


Abstract
The persistence of fetal hemoglobin in many patients with deletion type β thalassemias and the expression patterns of human globin genes in transgenic mice suggest that γ- to β-globin gene switching results primarily from competition of γ- and β-globin genes for interaction with the β-globin locus control region (LCR). To define regulatory sequences that are essential for the competitive advantage of the γ gene at early developmental stages, stable transgenic mouse lines were produced with LCR γ-β constructs containing deletions of γ 5′-flanking DNA. All constructs contained the full 22 kb LCR, a 4.1 kb β-globin gene and a γ-globin gene with 1348, 383, 202, 130, 72 or 52 bp of 5′-flanking sequence. Primer extension analysis of yolk sac, fetal liver and blood RNA from these lines demonstrated that a region between –202 and –130 of the human γ-globin gene promoter was required to suppress β-globin gene expression at early developmental stages. Four transcription factor binding sites within this region [GATA(p), Oct1, GATA(d) and CACCC] were mutated independently in LCR γ-β constructs and transgenic mouse lines were produced. Only the γ CACCC box mutation resulted in high levels of β-globin gene expression in early embryos. These results demonstrate that the CACCC box of the human γ-globin gene plays a critical role in human β-globin gene developmental specificity. The data also suggest that γ CACCC box binding factors mediate LCR–γ interactions which normally enhance γ-globin and suppress β-globin gene expression in fetal erythroid cells.
INTRODUCTION
In 1990 two groups postulated that competition of human globin gene family members for interaction with the powerful β-globin LCR is a critical feature of developmental control (1,2). This model was based on transgenic animal experiments demonstrating that developmental specificity is lost when single human β- or γ-globin genes are inserted downstream of the LCR. In these experiments LCR β and LCR γ transgenes were expressed at high levels in embryonic, fetal and adult erythroid cells. However, temporal specificity was restored when linked γ- and β-globin genes were inserted downstream of the LCR. The results were consistent with a model in which correct temporal regulation results from competition of individual globin gene family members for interaction with LCR sequences. Promoter and proximal enhancer binding factors synthesized in yolk sac, fetal liver and bone marrow influence these competitive interactions either positively or negatively and, subsequently, determine developmental specificity (3). Normal gene order in the β-globin locus also plays an important role in globin gene switching (4–7). Therefore, the context of the locus and the transcription factors that influence LCR–globin gene interactions function synergistically to regulate globin gene expression during development. Recent results have demonstrated that erythroid Krupple-like factor (EKLF) is a critical protein involved in the competitive advantage of the β-globin gene in adult erythroid cells. Overexpression of EKLF in fetal erythroid cells transfected with LCR γ-β constructs enhances β-globin gene expression over 1000-fold and mutation of the β-globin gene CACCC box strongly inhibits this effect (8). Also, a homozygous knockout mutation of EKLF in mice is lethal but results in persistent expression of human γ-globin transgenes in adult erythroid cells (9–13). These results demonstrate that EKLF is an essential regulatory factor for globin gene switching. The data presented below demonstrate that the CACCC box in the human γ-globin gene promoter is also critical for correct switching and suggest that EKLF-like proteins may be involved in the competitive advantage of the γ-globin gene in fetal erythroid cells.
MATERIALS AND METHODS
The LCR –1348 γ-β cosmid was constructed as described previously (14) except that a human wild-type β-globin gene was used instead of the human βS gene. The LCR –383, –202 and –52 γ-β cosmids were constructed as described above except that StuI (–383), ApaI (–202) and NaeI (–52) were used to create the γ-globin promoter deletions. The –130 and –72 deletions were created with primers beginning at –130 and –72 of the human γ-globin gene. These fragments were inserted into the LCR γ-β cosmid in place of the –1348 bp γ promoter.
The GATA(d), Oct1, GATA(p) and CACCC box mutations were constructed by megaprimer mutagenesis (15–17) using the oligonucleotides GATA(d) (5′-GGGCCCCTTCCCCACAGAATTCCAATGCAAATATCTGTCTGA-3′), Oct1 (5′-GGGCCCCTTCCCCACACTATCTCAGAATTCATATCTGTCTGAAACGGT-3′), GATA(p) (5′-CCACACTATCTCAATGCAAATGAATTCCTGAAACGGTTCCTGGCT-3′) and CACCC (5′-CGGTCCCTGGCTAAACTGAATTCATGGGTTGGCCAGCCTTGCCTTG-3′). EcoRI sites were substituted for transcription factor binding sites so that mutations could be authenticated in transgenic founder DNA by Southern blot hybridization.
Primer extensions were performed as described previously (1) except that the β primer corresponding to sequences from +78 to +98 of the human β-globin gene was 5′-CCACAGGGCAGTAACGGCAGA-3′. Bands representing correctly initiated human γ- and β-globin mRNA were quantitated with a Molecular Dynamics PhosphorImager.
RESULTS AND DISCUSSION
The competition model described above predicts that γ-globin gene regulatory sequences enhance the competitive advantage of the γ gene for LCR interactions in early development and, consequently, suppress β-globin gene expression. Deletion of these sequences should result in high β-globin gene expression in early embryos of mice that contain human LCR γ-β transgenes. To define these sequences, we constructed a set of 5′-deletions of the γ-globin gene promoter and linked these DNA fragments to the β-globin gene and then to LCR DNase I hypersensitive sites 1–5 (HS 1–5) (Fig. 1A). The end points of the γ-globin deletions in these cosmids were 1348, 383, 202, 130, 72 and 52 bp upstream of the γ-globin gene transcription start site. Transgenic mouse lines were produced with each of these constructs and human γ- and β-globin mRNA were quantitated in yolk sac, fetal liver and adult blood by primer extension. As indicated above, we expected that deletion of important γ-globin regulatory sequences would reduce the competitive advantage of the γ-globin gene for LCR interactions and, consequently, stimulate β-globin gene expression in the yolk sac. The data in Figure 1B and Table 1 demonstrate that LCR –1348, –383 and –202 γ-β constructs switch correctly during development. Little if any human β-globin mRNA was observed in the yolk sac and high levels of β-globin mRNA were observed in fetal liver and adult blood. However, LCR –130, –72 and –52 γ-β constructs did not switch correctly; high levels of β-globin mRNA were observed in the yolk sac as well as in fetal liver and adult blood. The ratio of human β-globin to γ-globin mRNA (β/γ) in the yolk sac of LCR –130, –72 and –52 γ-β animals was 500-fold greater than the β/γ values in LCR –1348, –383 and –202 γ-β animals (Table 1). These results demonstrate that sequences required for β-globin gene developmental specificity are located between –202 and –130 of the human γ-globin gene promoter.
Figure 1.


Human LCR γ-β globin gene constructs and primer extension analysis of transgenic mouse RNA. (A) LCR γ-β constructs with –1348, –383, –202, –130, –72 and –52 bp of γ 5′-flanking sequence were co-injected with a human LCR α-globin gene construct into fertilized mouse eggs and transgenic lines containing intact copies of the transgenes were established. All constructs contained a 22 kb human β-globin LCR fragment including DNase I hypersensitive sites 1–5 (HS 1–5) (29,30). (B) Primer extension analysis of 10.5 day yolk sac RNA (YS), 15.5 day fetal liver RNA (FL) and adult blood RNA (Bl) from transgenic mouse lines. Lanes 1 and 24, end-labeled markers; lanes 2 and 23, primer extensions of adult blood RNA from a transgenic mouse containing human LCR γ and LCR β transgenes; lanes 3 and 22, primer extensions of 10.5 day yolk sac RNA from normal control embryos. The results demonstrate that sequences between –202 and –130 of the human γ-globin gene promoter regulate β-globin gene developmental specificity.
Table 1. Human β/γ mRNA values in LCR γ-β mice.
| Construct | Yolk sac β/γ | No. of lines |
|---|---|---|
| LCR –1348 γ-β | <0.2 | 6 |
| LCR –383 γ-β | <0.2 | 5 |
| LCR –202 γ-β | <0.2 | 5 |
| LCR –130 γ-β | >500 | 3 |
| LCR –72 γ-β | >500 | 3 |
| LCR –52 γ-β | >500 | 5 |
Primer extension products from yolk sac of each transgenic line were quantitated on a phosphoimager and mean values for β/γ are listed.
Figure 2A illustrates the sequence of the human Aγ-globin gene promoter from –202 to –130. Two GATA-1 sequences and an Oct-1 binding site are located between –189 and –172 (18) and the γ CACCC box is located at –144 (19). To assess the role of these sequences in γ- to β-globin gene switching, each of these sequences was mutated by the substitution of 6 bp (Fig. 2B). Transgenic mouse lines were produced with each of these constructs and human γ- and β-globin mRNA were quantitated in yolk sac and fetal liver by primer extension. The data in Figure 2C and Table 2 demonstrate that GATA(distal), Oct1 and GATA(proximal) mutant γ-β constructs switched correctly during development; little human β-globin mRNA was observed in the yolk sac and high levels of β-globin mRNA were observed in fetal liver. However, β-globin gene developmental specificity was lost in the γ CACCC mutants; high levels of human β-globin mRNA were observed in the yolk sac as well as in fetal liver and adult blood. The results for five additional lines of LCR –1348 γ-β CACCC mutant mice are presented in Figure 3; all of these animals expressed high levels of human β-globin mRNA in yolk sac. β/γ values in yolk sac of the γ CACCC box mutant mice were 40-fold greater than wild-type controls or GATA(p) mice (Table 2). These results demonstrate that the γ CACCC box is essential for correct β-globin gene developmental specificity. The GATA(d) and Oct1 mutations resulted in slightly elevated β-globin levels in yolk sac; therefore, these γ-globin promoter sequences may play a minor role in γ- to β-globin gene switching.
Figure 2.

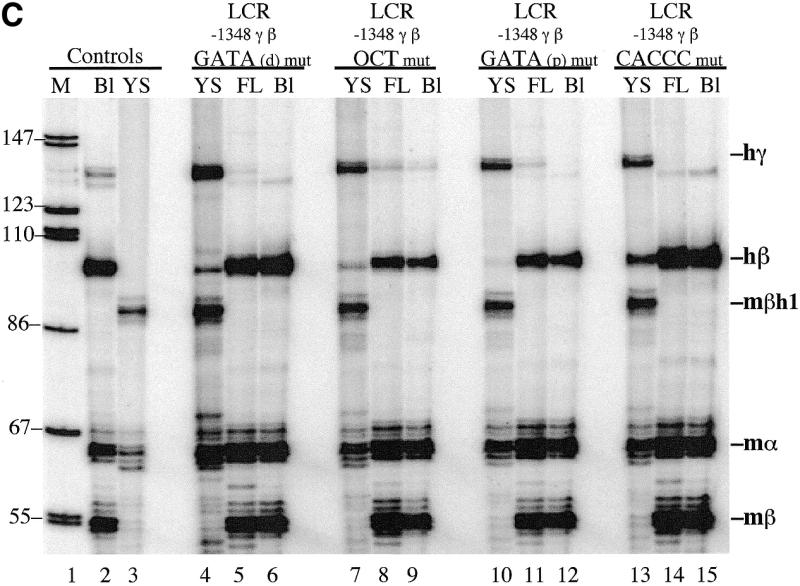
Human LCR γ-β globin gene constructs with γ promoter mutations and primer extension analysis of transgenic mouse RNA. (A) Human Aγ-globin gene promoter sequences from –202 to –130. The location of GATA(distal), Oct1, GATA(proximal) and CACCC box elements are marked above the sequence. (B) LCR γ-β constructs with GATA(distal), Oct1, GATA(proximal) and CACCC mutations in the human γ-globin gene promoter. (C) Primer extension analysis of 10.5 day yolk sac RNA (YS), 15.5 day fetal liver RNA (FL) and adult blood RNA (Bl) from transgenic mouse lines. The results demonstrate that the CACCC box of the human γ-globin gene promoter regulates β-globin gene developmental specificity.
Table 2. Human β/γ mRNA in yolk sac of LCR γ-β mutant mice.
| Constructs | β/γ fold increase | No. of lines |
|---|---|---|
| LCR –1348 γ-β wild type | 1× | 6 |
| LCR –1348 γ-β GATA(d) mut | 2× | 4 |
| LCR –1348 γ-β Oct1 mut | 2× | 4 |
| LCR –1348 γ-β GATA(p) mut | 1× | 6 |
| LCR –1348 γ-β CACCC mut | 40× | 8 |
Primer extension products from yolk sac of each transgenic line were quantitated and mean β/γ values were calculated. The mean for LCR –1348 γ-β wild-type mice was set at 1×.
Figure 3.
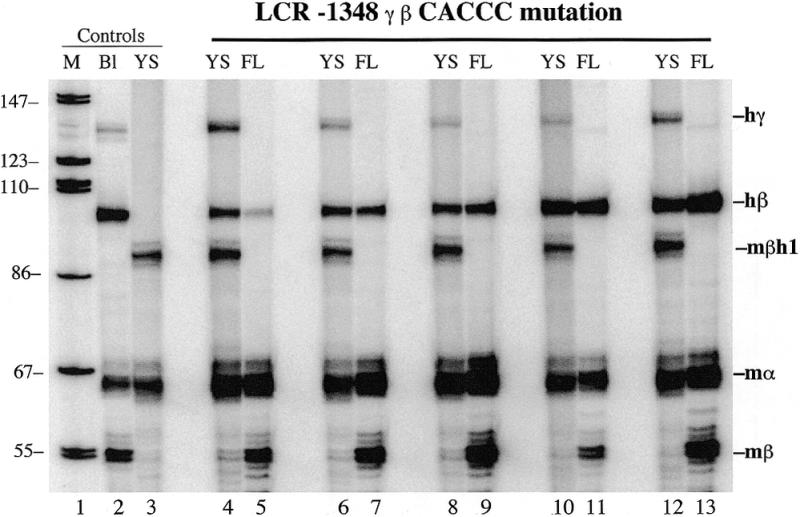
Primer extension analysis of 10.5 day yolk sac (YS) and 15.5 day fetal liver (FL) RNA from five additional LCR –1348 γ-β CACCC mutant transgenic mouse lines. The high levels of human β-globin RNA in the yolk sac confirm that the γ-globin gene CACCC box is essential for correct β-globin gene developmental specificity.
Jane et al. (20) previously tested HS2 γ-β globin gene constructs in K562 cells and demonstrated that γ-globin gene promoter deletions resulted in high levels of β-globin gene expression. In this assay enhanced β-globin expression was observed only after deletion to –53. A sequence designated the stage selector element (SSE) between –53 and –35 was required for correct developmental specificity and this element is analogous to a SSE defined earlier in the chicken β-globin gene promoter (21). Amrolia et al. (22) also described an element in the human γ-globin 5′-untranslated region that was required for correct developmental specificity in the K562 assay. The discrepancies in results obtained in K562 cells and transgenic mice suggest that different sequence elements are defined in the two systems. However, mutations in the γ SSE may affect switching in LCR γ-β transgenic mice, but these experiments have not been reported. Recently, Anderson et al. (23) and Sabatino et al. (24) have demonstrated that deletion of the γ-globin gene promoter or substitution of the γ promoter with a spectrin promoter in LCR γ-β constructs results in β-globin disregulation in transgenic mice. These results and recent data on the effect of γ-globin gene CACCC mutations in HEL and K562 cells (25) are consistent with the data presented here.
Recently Bulger and Groudine (26) proposed a mechanism for LCR–globin gene interactions that involves linking rather than looping. In this model the LCR promotes the formation of a chain of transcriptional facilitator proteins that extend along the chromatin fiber between the LCR and globin gene family members. When the chain reaches the γ-globin promoter in human fetal liver or mouse yolk sac, the facilitator enhances binding of a transcription factor(s) that activates γ-globin gene expression. The regulatory protein also acts as a boundary that inhibits further chain extension; therefore, the downstream β-globin gene is inactive. The model predicts that mutation of a critical γ-globin gene promoter element would permit extension of the chain in embryonic/fetal erythroid cells and, subsequently, activate β-globin gene expression prematurely. The data from LCR –1348 γ-β CACCC mutant mice described above demonstrate that mutation of the γ-globin gene CACCC box results in high level expression of the human β-globin gene in embryonic erythroid cells; therefore, these results are consistent with the LCR–globin linking model. However, the results do not preclude a looping mechanism in which the γ-globin gene with a promoter mutation in the CACCC box loses competitive advantage for looping interactions with the LCR and the β-globin gene is consequently expressed at high levels in early development.
In summary, we have demonstrated that an intact human γ-globin gene CACCC box is essential for correct human β-globin gene regulation in transgenic mice. When this site is mutated, the β-globin gene is expressed at high levels in early erythroid cells. The results suggest that a transcription factor in mouse yolk sac/human fetal liver normally binds to the γ CACCC box and provides the γ gene with a competitive advantage for LCR interactions through a linking or looping mechanism. Recently an EKLF-like protein that binds to the γ CACCC box was identified in human fetal liver (27). Further characterization of the activity of this protein and other EKLF-like family members should provide important insights into globin gene switching. Overexpression of these factors in adult erythroid cells may provide a mechanism for reactivating the human γ-globin gene in patients with β-thalassemia and sickle cell disease. If HbF (α2γ2) levels can be increased from normal levels of 0.5–1.0% to 9% or more, a significant improvement in disease prognosis would be possible (28).
Acknowledgments
ACKNOWLEDGEMENT
This work was supported by a grant from the NIH National Heart, Lung and Blood Institute.
REFERENCES
- 1.Behringer R.R., Ryan,T.M., Palmiter,R.D., Brinster,R.L. and Townes,T.M. (1990) Genes Dev., 4, 380–389. [DOI] [PubMed] [Google Scholar]
- 2.Enver T., Raich,N., Ebens,A.J., Papayannopoulou,T., Costantini,F. and Stamatoyannopoulos,G. (1990) Nature, 344, 309–313. [DOI] [PubMed] [Google Scholar]
- 3.Townes T.M. and Behringer,R.R. (1990) Trends Genet., 6, 219–223. [DOI] [PubMed] [Google Scholar]
- 4.Hanscombe O., Whyatt,D., Fraser,P., Yannoutsos,N., Greaves,D., Dillon,N. and Grosveld,F. (1991) Genes Dev., 5, 1387–1394. [DOI] [PubMed] [Google Scholar]
- 5.Peterson K.R. and Stamatoyannopoulos,G. (1993) Mol. Cell. Biol., 13, 4836–4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dillon N., Trimborn,T., Strouboulis,J., Fraser,P. and Grosveld,F. (1997) Mol. Cell, 1, 131–139. [DOI] [PubMed] [Google Scholar]
- 7.Tanimoto K., Liu,Q., Bungert,J. and Engel,J.D. (1999) Nature, 398, 344–348. [DOI] [PubMed] [Google Scholar]
- 8.Donze D., Townes,T.M. and Bieker,J.J. (1995) J. Biol. Chem., 270, 1955–1959. [DOI] [PubMed] [Google Scholar]
- 9.Perkins A.C., Sharpe,A.H. and Orkin,S.H. (1995) Nature, 375, 318–322. [DOI] [PubMed] [Google Scholar]
- 10.Nuez B., Michalovich,D., Bygrave,A., Ploemacher,R. and Grosveld,F. (1995) Nature, 375, 316–318. [DOI] [PubMed] [Google Scholar]
- 11.Perkins A.C., Gaensler,K.M. and Orkin,S.H. (1996) Proc. Natl Acad. Sci. USA, 93, 12267–12271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wijgerde M., Gribnau,J., Trimborn,T., Nuez,B., Philipsen,S., Grosveld,F. and Fraser,P. (1996) Genes Dev., 10, 2894–2902. [DOI] [PubMed] [Google Scholar]
- 13.Lim S.K., Bieker,J.J., Lin,C.S. and Costantini,F. (1997) Blood, 90, 1291–1299. [PubMed] [Google Scholar]
- 14.Ryan T.M., Ciavatta,D.J. and Townes,T.M. (1997) Science, 278, 873–876. [DOI] [PubMed] [Google Scholar]
- 15.Sarkar G. and Sommer,S.S. (1990) Biotechniques, 8, 404–407. [PubMed] [Google Scholar]
- 16.Barik S. and Galinski,M.S. (1991) Biotechniques, 10, 489–490. [PubMed] [Google Scholar]
- 17.Aiyar A. and Leis,J. (1993) Biotechniques, 14, 366–369. [PubMed] [Google Scholar]
- 18.Martin D.I., Tsai,S.F. and Orkin,S.H. (1989) Nature, 338, 435–438. [DOI] [PubMed] [Google Scholar]
- 19.Efstratiadis A., Posakony,J.W., Maniatis,T., Lawn,R.M., O’Connell,C., Spritz,R.A., DeRiel,J.K., Forget,B.G., Weissman,S.M., Slightom,J.L., Blechl,A.E., Smithies,O., Baralle,F.E., Shoulders,C.C. and Proudfoot,N.J. (1980) Cell, 21, 653–668. [DOI] [PubMed] [Google Scholar]
- 20.Jane S.M., Ney,P.A., Vanin,E.F., Gumucio,D.L. and Nienhuis,A.W. (1992) EMBO J., 11, 2961–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi O.R. and Engel,J.D. (1988) Cell, 55, 17–26. [DOI] [PubMed] [Google Scholar]
- 22.Amrolia P.J., Cunningham,J.M., Ney,P., Nienhuis,A.W. and Jane,S.M. (1995) J. Biol. Chem., 270, 12892–12898. [DOI] [PubMed] [Google Scholar]
- 23.Anderson K.P., Lloyd,J.A., Ponce,E., Crable,S.C., Neumann,J.C. and Lingrel,J.B. (1993) Mol. Biol. Cell, 4, 1077–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sabatino D.E., Cline,A.P., Gallagher,P.G., Garrett,L.J., Stamatoyannopoulos,G., Forget,B.G. and Bodine,D.M. (1998) Mol. Cell. Biol., 18, 6634–6640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sargent T.G., DuBois,C.C., Buller,A.M. and Lloyd,J.A. (1999) J. Biol. Chem., 274, 11229–11236. [DOI] [PubMed] [Google Scholar]
- 26.Bulger M. and Groudine,M. (1999) Genes Dev., 13, 2465–2477. [DOI] [PubMed] [Google Scholar]
- 27.Asano H., Li,X.S. and Stamatoyannopoulos,G. (1999) Mol. Cell. Biol., 19, 3571–3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Platt O.S., Brambilla,D.J., Rosse,W.F., Milner,P.F., Castro,O., Steinberg,M.H. and Klug,P.P. (1994) N. Engl. J. Med., 330, 1639–1644. [DOI] [PubMed] [Google Scholar]
- 29.Ryan T.M., Behringer,R.R., Martin,N.C., Townes,T.M., Palmiter,R.D. and Brinster,R.L. (1989) Genes Dev., 3, 314–323. [DOI] [PubMed] [Google Scholar]
- 30.Ryan T.M., Behringer,R.R., Townes,T.M., Palmiter,R.D. and Brinster,R.L. (1989) Proc. Natl Acad. Sci. USA, 86, 37–41. [DOI] [PMC free article] [PubMed] [Google Scholar]