

Abstract
Background
Diabetic retinopathy (DR) remains a major cause of sight loss worldwide, despite new therapies and improvements in the metabolic control of people living with diabetes. Therefore, DR creates a physical and psychological burden for people, and an economic burden for society. Preventing the development and progression of DR, or avoiding the occurrence of its sight‐threatening complications is essential, and must be pursued to save sight. Fenofibrate may be a useful strategy to achieve this goal, by reversing diabetes’ effects and reducing inflammation in the retina, as well as improving dyslipidaemia and hypertriglyceridaemia.
Objectives
To investigate the benefits and harms of fenofibrate for preventing the development and progression of diabetic retinopathy in people with type 1 (T1D) or type 2 diabetes (T2D), compared with placebo or observation.
Search methods
We searched CENTRAL, MEDLINE, Embase, and three trials registers (February 2022).
Selection criteria
We included randomised controlled trials (RCTs) that included people with T1D or T2D, when these compared fenofibrate with placebo or with observation, and assessed the effect of fenofibrate on the development or progression of DR (or both).
Data collection and analysis
We used standard Cochrane methods for data extraction and analysis.
Our primary outcome was progression of DR, a composite outcome of 1) incidence of overt retinopathy for participants who did not have DR at baseline, or 2) advancing two or more steps on the Early Treatment Diabetic Retinopathy Study (ETDRS) severity scale for participants who had any DR at baseline (or both), based on the evaluation of stereoscopic or non‐stereoscopic fundus photographs, during the follow‐up period. Overt retinopathy was defined as the presence of any DR observed on stereoscopic or non‐stereoscopic colour fundus photographs.
Secondary outcomes included the incidence of overt retinopathy, reduction in visual acuity of participants with a reduction in visual acuity of 10 ETDRS letters or more, proliferative diabetic retinopathy, and diabetic macular oedema; mean vision‐related quality of life, and serious adverse events of fenofibrate.
We used GRADE to assess the certainty of evidence.
Main results
We included two studies and their eye sub‐studies (15,313 participants) in people with T2D. The studies were conducted in the US, Canada, Australia, Finland, and New Zealand; follow‐up period was four to five years. One was funded by the government, the other by industry.
Compared to placebo or observation, fenofibrate likely results in little to no difference in progression of DR (risk ratio (RR) 0.86; 95% confidence interval (CI) 0.60 to 1.25; 1 study, 1012 participants; moderate‐certainty evidence) in a population with and without overt retinopathy at baseline. Those without overt retinopathy at baseline showed little or no progression (RR 1.00, 95% CI 0.68 to 1.47; 1 study, 804 participants); those with overt retinopathy at baseline found that their DR progressed slowly (RR 0.21, 95% CI 0.06 to 0.71; 1 study, 208 people; test for interaction P = 0.02).
Compared to placebo or observation, fenofibrate likely resulted in little to no difference in either the incidence of overt retinopathy (RR 0.91; 95% CI 0.76 to 1.09; 2 studies, 1631 participants; moderate‐certainty evidence); or the incidence of diabetic macular oedema (RR 0.39; 95% CI 0.12 to 1.24; 1 study, 1012 participants; moderate‐certainty evidence).
The use of fenofibrate increased severe adverse effects (RR 1.55; 95% CI 1.05 to 2.27; 2 studies, 15,313 participants; high‐certainty evidence).
The studies did not report on incidence of a reduction in visual acuity of 10 ETDRS letters or more, incidence of proliferative diabetic retinopathy, or mean vision‐related quality of life.
Authors' conclusions
Current, moderate‐certainty evidence suggests that in a mixed group of people with and without overt retinopathy, who live with T2D, fenofibrate likely results in little to no difference in progression of diabetic retinopathy. However, in people with overt retinopathy who live with T2D, fenofibrate likely reduces the progression.
Serious adverse events were rare, but the risk of their occurrence was increased by the use of fenofibrate.
There is no evidence on the effect of fenofibrate in people with T1D. More studies, with larger sample sizes, and participants with T1D are needed. They should measure outcomes that are important to people with diabetes, e.g. change in vision, reduction in visual acuity of 10 ETDRS letters or more, developing proliferative diabetic retinopathy; and evaluating the requirement of other treatments, e.g. injections of anti‐vascular endothelial growth factor therapies, steroids.
Keywords: Humans; Diabetes Mellitus, Type 1; Diabetes Mellitus, Type 2; Diabetes Mellitus, Type 2/complications; Diabetes Mellitus, Type 2/drug therapy; Diabetic Retinopathy; Diabetic Retinopathy/drug therapy; Fenofibrate; Fenofibrate/adverse effects; Macular Edema; Macular Edema/drug therapy; Macular Edema/etiology; Retinal Diseases
Plain language summary
Is fenofibrate effective for diabetic retinopathy?
What was the aim of this review?
The aim of this review was to find out whether fenofibrate prevents people with either type 1 (T1D) or type 2 (T2D) diabetes from developing diabetic retinopathy (DR), or if they already had DR, whether it slows its progression, when compared with placebo or observation.
Key messages
‐ overall, fenofibrate likely made little to no difference in the progression of DR when compared with placebo (moderate‐certainty evidence)
‐ for people with DR, their DR likely progressed slowly when they took fenofibrate (moderate‐certainty evidence)
‐ although rare, side effects increased when people took fenofibrate (high‐certainty evidence)
‐ more studies are needed; for example, studies that include people with type 1 diabetes, studies that take into account other treatments that people received, and importantly, studies that include outcomes that are important to people living with diabetes
What was studied in the review?
DR, a condition that occurs when the blood vessels in the back of your eye develop problems, is a major cause of sight loss worldwide and a burden to society. Preventing its occurrence, and if present, slowing or preventing its progression must be pursued to save sight. This review summarised the evidence about whether fenofibrate may be useful for this purpose (when compared to placebo or observation).
What are the main results of the review?
We found two studies. In total, they included 15,313 people with T2D, who were followed for four or five years. The studies were conducted in the US, Canada, Australia, Finland, and New Zealand. One was funded by the government, the other by industry.
For people with T2D, when those who may or may not have had DR were studied together, moderate‐certainty evidence suggested that fenofibrate likely made little to no difference in the progression of DR when compared with placebo. However, when people with DR were studied on their own, the evidence suggested that their DR progressed slowly when they were taking fenofibrate. Serious adverse events were rare, but the risk of their occurrence increased for those who took fenofibrate (high‐certainty evidence).
More studies are needed. For example, studies that include people with type 1 diabetes, and importantly, studies that include outcomes that are important to people living with diabetes, such as the number of people who experience a change in vision or sight loss, develop proliferative diabetic retinopathy (growth of new blood vessels), or require injections of anti‐vascular endothelial growth factor therapies, or steroids. Health‐related and vision‐related quality of life measures, acceptability of the treatment to people using it, and costs of the treatment should be also included.
How up‐to date is this review?
The review authors searched for studies published up to 1 February 2022.
Summary of findings
Summary of findings 1. Fenofibrate for diabetic retinopathy.
| Fenofibrate compared to placebo or observation for diabetic retinopathy | ||||||
| Patient or population: people with type 2 diabetes Setting: hospital settings Intervention: fenofibrate Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with fenofibrate | |||||
| Progression of DRa | Study population | RR 0.86 (0.60 to 1.25) |
1012c,d (1 RCT) | ⊕⊕⊕⊝ Moderatee |
Fenofibrate likely resulted in little to no difference in progression of DR (main analysis). Subgroup analysis, separating those with and without overt retinopathy at baseline, suggested a difference in progression (RR 1.00, 95% CI 0.68 to 1.47; 804 people without overt retinopathy) and (RR 0.21, 95% CI 0.06 to 0.71; 208 people with overt retinopathy; test for interaction P = 0.02) |
|
| 118 per 1000c | 96 per 1000d | |||||
| Incidence of overt retinopathya,b | Study population | RR 0.91 (0.76 to 1.09) | 1580 (1631c) (2 RCTs) | ⊕⊕⊕⊝ Moderatee |
||
| 223 per 1000 (216 per 1000c) |
203 per 1000
(169 to 243) (199 per 1000c) |
|||||
| Incidence of a reduction in visual acuity of 10 ETDR letters or more | Not reported | |||||
| Incidence of PDR | Not reported | |||||
| Incidence of DMOa | Study population | RR 0.39 (0.12 to 1.24) |
850 (1012c) (1 RCT) | ⊕⊕⊕⊝ Moderatee | ||
| 24 per 1000 (20 per 1000c) |
9 per 1000 (3 to 29) (8 per 1000c) |
|||||
| Mean vision‐related quality of life | Not reported | |||||
| SAEa | Study population | RR 1.55 (1.05 to 2.27) | 15226 (15313c) (2 RCTs) | ⊕⊕⊕⊕ High | |
|
| 6 per 1000 (5 per 1000c) |
9 per 1000
(6 to 13) (8 per 1000c) |
|||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; DMO: diabetic macular oedema; DR: diabetic retinopathy; ETDR: the Early Treatment Diabetic Retinopathy Study; OR: odds ratio; PDR: proliferative diabetic retinopathy; RCT: randomised controlled trial; RR: risk ratio; SAE: serious adverse events | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect | ||||||
athe data at 5 years bthe data at 4 years cCalculated with the number of randomised participants dIn 1012 participants, there were 105 (20.5%) in the fenofibrate group and 103 (20.6%) in the placebo group with overt retinopathy at baseline. eDowngraded one level for imprecision; sample sizes were less than the optimal information size, and the confidence intervals were wide and included no effect.
Background
Description of the condition
Diabetic retinopathy (DR) is a neurovascular complication of diabetes mellitus, initiated by chronically high blood sugar levels. Cells of the neurovascular unit, including endothelial cells, pericytes, glial cells, and resident and circulating immune cells, are affected by the disease, with subsequent alterations in permeability and blood perfusion to the retina, resulting in retinal leakage and ischaemia (Stitt 2016). Depending on the extension of capillary loss, among other factors, this deficiency or lack of blood supply may lead to the formation of what are called 'new vessels'. New vessels are newly formed, abnormally fragile, blood vessels that develop in an attempt to bring blood and nourishment to the retina. The presence of new vessels defines proliferative diabetic retinopathy (PDR), a sight‐threatening complication of DR (Evans 2014). New vessels can lead to sight loss as a result of them bleeding inside the eye (known as vitreous haemorrhage), or as a result of the formation of scarring tissue that accompanies them (so‐called fibrovascular membranes), which can contract and detach the retina. As the blood vessels become weakened by the reduced number of cells, blood and fluid contained in them may leak out, leading to retinal oedema (accumulation of fluid in the retina). When fluid accumulates in the centre of the retina, the macula, diabetic macular oedema (DMO) ensues (Tan 2017). Besides vascular degeneration, loss of neural and supporting cells (glial cells) in the retina (neurodegeneration) occurs in DR, which also has an impact on vision.
One study estimated that globally, approximately 103 million people may have DR, and 29 million people may have sight‐threatening stages of DR (Teo 2021). The study estimated that by 2045, 161 million people would have DR, and 45 million would have sight‐threatening stages of DR. In addition to constituting a psychological and physical burden to the individual, DR also bears an economic burden to society. Several studies in Europe, US, and Asia have recently reported an association of higher medical costs with DR (Heintz 2010; Romero‐Aroca 2016; Schmier 2009; Woung 2010; Zhang 2017). The total healthcare costs of DR in Sweden are up to approximately EUR 9.9 million per year, or EUR 106,000 per 100,000 inhabitants, when one considers a 4.8% prevalence of diabetes (Heintz 2010). A study in Singapore reported that the presence and severity of DR was associated with increased direct medical costs (Zhang 2017).
Description of the intervention
Strict control of blood glucose levels and blood pressure is essential to reduce the risk of sight loss from complications of DR, namely DMO, macular ischaemia, and PDR, but is often difficult to achieve. In some people with diabetes, sight‐threatening complications may still occur, even if glucose levels and blood pressure are controlled. Laser photocoagulation, intravitreal injections of anti‐vascular endothelial growth factor (VEGF) drugs, and corticosteroids are used to treat DMO and PDR (Duh 2017; Evans 2014; Gross 2015; McCulloch 2017; Virgili 2018). These therapeutic modalities, although sight‐saving in many cases, have inherent risks; and despite them, visual loss can still occur in a proportion of people who are unresponsive to them. Therapeutic strategies to prevent the development of the end‐stage complications of DR would be expected to be more fruitful to save sight.
Fenofibrate, a fibrate indicated for the treatment of mixed dyslipidaemia and hypertriglyceridaemia, came to the market in 1975, and is widely used (Blane 1989; Guay 1999). Its cost is low. The main clinical effects are mediated through peroxisome proliferator‐activated receptor (PPAR)‐alpha activation, and consist of a moderate reduction in total cholesterol and low‐density lipoprotein cholesterol (LDL‐C) levels, a marked reduction in triglycerides (TG), and an increase in high‐density lipoprotein cholesterol (HDL‐C).
How the intervention might work
PPAR‐alpha is highly expressed in tissues with high mitochondrial and peroxisomal fatty‐acid beta‐oxidation rates, such as the retina (Ciudin 2013 ). It has been reported that PPAR‐alpha is downregulated in the retinas of both type 1 and type 2 experimental diabetic models, and that high glucose is a cause of PPAR‐alpha downregulation (Hu 2013). PPAR‐alpha knockout mice develop vascular leakage, leukostasis, pericyte loss, capillary degeneration, and overexpression of inflammatory markers, all features observed in DR in humans (Hu 2013). Therefore, fenofibrate may help reverse the effects of diabetes in the retina. Other reported mechanisms through which fenofibrate may ameliorate DR include modulating Nrf2 signalling and NLRP3 inflammasome activation, and by cytochrome P450 epoxygenase (CYP)2C inhibition (Gong 2016; Liu 2017).
Why it is important to do this review
The number of people suffering from DR, as well as the number of people with diabetes are increasing worldwide (Teo 2021). As described above, laser photocoagulation, anti‐VEGFs, and steroids are used routinely for the treatment of established DMO and PDR, but not to prevent their occurrence or to prevent the development and progression of DR (Aiello 2010; Boyer 2014; Gross 2015; Sivaprasad 2017; Virgili 2018). Fenofibrate may be useful for this purpose.
Objectives
To investigate the benefits and harms of fenofibrate for preventing the development and progression of diabetic retinopathy in people with type 1 or type 2 diabetes, compared with placebo or observation.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials (RCTs). We planned to include ongoing or unpublished studies. We excluded post‐trial follow‐up studies.
Types of participants
Participants were people diagnosed with type 1 or type 2 diabetes (T1D; T2D). We included those who both did not have retinopathy, or who had non‐proliferative diabetic retinopathy (NPDR) at baseline.
We excluded studies that only included participants with established complications of diabetic retinopathy (DR, i.e. diabetic macular oedema (DMO) and proliferative diabetic retinopathy (PDR)). We included studies randomising participants with or without complications of DR (i.e. DMO or PDR) if the proportion of people with complications was low (i.e. less than 10%), or if data for people without complications were presented separately.
Types of interventions
Intervention: fenofibrate (any dose or regimen)
Comparison: placebo or observation
Types of outcome measures
Studies were included even if no outcome data were available, unless it was clear that none of the following outcomes were measured.
Primary outcomes
Progression of diabetic retinopathy
Progression of diabetic retinopathy was considered a composite outcome of: 1) incidence of overt retinopathy for participants who did not have DR at baseline, or 2) advancing two or more steps for participants who had any DR at baseline in the Early Treatment Diabetic Retinopathy Study (ETDRS) severity scale, based on evaluation of stereoscopic or non‐stereoscopic fundus photographs, during the follow‐up period, or both (ETDRS 1991). Overt retinopathy was defined as the presence of any DR observed on stereoscopic or non‐stereoscopic colour fundus photographs.
Secondary outcomes
Incidence of overt retinopathy
Mean change in visual acuity
Incidence of a reduction in visual acuity of 10 ETDRS letters or more
Incidence of PDR
Incidence of DMO
Additional treatments for DR (any laser, defined as any laser treatment including focal or grid, panretinal photocoagulation (PRP), or both; focal or grid laser and PRP (separately); anti‐vascular endothelial growth factor (VEGFs), steroids, vitrectomy, other)
Mean vision‐related quality of life
Incremental cost per Quality Adjusted Life years (QALY) gained
Acceptability of the treatment
Discontinuation of the treatment
Adverse effects (serious adverse events (SAE))
Adverse effects (rhabdomyolysis)
Adverse effects (hepatic disorder, i.e. alanine aminotransferase elevated three times more than upper limit of normal)
Adverse effects (pancreatitis)
Adverse effects (Stevens‐Johnson Syndrome)
Adverse effects (others defined by original study authors)
Search methods for identification of studies
Electronic searches
The Cochrane Eyes and Vision Information Specialist searched the following databases for randomised controlled trials and controlled clinical trials. There were no restrictions on language or year of publication. The date of the search was 1 February 2022.
Cochrane Central Register of Controlled Trials (CENTRAL; 2022, Issue 2; which contains the Cochrane Eyes and Vision Trials Register) in the Cochrane Library (searched 1 February 2022; Appendix 1);
MEDLINE Ovid (1946 to 1 February 2022; Appendix 2);
Embase Ovid (1980 to 1 February 2022; Appendix 3);
ISRCTN registry (www.isrctn.com/editAdvancedSearch; searched 1 February 2022; Appendix 4);
US National Institutes of Health Ongoing Trials Register ClinicalTrials.gov (www.clinicaltrials.gov; searched 1 February 2022; Appendix 5);
World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP; www.who.int/ictrp; searched 1 February 2022; Appendix 6).
Searching other resources
Two review authors (SYK, YK) independently searched the reference lists of identified clinical trials.
Data collection and analysis
Data extraction was undertaken using a previously piloted Excel data extraction sheet and Covidence.
Selection of studies
Two of three review authors (KI, SYK, SK) independently screened search results; discrepancies were resolved through discussion. We screened the list of titles and abstracts, and classified records as potentially eligible or not eligible. We obtained the full‐text articles of all potentially eligible studies, which were independently reviewed by two reviewers (SYK, YK), who classified them as eligible or not eligible. Disagreements were resolved through discussion with other authors (KI, SK, NW, NL). We gave the primary reasons for exclusion in the Characteristics of excluded studies table.
Data extraction and management
Two of four review authors (SYK, NL, SK, YK) independently extracted data from trial reports and entered the data into Review Manager 5 (RevMan 5) and RevMan Web (Review Manager 2020; RevMan Web 2023). We resolved any discrepancies in data extraction through discussion. If we could not reach consensus, we consulted another review author (NW). When information in the full‐text article was insufficient, we contacted the corresponding author of the original trial to request additional information. We used a data collection form, which we piloted prior to its use (Appendix 7). We planned to obtain English translations of any trials reported in non‐English. However, none of the eligible studies were written in other languages. Therefore, translations were not needed. We obtained the data on outcomes specified in Types of outcome measures. For dichotomous outcomes, we collected data on the number of events and total participants randomised and followed in each trial arm. For continuous outcomes, we collected data on the mean and standard deviation or median and interquartile range in each trial arm.
Assessment of risk of bias in included studies
Two of four review authors (SYK, NL, SK, YK) independently assessed study quality, study limitations and the extent of potential bias by using the Cochrane RoB 1 tool, described in Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2017). We considered the following domains.
Sequence generation (selection bias)
Allocation concealment (selection bias)
Masking (blinding) of participants, personnel (performance bias)
Masking (blinding) of outcomes assessors (detection bias)
Incomplete outcome data (attrition bias)
Selective outcome reporting (reporting bias)
Other ‐ other threats to validity
For each domain, we judged whether the trial authors made sufficient attempts to minimise bias in their study design. We made judgements using three measures: high, low, and unclear risk of bias. We recorded this judgement in the risk of bias tables and presented a summary risk of bias figure.
Measures of treatment effect
We measured treatment effect according to the data types described in Chapter 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2022).
Dichotomous data
Variables in this group included the primary outcome, progression of DR, and the following secondary outcomes: incidence of overt retinopathy, incidence of a reduction in visual acuity of 10 ETDRS letters or more, incidence of PDR, incidence of DMO, additional treatments for DR, acceptability of the treatment, discontinuation of the treatment, and adverse effects. We reported dichotomous variables as risk ratios (RRs) with 95% confidence intervals (CIs).
Continuous data
We planned to report continuous variables, including differences between groups for mean change in visual acuity, quality of life scores, and incremental cost per QALY gained as mean difference with 95% CI (if normally distributed) or median and interquartile range (if not normally distributed). We planned to calculate the standardised mean difference (SMD) when trials used different scales for the same outcome measure.
Unit of analysis issues
Trials reporting one eye per person
When trials included outcomes based on one eye per person, there were no issues regarding unit of analysis. We documented how the trials selected the included eye.
Trials reporting two eyes per person
Ideally, these studies are adjusted for within‐person correlation. We planned to collect data on the measure of effect and confidence interval and enter this into RevMan 5 or RevMan Web using the generic inverse variance method (Review Manager 2020; RevMan Web 2023). If trials reported both eyes without this adjustment, we planned to use the data and discuss the implications in the interpretation. When the results per person were reported using a scale that considered both eyes, we used these results, since in this case, there were no issues regarding the unit of analysis.
Dealing with missing data
We documented if loss to follow‐up was high (over 20%), or unbalanced between treatment groups, as a potential source of attrition bias. We used data as reported in the trial publications, including any imputation for missing data.
Assessment of heterogeneity
We assessed heterogeneity between trials by visual inspection of forest plots, and by formal statistical tests of heterogeneity (Chi² test (Deeks 2022)).
Assessment of reporting biases
We searched both registered trials and published trials. We contacted researchers of the unpublished trials to provide data related to outcomes in this review, though we found that these trials were ongoing studies.
Data synthesis
We performed statistical analyses according to guidance from Cochrane Eyes and Vision. We pooled data using a fixed‐effect model. When we conducted meta‐analysis using data measured with different scales, we described the scales’ characteristics.
Subgroup analysis and investigation of heterogeneity
We presented subgroup analysis undertaken in the included RCTs, but we did not undertake any subgroup analysis as part of the current review, as this was not possible.
Sensitivity analysis
We did not conduct any sensitivity analysis due to insufficient number of trials.
Summary of findings and assessment of the certainty of the evidence
We planned to report absolute risks and measures of effect in a summary of findings table, and provide an overall assessment of the certainty of the evidence for each outcome using the GRADE system (GRADEpro GDT ). Two review authors (SYK, YK) independently undertook the GRADE assessment. Discrepancies were resolved by discussion. If we could not reach consensus, we consulted another review author (NW, NL).
We included these outcomes in the summary of findings table. We reported the results at three years.
Progression of DR
Incidence of overt retinopathy
Incidence of a reduction in visual acuity of 10 ETDRS letters or more
Incidence of PDR
Incidence of DMO
Mean vision‐related quality of life
Adverse effects (SAE)
Results
Description of studies
Results of the search
The electronic searches yielded a total of 423 records (Figure 1). After removing 121 duplicates, the Cochrane Information Specialist (CIS) screened the remaining 302 records and removed 204 records that were not relevant to the scope of the review. We screened the remaining 98 records and obtained the full‐text reports of 34 records for further assessment.
1.
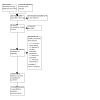
Study selection flow diagram
We included two studies (in 10 reports), excluded 18 studies (in 19 reports), and identified three ongoing studies (in 5 reports).
Included studies
Study design
We included two randomised controlled trials, both of which had an eye sub‐study (ACCORD‐Lipid; FIELD).
We included two studies, the Action to control cardiovascular risk in diabetes lipid trial (ACCORD‐Lipid) and the Fenofibrate intervention and event lowering in diabetes study (FIELD (ACCORD‐Lipid; FIELD)). Both were multicentre, double‐masked, placebo‐controlled RCTs, and each of them had an ophthalmological sub‐study (ACCORD eye study of ACCORD‐Lipid (ACCORD Eye Lipid), and FIELD ophthalmology sub‐study). We describe the characteristics of these studies, including those of the eye sub‐studies, in more detail below. Regarding eligibility criteria for the eye sub‐studies, ACCORD Eye Lipid added one exclusion criterion to ACCORD‐Lipid’s criteria: history of proliferative diabetic retinopathy (PDR) that had been treated with laser photocoagulation or vitrectomy. FIELD ophthalmology sub‐study added the following eligibility criteria to those of the FIELD main trial: two‐field colour fundus photographs of both eyes had to show no evidence of PDR, severe non‐proliferative diabetic retinopathy (NPDR), clinically significant DMO, or indication for, or evidence of, a history of laser treatment at a screening examination (this was done during the placebo run‐in phase). Additionally, there were a number of other exclusions based on the presence of other ocular pathology or 'technical problems' (not specified which ones). ACCORD‐Lipid was conducted in the United States and Canada. FIELD was conducted in Australia, Finland, and New Zealand. The follow‐up period of ACCORD‐Lipid was 4.7 years; FIELD was 5 years. The sample size of ACCORD‐Lipid was calculated based on the primary outcome, which was not diabetic retinopathy (DR), but the number of participants included was lower than that required, based on the investigator’s sample size calculation. For ACCORD Eye Lipid, a sample size calculation for the primary composite outcome related with advancing DR was provided, but like the full trial, this was not met. The sample size of FIELD was calculated based on each primary outcome, which were not DR, while in the FIELD ophthalmology sub‐study, the sample size calculation was not given. The unit of assessment for the outcomes from ACCORD‐Lipid, ACCORD Eye Lipid, and FIELD was the person. Outcomes reported in the FIELD ophthalmology sub‐study were from worse affected eye, or right eye when both eyes were equally affected. ACCORD‐Lipid was publicly funded, while FIELD was sponsored by industry.
Participants
See Characteristics of included studies tables (ACCORD‐Lipid; FIELD). Combined, the two studies included 15,313 participants (ACCORD‐Lipid: 5518, 36.0%; FIELD: 9795, 64.0%), with a predominance of males (total: 9962; 65.1%; ACCORD‐Lipid: 3824, 38.4%; FIELD: 6138, 61.6%), and Caucasians (total: 12,867; 84.0%; ACCORD‐Lipid: 3774, 29.3%; FIELD: 9093, 70.7%). The average age of participants was 62 years (ACCORD‐Lipid: 62.3 ± 6.8 (mean ± SD); FIELD: 62.2 (SD was not specified in the original article)). Eye sub‐studies included 2930 participants in total (ACCORD Eye Lipid: 1918, 65.6%; FIELD ophthalmology sub‐study: 1012, 34.5%). The criteria for selection of participants for the sub‐studies were as follows.
All ACCORD‐Lipid participants were recruited, and were assessed for eligibility for the ACCORD Eye Lipid sub‐study, using the baseline information obtained in ACCORD‐Lipid. Those who seemed eligible, were screened for eligibility. Informed consent was obtained from each participant specifically for the ACCORD Eye Lipid sub‐study, and recruited.
For FIELD, consents for the ophthalmology sub‐study were obtained from only 22 FIELD sites’ participants, not all FIELD participants. They were assessed for eligibility during the placebo run‐in phase.
All participants in both studies had T2D.
Intervention
Both studies used fenofibrate as the intervention and placebo as the control, although different doses were used. The dose of fenofibrate in ACCORD‐Lipid was 160 mg/day; in FIELD it was 200 mg/day. ACCORD‐Lipid had intensive glycaemic control (HbA1c target < 6.0%) or standard therapy (7.0% ≤ HbA1c target ≤ 7.9%) arms to evaluate other interventions (tight glycaemic control) with a 2‐by‐2 factorial design. In ACCORD‐Lipid, all participants received nutrition and physical activity counselling, a recommendation to use aspirin daily, and simvastatin 20 mg to 40 mg/day. Additionally, if participants had an additional risk factor for CVD, using an angiotensin‐converting enzyme inhibitor was recommended. Current smokers received smoking cessation counselling. Participants’ personal physicians received information about current guidelines for lipids and blood pressure management. In FIELD, all participants underwent an initial run‐in period of 16 weeks before randomisation, consisting of 4 weeks with only diet advice, 6 weeks with single‐blind placebo, and 6 weeks with single‐blind fenofibrate. Their intention for the run‐in period was to allow people time to discuss long‐term participation with their families and their usual doctors, and for evaluation of the benefits of fenofibrate treatment on a background of recommended dietary advice. The active run‐in period was to also determine to what extent any long‐term clinical benefits of treatment correlated with the short‐term effects of the drug to modify different lipid fractions.
Primary Outcome
Only the FIELD ophthalmology sub‐study reported the incidence of overt retinopathy and the incidence of participants with overt retinopathy at baseline advancing two or more steps in the ETDRS severity scale separately.
Secondary Outcomes
The following outcomes were reported and obtained from the main ACCORD‐Lipid trial: discontinuation of the treatment, severe adverse events (SAE), hepatic disorder, pulmonary embolism, and deep‐vein thrombosis. The ACCORD Eye Lipid sub‐study reported on incidence of overt retinopathy, and additional treatments for DR including focal/grid laser and PRP. FIELD reported additional treatment for DR, including any laser; additional treatments for DR, including focal/grid laser and PRP; discontinuation of the treatment; SAE; rhabdomyolysis; hepatic disorder; pancreatitis; pulmonary embolism; myositis; renal disease needing dialysis; and deep‐vein thrombosis. The FIELD ophthalmology sub‐study reported incidence of overt retinopathy, incidence of DMO, and additional treatment for DR, including vitrectomy. Following our protocol, we described all adverse events and SAE authors presented in their trials.
Neither study reported on the following outcomes: mean change in visual acuity; incidence of a reduction in visual acuity of 10 ETDRS letters or more; incidence of PDR; additional treatments for DR, including anti‐vascular endothelial growth factor (VEGFs), steroids, and others; mean vision‐related quality of life; incremental cost per QALY gained; or acceptability of the treatment.
Excluded studies
We excluded 18 studies (19 reports): 10 studies had an irrelevant study design, five studies (6 reports) had an irrelevant population, two measured irrelevant outcomes, and one study had an irrelevant intervention. See Characteristics of excluded studies for details.
Ongoing studies
We identified three ongoing studies (in 5 reports). We will assess and include them, as indicated, in future updates (FAME 1 EYE; NCT03439345; NCT04661358).
Risk of bias in included studies
Risk of bias of included studies are summarised in Figure 2 and Figure 3. ACCORD‐Lipid's risk of bias was low for all domains, and ACCORD Eye Lipid’s risk of bias was low for all domains. FIELD's risk of bias was low for all domains. For the FIELD ophthalmology sub‐study, the risk of bias for the domain of selecting reporting was unclear; the other domains were at low risk of bias.
2.

Risk of bias graph: review authors' judgements about each risk of bias item, presented as percentages across all included studies
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study
For sequence generation, randomisation was undertaken using permuted blocks in ACCORD‐Lipid, and using a dynamic allocation method centrally in FIELD.
Therefore, the method of both trials’ sequence generation was adequate.
Allocation
Both trials reported an adequate method of randomisation; one used permuted blocks and the other used a central computer system and dynamic allocation method.
Blinding
In both trials’ participants, research personnel, and outcome assessors were masked to treatment allocation. Matching placebo was used in control groups.
Incomplete outcome data
In ACCORD‐Lipid, data were missing for 56 participants (1.0%). This was a low proportion, but they did not provide the reasons for the missing data. We contacted the corresponding author of ACCORD Eye Lipid, who provided information on the number of participants whose data at baseline and 4‐year follow‐up did not exist (325 (16.9%)); therefore, risk of bias was judged to be low. In addition, 153 out of 325 were in the fenofibrate group (16.0%); 172 were in the placebo group (17.9%). Missing data were balanced between the groups. In FIELD, the number of missing data was 31 (0.31%); 9/31 withdrew their consents, and 22 were not followed up. The proportions were low, but they did not describe the reasons. In the FIELD ophthalmology sub‐study, three participants withdrew their consents, and 124 were not followed up. Therefore, 127 (12.5%) participants were missing; the proportion was low, considering the number of outcomes’ incidences. In addition, 67 of 127 were in the fenofibrate group (13.1%); 60 were in the placebo group (12.0%). The number missing was balanced between the groups. Therefore, we decided low risk for the FIELD ophthalmology sub‐study.
Selective reporting
In ACCORD‐Lipid, the authors stated their outcomes in the published protocol and reported all outcomes as defined (ACCORD‐Lipid). Therefore, the risk of bias for this domain was considered low. In ACCORD Eye Lipid's protocol, we found that one outcome (change in visual acuity at four years compared with baseline) was different from that reported in the manuscript presenting the results. In the published protocol for the ACCORD Eye Lipid sub‐study, the outcomes to be evaluated were: moderate vision loss or loss of 3 lines on the logarithmic minimum angle of resolution (LogMAR) visual acuity charts, legal blindness: 20/160 or worse, and severe vision loss of 5/200 or worse, all from baseline to year four. In contrast, in the main manuscript, they presented the results of the following outcomes instead: moderate vision loss, development of vision of 20/50 or worse from baseline, development of 20/200 or worse from baseline, worsening of ≥ 15 letters of visual acuity score, all from baseline to year four. However, this selective reporting did not affect the outcomes evaluated in our review, thus, we considered the risk of bias for ACCORD for the selective reporting domain to be low.
In FIELD, the authors stated their outcomes in the published protocols and reported them all, therefore, we classified the risks of bias as low (FIELD). We did not find a published protocol for the FIELD ophthalmology sub‐study, so it was not clear if all outcomes prespecified for this study were reported; thus, we considered the risk of bias for this domain was unclear.
Other potential sources of bias
No other potential sources of bias were identified for ACCORD‐Lipid or ACCORD Eye Lipid. It was not a cluster‐randomised trial or cross‐over trial. Baseline imbalance did not occur. Allocation concealment was adequate. No differential diagnostic activity was found. The vanguard phase did not affect the comparison. The risk of bias was graded low. In addition, ACCORD‐Lipid was not funded by industry, but publicly funded. The study drugs were donated by the manufacturer, but they did not participate in the study design or conduct of the trial; neither data accrual or analysis, or manuscript preparation.
Regarding the FIELD or FIELD ophthalmology sub‐study, we did not identify another potential source of bias, thus, the risk of bias for this domain was also low. Their methods in considering other potential source of bias were adequate. There was the run‐in period, though it did not affect the randomisation. However, the FIELD and FIELD ophthalmology sub‐study were sponsored by industry. Representatives of industry (i.e. sponsors) without voting rights attended meetings of the management committee. In the writing committee, some members had conflicts of interest with the sponsor. Both the writing committee and study management committee took part in the writing of the manuscript, and in making the decision to submit the manuscript for publication.
Effects of interventions
See: Table 1
See Table 1.
ACCORD‐Lipid randomised 2765 participants to fenofibrate and 2753 participants to placebo. The mean follow‐up was 4.7 years. In the eye sub‐study of ACCORD‐Lipid, 959 participants were randomised to fenofibrate and 959 to placebo. FIELD randomised 4895 participants to fenofibrate and 4900 to placebo. The median follow‐up was 5 years. In the FIELD ophthalmology sub‐study, 512 participants were randomised to fenofibrate and 500 to placebo.
ACCORD‐Lipid's data were collected from published studies and further information was provided by the authors; FIELD’s data were collected from published studies.
The ACCORD Eye Lipid and FIELD ophthalmology sub‐study used the ETDRS scale for DR severity. However, the ACCORD Eye Lipid sub‐study used the ETDRS retinopathy severity scale for the person, in which both eyes are assessed and severity considers the retinopathy in both eyes. Steps ranged from 1 to 17, with more severe DR being given higher numbers. FIELD, however, graded the retinopathy using the ETDRS grading of the more severely affected eye (or of the right eye if both eyes were equally affected). The scale they used ranged from 1 to 13, with higher numbers given as the severity of DR increased.
We conducted meta‐analysis for the following outcomes: incidence of overt retinopathy, additional treatments for DR including focal/grid laser and PRP, discontinuation of the treatment, and adverse effects including SAE, hepatic disorder, deep‐vein thrombosis, and pulmonary embolism, as both trials provided data on these outcomes. We found no substantial heterogeneity in the outcomes we meta‐analysed (I² = 0%), except discontinuation of the treatment (I² = 87%) and adverse effects (hepatic disorder I² = 82%). Meta‐analysis was not possible for any of the other outcomes, including our primary outcome.
Following factors of the sensitivity analysis or subgroup analysis, we also described the results of the included studies, if applicable.
Fenofibrate compared to placebo or observation
Progression of diabetic retinopathy (DR)
Overall, fenofibrate likely resulted in little to no difference in the progression of DR at five years (risk ratio (RR) 0.86, 95% confidence interval (CI) 0.60 to 1.25; 1 study, 1012 participants; Analysis 1.1; moderate‐certainty evidence). We downgraded one level for imprecision, since the sample size was less than the optimal information size (OIS), and crossed the line of no effect (Guyatt 2011; Schünemann 2022).
1.1. Analysis.

Comparison 1: Fenofibrate vs placebo (5 year), Outcome 1: Progression of diabetic retinopathy
The FIELD ophthalmology sub‐study reported that of those with overt retinopathy at baseline, 2.9% (3/105) of the fenofibrate group and 13.6% (14/103) of the placebo group progressed two or more stages in the ETDRS scale (RR 0.21, 95% CI 0.06 to 0.71; 1 study, 208 people; test for interaction P = 0.02; Analysis 1.1). In subgroup analysis, those without overt retinopathy at baseline showed little or no progression (RR 1.00, 95% CI 0.68 to 1.47; 1 study, 804 participants).
Incidence of overt retinopathy
The ACCORD Eye Lipid sub‐study reported this outcome at four years; the FIELD ophthalmology sub‐study at five years. In ACCORD Eye Lipid, 28.0% (120/429) of participants in the fenofibrate group and 31.9% (127/398) of participants in the placebo group developed this outcome at four years. In the FIELD ophthalmology sub‐study, 11.3% (46/407) of participants in the fenofibrate group and 11.3% (45/397) of participants in the placebo group developed this outcome at five years. Fenofibrate likely resulted in little to no difference in the incidence of overt retinopathy (RR 0.91, 95% CI 0.76 to 1.09; 2 studies, 1580 participants; Analysis 1.2; moderate‐certainty evidence). We downgraded one level for imprecision.
1.2. Analysis.

Comparison 1: Fenofibrate vs placebo (5 year), Outcome 2: Incidence of overt retinopathy
Excluding the industry‐funded FIELD ophthalmology sub‐study, the risk ratio was 0.88 (95% CI 0.72 to 1.09). On the other hand, excluding the study in which this outcome was measured at four years (rather than at five years, as stated in our protocol, i.e. excluding the ACCORD Eye Lipid trial), the risk ratio was 1.00 (95% CI 0.68 to 1.47).
Incidence of DMO
The FIELD ophthalmology sub‐study reported this outcome at five years; 0.8% (4/512) of participants in the fenofibrate group and 2.0% (10/500) of participants in the placebo group developed DMO. Fenofibrate likely resulted in little to no difference in the incidence of DMO (RR 0.39, 95% CI 0.12 to 1.24; 1 study, 850 participants; moderate‐certainty evidence; Analysis 1.3). We downgraded one level for imprecision.
1.3. Analysis.

Comparison 1: Fenofibrate vs placebo (5 year), Outcome 3: Incidence of DMO
Additional treatments for DR (any laser)
Only FIELD reported this outcome. In FIELD, 3.6% (178/4895) of participants in the fenofibrate group and 5.2% (253/4900) of participants in the placebo group received any laser treatment (including focal/grid, PRP, or both). Fenofibrate reduced the requirement for any laser when compared with placebo (RR 0.70 95%CI 0.58 to 0.85; 1 study, 9764 participants; Analysis 1.4).
1.4. Analysis.

Comparison 1: Fenofibrate vs placebo (5 year), Outcome 4: Additional treatments for diabetic retinopathy (any laser)
Additional treatments for DR (focal/grid laser)
Both ACCORD Eye Lipid (at four years) and FIELD (at five years) reported this outcome. In ACCORD Eye Lipid, 2% (19/959) of participants in the fenofibrate group and 2.7% (26/959) of participants in the placebo group required focal/grid laser. In FIELD, 2.3% (115/4895) of participants in the fenofibrate group and 3.4% (167/4900) of participants in the placebo group required this treatment. Fenofibrate reduced the requirement for focal/grid laser (RR 0.69, 95% CI 0.56 to 0.86; 2 studies, 11,358 participants; Analysis 1.5).
1.5. Analysis.

Comparison 1: Fenofibrate vs placebo (5 year), Outcome 5: Additional treatments for diabetic retinopathy (focal/grid laser)
Excluding the industry‐funded FIELD Ophthalmology sub‐study, the risk ratio was 0.71 (95% CI 0.40 to 1.28). On the other hand, excluding the study with this outcome measured at four years (rather than five, as established in our protocol, i.e. the ACCORD Eye Lipid trial) the risk ratio was 0.69 (95% CI 0.55 to 0.87).
Additional treatments for DR (PRP)
Both ACCORD Eye Lipid and FIELD reported this outcome. In ACCORD Eye Lipid at four years, 0.8% (8/959) of participants in the fenofibrate group and 1.6% (15/959) of participants in the placebo group required PRP. In FIELD at five years, 1.5% (75/4895) of participants in the fenofibrate group and 2.2% (108/4900) of participants in the placebo group required PRP. Fenofibrate reduced the requirement of PRP (RR 0.67, 95% CI 0.51 to 0.89; 2 studies, 11,347; Analysis 1.6).
1.6. Analysis.

Comparison 1: Fenofibrate vs placebo (5 year), Outcome 6: Additional treatments for diabetic retinopathy (panretinal photocoaculation)
Excluding the industry‐funded FIELD study, the risk ratio was 0.52 (95% CI 0.22 to 1.22). Excluding the trial in which this outcome was measured at four years (rather than at five, as established in our protocol, i.e. the ACCORD Eye Lipid trial), the risk ratio was 0.70 (95% CI 0.52 to 0.93).
Additional treatments for DR (vitrectomy)
Only the FIELD ophthalmology sub‐study reported this outcome. In FIELD, 0.4% (2/512) of participants in the fenofibrate group and 0.2% (1/500) of participants in the placebo group required vitrectomy. Fenofibrate may result in little to no difference in the requirement of vitrectomy (RR 1.96 95% CI 0.18 to 21.56; 1 study, 850 participants; Analysis 1.7; moderate‐certainty evidence). We downgraded for imprecision because the CIs were wide, with few events.
1.7. Analysis.

Comparison 1: Fenofibrate vs placebo (5 year), Outcome 7: Additional treatments for diabetic retinopathy (vitrectomy)
Discontinuation of the treatment
Both ACCORD‐Lipid and FIELD reported this outcome. In ACCORD‐Lipid, 22.7% (628/2765) of participants in the fenofibrate group and 18.7% (516/2753) of participants in the placebo group discontinued treatment with fenofibrate during the trial. In FIELD, 19.5% (954/4895) of participants in the fenofibrate group and 19.4% (950/4900) of participants in the placebo group discontinued treatment with fenofibrate during the trial. Fenofibrate likely increased discontinuation of the treatment (RR 1.08, 95%CI 1.01 to 1.15; 2 studies, 15,226 participants; with heterogeneity, I2 = 87%; Analysis 1.8).
1.8. Analysis.

Comparison 1: Fenofibrate vs placebo (5 year), Outcome 8: Discontinuation of the treatment
Excluding the industry‐funded FIELD study, the risk ratio was 1.21 (95% CI 1.09 to 1.34). Both studies were conducted with adequate methodology and reported outcomes at five years.
Adverse effects (serious adverse events (SAE))
Both ACCORD‐Lipid and FIELD reported this outcome at five years. In ACCORD‐Lipid, 1.0% (27/2765) of participants in the fenofibrate group and 0.7% (18/2753) of participants in the placebo group developed SAE. In FIELD, 0.8% (38/4895) of participants in the fenofibrate group and 0.5% (24/4900) of participants in the placebo group developed SAE. While SAE were rare, the risk of their occurrence increased with the use of fenofibrate (RR 1.55, 95% CI 1.05 to 2.27; 2 studies, 15,226 participants; high‐certainty evidence; Analysis 1.9).
1.9. Analysis.

Comparison 1: Fenofibrate vs placebo (5 year), Outcome 9: Adverse effects (serious adverse event)
Excluding the industry‐funded FIELD study, the risk ratio was 1.49 (95% CI 0.82 to 2.70).
Adverse effects (rhabdomyolysis)
FIELD reported this outcome. In the fenofibrate group, 0.1% (3/4895) of participants and in the placebo group, 0.0% (1/4900) of participants developed rhabdomyolysis. Data suggested that fenofibrate might result in little to no difference in the development of rhabdomyolysis (RR 3.00 95% CI 0.31 to 28.87; 1 study, 9764 participants; Analysis 1.10). However, due to the rarity of this complication and the very wide 95% CI, this result is uncertain.
1.10. Analysis.

Comparison 1: Fenofibrate vs placebo (5 year), Outcome 10: Adverse effects (rhabdomyolysis)
Adverse effects (hepatic disorder)
Both ACCORD‐Lipid and FIELD reported this outcome. In ACCORD‐Lipid, 1.9% (52/2765) of participants in the fenofibrate group and 1.5% (40/2753) of participants in the placebo group developed this outcome. In FIELD, 0.4% (22/4895) of participants in the fenofibrate group and 0.8% (38/4900) of participants in the placebo group developed this outcome. Fenofibrate likely resulted in little to no difference in the development of hepatic disorder (RR 0.95 95% CI 0.69 to 1.32; 1 study, 15,226 participants; with heterogeneity, I2 = 82%; Analysis 1.11).
1.11. Analysis.

Comparison 1: Fenofibrate vs placebo (5 year), Outcome 11: Adverse effects (hepatic disorder)
Excluding the industry‐funded FIELD study, the risk ratio was 1.29 (95% CI 0.86 to 1.95).
Adverse effects (pancreatitis)
FIELD reported this outcome. In FIELD, 0.8% (40/4895) of participants in the fenofibrate group and 0.5% (23/4900) of participants in the placebo group developed pancreatitis. Fenofibrate increased the development of pancreatitis (RR 1.74 95% CI 1.04 to 2.90; 1 study, 9764 participants; Analysis 1.12).
1.12. Analysis.

Comparison 1: Fenofibrate vs placebo (5 year), Outcome 12: Adverse effects (pancreatitis)
Adverse effects (pulmonary embolism)
Both ACCORD‐Lipid and FIELD reported this outcome. In ACCORD‐Lipid, 0.0% (0/2765) of participants in the fenofibrate group and 0.0% (0/2753) of participants in the placebo group developed this outcome. In FIELD, 1.1% (53/4895) of participants in the fenofibrate group and 0.7% (32/4900) of participants in the placebo group developed this outcome. Fenofibrate likely increased the development of pulmonary embolism (RR 1.66 95% CI 1.07 to 2.57; 2 studies, 15,226 participants; Analysis 1.13).
1.13. Analysis.

Comparison 1: Fenofibrate vs placebo (5 year), Outcome 13: Adverse effects (pulmonary embolism)
Excluding the industry‐funded FIELD study, fenofibrate resulted in little to no difference in pulmonary embolism, because no one in either group in ACCORD‐Lipid developed this outcome.
Adverse effects (myositis)
FIELD reported this outcome. In FIELD, 0.0% (2/4895) of participants in the fenofibrate group and 0.0% (1/4900) of participants in the placebo group developed this outcome. Data suggested that fenofibrate might result in little to no difference in the development of myositis (RR 2.00 95% CI 0.18 to 22.08; 1 study, 9764 participants; Analysis 1.14).
1.14. Analysis.

Comparison 1: Fenofibrate vs placebo (5 year), Outcome 14: Adverse effects (myositis)
Adverse effects (renal disease needing dialysis)
Only FIELD reported this outcome. In FIELD, 0.3% (16/4895) of participants in the fenofibrate group and 0.4% (21/4900) of participants in the placebo group developed this outcome. Fenofibrate resulted in little to no difference in the development of renal disease needing dialysis (RR 0.76 95% CI 0.40 to 1.46; 1 study, 9764 participants; Analysis 1.15).
1.15. Analysis.

Comparison 1: Fenofibrate vs placebo (5 year), Outcome 15: Adverse effects (renal disease needing dialysis)
Adverse effects (deep‐vein thrombosis)
Both ACCORD‐Lipid and FIELD reported this outcome. In ACCORD‐Lipid, 0% (0/2765) of participants in the fenofibrate group and 0% (0/2753) of participants in the placebo group developed this outcome. In FIELD, 1.4% (67/4895) of participants in the fenofibrate group and 1.0% (48/4900) of participants in the placebo group developed this outcome. Fenofibrate resulted in little to no difference in the development of deep vein thrombosis (RR 1.40 95% CI 0.97 to 2.02; 2 studies, 15,226 participants; Analysis 1.16).
1.16. Analysis.

Comparison 1: Fenofibrate vs placebo (5 year), Outcome 16: Adverse effects (deep‐vein thrombosis)
Excluding the industry‐funded FIELD study, fenofibrate resulted in little to no difference in deep‐vein thrombosis, because no one in either group of ACCORD‐Lipid developed this outcome.
Discussion
Summary of main results
We included two randomised controlled trials (RCTs; N = 15,226), each of which included an eye sub study (ACCORD‐Lipid; FIELD).
Moderate‐certainty evidence from one sub‐study found that fenofibrate likely resulted in little to no difference in the progression of diabetic retinopathy (DR) with a mixed population (with and without overt retinopathy), but likely resulted in slow progression of DR in a population with overt retinopathy at baseline.
Moderate‐certainty evidence found that fenofibrate likely resulted in little to no difference in the incidence of overt retinopathy (two studies) or diabetic macular oedema (DMO; one study).
High‐certainty evidence found that fenofibrate increased serious adverse events overall.
However, only the FIELD ophthalmology sub‐study reported on our primary outcome, progression of DR. Thus, meta‐analysis was not possible. The FIELD ophthalmology sub‐study reported that in a mixed group of people with and without overt retinopathy, fenofibrate likely resulted in little to no difference in progression of DR. Because of the small sample size (N = 850) compared with the optimal information size (OIS), this finding should be interpreted cautiously (Guyatt 2011; Schünemann 2022). The degree of certainty was moderate. However, subgroup analysis by the presence of overt retinopathy at baseline suggested a difference in progression. For the secondary outcome, incidence of overt retinopathy, we conducted meta‐analysis and found that compared to placebo or observation, fenofibrate likely resulted in little to no difference in the incidence of overt retinopathy. Because of the imprecision due to the small sample size (N = 1580) compared with the OIS, this result should be also interpreted with caution, as we assessed the certainty of the evidence to be moderate.
For the incidence of DMO, fenofibrate likely resulted in little to no difference, but here, due to the imprecision because of the small sample size (N = 850) compared with the OIS, we assessed the certainty of the evidence as moderate; yet again, results should be interpreted with caution. Fenofibrate reduced the requirement of any laser, focal/grid laser, and panretinal photocoagulation. Fenofibrate might result in little to no difference in the need for vitrectomy. With imprecision due to the small sample size (N = 850) and few events (n = 3), this result should also be interpreted cautiously. Fenofibrate likely increased discontinuation of the treatment.
Regarding adverse effects, the use of fenofibrate increased severe adverse events with high‐certainty evidence. The use of fenofibrate also increased pancreatitis, and likely increased pulmonary embolism. Fenofibrate likely resulted in little to no difference in the development of hepatic disorder and might result in little to no difference in the development of rhabdomyolysis or myositis. Fenofibrate resulted in little to no difference in the development of renal disease needing dialysis and deep‐vein thrombosis.
Neither ACCORD‐Lipid nor FIELD examined any of the other outcomes specified in our review, including mean change in visual acuity, incidence of a reduction in visual acuity of 10 ETDRS letters or more, incidence of proliferative diabetic retinopathy, additional treatments for DR including anti‐vascular endothelial growth factor (VEGFs) or steroids, mean vision‐related quality of life, incremental cost per quality‐adjusted life year gained, acceptability of the treatment, or adverse effects (Steven‐Johnson syndrome). At present, there are no other preventive measures besides glycaemia, blood pressure, and lipid control that could potentially reduce the risk of progression and complications of DR. Therefore, new prophylactic strategies are needed. Recent trials have demonstrated that intravitreal anti‐VEGF use in eyes with moderately severe and severe non‐proliferative DR may lead to an improvement in retinopathy levels, measured using the diabetic retinopathy severity scale (Brown 2021; Maturi 2021). Further evidence is required to support the use of fenofibrate in people with, or at risk of developing DR. LENS, FAME 1 eye, and Fenofibrate for prevention of DR worsening studies will hopefully provide this evidence (FAME 1 EYE; NCT03439345; NCT04661358).
Overall completeness and applicability of evidence
We included two studies conducted in the US, Canada, Australia, Finland, and New Zealand. Participants were 40 to 79 years old, with a higher proportion of males (65.1%) and Caucasians (84.0%); all had type 2 diabetes (T2D). Participants were similar in age and gender in both trials. We are not confident that the results are generalisable to people of other races or ages, or those with type 1 diabetes (T1D), without further evidence from new studies.
No studies examined the: mean change in visual acuity, incidence of a reduction in visual acuity of 10 ETDRS letters or more, incidence of proliferative diabetic retinopathy (PDR), additional treatments for DR, mean vision‐related quality of life, incremental cost per quality adjusted life years (QALY) gained, acceptability of the treatment, or adverse effects (Steven‐Johnson syndrome).
Regarding ongoing trials, LENS’s participants had any diabetes mellitus except gestational diabetes, FAME 1 eye’s participants had T1D, and Fenofibrate for Prevention of DR Worsening’s participants were either T1D or T2D. We await publication of the data from their outcomes, progression of DR, incidence of DMO, additional treatments including laser, anti‐VEGFs, steroid, and vitrectomy, and visual acuity from three ongoing trials, cost‐effectiveness from LENS, and health‐related quality of life from LENS and FAME 1 eye.
On applicability of the data: fenofibrate likely results in little to no difference in the progression of diabetic retinopathy, but not in a group of people with overt retinopathy at baseline (FIELD sub‐study); and reduces the requirement for any laser treatment (FIELD), and for laser treatment including focal/grid and panretinal photocoagulation (ACCORD‐Lipid sub‐study; FIELD sub‐study). However, these findings should be interpreted with caution, as stated above. In addition, fenofibrate increases severe adverse events, pancreatitis, and pulmonary embolism.
Quality of the evidence
We included two high‐quality RCTs comprising a total of 15,313 participants (ACCORD‐Lipid: 5518; ACCORD Eye Lipid: 1918; FIELD: 9795; FIELD ophthalmology sub‐study: 1012). These were multi‐centre RCTs, using matching placebo, with appropriate sequence generation and allocation concealment. We assessed ACCORD‐Lipid as low risk of bias. The sample size of ACCORD‐Lipid was calculated based on the primary outcome, which was not DR. Despite its large sample size, the number of participants included was lower than required, based on the investigator’s sample size calculation (ACCORD‐Lipid). In ACCORD Eye Lipid, a sample size calculation for the primary composite outcome, related with advancing DR, was provided, but similar to the full trial, this was not met. We detected selective reporting bias, but it did not affect the outcomes evaluated in our review, therefore, we considered the risk of bias for ACCORD Eye lipid for the selective reporting domain to be low. We considered FIELD to have a low risk of bias and large sample size (FIELD). The sample size of FIELD was calculated based on each primary outcome, none of which were DR. FIELD was supported by industry, which took part in the design of the trial, the writing of the manuscripts, and presenting of the results of the trial. In the FIELD ophthalmology sub‐study, the sample size calculation was not given. The FIELD ophthalmology sub‐study had no published protocol, and its reporting bias was unclear. The bias of other domains was adequate.
Results for the outcomes from the eye sub‐studies for both trials should be interpreted with caution (Table 1). We assessed the certainty of the evidence for the progression of DR as moderate, because of imprecision due to small sample size (N = 850) compared with the OIS (Guyatt 2011; Schünemann 2022). We assessed moderate‐certainty evidence for the incidence of overt retinopathy, downgrading due to imprecision related to small sample size (N = 1580) comparing with the OIS, and moderate‐certainty evidence for incidence of DMO, due to imprecision related to small sample size (N = 850).
We found no reason to downgrade the certainty of the evidence for additional treatment for DR (any laser (FIELD)), or additional treatment for DR (focal/grid laser and PRP (ACCORD‐Lipid sub‐study; FIELD)), therefore, both of these outcomes were supported by high‐certainty evidence. Moderate‐certainty evidence supported the need for additional treatments for DR (vitrectomy), downgraded for imprecision due to only 850 participants and few events (n = 3 (FIELD sub‐study).
For discontinuation of treatment, we included data at five years from both studies, with adequate study designs and a large sample size (N = 15,226), but we detected inconsistency (I2 = 87%). We found no reasons to downgrade the certainty of evidence for severe adverse events, because of adequate study designs and a large sample size (N = 15,226). There was high‐certainty evidence that fenofibrate increases the risk of severe adverse events overall, and deep‐vein thrombosis (N = 15226) in particular. Other adverse effects reported few events, rhabdomyolysis (4/9764), myositis (3/9764); and heterogeneity between studies, hepatic disorder (N = 15,226), pulmonary embolism (N = 15,226). Only FIELD reported rhabdomyolysis (N = 9764), pancreatitis (N = 9764), and renal disease needing dialysis (N = 9764), thus reducing the sample size.
Potential biases in the review process
We followed Cochrane guidelines to undertake this review. None of the authors of this review have any potential conflicts of interest to report. Therefore, there should be no potential bias introduced in this review.
We did introduce several changes in the methodology for this review compared with our plan at the protocol stage (Inoue 2019). We made these changes before initiating the literature searches and data extraction. All changes are detailed in the Differences between protocol and review section.
Agreements and disagreements with other studies or reviews
Two other systematic reviews have been published that evaluated the effects of fenofibrate on DR. There were some differences between them and our review.
Czupryniak 2016 set out to estimate the effects of micronized fenofibrate alone or with a statin on microvascular complications (retinopathy, nephropathy, or neuropathy) in people with T2D. They searched PubMed between January 1990 and November 2015. They included ACCORD‐Lipid, FIELD, their sub‐studies, and the MacuFen study (Massin 2014). They reported results similar to our review, that fenofibrate reduced the incidence of advancing two or more steps in the ETDRS scale in people with overt retinopathy; the composite outcome of advancing three or more steps in the ETDRS scale; the need for laser treatment for DMO and proliferative DR, and the lack of progression of DR in those without overt retinopathy at baseline. The real difference came from their inclusion of the MacuFen study, which found that fenofibrate reduced total macular volume in participants with DMO. Our review found little or no difference in the incidence of DMO, but we excluded those with DMO at baseline.
Elkjaer 2020 examined whether systemic treatments would prevent or delay the progression of DR in people with diabetes. The treatments included fenofibrate, intensive glycaemic control, medications to reduce blood pressure, combination treatment, and others, which covered a wider scope than our review. They searched for prospective studies, including RCTs, written in English, in PubMed and Embase, without limiting the type of diabetes or systematic treatments. They included 13 studies, two of which covered fenofibrate (ACCORD‐Lipid; FIELD). They also reported that fenofibrate only reduced progression of DR in participants with overt retinopathy; it reduced the need for laser treatment, the risk of a two‐step progression of DR grade, DMO, or laser treatment, when compared with placebo.
Su 2019 published the protocol of a systematic review investigating the effects of fenofibrate on people with DR. They plan to search for RCTs in CENTRAL, PubMed, Embase, CINAHI, ACMD, CBM, CNKI, VIP, and WANG‐FANG without limitations on the study period.
Authors' conclusions
Implications for practice.
Current, moderate‐certainty evidence suggests that in a mixed group of people with and without overt retinopathy, who live with type 2 diabetes (T2D), fenofibrate likely results in little to no difference in progression of diabetic retinopathy (DR). However, in people with overt retinopathy who live with T2D, fenofibrate likely reduces the progression.
Serious adverse events were rare, but the risk of their occurrence was increased by the use of fenofibrate.
There is no evidence on the effect of fenofibrate in people with type 1 diabetes (T1D).
Implications for research.
Further studies are needed to determine the possible beneficial effects of fenofibrate in people living with diabetes.
Participants in the randomised controlled trials (RCTs) included in this review had all T2D. Therefore, research is needed to determine the effect of fenofibrate in people with T1D.
Only one sub‐study contributed data on progression of diabetic retinopathy (DR), incidence of diabetic macular oedema (DMO), and additional treatments for DR (especially vitrectomy), therefore, the number of participants was small compared with the optimal information size (OIS). Although two sub‐studies contributed data on the incidence of overt retinopathy, the number of participants was still small compared with the OIS on this outcome. To establish high‐certainty evidence, future studies should be powered appropriately.
Future studies should consider evaluating other important outcomes, e.g. other measures of visual acuity (e.g. mean change in visual acuity, and proportion of people experiencing a reduction in visual acuity of 10 ETDRS letters or more, i.e. the Early Treatment Diabetic Retinopathy Study); the incidence of proliferative diabetic retinopathy; use of more recently introduced treatments for complications of DR, including anti‐vascular endothelial growth factor and steroids; health‐related and vision‐related quality of life; cost‐effectiveness; and acceptability of the treatment. Involving people living with diabetes in the design of future trials is essential to ensure that outcomes that are important to people with the disease are included.
History
Protocol first published: Issue 4, 2019
Acknowledgements
We would like to thank the following:
Iris Gordon for creating and executing electronic search strategies for this review
Dr Emily Chew for input to this review’s protocol and for kindly providing us with additional information required for the assessment of the ACCORD trials
Leslie Choi, Evidence Synthesis Development Editor (Cochrane Central Executive Team) for comments on the review
Katie Curran, MPH, PhD for comments on the review
Chiara M. Eandi, Department of Surgical Sciences, University of Torino, Italy for comments on the review
Dr Toshi A Furukawa for his input to this review’s protocol
Dr Jennifer Evans for her input to this review’s protocol and review
Anupa Shah, Managing Editor for CEV for her assistance throughout the editorial process
Victoria Pennick for her assistance in copy editing
FAME 1 Eye study team and Sarah Howard for providing information of ongoing trials
We thank Hsin‐wen Wu for translating Cui 2018
We thank Chin Han Tan for full‐text screening and Yan Luo, Hsin‐wen Wu, and Nyurguyana Grigoryeva for extracting data.
Appendices
Appendix 1. CENTRAL search strategy
#1 MeSH descriptor: [Diabetic Retinopathy] explode all trees #2 (diabet* or proliferative or non‐proliferative) near/4 retinopath* #3 diabet* near/3 (eye* or vision or visual* or sight*) #4 retinopath* near/3 (eye* or vision or visual* or sight*) #5 DR near/3 (eye* or vision or visual* or sight*) #6 #1 or #2 or #3 or #4 or #5 #7 MeSH descriptor: [Fenofibrate] this term only #8 fenofibrate or phenofibrate #9 antara or controlip or durafenat or fenoglide or fenobeta or fenofanton or lipofen or lipanthyl or lipantil or liparison or livesan or lofibra or normalip or procetofen or procetofene or secalip or supralip or tricor or triglide #10 #7 or #8 or #9 #11 #6 and #10
Appendix 2. MEDLINE Ovid search strategy
1. randomized controlled trial.pt. 2. random$.ab,ti. 3. placebo.ab,ti. 4. dt.fs. 5. trial.ab,ti. 6. (group or groups).ab,ti. 7. or/1‐6 8. exp animals/ 9. exp humans/ 10. 8 not (8 and 9) 11. 7 not 10 12. exp Diabetic Retinopathy/ 13. ((diabet$ or proliferative or non‐proliferative) adj4 retinopath$).tw. 14. diabetic retinopathy.kw. 15. (diabet$ adj3 (eye$ or vision or visual$ or sight$)).tw. 16. (retinopath$ adj3 (eye$ or vision or visual$ or sight$)).tw. 17. (DR adj3 (eye$ or vision or visual$ or sight$)).tw. 18. or/12‐17 19. Fenofibrate/ 20. (fenofibrate or phenofibrate).tw. 21. (antara or controlip or durafenat or fenoglide or fenobeta or fenofanton or lipofen or lipanthyl or lipantil or liparison or livesan or lofibra or normalip or procetofen or procetofene or secalip or supralip or tricor or triglide).tw. 22. or/19‐21 23. 18 and 22 24. 11 and 23
The search filter for trials at the beginning of the MEDLINE strategy is from the published paper by Glanville 2006.
Appendix 3. Embase Ovid search strategy
1. exp randomized controlled trial/ 2. exp randomization/ 3. exp double blind procedure/ 4. exp single blind procedure/ 5. random$.tw. 6. or/1‐5 7. (animal or animal experiment).sh. 8. human.sh. 9. 7 and 8 10. 7 not 9 11. 6 not 10 12. exp clinical trial/ 13. (clin$ adj3 trial$).tw. 14. ((singl$ or doubl$ or trebl$ or tripl$) adj3 (blind$ or mask$)).tw. 15. exp placebo/ 16. placebo$.tw. 17. random$.tw. 18. exp experimental design/ 19. exp crossover procedure/ 20. exp control group/ 21. exp latin square design/ 22. or/12‐21 23. 22 not 10 24. 23 not 11 25. exp comparative study/ 26. exp evaluation/ 27. exp prospective study/ 28. (control$ or prospectiv$ or volunteer$).tw. 29. or/25‐28 30. 29 not 10 31. 30 not (11 or 23) 32. 11 or 24 or 31 33. exp Diabetic Retinopathy/ 34. ((diabet$ or proliferative or non‐proliferative) adj4 retinopath$).tw. 35. diabetic retinopathy.kw. 36. (diabet$ adj3 (eye$ or vision or visual$ or sight$)).tw. 37. (retinopath$ adj3 (eye$ or vision or visual$ or sight$)).tw. 38. (DR adj3 (eye$ or vision or visual$ or sight$)).tw. 39. or/33‐38 40. Fenofibrate/ 41. (fenofibrate or phenofibrate).tw. 42. (antara or controlip or durafenat or fenoglide or fenobeta or fenofanton or lipofen or lipanthyl or lipantil or liparison or livesan or lofibra or normalip or procetofen or procetofene or secalip or supralip or tricor or triglide).tw. 43. or/40‐42 44. 39 and 43 45. 32 and 44
Appendix 4. ISRCTN search strategy
(fenofibrate OR phenofibrate OR tricor) AND diabetic retinopathy
Appendix 5. ClinicalTrials.gov search strategy
(fenofibrate OR phenofibrate OR tricor) AND (diabetic retinopathy)
Appendix 6. WHO ICTRP search strategy
diabetic retinopathy = Condition AND fenofibrate OR phenofibrate OR tricor = Intervention
Appendix 7. Data on study characteristics
| Mandatory items | Optional items | |
| Methods | ||
| Study design |
Exclusions after randomisation Losses to follow‐up Number randomised/analysed How were missing data handled? e.g. available case analysis, imputation methods Reported power calculation (Y/N), if yes, sample size and power Unusual study design/issues |
Eyes or Unit of randomisation/ unit of analysis |
| Participants | ||
| Two eyes included in study, both eyes received same treatment, briefly specify how analysed (best/worst/average/both and adjusted for within‐person correlation/both and not adjusted for within person correlation) and specify if mixture one eye and two eye | ||
| Country | ||
| Setting Ethnic group Equivalence of baseline characteristics (Y/N) |
Total number of participants | This information should be collected for total study population recruited into the study. If these data are only reported for the people who were followed up, please indicate. |
| Number (%) of men and women | ||
| Average age and age range | ||
| Inclusion criteria | ||
| Exclusion criteria | ||
| Interventions | ||
| Intervention (N = ) Comparator (N = ) |
|
|
| Outcomes | ||
| Primary and secondary outcomes as defined in study reports |
List outcomes Adverse events reported (Y/N) |
|
| Notes | ||
| Date conducted | Specify dates of recruitment of participants mm/yr to mm/yr | |
| Full study name: (if applicable) Reported subgroup analyses (Y/N) Were trial investigators contacted? |
||
| Sources of funding | ||
| Declaration of interest | ||
Data and analyses
Comparison 1. Fenofibrate vs placebo (5 year).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Progression of diabetic retinopathy | 1 | 1012 | Risk Ratio (IV, Fixed, 95% CI) | 0.86 [0.60, 1.25] |
| 1.1.1 With overt retinopathy at baseline | 1 | 208 | Risk Ratio (IV, Fixed, 95% CI) | 0.21 [0.06, 0.71] |
| 1.1.2 Without overt retinopathy at baseline | 1 | 804 | Risk Ratio (IV, Fixed, 95% CI) | 1.00 [0.68, 1.47] |
| 1.2 Incidence of overt retinopathy | 2 | 1580 | Risk Ratio (IV, Fixed, 95% CI) | 0.91 [0.76, 1.09] |
| 1.3 Incidence of DMO | 1 | 850 | Risk Ratio (IV, Fixed, 95% CI) | 0.39 [0.12, 1.24] |
| 1.4 Additional treatments for diabetic retinopathy (any laser) | 1 | 9764 | Risk Ratio (IV, Fixed, 95% CI) | 0.70 [0.58, 0.85] |
| 1.5 Additional treatments for diabetic retinopathy (focal/grid laser) | 2 | 11358 | Risk Ratio (IV, Fixed, 95% CI) | 0.69 [0.56, 0.86] |
| 1.6 Additional treatments for diabetic retinopathy (panretinal photocoaculation) | 2 | 11347 | Risk Ratio (IV, Fixed, 95% CI) | 0.67 [0.51, 0.89] |
| 1.7 Additional treatments for diabetic retinopathy (vitrectomy) | 1 | 850 | Risk Ratio (IV, Fixed, 95% CI) | 1.96 [0.18, 21.56] |
| 1.8 Discontinuation of the treatment | 2 | 15226 | Risk Ratio (IV, Fixed, 95% CI) | 1.08 [1.01, 1.15] |
| 1.9 Adverse effects (serious adverse event) | 2 | 15226 | Risk Ratio (IV, Fixed, 95% CI) | 1.55 [1.05, 2.27] |
| 1.10 Adverse effects (rhabdomyolysis) | 1 | 9764 | Risk Ratio (IV, Fixed, 95% CI) | 3.00 [0.31, 28.87] |
| 1.11 Adverse effects (hepatic disorder) | 2 | 15226 | Risk Ratio (IV, Fixed, 95% CI) | 0.95 [0.69, 1.32] |
| 1.12 Adverse effects (pancreatitis) | 1 | 9764 | Risk Ratio (IV, Fixed, 95% CI) | 1.74 [1.04, 2.90] |
| 1.13 Adverse effects (pulmonary embolism) | 2 | 15226 | Risk Ratio (IV, Fixed, 95% CI) | 1.66 [1.07, 2.57] |
| 1.14 Adverse effects (myositis) | 1 | 9764 | Risk Ratio (IV, Fixed, 95% CI) | 2.00 [0.18, 22.08] |
| 1.15 Adverse effects (renal disease needing dialysis) | 1 | 9764 | Risk Ratio (IV, Fixed, 95% CI) | 0.76 [0.40, 1.46] |
| 1.16 Adverse effects (deep‐vein thrombosis) | 2 | 15226 | Risk Ratio (IV, Fixed, 95% CI) | 1.40 [0.97, 2.02] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
ACCORD‐Lipid.
| Study characteristics | ||
| Methods |
Types of study: parallel‐group randomised controlled trial Number of exclusion after randomisation: fenofibrate: 0 (substudy: NI), placebo: 0 (substudy: NI) Losses to follow‐up: fenofibrate: 27 (substudy: 153), placebo: 29 (substudy: 172) Number randomised: fenofibrate: 2765 (substudy: 959), placebo: 2753 (substudy: 959) Number analysed: fenofibrate: 2765 out of 2765 (substudy: 806 out of 959), placebo: 2753 out of 2753 (substudy: 787 out of 959) The method of handling missing data: NI Power calculation conducted prior to the commencement of the study: yes Planned sample size by power calculation: 5800 (substudy: NI) Planned power: the trial had 87% power to detect an observed 20% reduction in the primary outcome (substudy: the trial had 91% power to detect an observed 20% reduction in the primary outcome) Planned primary time point the trialists had defined (year/month /date): participants will be treated and followed for 4 to 8 years (approximate mean, 5.6 years); (substudy: participants were evaluated at 4‐year follow‐up with 7FIELD ETDRS images. Information was collected annually about whether laser or vitrectomy performed; visual acuity done every 2 years to determine moderate visual loss, defined as worsening in either eye of 3 or more ETDRS lines on ETDRS VA chart.) Other specific addition of statistical methods: NI Another intervention: yes Unit of randomisation: person Unit of analysis: person |
|
| Participants |
Countries where the participants were recruited: the US and Canada Single centre or multicentre: multicentre Setting: clinical sites Number of recruiting centres: 77 Baseline characteristics fenofibrate
substudy:
placebo
substudy:
Equivalence of baseline characteristics: yes Inclusion criteria:
Exclusion criteria:
In the substudy, one more criteria was added:
|
|
| Interventions |
Intervention Characteristics fenofibrate
placebo
Another intervention for both groups
|
|
| Outcomes | Primary outcome
(substudy:
Secondary outcome
(substudy:
Outcomes reported in manuscript:
(substudy:
Adverse effects: reported
Unit of measure: person Planned length of follow‐up: participants to be treated and followed for 4 to 8 years (approximate mean 5.6 years) Actual length of follow‐up: the mean duration of follow‐up for the primary outcome was 4.7 years. The study report only provided the mean. |
|
| Identification |
Full study name: The action to control cardiovascular risk in diabetes lipid trial (substudy: The action to control cardiovascular risk in diabetes eye study) Clinical trial registration number and name of register: ClinicalTrials.gov number NCT00000620 for the ACCORD study (substudy: NCT00542178 for the ACCORD Eye study) Authors name: ACCORD study group (substudy: Emily Y. Chew; ACCORD Eye study group) Institution: the Department of Medicine, Columbia University College of Physicians and Surgeons (substudy: National Eye Institute) Email: hng1@columbia.edu (substudy: echew@nei.nih.gov) Address: the Department of Medicine, Columbia University College of Physicians and Surgeons, Rm. PH10‐305, New York, NY 10032 (substudy: the National Institutes of Health, Bldg. 10, Clinical Research Center, Rm. 3‐2531, 10 Center Dr., Mail Stop Center 1204, Bethesda, MD 20892) |
|
| Notes |
Date of enrolment of the first participant: early 2001 (substudy: October 2003) Date of the final follow‐up date of the last participant: June 2009 (substudy: June 2009) Source of funding: the National Heart, Lung, and Blood Institute, the National Institutes of Health, the National Institute of Diabetes and Digestive and Kidney Diseases, the National Eye Institute, the National Institute on Aging, and the Centers for Disease Control and Prevention. General Clinical Research Centers provided support at many sites. These companies donated study medications, equipment, or supplies: Abbott Laboratories, Amylin Pharmaceutical, AstraZeneca Pharmaceuticals, Bayer HealthCare, Closer Healthcare, GlaxoSmithKline Pharmaceuticals, King Pharmaceuticals, Merck, Novartis Pharmaceuticals, Novo Nordisk, Omron Healthcare, Sanofi‐Aventis U.S., and Takeda Pharmaceuticals. Sub‐group analyses reported by the authors: yes Were trial investigators contacted? We contacted and received missing data for the substudy Declaration of interest: Dr. Goff ‐ grant support or pending grant support from Merck, and money for serving as a data and safety monitoring board member for a trial of a diabetes medication from Takeda Dr. Cushman ‐ consulting fees from Novartis, Takeda, Sanofi‐Aventis, Bristol‐Myers Squibb, King Pharmaceuticals, Daiichi–Sankyo, Gilead, Theravance, Pharmacopeia, and Sciele, and grant support or pending grant support from Novartis, GlaxoSmithKline, and Merck Dr. Ginsberg ‐ advisory fees from Merck, Merck–Schering Plough, and Bristol‐Myers Squibb–AstraZeneca; consulting fees from Merck, Abbott–AstraZeneca, Bristol‐Myers Squibb, Roche, Isis–Genzyme, GlaxoSmithKline, Novartis, Pfizer, and Regeneron–Sanofi‐Aventis; grant support or pending grant support from Merck, Isis–Genzyme, Roche, and AstraZeneca; payment for development of education presentations from Pfizer; and payment for travel and accommodation expenses from all these companies Dr. Ela ‐ payment for development of education presentations from Pfizer, Abbott Pharmaceuticals, and Merck–Schering Plough Dr. Gerstein ‐ consulting fees from Sanofi‐Aventis, GlaxoSmithKline, Eli Lilly, Novo Nordisk, AstraZeneca, Bristol‐Myers Squibb, Roche, Medtronic, Merck, Bayer, Bioavail, and Jansen Ortho; grant support or pending grant support from Sanofi‐Aventis, GlaxoSmithKline, Novo Nordisk, Merck, Pronova, and Roche; honoraria from Sanofi‐Aventis, GlaxoSmithKline, Solvay, Boehringer Ingelheim, Servier, Bayer, Eli Lilly, Novo Nordisk, and Takeda; and payment for travel and accommodation expenses from all these companies Dr. Schubart ‐ participated in trials sponsored by Sanofi‐Aventis, Merck, and Johnson & Johnson No other potential conflict of interest relevant to this article was reported. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was performed centrally on the trial’s website with the use of permuted blocks to maintain concealment of study‐group assignments |
| Allocation concealment (selection bias) | Low risk | Randomization was performed centrally on the trial’s website with the use of permuted blocks to maintain concealment of study‐group assignments. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | They used placebo; this study was a fully masked randomised trial. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was a fully masked randomised trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The proportion of loss to follow up was low and balanced. We did not find the reason. |
| Selective reporting (reporting bias) | Low risk | In ACCORD Eye Lipid, the visual outcomes reported in the published protocol are different to those listed and presented in the main sub‐study results manuscript. However, as this change does not affect the outcomes we were investigating in our review, we considered the risk of bias for the domain selective reporting to be low. |
| Other bias | Low risk | None |
FIELD.
| Study characteristics | ||
| Methods |
Types of study: parallel‐group randomised controlled trial Number of exclusion after randomisation: fenofibrate: 0 (substudy: 0), placebo: 0 (substudy: 0) Losses to follow‐up: fenofibrate:12; 4 more withdrew consent (substudy: 67; none withdrew consent), placebo: 10; 5 more withdrew consent (substudy: 57; 3 more withdrew consent) Number randomised: fenofibrate: 4895 (substudy: 512), placebo: 4900 (substudy: 500) Number analysed: fenofibrate: 4852 out of 4895 (substudy: 429 out of 512); placebo: 4856 out of 4900 (primary outcome was assessed) (substudy: 421 out of 500 ) The method of handling missing data: NI Power calculation conducted prior to the commencement of the study: yes (substudy: NI) Planned sample size by power calculation: 9795 (substudy: NI) Planned power: the trial had 80% power to detect a 22% reduction in CHD events. This also provide 90% power to detect a 25% relative reduction in CHD events. (substudy: NI) Planned primary time point the trialists had defined (year/month /date): this outcome could occur anytime during the minimum follow‐up time of 5 years (60 months) (substudy: planned period was 5 years (60 months) on average) Other specific addition of statistical methods: NI Another intervention: no Unit of randomisation: person Unit of analysis: person (substudy: eye) |
|
| Participants |
Countries where the participants were recruited: Australia, Finland, and New Zealand Single centre or multi centres: multicentre Setting: hospital clinics and community base sources Number of recruiting centres: 63 (substudy: 22) Baseline characteristics fenofibrate
substudy
placebo
substudy
Equivalence of baseline characteristics: yes Inclusion criteria:
In the substudy, one more criteria was added:
Exclusion criteria:
In the substudy, one more criteria was added:
|
|
| Interventions | fenofibrate
placebo
Another intervention for both groups There was a run‐in phase for all participants that consisted of three periods before randomisation.
|
|
| Outcomes | Primary outcome:
substudy:
Secondary outcomes:
substudy:
Tertiary outcomes:
Outcomes reported in manuscript:
substudy:
Adverse effects: reported
Unit of measure: person (substudy: worst eye) Planned length of follow‐up: 5 years Actual length of follow‐up: reported a median of 5 years |
|
| Identification |
Full study name: The fenofibrate intervention and event lowering in diabetes study (substudy: ophthalmology substudy) Clinical trial registration number and name of register: International standard randomised controlled trial, number ISRCTN64783481 Authors name: AC Keech Institution: NHMRC Clinical Trials Centre, University of Sydney Email: tony@ctc.usyd.edu.au Address: FIELD study, NHMRC Clinical Trials Centre, University of Sydney, Building F, 88 Mallet Street, Camperdown, NSW 2050, Australia |
|
| Notes |
Date of enrolment of the first participant: NI Date of the final follow‐up date of the last participant: NI Source of funding: grant from Laboratoires Fournier SA, Dijon, France, and the National Health and Medical Research Council of Australia Subgroup analyses reported by the authors: yes Were trial investigators contacted?: yes, but they did not reply. Declaration of interest: some members of the writing committee (ACK, PM, PAS, JO’D, TMED, M‐RT, RJS, LTL, MCdE, PGC) had the costs of participation in scientific meetings and/or contributions to advisory boards, or doing other research reimbursed by the pharmaceutical industry. ACK is a listed applicant on a patent application in relation to some findings contained in this scientific report. DCC is an employee of the study sponsor. MSM, EW, AM, RLO’C, and DT have no conflict of interest to declare. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was carried out using a dynamic allocation method. |
| Allocation concealment (selection bias) | Low risk | Randomisation was done by central computer, using a dynamic allocation method with stratification for important prognostic factors, including age, sex, previous myocardial infarction, lipid levels, and urinary albumin concentration. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants took micronised fenofibrate 200 mg once daily or matching placebo. Members of the trial’s independent safety and data monitoring committee and the unblinded statistician were the only personnel to view data by treatment allocation. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Members of the trial’s independent safety and data monitoring committee and the unblinded statistician were the only personnel to view data by treatment allocation. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The proportion of missing data was low and balanced between groups. However, we did not find the reason. |
| Selective reporting (reporting bias) | Low risk | We were unable to find a published protocol for the FIELD ophthalmology substudy. Thus, we considered the risk of bias for selective reporting of the FIELD ophthalmology substudy to be unclear, as a result. |
| Other bias | Low risk | None |
ADA: American Diabetes Association; BMI: body mass index; BP: blood pressure; CVD: cardiovascular disease; DMO: diabetic macular oedema; DR: diabetic retinopathy; ECG: electrocardiography; ETDRS: the early treatment diabetic retinopathy study; HDL‐C: high density lipoprotein cholesterol; HbA1c: glycated haemoglobin A1C; IQR: interquartile range; LDL‐C: low density lipoprotein cholesterol; MI: myocardial infarction; NA: not available; NI: no information; NPDR: non‐proliferative diabetic retinopathy; NYHA: New York Heart Association; PDR: proliferative diabetic retinopathy; PRP: pan retinal photocoagulation; SD: standard deviation; T‐chol: total cholesterol; TG: triglycerides; T1D: type 1 diabetes; T2D: type 2 diabetes
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| ACCORDION 2016 | Irrelevant intervention |
| ACTRN 12618000592246 | Irrelevant population |
| Borona 2021 | Irrelevant population |
| Bronson 2010 | Irrelevant study design |
| Cui 2018 | Irrelevant population |
| Elam 2011 | Irrelevant study design |
| Fazio 2009 | Irrelevant study design |
| Feher 2005 | Irrelevant study design |
| FIELD 2008 | Irrelevant study design |
| Fuessl 2008 | Irrelevant study design |
| Grigoryeva 2011 | Irrelevant study design |
| Massin 2014 | Irrelevant population |
| Matthews 2011 | Irrelevant study design |
| NCT04140201 | Irrelevant outcomes |
| NCT04885153 | Irrelevant outcomes |
| O'Connor 2011 | Irrelevant study design |
| Srinivasan 2018 | Irrelevant population |
| Valentine 2013 | Irrelevant study design |
Characteristics of ongoing studies [ordered by study ID]
FAME 1 EYE.
| Study name | The fenofibrate and microvascular events in type 1 diabetes eye: a randomised trial to evaluate the efficacy on retinopathy and safety of fenofibrate in adults with type 1 diabetes. A multicentre double‐blind placebo‐controlled study in Australia and internationally |
| Methods |
Types of study: parallel‐group randomised controlled trial Number of exclusion after randomisation: Losses to follow‐up: fenofibrate:, placebo: Number randomised: fenofibrate:, placebo: Number analysed: fenofibrate: out of, placebo: out of The method of handling missing data: Power calculation conducted prior to the commencement of the study (yes or no): Planned sample size by power calculation: 450 (NI regarding power calculation) Planned power: Planned primary time point the trialists had defined (year/month /date): 36 months Other specific addition of statistical methods: Another intervention (yes/no): Unit of randomisation: person Unit of analysis: |
| Participants |
Where the participants were recruited: Hong Kong, New Zealand, and Australia Single centre or multicentre: multicentre Setting: clinic Number of recruiting centres: 21 Baseline Characteristics fenofibrate
placebo
Equivalence of baseline characteristics: Inclusion criteria:
Exclusion criteria:
|
| Interventions |
Intervention Characteristics fenofibrate
placebo
Another intervention for both group (yes/no): |
| Outcomes | Primary outcome:
Secondary outcomes:
Outcomes reported in manuscript: Adverse effects: Unit of measure: Planned length of follow up: 36 months Actual length of follow up: |
| Starting date | March 2016 |
| Contact information |
Name: Liping Li Institution: NHMRC Clinical Trials Centre, The University of Sydney Email: fame1eye@ctc.usyd.edu.au Address: Medical Foundation Building 92‐94 Parramatta Road Camperdown NSW 2050 |
| Notes |
Date of enrolment of the first participant: November 2016 Date of the final follow‐up date of the last participant (if any): Source of funding: NHMRC Clinical Trials Centre, University of Sydney (Australia), Mylan EPD Europe, National Health and Medical Research Council (Australia), Juvenile Diabetes Research Foundation (Australia) Were trial investigators contacted? yes Declaration of interest: |
NCT03439345.
| Study name | A randomised placebo‐controlled trial of fenofibrate to prevent progression of non‐proliferative retinopathy in diabetes |
| Methods |
Types of study: parallel‐group randomised controlled trial Number of exclusion after randomisation: Losses to follow‐up: fenofibrate:, placebo: Number randomised: fenofibrate:, placebo: Number analysed: fenofibrate: out of, placebo: out of The method of handling missing data: Power calculation conducted prior to the commencement of the study (yes or no): Planned sample size by power calculation: 1151 (NI regarding power calculation) Planned power: Planned primary time point the trialists had defined (year/month /date): this outcome could occur anytime during the minimum follow‐up time of 5 years (60 months) Other specific addition of statistical methods: Another intervention (yes/no): no Unit of randomisation: person Unit of analysis: |
| Participants |
Countries where the participants were recruited: United Kingdom Single centre or multicentre: multicentre Setting: National Health Service (NHS) Number of recruiting centres: 16 Baseline Characteristics fenofibrate
placebo
Equivalence of baseline characteristics: yes Inclusion criteria:
Exclusion criteria:
|
| Interventions | Fenofibrate
Placebo
|
| Outcomes | Primary outcome
Secondary outcomes
Outcomes reported in manuscript: Adverse effects: Unit of measure: Planned length of follow‐up: 48 months Actual length of follow‐up: |
| Starting date | July 2018 |
| Contact information |
Authors name: David Preiss Institution: LENS trial, University of Oxford Email: lens@ndph.ox.ac.uk Address: LENS trial, CTSU Richard Doll Building, University of Oxford Roosevelt Drive OXFORD, OX3 7LF |
| Notes |
Date of enrolment of the first participant: 23 July 2018 Date of the final follow‐up date of the last participant: NI Source of funding: National Institute for Health Research (United Kingdom), NHS Scotland Diabetic Retinopathy Screening Collaborative, University of Aberdeen, University of Dundee, University of Edinburgh, and University of Glasgow Were trial investigators contacted?: yes Declaration of interest: |
NCT04661358.
| Study name | A Randomized Clinical Trial Evaluating Fenofibrate for Prevention of Diabetic Retinopathy Worsening |
| Methods |
Types of study: parallel‐group randomised controlled trial Number of exclusion after randomisation: Losses to follow‐up: fenofibrate:, placebo: Number randomised: fenofibrate:, placebo: Number analysed: fenofibrate: out of, placebo: out of The method of handling missing data: Power calculation conducted prior to the commencement of the study (e.g. yes or no): Planned sample size by power calculation: 910 (NI regarding power calculation) Planned power: Planned primary time point the trialists had defined (year/month /date): the time flame is 4 years (48 months). Other specific addition of statistical methods: Another intervention (yes/no): no Unit of randomisation: person Unit of analysis: |
| Participants |
Countries where the participants were recruited: the United States Single centre or multicentre: multicentre Setting: clinics, institution, or university Number of recruiting centres: 42 Baseline characteristics fenofibrate
placebo
Equivalence of baseline characteristics: yes Inclusion criteria:
Exclusion criteria:
|
| Interventions | Fenofibrate
Placebo
|
| Outcomes | Primary outcome
Secondary outcomes
Outcomes reported in manuscript Adverse effects: NI Unit of measure: Planned length of follow‐up: 4 years Actual length of follow‐up: |
| Starting date | March 2021 |
| Contact information |
Authors name: Emily Y Chew Institution: Jaeb Center for Health Research Email: drcrnet@jaeb.org Address: NI Declaration of interest: NI |
| Notes |
ALT: alanine aminotransferase; AST: aspartate aminotransferase; CK: creatine kinase; CST: central subfield thickness; DMO: diabetic macular oedema; DR: diabetic retinopathy; eGFR: estimated glomerular filtration rate; ETDRS: the early treatment diabetic retinopathy study severity scale; HbA1c: glycated Hemoglobin A1C; HDL‐C: high density lipoprotein cholesterol; IQR: interquartile range; LDL‐C: low density lipoprotein cholesterol; NI: no information; NPDR: non‐proliferative diabetic retinopathy; OCT: optical coherence tomography; SD: standard deviation; TG: triglycerides; T1D: type 1 diabetes; T2D: type 2 diabetes; PDR: proliferative diabetic retinopathy; PRP: pan‐retinal photocoagulation; ULN: the upper limit of normal; VEGF: vascular endothelial growth factor
Differences between protocol and review
The following changes were made to the original protocol (Inoue 2019).
We limited the inclusion of studies to RCTs whose intervention group's participants took fenofibrate throughout the trial.
We changed the inclusion criteria of participants. In our original protocol, participants were people diagnosed with T1D or T2D, and we included those who did not have retinopathy or who had DR at baseline. We did not mention we would exclude people with the established complications of DR (i.e. DMO/PDR). In the final protocol we followed, we excluded studies including only participants with established complications of DR, which were evaluating the effect of fenofibrate on the established complications (rather than on preventing them). Studies randomising participants with complications were included in this review if only a small proportion of participants had established complications of DR at baseline (i.e. less than 10%), or if data for people without complications were presented separately and could be extracted.
We searched electronic databases for the European Association for the Study of Diabetes (EASD) congress and the European Association for the Study of Diabetes Eye Complications Study Group (EASDEC) congress from 1990 to the present instead of handsearching.
We changed some of secondary outcomes’ names, proportion of participants with a reduction in visual acuity of 10 letters or more, and proportion of participants in which treatment is discontinued to incidence of a reduction in visual acuity of 10 ETDRS letters or more, and discontinuation of the treatment.
Our original protocol established that all outcomes would be investigated at one, three, and five years. We changed the protocol to include outcomes measured at 3 ± 1, and 5 ± 1 years, instead of 3 years ± 6 months and 5 years ± 6 months, and we included the 4‐year data with the 5‐year data.
Four reviewers (SYK, NL, SK, YK) engaged in data extraction, and assessment of risk bias in included studies, and data management using Covidence, Excel, RevMan 5, RevMan Web.
In unit of analysis, we included results that reported using per person (as done in ACCORD‐Lipid using the ETDRS scale which considers the grading of DR from both eyes), and the worse eye or right eye when both eyes had the same retinopathy severity, for ocular outcomes (i.e. incidence of overt retinopathy and additional treatments for DR (focal/grid and PRP)) in conducting meta‐analysis.
Though we did not conduct subgroup analysis due to insufficient numbers of trials, we changed the definition of with or without overt retinopathy at baseline (ETDRS scale; Final Retinopathy Severity Scale for Persons of step 3 or less, or step 4 or greater (i.e. step 3 suggests the existence of microaneurysms detected in both eyes; step 4 suggests the existence of mild NPDR in one eye)) in subgroup analysis to that of with or without overt retinopathy at baseline (we used the original study authors' definitions) in subgroup analysis.
Reviewers (SYK, NL, YK, NW) agreed and prepared the summary of findings.
Contributions of authors
IK produced the first draft of the protocol; NL edited the subsequent draft
SYK, NL, SK, and NW reviewed and commented on the protocol draft
SYK, SK, and KI screened abstracts
SYK and YK reviewed full texts and identified eligible studies
SYK, NL, SK, and YK extracted data and estimated risk of bias
SYK, YK, NW, NL were involved in estimation of GRADE
SYK produced the first draft of the manuscript
NL edited the subsequent draft; NL, IK, SK, YK and NW reviewed and commented on the subsequent draft. All authors approved the final manuscript for submission.
Sources of support
Internal sources
-
None, Other
None
External sources
-
Public Health Agency, UK
The HSC Research and Development (R&D) Division of the Public Health Agency funds the Cochrane Eyes and Vision editorial base at Queen's University Belfast.
-
Queen's University Belfast, UK
Gianni Virgili, Co‐ordinating Editor for Cochrane Eyes and Vision’s work is funded by the Centre for Public Health, Queen’s University of Belfast, Northern Ireland.
Declarations of interest
SYK: none
NL: none
YK: none
SK: none
KI: none
NW: none related to the present review
New
References
References to studies included in this review
ACCORD‐Lipid {published data only}
- ACCORD Study Group, ACCORD Eye Study Group, Chew EY, Ambrosius WT, Davis MD, Danis RP, et al. Effects of medical therapies on retinopathy progression in type 2 diabetes. New England Journal of Medicine 2010;363(3):233-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ACCORD Study Group, Buse JB, Bigger JT, Byington RP, Cooper LS, Cushman WC, Friedewald WT, et al. Action to control cardiovascular risk in diabetes (ACCORD) trial: design and methods. American Journal of Cardiology 2007;99(12):S21-33. [DOI: 10.1016/j.amjcard.2007.03.003] [DOI] [PubMed] [Google Scholar]
- ACCORD Study Group, Ginsberg HN, Elam MB, Lovato LC, Crouse JR 3rd, Leiter LA, Linz P, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. New England Journal of Medicine 2010;362(17):1563-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew EY, Ambrosius WT, Howard LT, Greven CM, Johnson S, Danis RP, et al. Rationale, design, and methods of the action to control cardiovascular risk in diabetes eye study (ACCORD-EYE). American Journal of Cardiology 2007;99(Suppl 12 ):S103-11. [DOI] [PubMed] [Google Scholar]
- Chew EY, Davis MD, Danis RP, Lovato JF, Perdue LH, Greven C, et al. The effects of medical management on the progression of diabetic retinopathy in persons with type 2 diabetes: the Action to Control Cardiovascular Risk in Diabetes (ACCORD) eye study. Ophthalmology 2014;121(12):2443‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
FIELD {published data only}
- D'Emden M, Li LP, Zannino D, Best J, Keech AC, on behalf of the FIELD Study Investigators. Effect of fenofibrate on cardiovascular events and mortality in women with type 2 diabetes: results from the fenofibrate intervention and event lowering in diabetes (FIELD) study. Diabetes 2009;58(Suppl 1A):A178. [Google Scholar]
- FIELD Study Investigators. The need for a large-scale trial of fibrate therapy in diabetes: the rationale and design of the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study. Cardiovascular Diabetology 2004;3(9):1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ISRCTN64783481. Fenofibrate intervention and event lowering in diabetes. www.isrctn.com/ISRCTN64783481? (first registered 19 October 2004). [DOI: 10.1186/ISRCTN64783481] [DOI]
- Keech A, Simes RJ, Barter P, Best J, Scott R, Taskinen MR, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet 2005;366(9500):1849-61. [DOI] [PubMed] [Google Scholar]
- Keech AC, Mitchell P, Summanen PA, O'Day J, Davis TM, Moffitt MS, et al. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): a randomised controlled trial. Lancet 2007;370(9600):1687‐97. [DOI] [PubMed] [Google Scholar]
- Scott R, Best J, Forder P, Taskinen MR, Simes J, Barter P, et al. Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study: baseline characteristics and short-term effects of fenofibrate. Cardiovascular Diabetology 2005;4(13):1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
ACCORDION 2016 {published data only}
- Action to Control Cardiovascular Risk in Diabetes Follow-On (ACCORDION) Eye Study Group and the Action to Control Cardiovascular Risk in Diabetes Follow-On (ACCORDION) Study Group. Persistent effects of intensive glycemic control on retinopathy in type 2 diabetes in the action to control cardiovascular risk in diabetes (ACCORD) follow-on study. Diabetes Care 2016;39(7):1089-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
ACTRN 12618000592246 {published data only}
- ACTRN 12618000592246. A randomised multi-centre placebo controlled trial of fenofibrate for treatment of diabetic macular oedema with economic evaluation (FORTE Study). anzctr.org.au/Trial/Registration/TrialReview.aspx?ACTRN=12618000592246 (first received 17 April 2018). [ANZCTR: 12618000592246]
Borona 2021 {published data only}
- Bonora BM, Albiero M, Morieri Ml, Cappellari R, Amendolagine FI, Mazzucato M, et al. Fenofibrate increases circulating haematopoietic stem cells in people with diabetic retinopathy: a randomised, placebo-controlled trial. Diabetologia 2021;64:2334-44. [DOI] [PubMed] [Google Scholar]
Bronson 2010 {published data only}
- Bronson DL. Intensifying glucose control and adding fenofibrate to simvastatin each reduced progression of retinopathy in type 2 diabetes. Annals of Internal Medicine 2010;153(10):JC5-10. [DOI] [PubMed] [Google Scholar]
Cui 2018 {published data only}
- Cui Y, Li XD. Efficacy of fenofibrate combined with 23G minimally invasive vitrectomy for diabetic retinopathy. International Eye Science 2018;18(12):2155-9. [Google Scholar]
Elam 2011 {published data only}
- Elam M, Lovato L, Ginsberg H. The ACCORD-Lipid study: implications for treatment of dyslipidemia in type 2 diabetes mellitus. Clinical Lipidology 2011;6(1):9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Fazio 2009 {published data only}
- Fazio S. More clinical lessons from the FIELD study. Cardiovascular Drugs and Therapy 2009;23(3):235-41. [DOI] [PubMed] [Google Scholar]
Feher 2005 {published data only}
- Feher MD, Elkeles RS. Fenofibrate in type 2 diabetes: the FIELD study. British Journal of Diabetes and Vascular Disease 2005;5(6):330‐3. [Google Scholar]
FIELD 2008 {published data only}
- Fenofibrate reduces laser treatment needs in patients with diabetic retinopathy. Australian Journal of Pharmacy 2008;89(1061):77.
Fuessl 2008 {published data only}
- Fuessl HS. Fenofibrate reduces the need for laser treatment for diabetic retinopathy. MMW-Fortschritte der Medizin 2008;150(13):22. [Google Scholar]
Grigoryeva 2011 {published data only}
- Grigoryeva N, Shklyarov E, Shadrichev F, Kryaneva O. Fenofibrate effect on diabetic retinopathy. European Journal of Ophthalmology. 2011;21(3):344. [Google Scholar]
Massin 2014 {published data only}
- Massin P, Peto T, Ansquer JC, Aubonnet P. Effects of fenofibric acid on diabetic macular edema: the MacuFen study. Ophthalmic Epidemiology 2014;21(5):307‐17. [DOI] [PubMed] [Google Scholar]
- Massin P, Peto T, Le-Malicot K, Ansquer J. Effects of fenofibric acid on diabetic macular edema measured by optical coherence tomography. European Journal of Ophthalmology 2012;22(3):518. [Google Scholar]
Matthews 2011 {published data only}
- Matthews DR. Fenofibrate and statin therapy, compared with placebo and statin, slows the development of retinopathy in type 2 diabetes patients of 10 years duration: the ACCORD study. Evidence-Based Medicine 2011;16(2):45‐6. [DOI] [PubMed] [Google Scholar]
NCT04140201 {published data only}
- NCT04140201. Effect of lipid lowering agents on diabetic retinopathy and cardiovascular risk of diabetic patients. clinicaltrials.gov/ct2/show/study/NCT04140201 (first received 25 October 2019).
NCT04885153 {published data only}
- NCT04885153. Effects of oral fenofibrate on retinal thickness and macular volume. clinicaltrials.gov/ct2/show/NCT04885153 (first received 13 May 2021).
O'Connor 2011 {published data only}
- O'Connor PJ, Ismail-Beigi F. Near-normalization of glucose and microvascular diabetes complications: data from ACCORD and ADVANCE. Therapeutic Advances in Endocrinology and Metabolism 2011;2(1):17‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Srinivasan 2018 {published data only}
- Srinivasan S, Hande P, Shetty J, Murali S. Efficiency of fenofibrate in facilitating the reduction of central macular thickness in diabetic macular edema. Indian Journal of Ophthalmology 2018;66(1):98‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Valentine 2013 {published data only}
- Valentine WJ, Pollock RF, Carr E, Aubonnet P, Mitchell P, Keech A. Evaluating the cost-utility of fenofibrate treatment of diabetic retinopathy in Australia. Value in Health 2013;16(7):A442. [Google Scholar]
References to ongoing studies
FAME 1 EYE {unpublished data only}
- ACTRN 12611000249954. The fenofibrate and microvascular event eye (FAME 1 EYE) trial in adults with type 1 diabetes mellitus: a randomised trial in adults with type 1 diabetes mellitus evaluating the effects of daily oral fenofibrate compared with placebo on macular thickness and volume. anzctr.org.au/Trial/Registration/TrialReview.aspx?ACTRN=12611000249954 (first received 7 March 2011).
- NCT01320345. The fenofibrate and microvascular events in type 1 diabetes eye (FAME 1 EYE) [A randomised trial to evaluate the efficacy on retinopathy and safety of fenofibrate in adults with type 1 diabetes. A multicentre double-blind placebo-controlled study in Australia and internationally.]. clinicaltrials.gov/ct2/show/NCT01320345 (first received 22 March 2011).
NCT03439345 {published and unpublished data}
- NCT03439345. Lowering events in non-proliferative retinopathy in Scotland [A randomised placebo-controlled trial of fenofibrate to prevent progression of non-proliferative retinopathy in diabetes]. clinicaltrials.gov/ct2/show/nct03439345 (first received 20 February 2018).
- Preiss D, Armitage J, Olson J, Scotland G, Leese G, Colhoun H, et al. LENS-a clinical trial embedded in routine clinical practice to reduce the burden of diabetic eye disease. Trials 2017;18 (Suppl 1):56. [DOI: 10.1186/s13063-017-1902-y] [DOI] [Google Scholar]
NCT04661358 {unpublished data only}
- NCT04661358. Fenofibrate for prevention of DR worsening (protocol AF) [A randomized clinical trial evaluating fenofibrate for prevention of diabetic retinopathy worsening]. clinicaltrials.gov/ct2/show/NCT04661358 (first received 10 December 2020).
Additional references
Aiello 2010
- Aiello LP, Edwards AR, Beck RW, Bressler NM, Davis MD, Ferris F, et al. Factors associated with improvement and worsening of visual acuity 2 years after focal/grid photocoagulation for diabetic macular edema. Ophthalmology 2010;117(5):946-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Blane 1989
- Blane G F. Review of European clinical experience with fenofibrate. Cardiology 1989;76(Suppl 1):1-13. [DOI] [PubMed] [Google Scholar]
Boyer 2014
- Boyer DS, Yoon YH, Belfort R Jr, Bandello F, Maturi RK, Augustin AJ. Three-year, randomized, sham-controlled trial of dexamethasone intravitreal implant in patients with diabetic macular edema. Ophthalmology 2014;121(10):1904-14. [DOI] [PubMed] [Google Scholar]
Brown 2021
- Brown DM, Wykoff CC, Boyer D, Heier JS, Clark WL, Emanuelli A, et al. Evaluation of intravitreal aflibercept for the treatment of severe nonproliferative diabetic retinopathy: results from the PANORAMA randomized clinical trial. JAMA Ophthalmology 2021;139(9):946-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ciudin 2013
- Ciudin A, Hernandez C, Simo R. Molecular implications of the PPARs in the diabetic eye. PPAR Research 2013;2013:686525. [DOI: 10.1155/2013/686525] [DOI] [PMC free article] [PubMed] [Google Scholar]
Covidence [Computer program]
- Covidence. Version accessed 28 February 2022. Melbourne, Australia: Veritas Health Innovation. Available at www.covidence.org.
Czupryniak 2016
- Czupryniak L, Joshi SR, Gogtay JA, Lopez M. Effect of micronized fenofibrate on microvascular complications of type 2 diabetes: a systematic review. Expert Opinion on Pharmacotherapy 2016;17(11):1463-73. [DOI] [PubMed] [Google Scholar]
Deeks 2022
- Deeks JJ, Higgins JP, Altman DG, editor(s) on behalf of the Cochrane Statistical Methods Group. Chapter 10: Analysing data and undertaking meta-analyses. In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s), Cochrane Handbook for Systematic Reviews of Interventions version 6.3 (updated February 2022), Cochrane, 2022. Available at www.training.cochrane.org/handbook.
Duh 2017
- Duh EJ, Sun JK, Stitt AW. Diabetic retinopathy: current understanding, mechanisms, and treatment strategies. JCI Insight 2017;2(14):pii: 93751. [DOI: 10.1172/jci.insight.93751] [DOI] [PMC free article] [PubMed] [Google Scholar]
Elkjaer 2020
- Elkjaer AS, Lynge SK, Grauslund J. Evidence and indications for systemic treatment in diabetic retinopathy: a systematic review. Acta Ophthalmologica 2020;98(4):329-36. [DOI] [PubMed] [Google Scholar]
ETDRS 1991
- Early Treatment Diabetic Retinopathy Study Research Group. Fundus photographic risk factors for progression of diabetic retinopathy. ETDRS report number 12. Ophthalmology 1991;98(Suppl 5 ):823-33. [PubMed] [Google Scholar]
Evans 2014
- Evans JR, Michelessi M, Virgili G. Laser photocoagulation for proliferative diabetic retinopathy. Cochrane Database of Systematic Reviews 2014, Issue 11. Art. No: CD011234. [DOI: 10.1002/14651858.CD011234.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Glanville 2006
- Glanville JM, Lefebvre C, Miles JN, Camosso-Stefinovic J. How to identify randomized controlled trials in MEDLINE: ten years on. Journal of the Medical Library Association 2006;94(2):130-6. [PMC free article] [PubMed] [Google Scholar]
Gong 2016
- Gong Y, Shao Z, Fu Z, Edin ML, Sun Y, Liegl RG, et al. Fenofibrate inhibits cytochrome P450 epoxygenase 2C activity to suppress pathological ocular angiogenesis. EBioMedicine 2016;13:201-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADEpro GDT [Computer program]
- GRADEpro GDT. Version accessed 30 June 2022. Hamilton (ON): McMaster University (developed by Evidence Prime). Available at gradepro.org.
Gross 2015
- Gross JG, Glassman AR, Jampol LM, Inusah S, Aiello LP, Antoszyk AN, et al. Panretinal photocoagulation vs intravitreous ranibizumab for proliferative diabetic retinopathy: a randomized clinical trial. JAMA 2015;314(20):2137-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Guay 1999
- Guay RP. Micronized fenofibrate: a new fibric acid hypolipidemic agent. Annals of Pharmacotherapy 1999;33(10):1083-103. [DOI] [PubMed] [Google Scholar]
Guyatt 2011
- Guyatt GH, Oxman AD, Kunz R, Brozek J, Alonso-Coello P, Rind D, et al. GRADE guidelines 6. Rating the quality of evidence—imprecision. Journal of Clinical Epidemiology 2011;64(12):1283-93. [DOI] [PubMed] [Google Scholar]
Heintz 2010
- Heintz E, Wirehn AB, Peebo BB, Rosenqvist U, Levin LA. Prevalence and healthcare costs of diabetic retinopathy: a population-based register study in Sweden. Diabetologia 2010;53(10):2147-54. [DOI] [PubMed] [Google Scholar]
Higgins 2017
- Higgins JP, Altman DG, Sterne JA, editor(s). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Churchill R, Chandler J, Cumpston MS, editor(s), Cochrane Handbook for Systematic Reviews of Interventions version 5.2.0 (updated June 2017), Cochrane, 2017. Available from training.cochrane.org/handbook/archive/v5.2.
Hu 2013
- Hu Y, Chen Y, Ding L, He X, Takahashi Y, Gao Y, et al. Pathogenic role of diabetes-induced PPAR-alpha down-regulation in microvascular dysfunction. Proceedings of the National Academy of Sciences of the United States of America 2013;110(38):15401-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Liu 2017
Maturi 2021
- Maturi RK, Glassman AR, Josic K, Antoszyk AN, Blodi BA, Jampol LM, et al. Effect of intravitreous anti-vascular endothelial growth factor vs sham treatment for prevention of vision-threatening complications of diabetic retinopathy: the protocol W randomized clinical trial. JAMA Ophthalmology 2021;139(7):E1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
McCulloch 2017
- McCulloch DK, Nathan DM, Trobe J, Mulder JE. Diabetic retinopathy: pathogenesis. www.uptodate.com/contents/diabetic-retinopathy-pathogenesis (accessed 14 January 2019).
Review Manager 2020 [Computer program]
- Review Manager (RevMan). Version 5.4. The Cochrane Collaboration, 2020.
RevMan Web 2023 [Computer program]
- Review Manager Web (Rev Man Web). Version 5.3.1. The Cochrane Collaboration, 2023. Available at revman.cochrane.org.
Romero‐Aroca 2016
- Romero-Aroca P, la Riva-Fernandez S, Valls-Mateu A, Sagarra-Alamo R, Moreno-Ribas A, Soler N, et al. Cost of diabetic retinopathy and macular oedema in a population, an eight year follow up. BMC Ophthalmology 2016;16:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schmier 2009
- Schmier JK, Covert DW, Lau EC, Matthews GP. Medicare expenditures associated with diabetes and diabetic retinopathy. Retina 2009;29(2):199-206. [DOI] [PubMed] [Google Scholar]
Schünemann 2022
- Schünemann HJ, Higgins JPT, Vist GE, Glasziou P, Akl EA, Skoetz N, et al. Chapter 14: Completing ‘Summary of findings’ tables and grading the certainty of the evidence. In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.3 (updated February 2022). Cochrane, 2022. Available from training.cochrane.org/handbook.
Sivaprasad 2017
- Sivaprasad S, Prevost AT, Vasconcelos JC, Riddell A, Murphy C, Kelly J, et al. Clinical efficacy of intravitreal aflibercept versus panretinal photocoagulation for best corrected visual acuity in patients with proliferative diabetic retinopathy at 52 weeks (CLARITY): a multicentre, single-blinded, randomised, controlled, phase 2b, non-inferiority trial. Lancet 2017;389(10085):2193-203. [DOI] [PubMed] [Google Scholar]
Stitt 2016
- Stitt AW, Curtis TM, Chen M, Medina RJ, McKay GJ, Jenkins A, et al. The progress in understanding and treatment of diabetic retinopathy. Progress in Retinal and Eye Research 2016;51:156-86. [DOI] [PubMed] [Google Scholar]
Su 2019
- Su XJ, Han L, Qi YX, Liu HW. Efficacy of fenofibrate for diabetic retinopathy: a systematic review protocol. Medicine 2019;98(14):e14999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tan 2017
- Tan GS, Cheung N, Simó R, Cheung GC, Wong TY. Diabetic macular oedema. Lancet Diabetes and Endocrinology 2017;5(2):143-55. [DOI] [PubMed] [Google Scholar]
Teo 2021
- Teo ZL, Tham YC, Yu M, Chee ML, Rim TH, Cheung N, et al. Global prevalence of diabetic retinopathy and projection of burden through 2045: systematic review and meta-analysis. Ophthalmology 2021;128(11):1580-91. [DOI] [PubMed] [Google Scholar]
Virgili 2018
- Virgili G, Parravano M, Evans JR, Gordon I, Lucenteforte E. Anti-vascular endothelial growth factor for diabetic macular oedema: a network meta-analysis. Cochrane Database of Systematic Reviews 2018, Issue 10. Art. No: CD007419. [DOI: 10.1002/14651858.CD007419.pub6] [DOI] [PMC free article] [PubMed] [Google Scholar]
Woung 2010
- Woung LC, Tsai CY, Chou HK, Tsai MT, Tsai WH, Chou P, et al. Healthcare costs associated with progressive diabetic retinopathy among National Health Insurance enrollees in Taiwan, 2000-2004. BMC Health Service Research 2010;10:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhang 2017
- Zhang X, Low S, Kumari N, Wang J, Ang K, Yeo D, et al. Direct medical cost associated with diabetic retinopathy severity in type 2 diabetes in Singapore. PLoS One 2017;12(7):e0180949. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Inoue 2019
- Inoue K, Kataoka SY, Kawano S, Furukawa TA, Lois N, Watanabe N. Fenofibrate for diabetic retinopathy. Cochrane Database of Systematic Reviews 2019, Issue 4. Art. No: CD013318. [DOI: 10.1002/14651858.CD013318] [DOI] [PMC free article] [PubMed] [Google Scholar]