

Abstract
In mammals, the endometrium undergoes dynamic changes in response to estrogen and progesterone to prepare for blastocyst implantation. Two distinct types of endometrial epithelial cells, the luminal (LE) and glandular (GE) epithelial cells play different functional roles during this physiological process. Previously, we have reported that Notch signaling plays multiple roles in embryo implantation, decidualization, and postpartum repair. Here, using the uterine epithelial specific Ltf-iCre, we showed that Notch1 signaling over-activation in the endometrial epithelium caused dysfunction of the epithelium during the estrous cycle, resulting in hyper-proliferation. During pregnancy, it further led to dysregulation of estrogen and progesterone signaling, resulting in infertility in these animals. Using 3D organoids, we showed that over-activation of Notch1 signaling increased the proliferative potential of both LE and GE cells and reduced the difference in transcription profiles between them, suggesting disrupted differentiation of the uterine epithelium. In addition, we demonstrated that both canonical and non-canonical Notch signaling contributed to the hyper-proliferation of GE cells, but only the non-canonical pathway was involved with estrogen sensitivity in the GE cells. These findings provided insights into the effects of Notch1 signaling on the proliferation, differentiation, and function of the uterine epithelium. This study demonstrated the important roles of Notch1 signaling in regulating hormone response and differentiation of endometrial epithelial cells and provides an opportunity for future studies in estrogen-dependent diseases, such as endometriosis.
Keywords: Notch1 signaling, luminal epithelium, glandular epithelium, hyper-proliferation, estrogen sensitivity
Graphical Abstract
Over-activation of Notch1 signaling results in hyperproliferation of both LE and GE cells, weakens the differentiation features and function of GE, resulting in decreased LIF levels and impaired uterine receptivity. Moreover, the higher level of Notch1 signaling enhances the sensitivity of the GE to estrogen. In vitro culture, over-activation of Notch1 signaling increases the regeneration capacity of both LE and GE in organoid.
Introduction
The uterus is a critical female organ for reproduction in mammals, which supports the development and growth of the embryo and fetus by providing a proper micro-environment and nutrient supply [1]. The uterine endometrium is comprised of many cell types, including stromal cells, immune cells, endothelial cells, and epithelial cells. The epithelium can be further divided into two distinct types, the luminal epithelium (LE) and glandular epithelium (GE), and both undergo dynamic changes throughout the estrous cycle and pregnancy in response to the ovarian hormones, estrogen and progesterone [2, 3]. Estrogen produced from the developing follicles stimulates endometrial growth, and progesterone is responsible for converting the estrogen-primed endometrium into a receptive state [4]. A successful pregnancy requires a series of changes in the endometrium to establish contact with the embryo, including endometrial stromal cell proliferation, depolarization of LE, and secretions from the GE to facilitate embryo implantation [5, 6].
The LE and GE are different in location, morphology, physiological function, response to hormones, and differentiation potential [1]. The columnar LE cells cover the luminal surface and serve as a barrier until the establishment of pregnancy [7], during which the LE cells depolarize and develop an adherent phenotype to establish transient uterine receptivity for embryo implantation [8], which is associated with the down-regulation of the estrogen target gene Mucin1 (Muc1) [9]. On the other hand, GE cells that form endometrial glands are cuboidal, produce histotroph to support the developing embryos, and secrete many vital factors such as LIF to ensure successful implantation. Endometrial gland development in mammals typically begins in the early postnatal period, during which some LE cells proliferate, differentiate, and migrate into the uterine stroma, eventually forming tubular or branched uterine glands [10]. The key regulators for endometrial gland development include Wnt signaling, transcription factors such as Foxa2, the Distal-less homeobox (Dlx) gene family, estrogen receptor ESR1, and the pituitary hormone prolactin [11]. Additionally, our previous study also showed that Notch1 signaling is involved in gland initiation in mice [12].
Notch signaling is a highly conserved pathway across species and plays vitally important roles in cellular proliferation, apoptosis, and differentiation. In mammals, Notch signaling transmembrane receptors (Notch1-4) initiate their functions through the direct binding of cell-bound ligands (δ-like 1, 3, or 4 or Jagged 1 or 2) from adjacent cells, followed by the cleavage cascade of Notch involving the ADAM proteases and γ-secretase [13]. The cleaved Notch intracellular domain (NICD) then enters the nucleus and interacts with transcriptional repressor Recombination Signal Binding Protein (RBPJ) to regulate the Notch target transcription genes [13]. Besides the canonical cascade, Notch signaling can also be initiated non-canonically. Independent of RBPJ, NICD can interact with the NF-κB, mTOR, PTEN, AKT, Wnt, Hippo, or TGF-β pathways at the cytoplasmic and/or nuclear level to regulate the transcription of target genes [14].
The roles of Notch signaling in mammalian pregnancy have been investigated in many studies during the past decade. Uterine-specific knockout of Notch1 results in impaired decidualization in an artificial decidualization mouse model [15]. The decreased expression of Notch receptors and target genes in the eutopic endometrium from patients with endometriosis also contributes to the impairment of decidualization [16]. The deletion of Rbpj in the mouse uterus results in aberrant embryonic-uterine axis alignment and impaired decidual patterning at post-implantation stages, leading to substantial embryo loss [17, 18]. Further, the loss of Rbpj in the uterus impairs the postpartum endometrial repair process and leads to secondary infertility in mice in subsequent pregnancies. Previously, our study showed that activation of Notch1 signaling via overexpression of N1ICD in the mouse uterus driven by Pgr-Cre resulted in infertility due to altered uterine receptivity [12]. Further investigation showed that the decreased expression of Pgr in stromal cells due to hypermethylation of its promoter region by the Notch1-PU.1-Dnmt3b pathway is responsible for the dysfunction of estrogen and progesterone signaling in these mice [12]. Another reason that may contribute to the infertility phenotype of N1ICD overexpressing mice is the absence of uterine glands, suggesting that the activation of Notch1 signaling also affects the differentiation of epithelial cells. However, whether the defects in proliferation and differentiation of the uterine epithelium are due to a role of Notch signaling in the uterine stromal cells or a direct effect on the epithelium remains unknown.
In this study, we overexpressed the Notch1 intracellular domain (N1ICD) only in the endometrial epithelium driven by the Ltf-iCre. We show that the over-activation of Notch1 signaling in the uterine epithelium is sufficient to compromise uterine receptivity through hyper-proliferation of the luminal epithelium and the dysfunction of the endometrial glands. The higher level of active Notch1 increases the proliferative potential of the epithelial cells. Overactivation of Notch1 signaling alters the sensitivity of the GE to estrogen, resulting in hyper-proliferation, which may contribute to the development of estrogen-dependent diseases such as endometriosis and adenomyosis. This study demonstrates the critical roles of Notch1 signaling in the proliferation and differentiation of the endometrial epithelium during the estrous cycle and pregnancy.
Materials and Methods
Animals
All mice were housed in the SPF Animal Facility with the approval of the Institutional Animal Care and Use Committee of South China Agricultural University. Mice were maintained in a temperature- and light-controlled environment (12 h light and 12 h dark cycle) with free access to regular food and water. Ltfcre/+ and Rosa26 N1ICD/N1ICD mice were mated to generate Ltfcre/+ Rosa26N1ICD/+ (E-OEx) and Ltf+/+ Rosa26N1ICD/+ (Ctrl) in this study. Mature female mice (8–10 weeks) were mated with fertile males of the same strain to initiate pregnancy, and the day of vaginal plug was designated as 0.5 dpc. Mice were sacrificed at 0.5 dpc to 4.5 dpc, and the pregnancy was confirmed by flushing out embryos from the oviduct or uterine cavity at 0.5 dpc to 3.5 dpc, respectively, while the i.v. injection of Chicago Sky Blue was used to visualize the implantation sites at 4.5 dpc. The serum was collected for hormone measurements. The uteri were collected and weighed and then either snap-frozen in liquid nitrogen or fixed in 4% paraformaldehyde solution for histological analyses.
LIF administration
The rLIF administration was conducted following the method described by Zheng et al., [20]. In brief, 5 μg rLIF was injected i.p to the E-OEx mice at 10 a.m. and 3 p.m. on 3.5 dpc and the mice were sacrificed at 4.5 dpc to identify the implantation sites by injecting Chicago Sky Blue dye i.v. For the delayed implantation model, we followed a protocol described in a previous study [21]. In brief, after ovariectomy on the morning of 3.5 dpc, the mice were treated with exogenous progesterone via a s.c. injection to maintain the delay of embryo implantation. Two days later, the rLIF (5 μg/mouse) was injected to activate implantation. Mice without LIF injection (Delay) were used as negative controls. Three hours following the LIF injection, the uteri were collected for histological analyses.
Artificial Decidualization
Artificial decidualization was induced as previously described [22]. Briefly, female mice were ovariectomized and allowed 2 weeks for recovery. They were then injected with three daily s.c. injections of 100 ng E2 in 100 μl of sesame oil, followed by 2 days of no injection and then 3 days of s.c. injections of 6.7 ng of E2 together with 1 mg of P4. Two hours following the last injection, the left uterine horn received a mechanical stimulus to induce decidualization. Animals were sacrificed 3 days after the stimulation, and both horns were collected for analysis.
Isolation of LE and GE from Mice Uterine Tissue
Uterine tissues were removed and washed with normal saline and then digested in HBSS solution containing 0.2% Trypsin and 10% Dispase II (Roche BR) at 4 °C for 60 min and 37 °C for another 60 min. The intact LE separation was performed by the shear force generated by a syringe. Then, the remaining tissues were chopped into approximately 0.1 mm3 cubes and enzymatically digested in HBSS solution containing 0.06% Dispase II, 0.1% collagenase IV (Gibco, 17104-019), and 10% FBS (Gibco) with gentle shaking at 37°C for 30–40 min. The supernatant was passed through 70 μm and 13 μm cell mesh sequentially, and the glandular elements were collected from the upper portion of the 13 μm mesh by inverting it over a petri dish and a backwash.
Organoid Culture
After we isolated the LE and GE from mice uterine tissue, the fragments of LE and GE cells were washed in cold DMEM/F12 medium and passed through a 40 μm cell strainer to ensure single cell suspension. The isolated LE and GE single cells were resuspended in 75% Matrigel/25% DMEM/F12 (11039-021, Gibco), 7000 cells were resuspended in 45 μl Matrigel and cultured in DMEM/F12 supplemented with 1% Penicillin-Streptomycin (Sigma), 1% ITS (41400-045, Gibco), 2 mM L-glutamine (21051-024, Gibco), 1 mM Nicotinamide (N3376, Sigma), 2% B27 (17504-044, Gibco), 1% N2 (17502-048, Gibco), 50 ng/ml mouse EGF (315-09, Peprotech), 100 ng/ml FGF (450-33, Peprotech), 100 ng/ml Noggin (250-38, Peprotech), 200 ng/ml WNT-3A (315-02, Peprotech), 200 ng/ml R-Spondin-1 (120-38, Peprotech) and 0.5 mM A83-01 (TGFb/Alk inhibitor) (HY-10432, MCE). The medium was changed every 2–3 days. Whole-well images were captured and the number and diameter of formed organoids were determined using ImageJ to calculate the organoid-forming efficiency. The organoids were embedded in agarose and then fixed in 4% paraformaldehyde. After further embedded by paraffin, organoids were sectioned at 5 μm. Following deparaffinization and rehydration, sections were stained with H&E for histological analysis.
Total RNA Isolation and Real-Time PCR
Total RNA was isolated using TRIZOL reagent (TaKaRa). Then, cDNA was reverse transcribed using the HiSuperscript II kit (Vazyme) according to the manufacturer`s instructions. Real-time PCR reactions were performed using the SYBR qPCR Master Mix kit (Vazyme) on the BIORAD-CFX96 Real-Time System (Bio-Rad). The data were normalized to mouse Rpl19 or Rpl7 and analyzed using the ΔΔCt method. All primers for real-time PCR are listed in Table S1.
Histological Analysis and Immunohistochemistry.
Paraformaldehyde-fixed, paraffin-embedded uterine tissue was sectioned at 5 μm. Following deparaffinization and rehydration, sections were stained with H&E for histological analysis. For immunohistochemistry, antigen retrieval was performed in a microwave oven in pH 6.0 sodium citrate solution following rehydration. Endogenous peroxidase was blocked with 3% hydrogen peroxide for 15 min, and sections were sequentially incubated with 10% horse normal serum at 37 °C for 1 h. Primary antibody incubation was conducted at 4°C overnight, followed by incubation with HRP conjugated secondary Antibody (ORIGINE, Beijing, China) at 37 °C for 1 h. The sections were then stained with diaminobenzidine (DAB) (ORIGINE, Beijing, China) and counterstained with hematoxylin. The sections were then dehydrated and cover slipped. Antibodies against FOXA2 (Abcam, ab108422), Ki67 (AXI-BIO, CB111141), ESR1 (Santa, sc7207), PGR (Invitrogen, MA5-16393), Ac-TUBULIN (Abcam, ab179484), Cleaved-caspase3 (CST, 9661s) were used in this study. Digital H-Score of DAB staining was measured by Image J. The number of glands were counted in 10 continuous sections. Proliferation rate equals Ki67 positive cells/ total number of cells per gland.
Western blot
Tissues were lysed in RIPA Lysis Buffer (Yamei, China) and proteins were quantified using Pierce™ BCA Protein Assey Reagent (Thermo, USA). Total 10 μg proteins were loaded on 10% SDS-PAGE gel and transferred onto a nitrocellulose membrane. Blotting membranes were labeled with the NOTCH1 (Abcam, ab128076) and β-ACTIN (CST, 4967s) antibody and scanned by Tanon-5200 imager.
Measurement of Serum E2 and P4 Levels
Serum levels of E2 and P4 at 3.5 dpc of pregnancy were measured by enzyme immunoassays performed by the Shanghai Yanhui Biotechnology Co., Ltd.
Whole-mount Immunofluorescence
Whole-mount immunofluorescence was performed as previously described [10]. Briefly, uteri were dissected from mice and fixed in DMSO:Methanol (1:4). For immunofluorescence, uteri were rehydrated in 1:1 solution of Methanol:PBT (1% Triton X-100 in PBS) for 15 min, washed in PBT for 15 min and incubated in blocking solution (2% powdered milk in PBT) for 2 hours, at room temperature. Uteri were incubated with 1:500 concentration of primary antibodies CDH1 (M108, Takara Biosciences) and FOXA2 (Abcam, ab108422), diluted in blocking solution for 5 to 7 nights at 4°C following which they were washed with 1% PBT 4 to 6 times for 30 min each at room temperature. Uteri were then incubated with secondary antibodies Alexa Fluor Donkey anti-Rabbit 555 (Invitrogen, A31572) and Goat anti-Rat 647 (Invitrogen, A21247), and Hoechst (Sigma Aldrich, B2261) at 4 °C for 2 to 3 nights, followed by 4 to 6 washes of 30 min each with 1% PBT and dehydrated in methanol for 30 min. Uteri were then bleached in a solution of 3% H2O2 prepared in methanol overnight at 4 °C. Finally, the samples were washed in 100% methanol for 30 min and cleared in BABB (1:2, benzyl alcohol: benzyl benzoate) (Sigma-Aldrich, 108006, B6630).
Confocal Microscopy
Samples were imaged using a Leica TCS SP8 Confocal Microscope equipped with white-light laser and 10X air objective. The entire length and thickness of the uterine horn was imaged using the tile scan function. Individual Z stacks collected were 5–7μm apart. Images were merged using Leica software LASX version 3.5.5.
3D Reconstruction and Image Analysis
Image analysis was performed using commercial software Imaris v9.2.1 (Bitplane). The Leica confocal images (.LIF) files were imported into the Surpass mode of Imaris. Using the channel arithmetics function in Imaris (XT), the FOXA2 signal of glands was subtracted from the epithelial CDH1 signal to get the lumen-only signal. Using the Surface module of Imaris, the 3D surface of the lumen and glands was reconstructed using the lumen-only channel and FOXA2 channel respectively.
RNA-Seq and Data Analysis
The Trizol RNA Reagent (Takara, Dalian, China) was used to extract total RNA from the tissues. The concentration and integrity of the RNA were measured with the ND-1000 Nanodrop and the Agilent 2100 TapeStation (Novogene Bioinformatic Technology, Beijing, China), respectively. The quality control parameters used in this study were: A260/A280 ratio ⩾1.8, A260/A230 ratio ⩾2.0, and RNA integrity number ⩾8.0. The TruSeq RNA sample preparation kit (Illumina, San Diego, CA, USA) was used to generate cDNA libraries. RNA-sequencing was run on an Illumina HiSeq 2500 system. Raw data were processed with an in-house computational pipeline. Differentially expressed genes were chosen based on the criteria of fold change > 2 and FDR (false discovery rate) < 0.05. The RNA-seq raw data were deposited in Gene Expression Omnibus (GEO) with the accession number GSE206491. GO and KEGG analysis was performed by using the DAVID online tools. The cutoff for false discovery rate (FDR) was set at 0.05.
Statistical Analysis.
Statistical analyses were conducted using a two-tailed Student’s t-test for two-group data or two-way ANOVA with Tukey’s multiple comparisons to determine significance in multiple-group experiments. All data are presented as Mean±SEM. All statistical analyses were performed by GraphPad Prism 8.0 (GraphPad Software). P-value<0.05 was considered significant.
Results
1. Over-activation of Notch1 signaling in uterine epithelium impairs uterine receptivity and leads to infertility in mice.
The strategy to generate the Lactoferrin Cre (Ltf-iCre) driven, epithelium-specific, N1ICD overexpression mouse (E-OEx, Ltfcre/+Rosa26N1ICD/+) is shown in Fig. 1a. We first confirmed the overexpression of N1ICD by qPCR using a pair of primers that could specifically identify the intercellular domain of Notch1, together with a primer set that detected the co-transcribed Egfp. Both primer sets showed significantly increased signals in the E-OEx mice compared to the Ltf+/+Rosa26N1ICD/+ (Ctrl) mice (Fig. 1b). On the other hand, we also confirmed that the protein expression of NOTCH1 in E-OEx mice was higher than Ctrl mice (Fig. 1c). Further, the localization of GFP protein was detected in the uterine epithelium of E-OEx mice by immunostaining (Fig.S1a). Next, we tested the expression of Notch1 signaling target genes Hey1 and Hes5. Both target genes were up-regulated (Fig. 1b), indicating that the overexpression of N1ICD was functional. After confirming the activation of Notch1 signaling, we performed a fertility test using the E-OEx mice. In a 5-month breeding test, each Ctrl females (n = 4) produced 4.25±0.4 litters, with an average litter size of 8.5±0.3 pups (Table.S2). However, the E-OEx female mice (n = 4) were completely infertile (Table.S2). We then tested if embryo implantation and decidualization were impaired in the E-OEx mice. At 4.5 d postconception (dpc), there were no implantation sites in the E-OEx uterus, while the Ctrl mice showed 8.2±1.6 implantation sites (Fig. 1d). The histological staining showed blastocysts attached to the LE of Ctrl mice while in the E-OEx mice the blastocysts were found floating in the lumen, but not attached to the LE (Fig. 1e). However, comparable number of blastocyst-stage embryos were flushed out from the uterine lumen of both E-OEx and Ctrl mice at 3.5 dpc, without significant morphological differences (Fig. 1f), indicating that ovulation, fertilization, and early development of embryos was not impaired in the E-OEx mice. These data suggested that the uterine receptivity was impaired in E-OEx mice. Leukemia inhibitory factor (LIF) is a cytokine secreted from the uterine glands, and Lif was the first gene to be proven as a key factor for embryo implantation using gene knockout mouse models [23, 24]. To test if the failure of implantation is due to the loss of GE function, we tested the Lif expression. The results showed that the mRNA level of Lif was significantly decreased in the E-OEx uterus (Fig. 1g). However, LIF supplementation could not rescue implantation failure caused by the over-activation of Notch1 signaling (Fig. 1d&e). Though the activity of rLIF was confirmed by activation of phosphorylated STAT3 in a delayed implantation mouse model (Fig.S1c). These data suggested that factors other than the absence of LIF contribute to the impaired endometrial receptivity in the E-OEx mice.
Fig. 1.

Defective uterine receptivity in E-OEx mice. (a) Generation strategy for conditional overexpression of N1ICD in the epithelial endometrium (Epithelium-specific overexpressed N1ICD, E-OEx, Ltfcre/+Rosa26N1ICD/+). (b) The mRNA expression of Notch1 (N1ICD), Egfp, Hey1, and Hes5 in the Ctrl (Ltf+/+Rosa26N1ICD/+) and E-OEx mice (n=3, t test). (c) Comparison of the protein expression of NOTCH1 in Ctrl and E-OEx mice. (d) Implantation sites are shown as blue dots in Ctrl, E-OEx, E-OEx mice supplemented with LIF at 3.5 dpc (E-OEx-Lif), and E-OEx mice with Rbpj knockout (Ltfcre/+Rosa26N1ICD/+RbpjF/F, E-OEx-KO) mice at 4.5 dpc. The black arrow marks the implantation sites. (e) The histological staining of implantation sites. The black arrow marks the implantation sites, the red arrow marks the floating embryos. (f) Morphology and number of blastocysts flushed out of one uterine horn of Ctrl and E-OEx mice on 3.5 dpc (n=5 in Ctrl mice, n=6 in E-OEx mice, t test). (g) The mRNA level of Lif in the uterus from Ctrl and E-OEx mice on 3.5 dpc (n=3, t test). (h) Weight of stimulated horns and non-stimulated uterine horns in Ctrl and E-OEx mice. (i) Uterine morphology in response to an artificial decidualization stimulus in Ctrl and E-OEx mice. Left horns served as non-stimulated controls. (j-l) qPCR analysis of decidua markers Wnt4, Bmp2, and Prl8a2 in stimulated and non-stimulated uterine horns in Ctrl and E-OEx mice (n=3, t test). Black dotted lines mark the LE, red arrow mark the GE. Values are mean ± SEM.
Furthermore, E-OEx females showed no response to an artificial decidualization stimulus induced by intrauterine mechanical stimulation in an exogenous hormone-primed ovariectomized mice model (Fig.S1b), whereas Ctrl mice exhibited a well-defined decidual response (Fig. 1i). The uterine weight of the stimulated horn in Ctrl was much higher than the non-stimulated horn, but no difference was evident between the stimulated and non-stimulated horns in the E-OEx mice (Fig. 1h). In addition, expression of decidualization markers such as Wnt4, Bmp2, and Prl8a2 in the stimulated horn of Ctrl mice was significantly higher than that in the E-OEx mice (Fig. 1j–l). These data suggest that the receptivity and decidualization capacity of endometrium, but not the activity of blastocysts, was impaired in the E-OEx mice.
The ovarian hormones estrogen and progesterone are key for female reproductive physiological processes including pregnancy [25]. Thus, we determined the serum levels of estrogen and progesterone. We observed that at 3.5 dpc, the serum progesterone levels were comparable between E-OEx and Ctrl mice (Fig.S1d). However, the serum estrogen levels were slightly reduced in the E-OEx mice than in the Ctrl mice at 3.5 dpc (Fig.S1e). To test whether the lower serum estrogen level was responsible for the implantation failure, we mimicked the hormone levels during early pregnancy by exogenous injections of hormones in an ovariectomized mice model, as previously reported [25, 26] (Fig.S1f). The results showed that exogenous estrogen could not rescue embryo implantation in the E-OEx mice (Fig.S1g). These results, together with the failed artificial decidualization response in exogenous hormone-primed mice (Fig. 1h–l), confirmed that the receptivity failure in the E-OEx mice was not due to an ovarian factor. To determine whether the over-activation of Notch1 signaling induced an infertile phenotype via RBPJ, the transcription factor of the canonical Notch pathway, we generated N1ICD overactivation mice without RBPJ (Ltfcre/+Rosa26N1ICD/+RbpjF/F, E-OEx-KO). The result showed that loss of RBPJ rescued the implantation failure defects observed in the E-OEx mice (Fig. 1d&e), indicating that implantation failure resulting by N1ICD overexpression depends on the canonical Notch signaling.
2. Epithelial hyper-proliferation associates with dysregulation of estrogen and progesterone signaling in E-OEx mice.
Next, we studied the uterine morphology in the pre-implantation stage at 3.5 dpc. Histological analysis revealed that the endometrial lumen of the E-OEx mice has extensive branch structure that extends into stroma, while the lumen of Ctrl mice was much flatter (Fig. 2a). 3D imaging also demonstrated that the evenly arranged transverse folds in Ctrl mice [18] were not observed in the E-OEx mice, but bubble-like folds appeared instead (Fig. 2b). To explore if the proliferation or apoptosis status changed in endometrial epithelium, we performed KI67 and Cleaved-caspase3 (CC3) staining. In the Ctrl mice, proliferation was observed in the LE cells at 1.5 and 2.5 but not at 3.5 dpc (Fig. 2a, Fig.S2a, b). However, in the E-OEx mice, the proliferation rate of LE cells was lower at 1.5 and 2.5 dpc, but higher at 3.5 dpc when compared to the Ctrl mice (Fig. 2a, Fig.S2a, b, c). In contrast, the staining pattern of CC3 was similar in Ctrl and E-OEx mice (Fig.S2d). Furthermore, the altered proliferation pattern of the uterine epithelium was also rescued in E-OEx-KO mice, suggesting it was controlled by the canonical Notch1 signaling (Fig. 2a, S2a, b, c).
Fig. 2.

Overactivation of Notch1 signaling results in a dysregulated luminal epithelium via hormone-controlled cell proliferation. (a) H&E and Ki67 staining in the uterus of Ctrl, E-OEx, and E-OEx-KO mice on 3.5 dpc. Black dotted lines mark the LE, red arrow marks the GE, Blue font marks the location of Stroma (S). (b) 3D images of the uterine lumen of Ctrl and E-OEx females on 3.5 dpc. (c) The mRNA expression of progesterone (P4) target genes in the uterus of Ctrl and E-OEx mice on 3.5 dpc (n=3, t test). (d) The mRNA expression of and estrogen (E2) target genes in the uterus of Ctrl and E-OEx mice on 3.5 dpc (n=3, t test). (e) The immunostaining of ESR1, PR, and Ac-Tubulin in the uterus of Ctrl and E-OEx mice on 3.5 dpc. (f) Digital H-Score of ESR1 expression in luminal (LE) and glandular epithelium (GE) of Ctrl and E-OEx uterus shown in e. (g) Digital H-Score of PR expression in luminal (LE) and glandular epithelium (GE) of Ctrl and E-OEx uterus shown in e (n=3 for LE, n=9 for GE, Two-way ANOVA). Values are mean ± SEM. Scale bar = 100 μm
The harmonized actions of ovarian estrogen (17β-estradiol, E2) and progesterone (P4) regulate the proliferation of uterine epithelial cells in a spatiotemporal manner [27]. Therefore, we tested if the activation of these two signaling pathways were altered in the E-OEx mice at 3.5 dpc. The qPCR results showed that the epithelial E2-responsive genes Muc4 and Ltf were significantly increased in the E-OEx uterus, while epithelial P4-responsive genes Areg and Ihh were significantly decreased at 3.5 dpc (Fig. 2c, d). However, there was no significant difference in the expression levels of stromal E2 target genes Vegf and Fgf or P4 target genes Hoxa10 and Hand2 between Ctrl and E-OEx mice (Fig. 2c, d). These results suggested that the estrogen signaling response was over-activated, and the progesterone signaling was inhibited in the E-OEx mice. We then analyzed the expression of hormone receptors ESR1 and PGR. The ESR1 expression in the LE of Ctrl and E-OEx mice were comparable, but GE expression of ESR1 was significantly lower in the E-OEx mice when compared to the Ctrl mice (Fig. 2e, f). The expression of PGR in the GE was much higher in the E-OEx mice than in the Ctrl mice, but the PGR level in the LE of the E-OEx mice was similar to the Ctrl mice (Fig. 2e, g). The expression patterns of hormone receptors indicated that the changes in E2 and P4 signaling likely occurs in the GE but not the LE. Moreover, we found that the expression pattern of the ciliated cell marker, acetylated TUBULIN (Ac-TUBULIN) followed the same pattern. It was highly expressed in the GE of Ctrl uterus, but lower in the GE of E-OEx mice (Fig. 2e). These results suggested that the over-activated Notch1 signaling might affect the GE and LE differently. On the other hand, the expression level of both ESR1, PGR and Ac-TUBULIN were significantly different between LE and GE in the Ctrl mice, suggesting the difference in hormone response of the two types of epithelial cells (Fig. 2e–f). However, the differences in expression of these proteins between LE and GE were decreased or absent in the E-OEx mice (Fig. 2e–f), suggesting that the differences between LE and GE was attenuated by the over-activation of Notch1 signaling.
3. Notch1 signaling enhances cell proliferative potential in E-OEx driven organoids.
To investigate the different effects of Notch1 signaling on LE and GE, we obtained the LE and GE fractions separately from Ctrl and E-OEx mice using our LE-GE separating protocol developed based on Cunhàs epithelium isolation method [28, 29]. The morphology of intact LE fractions from E-OEx and Ctrl mice at 3.5 dpc is shown in Fig. 3a, the intact LE fractions of Ctrl mice were much flatter than that of E-OEx mice. We first confirmed the purity of the LE and GE fractions by testing the expression of LE, GE, and stromal cell (S) markers using qPCR. In both Ctrl and the E-OEx mice, the LE and GE fractions expressed a significantly lower level of S marker Hoxa10 compared to the S fraction (Fig.S3a). The LE marker Calb1 (Calbindin 1) was significantly higher in LE than the GE and S fractions (Fig. S3b). The GE marker Foxa2 (Forkhead box A2) [30] was significantly increased in the GE fraction than the LE and S fractions (Fig. 3b). These data indicated that the LE, GE, and S fractions we separated were highly purified. However, the LE of E-OEx mice also expressed Foxa2, albeit at a much lower than the GE fraction, while the Ctrl-LE did not (Fig. 3b), indicating the LE and GE were not fully differentiated in the E-OEx uterus. On the other hand, Spink3 (Serine peptidase inhibitor kazal type 3) serves as a GE marker only when the pregnancy is successfully established [31]. Herein, the expression of Spink3 was significantly higher in GE than LE in the Ctrl mice but not in E-OEx mice, which is consistent with the infertile phenotype of the E-OEx mice (Fig. 3c).
Fig. 3.

Effects of Notch1 signaling on proliferative potential of organoids derive from LE and GE. (a) Comparison of the isolated LE morphology of Ctrl and E-OEx mice. (b, c) mRNA expression of Spink3 and Foxa2 in LE, GE, and S isolated from Ctrl and E-OEx mice on 3.5 dpc (n>3, Two-way ANOVA). (d) Morphological and histological images of LE and GE- derived organoids from Ctrl and E-OEx mice following 3 days of culture. (e,f) The number (e) and diameter (f) of the LE and GE- derived organoids from Ctrl and E-OEx mice after 3 days of culture (n>3, Two-way ANOVA). (g) Quantification of the distribution of organoid sizes after 3 days in culture. (h) Morphological images of LE and GE- derived organoids after activation of N1ICD by adenovirus with Cre recombinase (Cre-Ad) following 5 days of culture. (i) Diameter of organoids in (h) after 5 days of culture (n>3, Two-way ANOVA). Values are mean ± SEM.
Notch1 signaling promotes the cell proliferative potential in organoids from human fallopian tube epithelium [32]. We therefore hypothesized that the over-activation of Notch1 signaling in the endometrium affected the proliferative potential of LE and GE cells. We performed 3D organoid culture using isolated LE and GE fractions at 3.5 dpc from Ctrl and E-OEx mice. After three days of culture, both E-OEx LE and GE- derived organoids grew well, while the growth of organoids derived from the LE and GE from Ctrl mice was minimal (Fig. 3d). The organoids derived from the E-OEx epithelium grew too fast to be cultured longer and the spherical structure began to collapse at day 4 (Fig. S3c). The diameter of LE and GE derived organoids from the E-OEx groups were approximately 3 and 4 times bigger than those from the Ctrl mice (Fig. 3d&e). The number of both LE and GE- derived organoids in the E-OEx mice were also significantly higher than those of the Ctrl mice (Fig. 3d&f). In addition, the number of larger organoids in the E-OEx organoids was higher than the Ctrl ones (Fig. 3g). Our results suggest that the over-activation of Notch1 signaling in the endometrial epithelium promotes the proliferative potential of both LE and GE cells. Surprisingly, our data also showed that the LE-derived organoids were much smaller than the GE- derived ones in the E-OEx group, suggesting a more potent effect of Notch1 signaling on cell proliferative potential in GE than LE, at least in cells isolated at 3.5 dpc (Fig. 3d–g). To confirm this observation, we used a second approach which demonstrated that both Ctrl-LE and GE fractions-derived organoids grow faster when we overexpressed N1ICD induced by transfecting Cre recombinase via adenovirus (Cre-Ad) during the organoid culture (Fig. 3h&i).
The Ctrl LE and GE organoids were cultured for 8 days in this study and showed good morphology at the end of culture (Fig.S3c). GE fractions-derived organoids grew faster than the LE- derived ones in both diestrus and estrus stages, indicating the proliferation potential of GE fraction was higher than the LE-fraction, regardless of stage of cycle (Fig.S3d).
4. Transcriptome changes in LE and GE caused by the over-activation of Notch1 signaling.
Next, we performed RNA sequencing (RNA-seq) analysis on LE and GE fractions isolated from Ctrl and E-OEx mice at 3.5 dpc, and the comparison diagram is shown in Fig. 4a. The Venn diagram shows the number of differentially expressed genes (DEG) among all 4 comparisons (Fig.S4a–e). Comparison 1 represented the DEGs between LE fractions from Ctrl and E-OEx mice at 3.5 dpc. Of the 4134 DEG’s, 2529 were increased and 1605 were decreased (Fig.S4a, e). Gene Ontology (GO) analysis showed that the DEGs in comparison 1 were enriched in biological processes for cell maturity, epithelial cell differentiation, cellular proliferation, gland development, and stem cell development (Fig. 4b). These transcriptomic data supported the proliferative phenotype in the LE of E-OEx mice compared to the Ctrl mice at 3.5 dpc (Fig. 2a). The clustered heatmap shows that most proliferation markers were expressed at a higher level in LE and GE of E-OEx mice compared to the Ctrl epithelium (Fig. 4e).
Fig. 4.
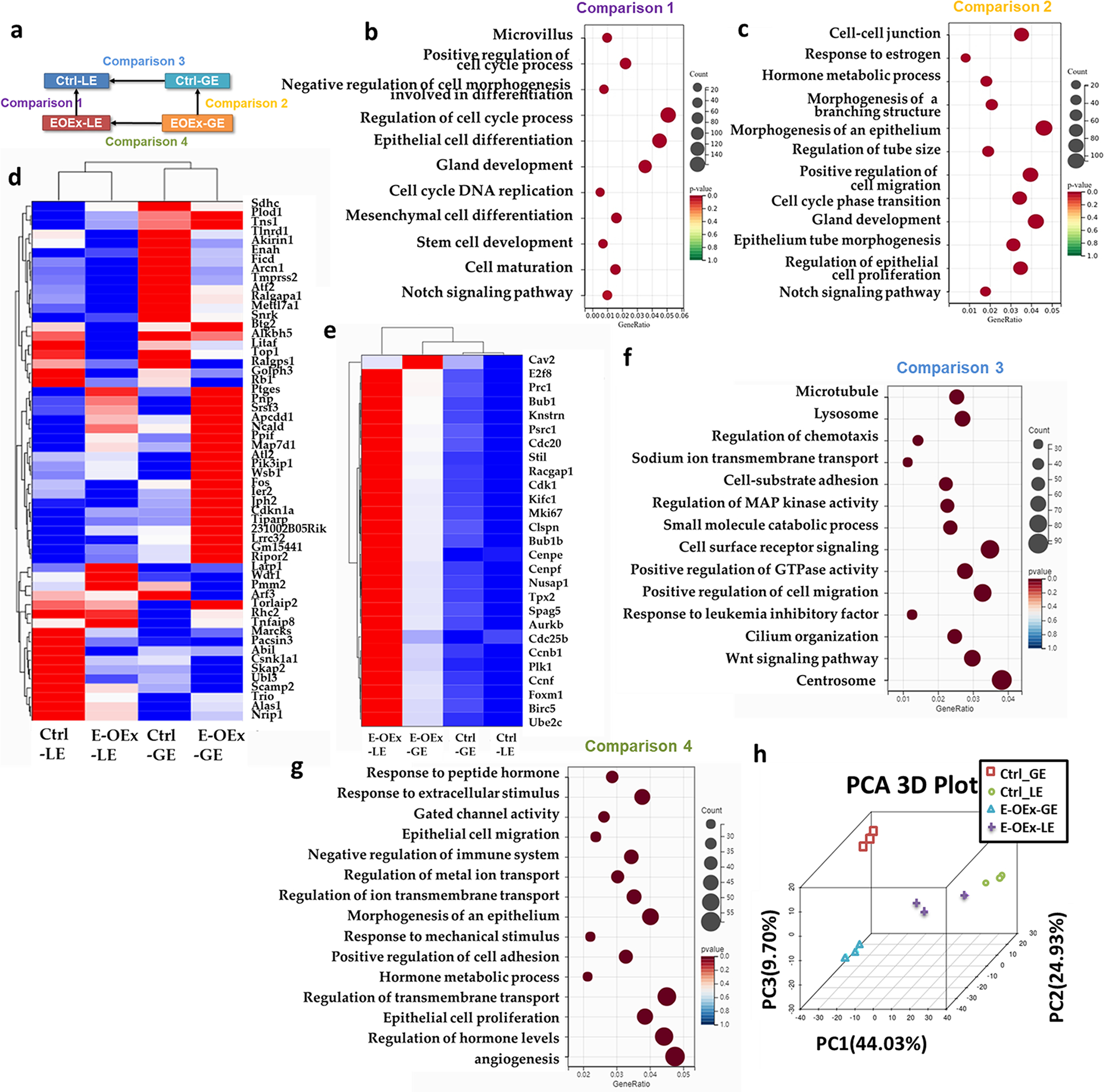
RNA-seq data analysis of LE and GE from Ctrl and E-OEx mice. (a) Diagram of the comparisons setup. (b) Gene ontology (GO) analysis of comparison 1. (c) Gene ontology (GO) analysis of comparison 2. (d) Clustered heatmap of estrogen-sensitive genes in Ctrl-LE, Ctrl-GE, E-OEx-LE, and E-OEx-GE. (e) Heatmap depicts the expression pattern proliferation associated genes in Ctrl-LE, Ctrl-GE, E-OEx-LE, and E-OEx-GE. (f) Gene ontology (GO) analysis of DEGs in comparison 3 but not 4. (g) Gene ontology (GO) analysis of DEGs in comparison 4 but not 3. (h) PCA analysis of Ctrl-LE, Ctrl-GE, E-OEx-LE, and E-OEx-GE.
In comparison 2, we enriched 3168 DEGs from GE fractions between Ctrl and E-OEx mice, of which 2146 were increased and 1022 were decreased (Fig.S4b). GO analysis found that the DEGs were enriched for cell-cell junctions, positive regulation of cell migration, hormone metabolic process, and, most importantly, response to estrogen (Fig. 4c). Hewitt et al. have described a group of direct E2 response genes in the uterus which respond to E2 treatment within 2 hours in the uterus from ovariectomized mice [33]. Therefore, we focused on the expression of these genes in our data to test if the E2 response is affected by the over-activation of Notch1 signaling. The clustered heatmap showed that the changes of estrogen-sensitive genes between Ctrl, GE and E-OEx GE were highly significant, consistent with the GO term “response to the estrogen” in comparison 2 (Fig. 4d).
LE and GE play different functional roles in the uterus [34]. To further understand the effects of Notch1 over-activation on the difference in response between LE and GE, we analyzed the DEGs between LE and GE with or without over-activation of the Notch1 signaling by overlapping the comparisons 3 and 4, which represented DEGs between LE and GE in the Ctrl and E-OEx mice, respectively (Fig.S4f). There were 3481 genes that changed significantly between the LE and GE in the Ctrl mice with the GO terms including cilium organization, Wnt signaling pathway, and response to LIF (Fig. 4f). In contrast, only 1970 genes were changed in E-OEx mice between the LE and the GE, with the GO terms related to hormone metabolic processes and response to hormones (Fig. 4g). Furthermore, the decreased number of DEGs between LE and GE in E-OEx mice compared to the Ctrl mice suggested a less differentiated status of epithelium in the E-OEx mice, consistent with the PCA analysis (Fig. 4h).
5. The GE is more sensitive to estrogen in the E-OEx mice.
During the estrous cycle, serum progesterone levels reach peak values at the diestrus stage and are lowest value at the proestrus stage, while serum estradiol levels are highest in the proestrus stage [35]. In response to E2 and P4, the endometrium undergoes extensive remodeling during each estrous cycle [36, 37]. We wondered if the disturbed proliferation status in response to E2 and P4 observed in early pregnancy in our E-OEx mice was already occurring during the estrous cycle. We detected the immunostaining of KI67 in both Ctrl and E-OEx mice during the estrous cycle (Fig. 5a). The digital H-Score suggested that the proliferation rate of GE was higher in E-OEx mice than in Ctrl mice at the diestrus stage compared to the estrus stage, but not during the metestrus (Fig. 5a, b). As a result of dynamic proliferation, the gland number changes during the estrous cycle in the Ctrl mice, which was highest at the estrus stage (Fig.S5a). However, there were no significant differences in E-OEx mice during the different stages of estrous (Fig.S5a). There were no significant differences in proliferation in LE between Ctrl and E-OEx mice at the diestrus and proestrus stages but were increased at the estrus and decreased during the metestrus stages (Fig. 5c). Considering the different estrogen levels during the different stages of the estrous cycle, and the GO term that the E-OEx-GE responds differently to the Ctrl GE, we hypothesized that the GE from E-OEx mice might be more sensitive to an estrogen response compared to the Ctrl GE.
Fig. 5.

The sensitized proliferation in response to E2 in the E-OEx mice. (a) Ki67 staining in the Ctrl and E-OEx uterus at different stages of the estrous cycle. (b) Digital H-Score of GE KI67 expression in a (n=12, Two-way ANOVA). (c) Digital H-Score of LE KI67 expression in a (n=3, Two-way ANOVA). (d) H&E staining of the uterus from ovariectomized Ctrl and E-OEx mice after being treated with sesame oil (Vehicle), lower (10 ng/0.1ml) and higher (100 ng/0.1 ml) E2, and E2 + P4 (100 ng/0.1 ml and 1 mg/0.1 ml). Black arrow showed the glands. Black dotted lines mark the LE, red arrow marks the GE, Blue fonts mark the location of Stroma (S). (e) Cell counts per gland in the uterus from Ctrl and E-OEx mice (n=12, Two-way ANOVA). Immunohistochemistry of the KI67 (f) and FOXA2 (g) of Ctrl, E-OEx, and E-OEx-KO uterus. (h) KI67 positive cells/total cell of GE in the uterus from Ctrl and E-OEx mice (n=12, Two-way ANOVA). (i) 3D imaging of the Ctrl and E-OEx uterus treated with Vehicle or 100ng/0.1ml E2, glands are stained with FOXA2. Values are mean ± SEM.
To test this hypothesis, we treated ovariectomized mice s.c. with sesame oil, E2 (10 ng/0.1 ml or 100 ng/0.1 ml), or E2 (100 ng/0.1 ml) plus P4 (1 mg/0.1 ml) for 24 h to confirm the different responses of Ctrl, E-OEx and E-OEx-KO mice to estrogen. The morphology and weight of the uterus in both Ctrl and E-OEx mice are shown in Fig.S5b&c. Histological staining of the uterus showed bigger glands in the E-OEx mice than in the Ctrl mice. The number of cells per gland in the E-OEx mice was significantly higher than that of Ctrl mice in all the hormone treatment groups except in the vehicle control (Fig. 5d&e). In the Ctrl mice, the cell number per gland was not affected by hormone treatment (Fig. 5d&e). However, in the E-OEx uterus, the number of cells per gland increased in the 10, 100 ng/0.1 ml E2 in a dose-dependent manner (Fig. 5d&e). In the E2+P4 group, the number of cells per gland was lower than E2 treatment group, but still higher than the vehicle group (Fig. 5d&e). The gland number of E-OEx mice was higher than Ctrl mice after being treated with hormones during the estrous cycle (Fig.S5d).
Next, we tested the immune localization of Ki67, and FOXA2 staining was used to identify glands on the adjacent sections stained for Ki67 in Ctrl, E-OEx, and the E-OEx-KO mice (Fig. 5f, g). The number of Ki67 positive cells in glands was significantly higher in the vehicle treated E-OEx mice compared to the Ctrl uterus, but in the E-OEx-KO mice, it was comparable to the Ctrl mice, suggesting that the increase in proliferation induced by the over-activation of Notch1 signaling in the vehicle treatment group was through the RBPJ dependent canonical cascade (Fig. 5h). When treated with a lower (10 ng/0.1 ml) and higher dose (100 ng/0.1 ml) of E2, the GE proliferation rate was increased in a dose-dependent manner in Ctrl mice, where it increased approximately 35% and 70% at the lower and higher doses, respectively (Fig. 5h). In contrast, the proliferation rate reached 100% at both the lower and higher doses in the E-OEx mice, and the deletion of RBPJ did not rescue the higher proliferation in the E-OEx-KO mice, demonstrating that the proliferation of GE cells in response to E2 was sensitized by the over-activation of Notch1 signaling in a RBPJ independent non-canonical manner (Fig. 5h). Furthermore, with P4 intervention, the GE proliferation rate of the Ctrl mice was significantly decreased compared to the E2 (100 ng/ml) only group, while there was no anti-estrogenic effect of P4 in the E-OEx and E-OEx-KO mice, suggesting that the antagonism of P4 to E2 was abolished when non-canonical Notch1 signaling was over-activated (Fig. 5f&h). Meanwhile, the proliferation rate in the LE showed no significant difference among the Ctrl, E-OEx, and E-OEx-KO mice in all the treatment groups, showing that the sensitized response to E2, and abolished antagonism of P4 by Notch1 signaling did not occur in LE (Fig.S5e). In addition, we performed 3D imaging using FOXA2 staining to show an integral picture of the glands in Ctrl and E-OEx mice. Each gland was separately identified by making surfaces and pseudocoloring them (Fig. 5i). The gland volume in Ctrl and E-OEx mice was increased by the E2 treatment, but the increase in gland volume was more pronounced in E-OEx mice (Fig.S5f). Collectively, our results support the hypothesis that the over-activation of Notch1 signaling sensitized GE to proliferate in response to E2 and further showed that the sensitization was through the non-canonical cascade.
Discussion
Embryo implantation is a key event in establishing pregnancy in mammals. Successful implantation requires dynamic remodeling of endometrium in response to the ovarian hormones E2 and P4 to initiate the appropriate cross-talk with the embryo [8, 38]. Previously, we have reported that the over-activation of Notch1 in the uterus under Pgr-Cre leads to implantation and decidualization failure [12]. However, the Pgr-Cre we used previously is active in both stromal and epithelial cells postnatally, which cannot differentiate whether the infertile phenotype caused by the over-activation of Notch1 signaling is through epithelial or stromal cells, or both. Furthermore, the postnatal activation of Pgr-Cre also leads to difficulties to clarify the developmental defects from the physiological response. In this study, using the Ltf-iCre that only activates in the epithelium of adult animals, we found that the over-activated canonical Notch1 signaling in epithelial cells during adulthood was sufficient to lead to infertility in mouse. Uterine receptivity was interrupted by the epithelial-specific over-activation of Notch1 signaling. The E-OEx mice failed to respond to the artificial decidualization stimulus, and this was associated with dysregulated epithelial proliferation, differentiation, and secretory functions, but not a development defect of the glands. The findings of this study further refines the cellular mechanisms by which overactivation of the Notch1 signaling pathway affects the receptive state of the uterus. Furthermore, in this study, we have demonstrated that the non-canonical Notch1 signaling contributed to the sensitivity of GE cells to estrogen, providing a novel possibility to understand the pathological mechanism of estrogen-dependent diseases such as endometriosis and adenomyosis. A summary diagram highlighting the findings in our study is shown in Figure 6.
Fig. 6.
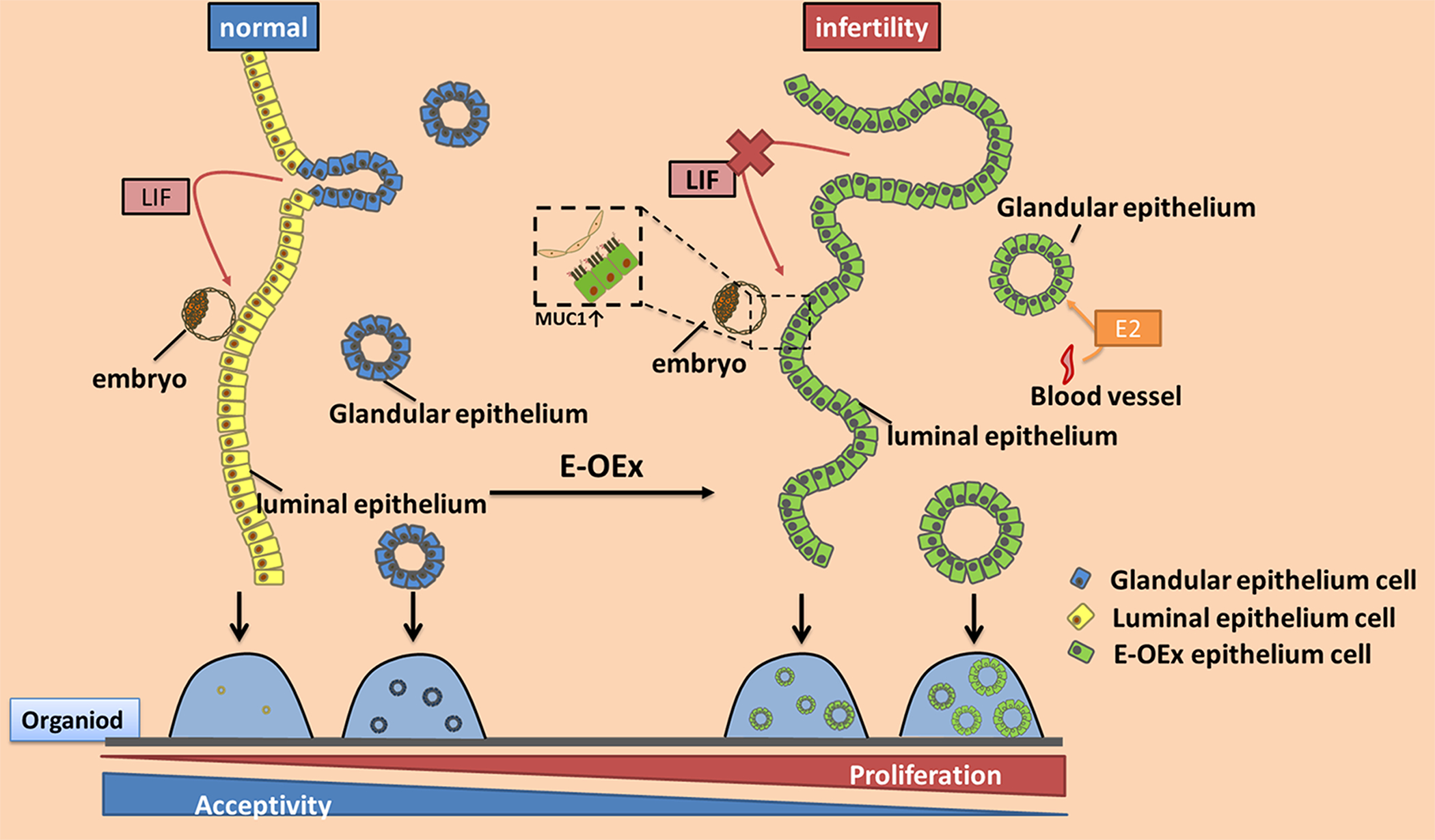
A diagrammatic summary of the results of this study. Over-activation of Notch1 signaling results in hyperproliferation of both LE and GE cells, weakens the differentiation features and function of GE, resulting in decreased LIF levels and impaired uterine receptivity. Moreover, the higher level of Notch1 signaling enhances the sensitivity of the GE to estrogen, which allows the GE cells to proliferate under lower estrogen levels during estrous cycle, and results in hyperproliferation of GE. Similarly, in the in vitro culture, over-activation of Notch1 signaling increases the regeneration capacity of both LE and GE in organoid.
The two types of endometrial epithelia, LE and GE, play different roles during embryo implantation. The LE proliferates under the control of E2, then the proliferation is inhibited on 3.5 dpc, and the LE undergoes depolarization in response to P4 in mice. This change in the LE is critical for the attachment and implantation of the embryos. Many studies have reported that the dysregulation in the cross-talk between epithelial estrogen and stromal progesterone signaling is associated with the hyper-proliferation of the LE cells and implantation failure [6, 39]. Previously, in the Pgr-Cre driven Notch1 over-activation model, we observed increased estrogen signaling and decreased progesterone signaling in both the epithelium and stroma of the uterus on 3.5 dpc [12]. A similar pattern of the two signaling pathways has been observed in this study. However, only the epithelial target genes of P4 are altered, and the stromal P4 target gene Hoxa10 expression is comparable between Ctrl and E-OEx mice, suggesting the changes in the epithelial estrogen and progesterone signaling is affected by the over-activation of Notch1 signaling directly in the epithelium, and does not involve epithelium-stromal crosstalk.
In mice, the development of glands begins postnatally, in coordination with the underlying stroma. After a series of processes which include budding, tubule formation, luminal epithelial curling, and branching, the single layer of epithelium from the postnatal uterus differentiates into the LE and GE [34]. The gland and its secretions are essential for pregnancy and play biological roles in uterine receptivity, survival, growth, and implantation of embryos, decidualization of stromal cells, and placental development [40]. In our previous study, the over-activation of Notch1 signaling in the postnatal period driven by Pgr-Cre resulted in the absence of uterine glands contributing to embryo implantation failure [12]. In the present study, the E-OEx uterus displays normal gland development, as the Ltf-iCre is only activated in adulthood. In mice, one of the most essential secretions of the uterine glands during embryo implantation is LIF, encoded by Lif, the first gene that was shown to be critical for embryo implantation in a knockout mouse model [41]. Supplementation of recombinant LIF protein can rescue the implantation failure in many studies, including gland-null mice induced by knocking out Foxa2 [30]. In the Pgr-driven Notch1 over-activation mice, we showed that the Lif expression is not changed, even though no glands are present in the uterus. Further investigation showed that the LIF protein is synthesized by the LE instead [12]. However, in Ltf-iCre driven mice, the expression of Lif mRNA is significantly decreased, indicating that the glands in these mice are not fully functional. Thus, our collective data shows that the over-activation of Notch1 signaling affects uterine gland development and function differently in the postnatal stage and adulthood: if Notch1 signaling is over-activated before the initiation of gland development, it prevents the development of glands but turns the LE to a GE-like phenotype; if it is over-activated after gland development, it abolishes the function of the glands. However, the supplementation of LIF protein cannot rescue embryo implantation in our E-OEx mice, suggesting other factors contribute to the receptivity phenotype.
Both LE and GE undergo regeneration during the estrous cycle or postpartum repair in adulthood. A recent study showed that the stem/progenitor cells for the regeneration are mainly from the epithelium and not the stroma [42]. The Lgr5+ or Axin2+ cells are thought to be the stem/progenitor cells, evidenced by both the in vivo mouse model and in vitro 3D cultured organoids. Many studies have shown that Notch1 signaling controls the regeneration and differentiation of the female reproductive tract epithelium. Continuous growth and differentiation of human fallopian tube organoids depends on Notch paracrine signaling [32]. At the transition zone of the cervix, the activation of Wnt signaling supports the growth of columnar epithelium, while Notch1 signaling guides the epithelium to be stratified [44]. In this study, the over-activation of Notch1 signaling increases the proliferative potential of both the LE or GE cells, suggesting that Notch1 signaling also contributes to the regeneration of epithelial cells in the uterus. However, our data show that the proliferative potential of LE cells is significantly lower than the GE cells in both the Ctrl and E-OEx mice, evidenced by the growth characteristics of the organoids. This could be either due to lower number of Lgr5 + progenitor cells or the decreased proliferation potential of LE regulated by the stromal P4 signaling via the inhibition of the epithelial estrogen signaling. Nonetheless, more detailed studies are needed to further understand the role of stem cells in LE regeneration. During early pregnancy, hyper-proliferation of LE is only observed at 3.5 dpc in the E-OEx mice. At 1.5 dpc and 2.5 dpc, proliferation in LE is lower in the E-OEx mice compared to the Ctrl mice. During estrus, proliferation in the E-OEx LE is higher in the estrus phase compared to the Ctrl but in the metestrus phase it is lower than the Ctrl. Given that LE cells of the E-OEx mice do not consistently show a higher rate of proliferation when compared to the controls the hyper-proliferation phenotype of the uterine lumen is only partially explained by the hyper-proliferation of the LE cells. On the other hand, the GE cells consistently show higher rates of proliferation in the E-OEx mice compared to the Ctrl mice both during the estrous cycle and during early pregnancy. Further, the LE fraction from E-OEx mice expresses some level of Foxa2, a GE marker. We hypothesize that a fraction of GE cells may lose their glandular character and become part of the LE through an unknown mechanism. Thus, the hyper-proliferated GE cells may contribute to the hyper-proliferation of the uterine luminal cells in the E-OEx mice. However, this hypothesis would require lineage labeling with a glandular marker and further investigation.
Previous studies demonstrated the interaction between estrogen and Notch1 signaling in the uterus. Uterine-specific knockout of Notch1 decreases the response of endometrium to estrogen and further leads to decreased cellular proliferation and the impairment of decidualization. On the other hand, over-activation of Notch1 in the uterus results in complete infertility, and Notch1 signaling up-regulates estrogen signaling because of the hypermethylation of the progesterone receptor [12]. In our study, the response of GE cells to estrogen is sensitized by the over-activation of Notch1 signaling, evidenced by the increased number of Ki67 positive cells. This process is mediated mainly by non-canonical Notch pathways, as the knockout of RBPJ, the transcription factor of canonical Notch1 signaling, cannot rescue the hyperproliferation of the GE cells. However, the canonical Notch1 signaling may also partially contribute to the proliferation, as evidenced by the reduced proliferation rate in the E-OEx-KO mice in all the hormonal conditions compared to the E-OEx mice. Endometriosis is an estrogen-dependent chronic inflammatory disease that affects women in their reproductive years and is associated with pelvic pain and infertility [45]. We have previously reported that the decreased Notch1 signaling contributes to the impaired decidualization of stromal cells in the eutopic endometrium of women with endometriosis [16]. However, NOTCH1 is significantly up regulated in the glandular epithelium (GE) of ectopic endometrium compared to the eutopic endometrium [46]. Therefore, the increased sensitivity of GE to estrogen by the over-activation of the Notch1 signaling might contribute to the growth of ectopic glandular epithelial cells in endometriosis. Adenomyosis is characterized by the presence of activated endometrial glands within the myometrium. The expression of NOTCH1 and its intracellular domain N1ICD is higher in the eutopic endometrium of women with adenomyosis [47], suggesting a similar mechanism might also contribute to the development of adenomyosis. Further exploration of the role of Notch1 signaling on epithelial cells responding to estrogen may be helpful in developing therapeutic approaches and improving the fertility outcome of infertile women with these diseases.
In summary, we have investigated the effects of epithelium-specific activation of Notch1 signaling on the function of the mouse uterus. We find that the over-activation of Notch1 signaling leads to infertility via dysregulated cellular proliferation, differentiation, and function of LE and GE in a RBPJ dependent manner. In addition, we demonstrate that higher Notch1 signaling activity increases the stemness of epithelial stem/progenitor cells and sensitizes the response of GE cells to estrogen through a non-canonical cascade. Further research on these mechanisms might explain the pathology observed in endometriosis and adenomyosis in human patients.
Supplementary Material
Acknowledgments
We thank South China Agricultural University for providing the Public Experimental Platform.
Funding Statement
This study is supported by the National Natural Science Foundation of China (31900601, 31771664) to RWS, the National Key Research and Development Program of China (2018YFC1004400) to RWS, NIH R01grant (HD099090) to ATF, and March of Dimes (5-FY20-209) and NIH R01grant (HD109152) to RA.
Footnotes
Conflict of Interest
The authors declare no competing financial interests.
Ethic Statement
Our studies did not include human participates, human data or human tissues. All mice were housed in the SPF Animal Facility with the approval of the Institutional Animal Care and Use Committee of South China Agricultural University.
Data Availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
References
- 1.Kelleher AM, Demayo FJ and Spencer TE Uterine Glands: Developmental Biology and Functional Roles in Pregnancy. Endocr Rev. 2019; 40: 1424–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wood GA, Fata JE, Watson KLM and Khokha R Circulating hormones and estrous stage predict cellular and stromal remodeling in murine uterus. Reproduction. 2007; 133: 1035–1044. [DOI] [PubMed] [Google Scholar]
- 3.Cha J, Sun X and Dey SK Mechanisms of implantation: strategies for successful pregnancy. Nat Med. 2012; 18: 1754–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Habiba M, Heyn R, Bianchi P, Brosens I and Benagiano G The development of the human uterus: morphogenesis to menarche. Hum Reprod Update. 2021; 27: 1–26. [DOI] [PubMed] [Google Scholar]
- 5.Feng D, Menger MD, Wang H and Laschke MW Luminal epithelium in endometrial fragments affects their vascularization, growth and morphological development into endometriosis-like lesions in mice. Dis Model Mech. 2014; 7: 225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kelleher AM, Milano-Foster J, Behura SK and Spencer TE Uterine glands coordinate on-time embryo implantation and impact endometrial decidualization for pregnancy success. Nature Communications. 2018; 9: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carson DD, Bagchi I, Dey SK, Enders AC, Fazleabas AT and Lessey BA et al. Embryo implantation. Dev Biol. 2000; 223: 217–237. [DOI] [PubMed] [Google Scholar]
- 8.Singh H and Aplin JD Adhesion molecules in endometrial epithelium: tissue integrity and embryo implantation. Journal of Anatomy. 2009; 215: 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shariati M, Niknafs B, Seghinsara AM, Shokrzadeh N and Alivand MR Administration of dexamethasone disrupts endometrial receptivity by alteration of expression of miRNA 223, 200a, LIF, Muc1, SGK1, and ENaC via the ERK1/2-mTOR pathway. J Cell Physiol. 2019; 234: 19629–19639. [DOI] [PubMed] [Google Scholar]
- 10.Arora R, Fries A, Oelerich K, Marchuk K, Sabeur K and Giudice LC et al. Insights from imaging the implanting embryo and the uterine environment in three dimensions. Development. 2016; 143: 4749–4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seishima R, Leung C, Yada S, Murad KBA, Tan LT and Hajamohideen A et al. Neonatal Wnt-dependent Lgr5 positive stem cells are essential for uterine gland development. Nature Communications. 2019; 10: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Su R, Strug MR, Jeong J, Miele L and Fazleabas AT Aberrant activation of canonical Notch1 signaling in the mouse uterus decreasesprogesterone receptor by hypermethylation and leads to infertility. Proceedings of the National Academy of Sciences. 2016; 113: 2300–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bray SJ Notch signalling in context. Nat Rev Mol Cell Biol. 2016; 17: 722–735. [DOI] [PubMed] [Google Scholar]
- 14.Zhou B, Lin W, Long Y, Yang Y, Zhang H and Wu K et al. Notch signaling pathway: architecture, disease, and therapeutics. Signal Transduction and Targeted Therapy. 2022; 7: 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Afshar Y, Jeong JW, Roqueiro D, Demayo F, Lydon J and Radtke F et al. Notch1 mediates uterine stromal differentiation and is critical for complete decidualization in the mouse. The FASEB Journal. 2011; 26: 282–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Su RW, Strug MR, Joshi NR, Jeong JW, Miele L and Lessey BA et al. Decreased Notch pathway signaling in the endometrium of women with endometriosis impairs decidualization. J Clin Endocrinol Metab. 2015; 100: E433–E442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang S, Kong S, Wang B, Cheng X, Chen Y and Wu W et al. Uterine Rbpj is required for embryonic-uterine orientation and decidual remodeling via Notch pathway-independent and -dependent mechanisms. Cell Res. 2014; 24: 925–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Madhavan MK, Demayo FJ, Lydon JP, Joshi NR, Fazleabas AT and Arora R Aberrant uterine folding in mice disrupts implantation chamber formation and alignment of embryo-uterine axes. Development. 2022; 149: v200300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strug MR, Su RW, Kim TH, Mauriello A, Ticconi C and Lessey BA et al. RBPJ mediates uterine repair in the mouse and is reduced in women with recurrent pregnancy loss. The FASEB Journal. 2018; 32: 2452–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng HT, Zhang HY, Chen ST, Li MY, Fu T and Yang ZM The detrimental effects of stress-induced glucocorticoid exposure on mouse uterine receptivity and decidualization. The FASEB Journal. 2020; 34: 14200–14216. [DOI] [PubMed] [Google Scholar]
- 21.Song H, Lim H, Das SK, Paria BC and Dey SK Dysregulation of EGF family of growth factors and COX-2 in the uterus during the preattachment and attachment reactions of the blastocyst with the luminal epithelium correlates with implantation failure in LIF-deficient mice. Mol Endocrinol. 2000; 14: 1147–1161. [DOI] [PubMed] [Google Scholar]
- 22.Tsai JH, Schulte M, O’Neill K, Chi MM, Frolova AI and Moley KH Glucosamine inhibits decidualization of human endometrial stromal cells and decreases litter sizes in mice. Biol Reprod. 2013; 89: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pawar S, Starosvetsky E, Orvis GD, Behringer RR, Bagchi IC and Bagchi MK STAT3 Regulates Uterine Epithelial Remodeling and Epithelial-Stromal Crosstalk During Implantation. Molecular Endocrinology. 2013; 27: 1996–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu W, Feng Z, Teresky AK and Levine AJ p53 regulates maternal reproduction through LIF. Nature. 2007; 450: 721–724. [DOI] [PubMed] [Google Scholar]
- 25.Paria BC, Lim H, Wang XN, Liehr J, Das SK and Dey SK Coordination of differential effects of primary estrogen and catecholestrogen on two distinct targets mediates embryo implantation in the mouse. Endocrinology. 1998; 139: 5235–5246. [DOI] [PubMed] [Google Scholar]
- 26.Cha JM and Dey SK Reflections on Rodent Implantation. Adv Anat Embryol Cell Biol. 2015; 216: 69–85. [DOI] [PubMed] [Google Scholar]
- 27.Kim H, Kim YS, Yoon JA, Yang SC, Park M and Seol D et al. Estrogen induces EGR1 to fine-tune its actions on uterine epithelium by controlling PR signaling for successful embryo implantation. The FASEB Journal. 2018; 32: 1184–1195. [DOI] [PubMed] [Google Scholar]
- 28.Bigsby RM, Cooke PS and Cunha GR A simple efficient method for separating murine uterine epithelial and mesenchymal cells. Am J Physiol. 1986; 251: E630–E636. [DOI] [PubMed] [Google Scholar]
- 29.Qixin Xu WZ L.L. Distinguish characters of luminal and glandular epithelium from mouse uterus using a novel enzyme-based separation method. BioRxiv. 2022; [DOI] [PubMed] [Google Scholar]
- 30.Kelleher AM, Peng W, Pru JK, Pru CA, Demayo FJ and Spencer TE Forkhead box a2 (FOXA2) is essential for uterine function and fertility. Proceedings of the National Academy of Sciences. 2017; 114: E1018–E1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen W, Han BC, Wang RC, Xiong GF and Peng JP Role of secretory protease inhibitor SPINK3 in mouse uterus during early pregnancy. Cell Tissue Res. 2010; 341: 441–451. [DOI] [PubMed] [Google Scholar]
- 32.Kessler M, Hoffmann K, Brinkmann V, Thieck O, Jackisch S and Toelle B et al. The Notch and Wnt pathways regulate stemness and differentiation in human fallopian tube organoids. Nat Commun. 2015; 6: 8989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hewitt SC, Deroo BJ, Hansen K, Collins J, Grissom S and Afshari CA et al. Estrogen Receptor-Dependent Genomic Responses in the Uterus Mirror the Biphasic Physiological Response to Estrogen. Molecular Endocrinology. 2003; 17: 2070–2083. [DOI] [PubMed] [Google Scholar]
- 34.Vue Z, Gonzalez G, Stewart CA, Mehra S and Behringer RR Volumetric imaging of the developing prepubertal mouse uterine epithelium using light sheet microscopy. Molecular Reproduction and Development. 2018; 85: 397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang M, Sun Y, Fu J, Song H, Xiao Z and Yang Q et al. mTORC1 signaling pathway integrates estrogen and growth factor to coordinate vaginal epithelial cells proliferation and differentiation. Cell Death Dis. 2022; 13: 862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin S Bipotent stem cells support the cyclical regeneration of endometrial epithelium of the murine uterus. Proceedings of the National Academy of Sciences. 2019; 116: 6848–6857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Q and Paria BC Importance of Uterine Cell Death, Renewal, and Their Hormonal Regulation in Hamsters that Show Progesterone-Dependent Implantation. Endocrinology. 2006; 147: 2215–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matsumoto L, Hirota Y, Saito-Fujita T, Takeda N, Tanaka T and Hiraoka T et al. HIF2α in the uterine stroma permits embryo invasion and luminal epithelium detachment. The Journal of Clinical Investigation. 2018; 128: 3186–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ye X Uterine Luminal Epithelium as the Transient Gateway for Embryo Implantation. Trends in Endocrinology & Metabolism. 2020; 31: 165–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kelleher AM, Burns GW, Behura S, Wu G and Spencer TE Uterine glands impact uterine receptivity, luminal fluid homeostasis and blastocyst implantation. Sci Rep. 2016; 6: 38078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stewart CL, Gadi I, Köntgen F, Abbondanzo SJ, Brunet LJ and Bhatt H et al. Blastocyst implantation depends on maternal expression of leukaemia inhibitory factor. Nature (London). 1992; 359: 76–79. [DOI] [PubMed] [Google Scholar]
- 42.Seishima R, Leung C, Yada S, Murad KBA, Tan LT and Hajamohideen A et al. Neonatal Wnt-dependent Lgr5 positive stem cells are essential for uterine gland development. Nature Communications. 2019; 10: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Syed SM, Kumar M, Ghosh A, Tomasetig F, Ali A and Whan RM et al. Endometrial Axin2+ Cells Drive Epithelial Homeostasis, Regeneration, and Cancer following Oncogenic Transformation. Cell Stem Cell. 2020; 26: 64–80. [DOI] [PubMed] [Google Scholar]
- 44.Chumduri C, Gurumurthy RK, Berger H, Dietrich O, Kumar N and Koster S et al. Opposing Wnt signals regulate cervical squamocolumnar homeostasis and emergence of metaplasia. Nature Cell Biology. 2021; 23: 184–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wallach EE and Schmidt CL Endometriosis: a reappraisal of pathogenesis and treatment. Fertility and Sterility. 1985; 44: 157–173. [DOI] [PubMed] [Google Scholar]
- 46.Song Y, Su RW, Joshi NR, Kim TH, Lessey BA and Jeong JW et al. Interleukin-6 (IL-6) Activates the NOTCH1 Signaling Pathway Through E-Proteins in Endometriotic Lesions. J Clin Endocrinol Metab. 2020; 105: 1316–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qi S, Zhao X, Li M, Zhang X, Lu Z and Yang C et al. Aberrant expression of Notch1/numb/snail signaling, an epithelial mesenchymal transition related pathway, in adenomyosis. Reprod Biol Endocrinol. 2015; 13: 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.