain atoms have been omitted.
Purification of HAP1 derivatives with a substitution at position 210
We have shown previously that the Asn-212 residue is essential for both efficient AP endonuclease activity and AP site recognition (26). In order to study the role of the Asp-210 residue in the enzymatic function of HAP1, the HAP1 cDNA was mutated to replace the codon for Asp-210 with one encoding a glutamate, an asparagine or an alanine residue. These mutant HAP1 proteins, designated HAP1–D210E, HAP1–D210N and HAP1–D210A, respectively, were overexpressed in E.coli and purified to apparent homogeneity using a combination of phosphocellulose and nickel chelate affinity chromatography (Fig. 3A). The mutant proteins were all fully soluble in E.coli and had chromatographic properties very similar to those of wild-type HAP1. These data, together with the findings that the mutant proteins were proficient at converting oxidised Jun protein to a form that could bind to an oligonucleotide containing a Jun recognition site (Fig. 3B), and alone could bind AP site-containing oligonucleotides (see Fig. 5 below), suggest that the overall tertiary structure of the mutant proteins was not grossly perturbed by substitution of the Asp-210 residue.
Figure 3.
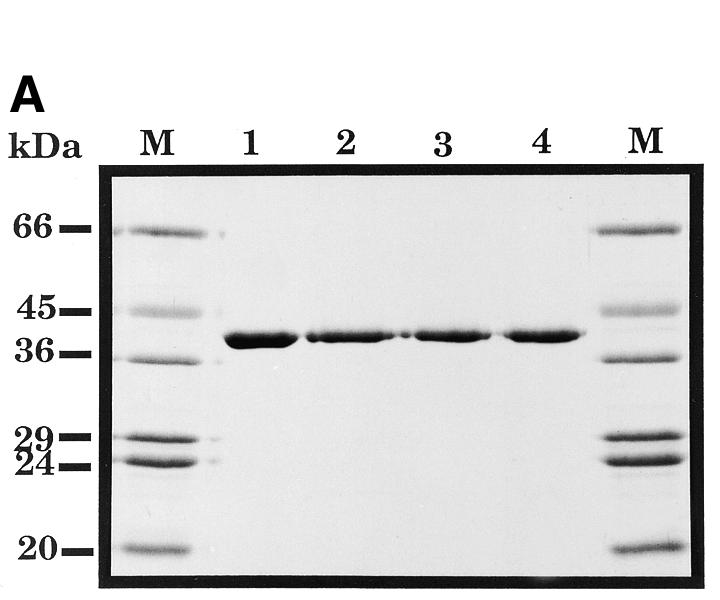
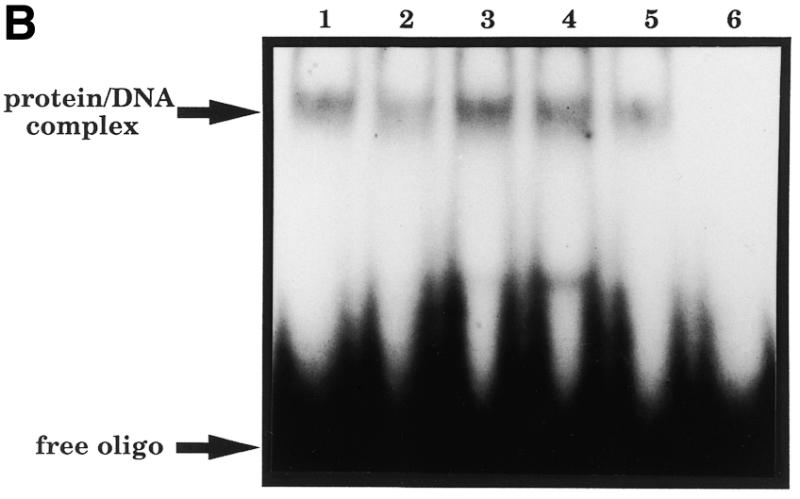
(A) Purification of site-specific mutant HAPI proteins with substitutions at position 210. A Coomassie blue-stained SDS–polyacrylamide gel is shown. Lane 1, wild-type HAP1; lane 2, HAP1–D210E; lane 3, HAP1–D210N; lane 4, HAP1–D210A. M, molecular mass standards, as indicated on the left. HAP1 has an apparent mass of 37 kDa. (B) Substitution of Asp-210 does not influence the ‘redox’ activity of HAP1. Oxidised Jun protein (100 nM) was incubated with 25 mM dithiothreitol (positive control, lane 1), or with 500 nM wild-type HAP1 (lane 2), HAP1–D210E (lane 3), HAP1–D210N (lane 4), HAP1–D210A (lane 5) or no additional factors (lane 6), and a band-shift assay was performed using a 32P-labelled oligonucleotide containing a consensus Jun binding site. The positions of the free oligonucleotide and the Jun–DNA complex are indicated on the left.
Figure 5.
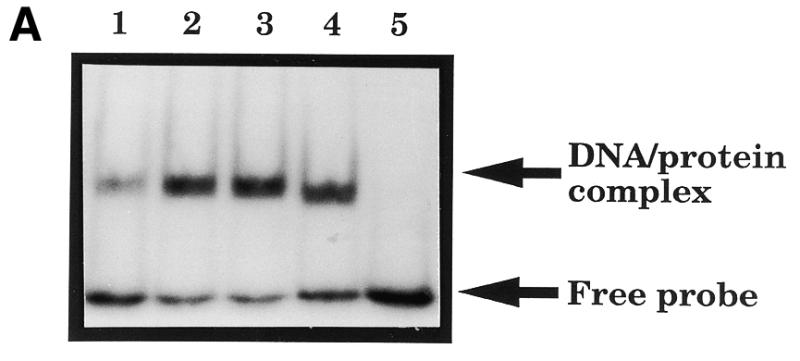
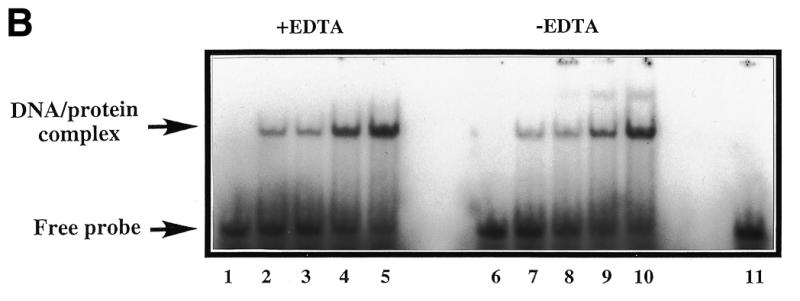

(A) A band-shift assay to show that substitution of residue 210 in HAP1 does not eliminate AP site-binding. 500 nM HAP1 (lane 1), HAP1–D210E (lane 2), HAP1–D210N (lane 3) or HAP1–D210A (lane 4) proteins were incubated in the presence of 4 mM EDTA with 0.5 ng 32P-labelled oligonucleotide containing a single AP site, and the products were separated on a polyacrylamide–TBE gel. In lane 5, the reaction mixture contained no HAP1 protein. The positions of the free oligonucleotide and the HAP1–DNA complex are shown on the right. (B) Binding of HAP1–D210N protein to a pBQ-dC-containing oligonucleotide in the presence of 5 mM EDTA (lanes 1–5) or in the absence of EDTA (lanes 6–10). Lanes 1 and 6, no HAP1; lanes 2 and 7, 62.5 ng HAP1–D210N; lanes 3 and 8, 125 ng HAP1–D210N; lanes 4 and 9, 250 ng HAP1–D210N; lanes 5 and 10, 500 ng HAP1–D210N. Lane 11 shows the position of the free probe. (C) Effect of substitution at position 210 of HAP1 on the rate of cleavage of a pBQ-dC-containing oligonucleotide. The time course of cleavage for wild-type HAP1 (squares) and HAP1–D210N (triangles) was measured using 180 ng protein, as described in Materials and Methods.
Asp-210 is essential for the AP endonuclease activity of HAP1
The AP endonuclease activity of the wild-type and mutant HAP1 proteins was studied using a substrate consisting of an oligonucleotide containing a single AP site, as described previously (11,12). Figure 4 shows that substitution of Asp-210 by Asn or Ala reduced the AP endonuclease activity of HAP1 to a level below the detection limit for this assay (<0.025 nmol AP sites cleaved/mg protein/min). In contrast, the HAP1–D210E protein retained a detectable level of AP endonuclease activity, although this derivative displayed a specific activity ~500-fold lower than that of wild-type HAP1 (Fig. 4).
Figure 4.

Effects of substitutions at position 210 on the AP endonuclease activity of HAP1. Data represent the rates of cleavage of an oligonucleotide containing a single AP site catalysed by wild-type HAP1 (open squares), HAP1–D210E (closed squares), HAP1–D210N (open triangles) and HAP1–D210A (closed diamonds). Note that the vertical axis is broken.
In order to quantify the effects on activity of substituting the Asp-210 residue with Glu, and to gain an insight into the precise role played by this residue, we determined the specificity constants (Kcat/Km) for the wild-type HAP1 and HAP1–D210E proteins using the AP site-containing oligonucleotide substrate. Table 1 shows that the Km values for AP site recognition by the HAP1 and HAP1–D210E proteins are very similar, while the Kcat for HAP1–D210E is reduced by >3000-fold compared with that for wild-type HAP1. Hence, the specificity constant for the HAP1–D210E protein is at least three orders of magnitude lower than that of the wild-type HAP1 protein.
Table 1. Kinetic analysis of HAP1–D210E protein.
| Protein | Kcat (min–1) | Km (nM) | Kcat/Km (min–1 nM–1) |
|---|---|---|---|
| HAP1 | 138 | 47 | 2.9 |
| HAP1–D210E | 3.6 × 10–2 | 57 | 6.3 × 10–4 |
Substitution of Asp-210 stabilises binding of HAP1 to an oligonucleotide containing either an AP site or a site-specific pBQ adduct
The kinetic data presented above strongly suggested that the Asp-210 residue is important for catalysis of phosphodiester bond cleavage without contributing significantly to AP site recognition by the HAP1 enzyme. To provide additional data to substantiate this point, we performed band-shift assays (gel retardation assays) initially using the AP site-containing oligonucleotide. Figure 5A shows that HAP1–D210E, HAP1–D210N and HAP1–D210A proteins were all proficient at binding the AP site-containing substrate. Indeed, the efficiency of binding by the mutant proteins to this substrate was consistently enhanced compared with that seen with wild-type HAP1 protein, suggesting that substitution of Asp-210 could be influencing the stability of the HAP1–substrate complex.
To analyse this further, and to address whether the Asp-210 substitution mutants could also bind stably to pBQ adduct-containing oligonucleotides, additional band-shift assays were performed. In an initial study of the pBQ adduct binding activity of wild-type and various mutant HAP1 proteins, we found that the HAP1–D210N protein could form a stable complex with an oligonucleotide containing a pBQ-dC residue, whereas wild-type protein could not (Fig. 5B and data not shown). Moreover the binding of HAP1–D210N protein to the pBQ-dC-containing oligonucleotide was protein concentration dependent and observed even in the absence of EDTA (Fig. 5B). This latter result was unexpected, given previous findings showing that wild-type HAP1 will only form a stable complex with an AP site-containing oligonucleotide in the presence of EDTA (which chelates the active metal ion and thus prevents catalysis and enzyme dissociation) (25,26,28,29).
Consistent with data presented above using AP site-containing substrates, the D210N protein was essentially inactive when tested for its ability to cleave a 25mer oligonucleotide containing a pBQ-dC residue (Fig. 5C). Taken together, these data strongly suggest that HAP1–D210N protein recognises AP sites and the pBQ-dC adduct efficiently, but fails to effect hydrolysis of the phosphodiester backbone. This conclusion is consistent with the kinetic data described above.
To analyse the relative efficiencies of HAP1–D210N binding to AP site-containing and pBQ-dC-containing oligonucleotides, competition experiments were performed. The data in Figure 6 indicate that addition of a 40-fold excess of the AP site-containing oligonucleotide was able to dramatically reduce binding of HAP1–D210N to the pBQ-dC oligonucleotide. In contast, an unlabelled pBQ-dC-containing 25mer duplex only partially competed for HAP1–D210N binding when a ≥80-fold molar excess of competitor DNA was used (Fig. 6). Moreover, the unmodified 25mer duplex competitor caused only a slight reduction of the extent of HAP1 binding to the pBQ-dC-containing oligonucleotide even when tested at a 240-fold molar excess (Fig. 6). These data suggest that HAP1–D210N has a higher binding affinity for an AP site than for a pBQ-dC substrate, and are consistent with our previous findings on the kinetics of catalysis of these substrates by wild-type HAP1.
Figure 6.
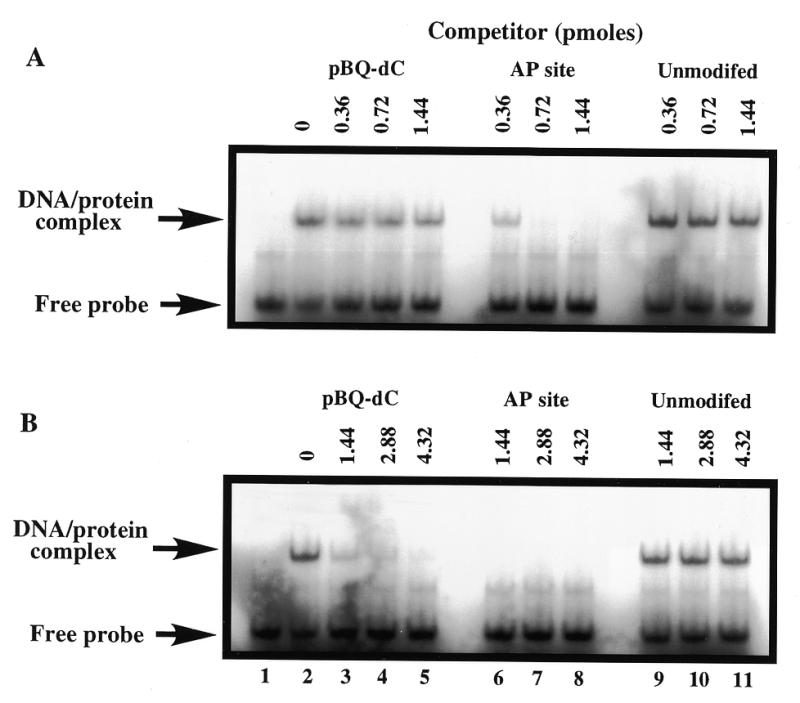
Specificity of HAP1–D210N protein binding to oligonucleotide duplex containing the pBQ-dC adduct. HAP1–D210N protein (34 nM) was incubated with 32P-end-labelled 25mer pBQ-dC oligonucleotide and a molar excess of oligonucleotide competitors (AP site-containing, pBQ-dC or unmodified; as indicated above the lanes). (A) Low range concentrations of competitors (20-, 40- and 80-fold molar excess) were used, while in (B), higher concentrations (80-, 160- and 240-fold molar excess) of the same competitors were used. Lane 1 in each panel contained binding buffer only, and lane 2 contained HAP1–D210N protein, but without any competitor added. All reactions contained 5 mM EDTA.
We further examined protein binding to different pBQ purine adducts since HAP1 protein has been shown previously to be capable of cleaving all three pBQ adducts in oligonucleotides, but with different efficiencies (pBQ-dC>>pBQ-dA/pBQ-dG) (36,37). In accordance with these cleavage rates, the pBQ-dC-containing duplex was also bound more efficiently than the pBQ-dA or pBQ-dG substrates (Fig. 7).
Figure 7.
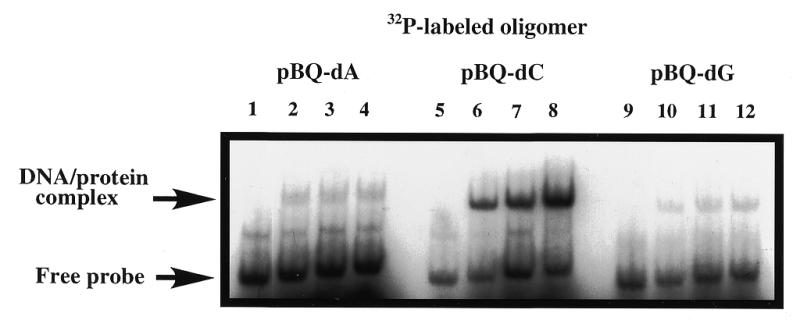
Differential binding of D210N to oligonucleotides with pBQ-dA, pBQ-dC or pBQ-dG adduct. All three pBQ adduct-containing 25mers were 32P-end-labelled and annealed to a common complementary strand (Materials and Methods). Binding reactions were carried out with increasing amounts of D210N protein in the presence of 5 mM EDTA. Lanes 1, 5 and 9, controls with binding buffer only. Lanes 2, 6 and 10 contained 125 ng HAP1–D210N; lanes 3, 7 and 11 contained 250 ng HAP1–D210N; lanes 4, 8 and 12 contained 500 ng HAP1–D210N. Arrows are as indicated in Figure 6.
DISCUSSION
We have shown that substitution of the Asp-210 residue of HAP1 with Ala, Asn or Glu strongly diminishes AP endonuclease activity but concomitantly stabilises substrate binding. These results are fully consistent with our enzyme kinetic data showing that the HAP1–D210E mutant has a similar Km value for AP site cleavage to that of wild-type HAP1, but a Kcat which is more than three orders of magnitude lower.
Previous work has shown that HAP1 can be induced to bind stably to AP site-containing oligonucleotides (or oligonucleotides with modified AP sites such as tetrahydrofuran) by chelation of the active site metal ion (25,26,28,29). In this way, the activity of HAP1 can be ‘trapped’ in a stable complex with DNA at a stage subsequent to substrate recognition, but prior to phosphodiester backbone incision. Hence, the HAP1 catalytic cycle can be viewed as being comprised of two steps that are separable both mechanistically and temporally. Re-addition of Mg2+ to the HAP1–DNA complex leads to rapid incision of the DNA backbone and consequent dissociation of the complex. Wilson et al. (38) determined the half-life of HAP1 complexes with tetrahydrofuran-containing DNA to be <15 s in the presence of Mg2+. These previous results strongly suggest that the stable complex of HAP1 bound to a pre-incised AP site is one step in the catalytic cycle that normally occurs too rapidly to be visualised using conventional methods such as band-shift assays. The data presented here show that substitution of Asp-210 by any of three other amino acids effectively mimics treatment of wild-type HAP1 with a metal chelating agent in that it creates an altered enzyme that retains the ability to recognise its substrate, but cannot perform the hydrolysis reaction that is required for phosphodiester backbone scission. Hence, the HAP1–D210N protein can bind stably to substrate oligonucleotides even in the absence of metal chelators, and provides a novel resource through which detailed analyses of the mode of HAP1 binding to its substrates can be performed. In particular, it should now be possible to use these mutants to further probe the structural basis for AP site recognition by HAP1.
Whilst this manuscript was in preparation, Erzberger and Wilson (39) presented evidence that substitution of Asp-210 with Ala or Asn did not affect binding of the mutant proteins to oligonucleotides containing AP site analogues, but did reduce catalytic activity. Our data, analysing both ‘regular’ AP sites and pBQ-dC adducts, are consistent with those of Erzberger and Wilson (39).
Structural studies (24,30) on HAP1 have revealed that Asp-210 is located in a pocket near the base of the HAP1 active site, and contributes to an extensive hydrogen-bonding network (Fig. 2). Based on the recent determination of co-crystal structures of HAP1 and synthetic AP site-containing oligonucleotides (30), it is likely that Asp-210 plays a central role in the hydrolysis reaction. It has been proposed that Asp-210 is directly involved in generating the hydroxyl nucleophile that attacks scissile P-O3′ bond. The role of the Asp-283/His-309 pair, which had previously been thought to create the nucleophile (23,24), is more likely to be in the alignment and polarisation of the target bond. Without detailed structural data on the mutants described here lacking Asp-210, it is not possible to be definitive about the precise structural effects of each amino acid substitution. Nevertheless, some predictions can be made. The recently proposed reaction mechanism relies on an elevated pKa for Asp-210 such that it can be protonated to generate the attacking hydroxyl nucleophile (30). This would result in the loss of one hydrogen bond between Asp-210 and the backbone amide of Asn-212 or Asn-68. This mechanism also necessitates deprotonation of Asp-210 such that activation of the nucleophile can take place in subsequent reaction cycles. Clearly, mutation of Asp-210 to Asn would prevent activation of the attacking nucleophile in the current proposed mechanism. Furthermore, it could result in loss of either the Asp-210/His-309 side-chain hydrogen bond, or the Asp-210/Asn-212 main-chain hydrogen bond. This would certainly cause His-309 to adopt an alternative conformation and could affect the positioning of the scissile phosphate. Mutation of Asp-210 to Ala would also be strongly deleterious in terms of the above reaction scheme, in that nucleophile activation could not take place and the active site hydrogen-bonding network would be severely disrupted. In contrast, mutation of Asp-210 to Glu could lead to retention of the hydrogen-bonding network and activation of the nucleophile, and indeed this mutant displays residual AP endonuclease activity. Even though Glu is one carbon–carbon bond longer, and so would perturb the conformation of the active site pocket, it could still allow the non-optimal activation of the nucleophile.
In summary, we have identified the Asp-210 residue in HAP1 as vital for the catalytic function of this enzyme. Substitution of this residue strongly diminishes AP endonuclease activity, whilst concomitantly stabilising the binding of HAP1 to its substrates. The identification of a HAP1 mutant that lacks the ability to cleave the phosphodiester backbone of DNA, but binds to substrate, should permit detailed studies of the general mechanism of substrate recognition by the HAP1 protein, as well as studies of the mode of specific recognition of chemically diverse substrates.
Acknowledgments
ACKNOWLEDGEMENTS
We thank group members for useful discussions, Dr A. Chenna for synthesis of the 25mer pBQ-dC, pBQ-dA and pBQ-dG-containing oligonucleotides, Dr D. Gillespie for the Jun protein, Drs S. Islam and M. Sternberg for the preparation of Figure 2 and Jenny Pepper for preparation of the manuscript. This work was supported by the Imperial Cancer Research Fund (UK), and by the grants CA 72079 and ES 07363 to B.S. from the National Institutes of Health, Bethesda, USA.
REFERENCES
- 1.Doetsch P.W. and Cunningham,R.P. (1990) Mutat. Res., 236, 173–201. [DOI] [PubMed]
- 2.Lindahl T. (1990) Mutat. Res., 238, 305–311. [DOI] [PubMed]
- 3.Lindahl T. (1993) Nature, 362, 709–715. [DOI] [PubMed]
- 4.Barzilay G. and Hickson,I.D. (1995) Bioessays, 17, 713–719. [DOI] [PubMed]
- 5.Wallace S.S. (1998) Radiat. Res., 150, S60–S79. [PubMed]
- 6.Robson C.N. and Hickson,I.D. (1991) Nucleic Acids Res., 19, 5519–5523. [DOI] [PMC free article] [PubMed]
- 7.Demple B., Herman,T. and Chen,D.S. (1991) Proc. Natl Acad. Sci. USA, 88, 11450–11454. [DOI] [PMC free article] [PubMed]
- 8.Demple B. and Harrison,L. (1994) Annu. Rev. Biochem., 63, 915–948. [DOI] [PubMed]
- 9.Rothwell D.G., Barzilay,G., Gorman,M., Morera,S., Freemont,P. and Hickson,I.D. (1997) Oncol. Res., 9, 275–280. [PubMed]
- 10.Hickson I.D., Gorman,M.A. and Freemont,P.S. (1999) In Nickoloff,J. and Hoekstra,M.F. (eds), DNA Damage and Repair. Humana Press, NJ, in press.
- 11.Barzilay G., Mol,C.D., Robson,C.N., Walker,L.J., Cunningham,R.P., Tainer,J.A. and Hickson,I.D. (1995) Nature Struct. Biol., 2, 561–567. [DOI] [PubMed]
- 12.Barzilay G., Walker,L.J., Robson,C.N. and Hickson,I.D. (1995) Nucleic Acids Res., 23, 1544–1550. [DOI] [PMC free article] [PubMed]
- 13.Chen D., Herman,T. and Demple,B. (1991) Nucleic Acids Res., 19, 5907–5914. [DOI] [PMC free article] [PubMed]
- 14.Seki S., Akiyama,K., Watanabe,S., Hatsushika,M., Ikeda,S. and Tsutsui,K. (1991) J. Biol. Chem., 266, 20797–20802. [PubMed]
- 15.Wilson D.M., Takeshita,M., Grollman,A.P. and Demple,B. (1995) J. Biol. Chem., 270, 16002–16007. [DOI] [PubMed]
- 16.Suh D., Wilson,D.M. and Povirk,L.F. (1997) Nucleic Acids Res., 25, 2495–2500. [DOI] [PMC free article] [PubMed]
- 17.Winters T.A., Weinfeld,M. and Jorgensen,T.J. (1992) Nucleic Acids Res., 20, 2573–2580. [DOI] [PMC free article] [PubMed]
- 18.Hang B., Chenna,A., Fraenkel-Conrat,H. and Singer,B. (1996) Proc. Natl Acad. Sci. USA, 93, 13737–13741. [DOI] [PMC free article] [PubMed]
- 19.Hang B., Rothwell,D.G., Sagi,J., Hickson,I.D. and Singer,B. (1997) Biochemistry, 36, 15411–15418. [DOI] [PubMed]
- 20.Xanthoudakis S., Miao,G.G., Wang,F., Pan,Y.C.E. and Curran,T. (1992) EMBO J., 11, 3323–3335. [DOI] [PMC free article] [PubMed]
- 21.Walker L.J., Robson,C.N., Black,E., Gillespie,D. and Hickson,I.D. (1993) Mol. Cell. Biol., 13, 5370–5376. [DOI] [PMC free article] [PubMed]
- 22.Xanthoudakis S., Miao,G.G. and Curran,T. (1994) Proc. Natl Acad. Sci. USA, 91, 23–27. [DOI] [PMC free article] [PubMed]
- 23.Mol C.D., Kuo,C.-F., Thayer,M.M., Cunningham,R.P. and Tainer,J.A. (1995) Nature, 374, 381–386. [DOI] [PubMed]
- 24.Gorman M.A., Morera,S., Rothwell,D.G., de La Fortelle,E., Mol,C.D., Tainer,J.A., Hickson,I.D. and Freemont,P.S. (1997) EMBO J., 16, 6548–6558. [DOI] [PMC free article] [PubMed]
- 25.Masuda Y., Bennett,R.A.O. and Demple,B. (1998) J. Biol. Chem., 273, 30360–30365. [DOI] [PubMed]
- 26.Rothwell D.G. and Hickson,I.D. (1996) Nucleic Acids Res., 24, 4217–4221. [DOI] [PMC free article] [PubMed]
- 27.Lucas J.A., Masuda,Y., Bennett,R.A.O., Strauss,N.S. and Strauss,P.R. (1999) Biochemistry, 38, 4958–4964. [DOI] [PubMed]
- 28.Erzberger J.P., Barsky,D., Scharer,O.D., Colvin,M.E. and Wilson,D.M. (1998) Nucleic Acids Res., 26, 2771–2778. [DOI] [PMC free article] [PubMed]
- 29.Masuda Y., Bennett,R.A.O. and Demple,B. (1998) J. Biol. Chem., 273, 30352–30359. [DOI] [PubMed]
- 30.Mol C.F., Izumi,T., Mitra,S. and Tainer,J.A. (2000) Nature, 403, 451–456. [DOI] [PubMed]
- 31.Landt O., Grunert,H.-P. and Hahn,U. (1990) Gene, 96, 125–128. [DOI] [PubMed]
- 32.Chenna A. and Singer,B. (1995) Chem. Res. Toxicol., 8, 865–874. [DOI] [PubMed]
- 33.Chenna A. and Singer,B. (1997) Chem. Res. Toxicol., 10, 165–171. [DOI] [PubMed]
- 34.Sági J., Hang,B. and Singer,B. (1999) Chem. Res. Toxicol., 12, 917–923. [DOI] [PubMed]
- 35.Rydberg B., Dosanjh,M.K. and Singer,B. (1991) Proc. Natl Acad. Sci. USA, 88, 6839–6842. [DOI] [PMC free article] [PubMed]
- 36.Hang B., Chenna,A., Sági,J. and Singer,B. (1998) Carcinogenesis, 19, 1339–1343. [DOI] [PubMed]
- 37.Singer,B. and Hang,B. (1999) In Singer,B. and Bartsch,H. (eds), Exocyclic DNA Adducts in Mutagenesis and Carcinogenesis. IARC Scientific Publications No. 150, IARC, Lyon, France, pp. 233–248.
- 38.Wilson D.M., Takeshita,M. and Demple,B. (1997) Nucleic Acids Res., 25, 933–939. [DOI] [PMC free article] [PubMed]
- 39.Erzberger J.P. and Wilson,D.M. (1999) J. Mol. Biol., 290, 447–457. [DOI] [PubMed]