


Abstract
An efficient insertional mutagenesis system has been developed for Schizosaccharomyces pombe based on linear PCR-generated cassettes containing selectable markers. It depends upon illegitimate recombination for integration into the genome. Various selectable markers of different sizes can be used to obtain sufficiently high transformation and integration frequencies. Based on Southern blotting, a single insertion is found in each strain and integration sites are broadly distributed in the genome. Sequence analysis of the insert junctions frequently reveals small regions of homology (4–10 bp) between the ends of the integrated cassette and the disrupted gene. The system has been used for simple genetic screens of various types and as a promoter trap for in-frame GFP fusions.
INTRODUCTION
In Saccharomyces cerevisiae there is efficient integration of homologous DNA into the genome (1). Linear DNA with free ends within the region of homology gives the maximum frequency of homologous integration (2), enabling targeted gene disruptions and tagging (1,3). Although the one-step gene disruption method (4) is routinely used to generate deletion strains, a faster, cloning-free alternative is now available. This uses PCR-amplified constructs based on amplifying selectable markers using primers terminating in genomic sequences. These primers may have homologous regions as short as 40 bp (5,6). Extensions of this technology include constructs carrying several types of protein tags, such as Myc or GFP (green fluorescent protein), that can be precisely fused with the target gene (7,8). In Schizosaccharomyces pombe, similar tools are available, but longer stretches (60–80 bp) of homology are required for targeted integration (9,10).
In the absence of homologous regions, illegitimate recombination is thought to occur at a much higher rate in S.pombe than in S.cerevisiae (1,11,12). Non-homologous DNA end joining, an event of illegitimate recombination, has been shown to occur more efficiently in S.pombe than in S.cerevisiae (11,12). In this paper, we show that this frequency in S.pombe is sufficiently high to be readily exploited as an insertional mutagenesis technique. Mutants selected or screened based on this method allow for the retrieval of the gene without the need for cloning by functional complementation. Since the S.pombe genome sequencing project is virtually completed (www.sanger.ac.uk ), mutants isolated by this technique can be quickly identified. We show that the technique has general applicability using various screens, selectable markers and tagged constructs.
MATERIALS AND METHODS
Strains and media
Schizosaccharomyces pombe strains used in this study are listed in Table 1. Media preparation and manipulations of cells were performed as described previously (13–16).
Table 1. Strain list.
| Strain | Genotype | Source |
|---|---|---|
| Q252 | ade6-704 h+ | Laboratory collection |
| Q1411 | ura4-D18 h– | Laboratory collection |
| Q1412 | ade1-D25 h+ | Laboratory collection |
| Q1620 | ade1-D25 ura4-D18 h90 | This study |
Molecular techniques
Standard molecular biology protocols were used (17,18). Table 2 shows the sequences of the oligo primers used to amplify the cassettes and for inverse PCR (19). The structures of the PCR cassettes used for insertional mutagenesis are presented in Figure 1.
Table 2. Oligo primers used for amplification of cassettes and inverse PCR.

a18 bp random extensions were added to the 5′-ends to generate the random tailed construct.
b,cNested primers that produce a 180 bp band shift of the inverse PCR product.
Figure 1.
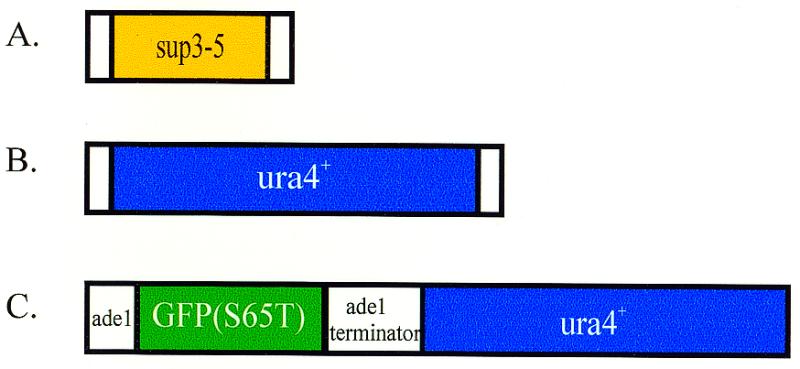
PCR cassettes used for insertional mutagenesis. (A) The sup3-5 gene and (B) the ura4+ gene (including coding and flanking sequences) were used as selectable markers. The 18 bp random extensions are indicated. (C) The GFP-containing cassette used to generate gene fusion mutants. The GFP gene is preceded by 150 bp of the ade1+ gene to protect the GFP ORF during integration and is followed by the ade1+ terminator. This construct was used in an ade1– ura4– deletion strain.
Amplification of the ura4+, ade1+ and sup3-5 PCR cassettes was performed in 100 µl reactions containing 60 pmol of each primer, a final MgCl2 concentration of 1.5 mM and 2.5 U of Taq polymerase (Promega or Pharmacia). The amplification conditions were as follows: an initial heat denaturation step of 95°C for 2 min, addition of Taq polymerase, followed by 35 cycles of denaturation (95°C for 60 s), primer annealing (55°C for 30 s) and extension (72°C for 90 s).
The amplification conditions of the larger GFP PCR cassette varied slightly from above. Specifically, 100 pmol of each primer, a final concentration of 1.5 mM MgCl2 and 2.8 U of high fidelity Taq polymerase (Roche Molecular Biochemicals) were used in 50 µl reactions. The PCR reactions were subjected to an initial heat denaturation step of 94°C for 4 min, followed by 35 cycles of amplification (five cycles of 94°C for 45 s, 50°C for 30 s, 68°C for 4 min; 30 cycles of 94°C for 45 s, 55°C for 30 s, 68°C for 4 min) and a final extension of 68°C for 7 min.
For inverse PCR (IPCR) (19), genomic DNA was isolated (16) and digested with restriction enzymes such as HhaI (Promega) and/or EcoRI (Promega) that do not cut within the ura4+ or GFP cassettes. The reaction was then heat inactivated at 65°C for 20 min, treated with phenol/chloroform and ethanol precipitated for at least 1 h at –20°C. The digested DNA from the ura4+ insertional strains and GFP fusion mutants was ligated at concentrations of 5 µg/ml (100 ng in 20 µl final volume) and 0.5 µg/ml (100 ng in 200 µl final volume), respectively. Three units of T4 DNA ligase (Promega) was added to each reaction, which was allowed to proceed for 16 h at 12°C. The ligation reaction was then heat inactivated at 65°C for 20 min. For the 20 µl reactions, the entire ligation was used directly in the IPCR reaction with no additional MgCl2. The 200 µl ligation reactions were treated with phenol/chloroform and ethanol precipitation prior to IPCR. The amplification conditions were as above except that a 5 min hot start and a longer extension time of 4 min were used for the ura4+ insertional experiments. A re-amplification using 2 µl of the reaction as template with the same temperature and time profiles was performed if no PCR product was present after the first round of amplification.
Transformation and selection of stable integrants
All transformations were by the lithium acetate method (20). Specifically, a 100 ml cell culture was grown to a density of 0.5–1.0 × 107 cells/ml in EMM low glucose (5 g/l) plus supplements at 30°C. The cells were then harvested by centrifugation (3000 r.p.m., 5 min), washed twice with 20 ml of 0.1 M lithium acetate (pH 4.9), resuspended in 0.1 M lithium acetate (pH 4.9) to a final density of 1 × 109 cells/ml and 100 µl aliquots were dispensed into microcentrifuge tubes. The cells were incubated at 25°C for 1 h. Transforming DNA (1/10 volume of a PCR reaction, 0.3–1.0 µg DNA) was mixed with 5 µl of 10 mg/ml sonicated salmon sperm DNA and added to each aliquot of cells. After a further 1 h incubation at 25°C, 290 µl of 50% PEG3350 (dissolved in 0.1 M lithium acetate, pH 4.9) was added and mixed by gentle pipetting. The cells were then incubated at 25°C for 1 h, heat shocked at 43°C for 15 min and allowed to recover at room temperature for 10 min. The cells were harvested by centrifugation (3000 r.p.m., 5 min) and the pellet resuspended in 10 ml of 0.5× YEA medium. This was followed by shaking for 1 h at 30°C. The cells were then harvested, resuspended in sterile water and plated onto selective media.
After 3–5 days at 30°C, transformants were maintained on non-selective medium for 1 week by replica plating once every 2 days on YEA. The transformants were then replica plated onto selective medium (EMM) and the stable integrants isolated as surviving colonies. Strains transformed with the ura4+ gene were checked for integration by demonstrating their sensitivity to 5-FOA. Routinely, 80–85% of the strains were integrants after 3 days growth on non-selective medium.
RESULTS AND DISCUSSION
High integration efficiencies are obtained by transformation of a variety of selectable markers of different sizes
An insertional mutagenesis system requires high transformation and integration efficiency of a non-homologous selectable marker. To determine whether this was achievable in S.pombe, we tested PCR products of various lengths (469–4000 bp) containing different selectable markers (ura4+ and sup3-5). Using the lithium acetate method (20), transformation efficiencies achieved with the ura4+ cassette were similar to a larger ura4+ cassette containing additional sequences of the GFP and ade1+ gene, altogether ranging from 882 to 7400 stable integrants/µg DNA in various experiments (mean ± SE, 3410 ± 79, n = 21). In contrast, the smaller sup3-5 cassette produced a mean transformation efficiency of 268 ± 34 stable integrants/µg DNA (n = 6). The low frequency of stable transformants exhibited by the sup3-5 cassette is presumably due to the lethal effects associated with expression of this nonsense suppressor tRNA (21). Although cells with single copies of sup3-5 grow well, it is known that multicopy expression is lethal (21). Any cell taking up multiple copies of the sup3-5 cassette prior to integration would display growth suppression and be lost. Similarly, it is known that unstable transformants contain multiple extrachromosomal copies of the selectable marker (22), which may account for the low numbers of unstable transformants with the sup3-5 cassette. This might explain the observed unstable transformant frequencies that appeared construct specific, ranging from ~85% of total transformants with the ura4+ cassettes and 15% of total transformants with the sup3-5 cassette. However, the unstable transformants do not interfere with insertional mutagenesis since they can be eliminated by maintaining the transformants on non-selective media for a few days (see Materials and Methods).
To determine whether another selectable marker could produce similar results, particularly with respect to the proportion of stable integrants, we also tried a cassette based on the ade1+ gene in an ade1-D25 background. The transformation efficiency of this cassette seemed comparable to the ura4+ cassette, as was the proportion of stable integrants, which was determined directly to be 15%. The ade1+ gene did not provide any particular advantage over the ura4+ gene and was not further pursued.
Two conclusions can be reached from these results. First, any type of selectable marker can be used to produce large numbers of stable illegitimate recombinants when transformed into a suitable S.pombe strain. Selectable markers that show multicopy toxicity, such as the sup3-5 gene, decrease the overall frequency of integrants. Second, the addition of other markers or tags that increase the size of the construct by >2-fold does not affect the transformation efficiency of this construct. This enables other types of selection schemes or sequence recovery mechanisms to be readily used.
Cassettes used for insertional mutagenesis must not have any homology to the genome
To ensure that all integration is illegitimate, there must be no homology to genomic sequences. The ura4+ cassette was transformed into the ura4-D18 strain, which is devoid of all cassette sequences (23). All integrants produced thus arose from illegitimate recombination events. However, the ade6-704 strain used for sup3-5 transformations contains a wild-type genomic copy of the sup3-5 gene. Genetic analysis of 50 stable transformants indicated that 2% of the integrations had occurred at or close to the sup3+ locus. The sup3-5 portion of the cassette showed >90% homology with other tRNASer-encoded genes such as sup9+ and sup12+, suggesting that integrations might also occur at those sites. Linkage of integrants to the latter two loci was not tested.
In addition, as part of the GFP experiments described below, we initially placed an upstream linker containing 164 bp of homology to the upstream region of the small heat shock protein gene hsp16, and devoid of stop codons, to protect the GFP open reading frame from degradation. The hsp16 sequence was chosen out of convenience since it was readily available in a plasmid. This cassette turned out to be unsuitable for insertional mutagenesis since homology of this length resulted in homologous integration with a frequency of 45% at the hsp16 locus. Sequence analysis of several insert junctions (discussed later) indicated that the construct had circularized in the cell and integrated by a single homologous recombination event. It is conceivable that circularization prior to integration may be a common feature of all introduced linear sequences. Overall, our data indicate that any selectable marker can be used for mutagenesis and should work equally well provided no strong sequence homology exists within the genome.
Integration occurs at broadly distributed points in the genome
Random or near random integration of the PCR cassette would be optimal for insertional mutagenesis. Apart from illegitimate recombination, it is possible that random integrations can also be promoted by short homologous degenerate sequences at the ends of the cassette. Since studies have reported that sequences as short as 29 and 40 bp of homology are sufficient to drive homologous recombination in S.cerevisiae and S.pombe, respectively (9,24), shorter regions may target at lower frequency. To investigate this, primers were synthesized to generate a PCR product containing the ura4+ gene flanked by 18 bp fully degenerate tails. This construct was then transformed and the efficiency compared to a tail-free ura4+ cassette. The 18 bp extension length was chosen since, theoretically, every possible 18 bp sequence would be represented in the 200 nmol of primer that was synthesized. Similar numbers of stable integrants were produced by transformation of the ura4+ cassette with (4197 ± 126 per µg DNA, n = 2) or without the random tails (3478 ± 573 per µg DNA, n = 2), indicating that the presence of these degenerate sequences did not affect the total number of integrants.
We next assayed the distribution of insertions in the genome amongst a population of stable integrants generated by the random tailed ura4+ cassette. Southern blot analysis of 28 stable integrants, pooled in samples of five or six strains and using the ura4+ construct as a probe, revealed 28 distinct EcoRI fragments (Fig. 2A), indicating that the integrations occurred at different positions in the genome. We also showed that each of the six strains in one of the pools contained a single insertion event (Fig. 2B). Based on the low probability of insertion events, the frequency of double integrants is expected to be low. In addition, Southern blots of three other mutant strains detected only one insertion (data not shown). Lastly, all strains subjected to detailed genetic analysis showed only one integration event.
Figure 2.
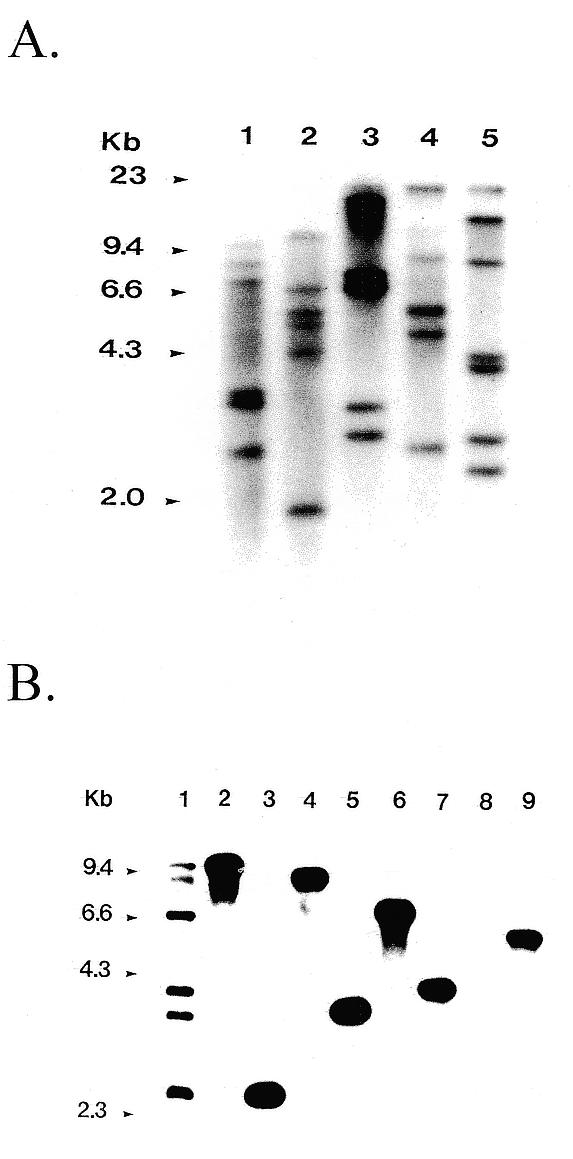
Southern analysis of stable ura4+ integrants. (A) Pools of either five (lanes 3 and 4) or six (lanes 1, 2 and 5) individual stable integrant strains (n = 28 in total) were grown and total genomic DNA was prepared, digested with EcoRI and analyzed by Southern blotting using ura4+ as a probe. Differences in band intensity within a lane reflect differing cell densities of individual strains in the cell cultures. The uppermost band in lanes 3–5 is undigested genomic DNA. (B) The six pooled integrant strains shown in lane 1 of (A) were analyzed individually. Southern blotting was performed as above except that total genomic DNA was prepared from individual strains. Lane 1, pooled DNA from all six strains; lanes 2–7, six individual integrant strains; lane 8, ura4-D18 strain; lane 9, wild type.
Integration events are broadly distributed in the genome. Several lines of evidence support this. First, intercrosses between three individual sup3-5 stable integrants (in an ade6-704 background) and 10 randomly chosen ones all produced adenine auxotrophs, indicating that for each group of 10 crosses none of the strains had integrated the cassette at the same locus. We were also able to retrieve insertional mutants from various screens (see later) and, furthermore, examination of the insert locations in the genome for some of the mutants where flanking genomic sequence was retrieved revealed a broad distribution on all three chromosomes (Fig. 3).
Figure 3.

Location of a number of integrants in the genome (arrowheads). Sequences retrieved from insertional mutant strains were localized by comparison to the physical maps at www.sanger.ac.uk . Centromeres are indicated by filled circles.
We have not performed an exhaustive analysis of multiple integration sites within a single gene following mutagenesis with constructs with or without the degenerate tails to assess whether the tailed constructs have a broader range of integration sites. This would require the collection of a large number of integrations within a single gene using each construct followed by sequencing of the insertion sites. For practical purposes of insertional mutagenesis our data suggest that there is no difference between the tailed and non-tailed constructs in terms of larger scale distribution or utility.
Isolation of cdr– mutants by insertional mutagenesis
The insertional mutagenesis system was applied in a screen for changed division response (cdr–) mutants. These are characterized by their inability to properly down-regulate their cell size upon nitrogen starvation (25,26). This particular mutant class was chosen for two reasons. First, the distinct cdr– phenotype is easily screened and we have extensive experience with it. Second, previous results from a chemical mutagenesis screen for cdr– mutants (26; unpublished data), where six major complementation groups were represented in a collection of 106 mutants, provided a comparison to determine whether there was bias in the types of mutants generated.
A PCR-generated construct containing the ura4+ gene flanked by degenerate tails was transformed into a ura4-D18 strain and the transformants were screened for the cdr– phenotype on minimal medium minus nitrogen plates (26). We recovered seven mutants representing alleles of four different cdr– genes in a total of 84 000 integrants for a frequency of ~1 in 12 000 overall. For the individual genes it ranged from one to three alleles, depending on the gene. The proportion of mutants isolated was comparable to the chemical mutagenesis screen (1.4 × 10–5). Although the number of insertional mutants recovered was small, three of the four cdr– genes were identical to complementation groups previously identified by chemical mutagenesis as determined by genetic analysis (16). This suggests that the integration events were well distributed in the genome. Mutants recovered included three insertional alleles of the protein kinase cdr2 (26–28), allowing for an assessment of the distributed sites of integration within a single gene (see below).
In addition, two other insertional mutagenesis screens were undertaken with similar success. A search for amiloride-resistant mutants recovered an insertional mutant allele of the car1+ gene amongst 21 780 integrants (29) and a screen for extragenic suppressors of the cdr1/nim1 kinase yielded an allele of wee1+ (26,30,31) and an allele of rad24+ (32) in a population of 25 000 stable integrants. These latter two loci were expected and isolated with a frequency similar to that seen in the cdr– insertional screen.
Examination of insertion sites from several of these mutants showed that two-thirds of the integrations occurred within the open reading frame while the remaining one-third was found proximal to the promoter sequences. This pattern of insertion is different from certain transposons, such as Ty1 and Tn916, which exhibit a high insertion preference for intergenic DNA (33,34). Our ability to retrieve several insertional mutants at a reasonable frequency and an expected distribution of known linkage groups in three separate screens demonstrates the general applicability of this technique.
Insertional mutagenesis using a GFP-containing cassette can be used as a promoter trap and a screen for protein targeting signals
The insertional mutagenesis technique can be extended to include the use of more complex cassettes. These can carry additional selectable markers for subsequent cloning or tags for identifying the protein product at the site of integration. We have designed and tested a cassette which generates a GFP fusion protein upon integration, thus allowing for selection of mutants based, first, on expression levels of the protein and, second, cellular localization. The cassette consists of the GFP gene (35,36) preceded by an upstream sequence devoid of genomic sequences and without stop codons in any frame. This upstream sequence was used to protect the GFP open reading frame during integration. The GFP gene was followed by a spacer and the ura4+ selectable marker.
Upon transformation into a ura4-D18 ade1-D25 strain, integrants were illuminated with 450–480 nm light to excite the GFP and monitored for colony fluorescence using a 500 nm long pass filter. Fluorescent strains were expected to be low in frequency since they were a result of a 1 in 6 chance of the cassette integrating in the correct frame and orientation as well as having it falling under control of a promoter that possessed sufficient strength for detection. In haploid cells, our detection system was able to recover fluorescent strains at ~0.5% of total stable integrants (233 GFP-expressing strains in a population of 46 027 stable integrants). This population included GFP fluorescence localized in the nucleus, nuclear periphery, cytoplasm and meiotic spores (Fig. 4). We have also integrated the GFP cassette into diploid cells to isolate fusion mutants of essential genes. A collection of these mutants remains to be analyzed. These results indicate that protein fusion mutants can be isolated at a reasonable frequency by insertional mutagenesis and that the use of a more sensitive detector, such as a cooled CCD camera or FACS, would increase the overall number of fluorescent-tagged integrants recovered.
Figure 4.
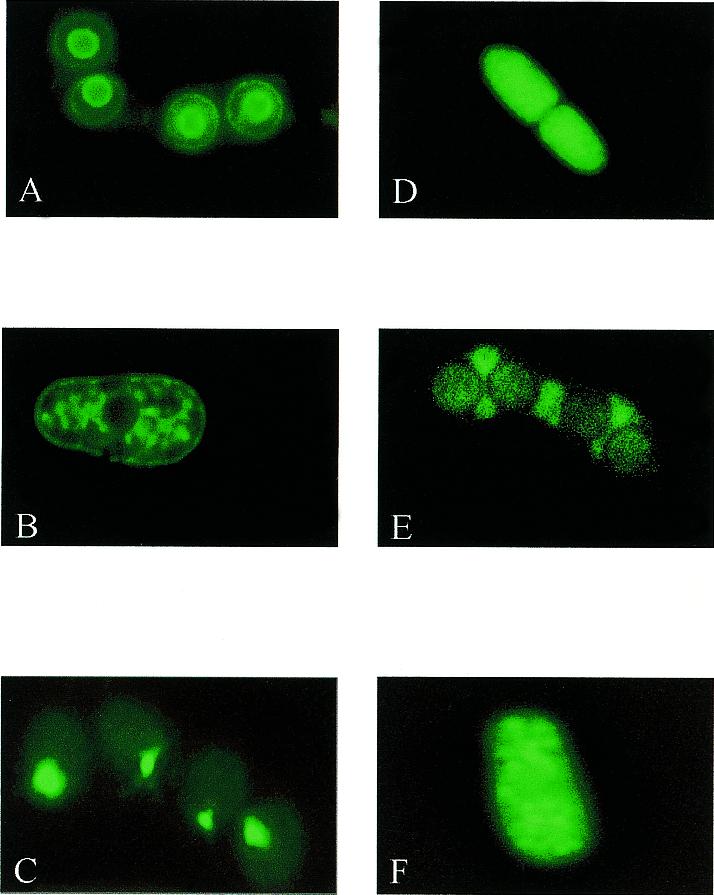
GFP fusion proteins displaying different cellular localizations. Living cells expressing GFP fusion proteins as genomic integrants were viewed by fluorescence microscopy. (A) lt13-26 (predominant staining in nuclear envelope and spore cytoplasm); (B) lt14-20 (nuclear envelope and ER); (C) lt1-4 (punctate staining proximal to nucleus in spores); (D) lt2-20 (cytoplasmic in septum-defective mutant); (E) lt3-2 (cytoplasmic in forming ascus); (F) lt2-3 (vacuolar). Images were taken with a Leica fluoresence microscope equipped with a high performance CCD camera (Sensicam) and Slidebook software (Intelligent Imaging Systems).
Insert junctions show small deletions of the gene and marker and no sequence specificity
We recovered and sequenced a number of insert junctions in order to examine the nature of any chromosomal rearrangements and possible sequence preferences for integration. Examination of the sequences at the insert junctions showed that the majority of integration events resulted in small deletions within the gene and at both termini of the cassette (Fig. 5). These deletions appeared to be variable and sequence non-specific (Fig. 5). Only one exception was seen, where in this case a large fragment (1.5 kb) of the chromosome was deleted (Fig. 5). This suggests that if the selectable marker(s) is sensitive to loss of a few base pairs (such as the case of GFP), then spacers are required to flank the selectable marker within the cassette for protection during integration.
Figure 5.

Sequence analysis of insert junctions in several mutants. Insertion of the ura4+ cassette (blue text) into genomic DNA (red and black text) is shown. (Left) Genomic sequences at upstream junction; (right) genomic sequences at downstream junction; (center) inserted ura4+ cassette sequences with terminal nucleotides and their positions (superscript) after integration. The red and black text flanked by deltas below the blue insert sequences are the genomic sequences deleted in each event. Nucleotide identity between the integrated construct and genomic DNA is displayed in red. Bottom line, the ura4+ cassette (blue) showing the terminal 13 nt at each end and the random tails (N18) upstream (left) and downstream (right). The top five mutants were generated by the ura4+ cassette flanked by degenerate tails while the bottom strain was mutagenized by the GFP cassette. Linker, upstream linker of the GFP cassette. The rad24-i1 strain contained a 1589 bp genomic deletion which included half of the 3′-end of the rad24+ gene (nucleotide positions +568 to +1283) and sequences downstream.
Small regions of homology are observed between the genomic DNA and the ends of the integrated cassette
Truncation of both ends of the integrating fragment in almost all cases hindered determination of whether the integration was actually dependent on any short (a few base pairs) homology found within the random tails. However, sequence analysis of an intact random tail present in the ssp1-i1 integrant showed that the four terminal nucleotides at the upstream end were homologous to the disrupted gene at the insertion junction (Fig. 5). The occurrence of microhomology in illegitimate recombination has been reported in S.cerevisiae and mammalian cells (37–39). Interestingly, there were also small regions of homology (4–10 bp) between the ends of the integrated cassette and the disrupted gene in almost all of the insertional mutants analyzed (Fig. 5). A portion of the homologous region was usually deleted in the disrupted gene (Fig. 5). The region of homology found in the genomic sequence of one of the insertional mutants (lt13-26) contained both termini of the integrated cassette. These observations suggest that in some cases circularization of the cassette results in small deletions of the termini (12) and that the short sequences remaining at or proximal to the end-joining promote integration at genomic sites containing microhomology to these sequences. The insertional mutant (rad24-i1) possessing a large genomic deletion is likely caused by integration of a linear cassette.
Our data suggest that the sites of genomic integration may be dependent upon microhomology at the upstream and downstream regions of the ura4+ gene in the cassette after truncation, since in most cases the entire random tails were deleted. However, integration of the cassette may still be broadly distributed in the genome because the sequence homologies seen in illegitimate recombination were very short and the degree of truncation of the ura4+ cassette was variable. Although only two cdr2– alleles were analyzed at the nucleotide level, they represent different insertion points in unrelated sequences in the gene (amino acid positions 369 and 426). While we have no proof that use of the degenerate tailed primers enhances the distribution of inserts, they are unlikely to detract. The broad applicability of this method provides a powerful tool for functional gene analysis in S.pombe.
Acknowledgments
ACKNOWLEDGEMENT
This work was supported by grants from the Natural Science and Engineering Research Council of Canada to P.G.Y.
REFERENCES
- 1.Hinnen A., Hicks,J.B. and Fink,G.R. (1978) Proc. Natl Acad. Sci. USA, 75, 1929–1933. [DOI] [PMC free article] [PubMed]
- 2.Orr-Weaver T.L., Szostak,J.W. and Rothstein,R.J. (1981) Proc. Natl Acad. Sci. USA, 78, 6354–6358. [DOI] [PMC free article] [PubMed]
- 3.Scherer S. and Davis,R.W. (1979) Proc. Natl Acad. Sci. USA, 76, 4951–4955. [DOI] [PMC free article] [PubMed]
- 4.Rothstein R.J. (1983) Methods Enzymol., 101, 202–211. [DOI] [PubMed]
- 5.Baudin A., Ozier-Kalogeropoulos,O., Denouel,A., Lacroute,F. and Cullin,C. (1993) Nucleic Acids Res., 21, 3329–3330. [DOI] [PMC free article] [PubMed]
- 6.Lorenz M.C., Muir,R.S., Lim,E., McElver,J., Weber,S.C. and Heitman,J. (1995) Gene, 158, 113–117. [DOI] [PubMed]
- 7.Wach A., Brachat,A., Pöhlmann,R. and Philippsen,P. (1994) Yeast, 10, 1793–1808. [DOI] [PubMed]
- 8.Wach A., Brachat,A., Alberti-Segui,C., Rebischung,C. and Philippsen,P. (1997) Yeast, 13, 1065–1075. [DOI] [PubMed]
- 9.Kaur R., Ingavale,S.S. and Bachhawat,A.K. (1997) Nucleic Acids Res., 25, 1080–1081. [DOI] [PMC free article] [PubMed]
- 10.Bahler J., Wu,J.-Q., Longtine,M.S., Shah,N.G., McKenzie,A.III, Steever,A.B., Wach,A., Philippsen,P. and Pringle,J.R. (1998) Yeast, 14, 943–951. [DOI] [PubMed]
- 11.Roth D. and Wilson,J.H. (1988) In Kucherlapati,R. and Smith,G. (eds), Genetic Recombination. American Society for Microbiology, Washington, DC, pp. 621–653.
- 12.Goedecke W., Pfeiffer,P. and Vielmetter,W. (1994) Nucleic Acids Res., 22, 2094–2101. [DOI] [PMC free article] [PubMed]
- 13.Mitchison J.M. and Creanor,J. (1971) Exp. Cell Res., 69, 244–247. [DOI] [PubMed]
- 14.Gutz H. and Doe,F.J. (1975) Mycologia, 67, 748–759. [PubMed]
- 15.Nurse P. (1975) Nature, 256, 547–551. [DOI] [PubMed]
- 16.Moreno S., Klar,A. and Nurse,P. (1991) Methods Enzymol., 194, 795–823. [DOI] [PubMed]
- 17.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 18.Ausubel F.M., Brent,R., Kingston,R.E., Moore,D.D., Seldman,J.G., Smith,J.G. and Struhl,K. (1994) Current Protocols in Molecular Biology. Greene Publishing Associates and John Wiley & Sons, New York, NY.
- 19.Ochman H., Gerber,A.S. and Hartl,D.L. (1988) Genetics, 120, 621–623. [DOI] [PMC free article] [PubMed]
- 20.Okazaki K., Okazaki,N., Kume,K., Jinno,S., Tanaka,K. and Okayama,H. (1990) Nucleic Acids Res., 18, 6485–6489. [DOI] [PMC free article] [PubMed]
- 21.Carr A.M., MacNeill,S.A., Hayles,J.A. and Nurse,P. (1989) Mol. Gen. Genet., 218, 41–49. [DOI] [PubMed]
- 22.Tatebayashi K., Kato,J. and Ikeda,K. (1994) Mol. Gen. Genet., 244, 111–119. [DOI] [PubMed]
- 23.Grimm C., Kohli,J., Murray,J. and Maundrell,K. (1988) Mol. Gen. Genet., 215, 81–86. [DOI] [PubMed]
- 24.Rothstein R. (1991) Methods Enzymol., 194, 281–306. [DOI] [PubMed]
- 25.Young P.G. and Fantes,P.A. (1984) In Skehan,P. and Friedman,S.J. (eds), Growth, Cancer and the Cell Cycle. Humana Press, Clifton, NJ, pp. 221–228.
- 26.Young P.G. and Fantes,P.A. (1987) J. Cell Sci., 88, 295–304. [DOI] [PubMed]
- 27.Breeding C.S., Hudson,J., Balasubramanian,M.K., Hemmingsen,S.M., Young,P.G. and Gould,K.L. (1998) Mol. Biol. Cell, 9, 3399–3415. [DOI] [PMC free article] [PubMed]
- 28.Kanoh J. and Russell,P. (1998) Mol. Biol. Cell, 9, 3321–3334. [DOI] [PMC free article] [PubMed]
- 29.Jia Z.P., McCullough,N., Wong,L. and Young,P.G. (1993) Mol. Gen. Genet., 241, 298–304. [DOI] [PubMed]
- 30.Russell P. and Nurse,P. (1987) Cell, 49, 569–576. [DOI] [PubMed]
- 31.Feilotter H., Nurse,P. and Young,P.G. (1991) Genetics, 127, 309–318. [DOI] [PMC free article] [PubMed]
- 32.Al-Khodairy F., Fotou,E., Sheldrick,K.S., Griffiths,D.J.F., Lehmann,A.R. and Carr,A.M. (1994) Mol. Cell. Biol., 5, 147–160. [DOI] [PMC free article] [PubMed]
- 33.Devine S.E. and Boeke,J.D. (1996) Genes Dev., 10, 620–633. [DOI] [PubMed]
- 34.Nelson K.E., Richardson,D.L. and Dougherty,B.A. (1997) Microb. Comp. Genomics, 2, 313–321. [DOI] [PubMed]
- 35.Heim R., Prasher,D.C. and Tsien,R.Y. (1994) Proc. Natl Acad. Sci. USA, 91, 12501–12504. [DOI] [PMC free article] [PubMed]
- 36.Cubitt A.B., Heim,R., Adams,S.R., Boyd,A.E., Gross,L.A. and Tsien,R.Y. (1995) Trends Biochem. Sci., 20, 448–455. [DOI] [PubMed]
- 37.Roth D.B. and Wilson,J.H. (1986) Mol. Cell. Biol., 6, 4295–4304. [DOI] [PMC free article] [PubMed]
- 38.Schiestl R.H. and Petes,T.D. (1991) Proc. Natl Acad. Sci. USA, 88, 7585–7589. [DOI] [PMC free article] [PubMed]
- 39.Schiestl R.H., Dominska,M. and Petes,T.D. (1993) Mol. Cell. Biol., 13, 2697–2705. [DOI] [PMC free article] [PubMed]