


Abstract
Vaccinia virus DNA topoisomerase catalyzes resolution of synthetic Holliday junctions in vitro. The mechanism entails concerted transesterifications at two recognition sites, 5′-CCCTT↓, that are opposed within a partially mobile four-way junction. Efficient resolution occurs on a junction with a 10 bp segment of branch mobility (5′-GCCCTTATCG) that extends 4 bp 3′ of the scissile phosphate. Here we report that resolution is decreased when branch mobility is limited to an 8 bp segment extending 2 bp 3′ of the cleavage site and then eliminated when branch mobility is confined to the 6 bp GCCCTT sequence 5′ of the scissile phosphate. We surmise that a spacer region 3′ of CCCTT is needed for simultaneous cleavage at two opposing sites at the junction. Branch mobility is not required for reaction chemistry at a junction, because topoisomerase cleaves a single CCCTT site in a non-mobile four-way junction where the scissile phosphate is at the crossover point. The junction resolvase activity of topoisomerase may be involved in forming the hairpin telomeres of the vaccinia genome.
INTRODUCTION
The eukaryotic type IB topoisomerase family includes nuclear topoisomerase I and the topoisomerases of vaccinia and other cytoplasmic poxviruses (1). These enzymes relax DNA supercoils by transiently breaking and rejoining one strand of the DNA duplex through a covalent DNA–(3′-phosphotyrosyl)-enzyme intermediate. A distinctive feature of the poxvirus topoisomerases is their sequence specificity in strand cleavage. Vaccinia topoisomerase binds duplex DNA and forms a covalent adduct at sites containing a pentapyrimidine element, 5′(C/T)CCTT↓, in the scissile strand (2). The T↓ residue (designated position +1) is linked via a 3′-phosphodiester bond to Tyr274 of the enzyme.
The catalytic domains of type IB topoisomerases and the λ Int family of site-specific recombinases (now called tyrosine recombinases) have a common tertiary structure, prompting the inference that they derive from a common ancestral DNA strand transferase (3–7). A constellation of conserved amino acids catalyzes attack of the tyrosine nucleophile on the scissile phosphate. Type IB topoisomerases and tyrosine recombinases consist of two domains that form a C-shaped clamp around duplex DNA.
The mechanistic and evolutionary connections between topoisomerase IB and site-specific recombinases lends currency to the long-standing hypothesis that eukaryotic topoisomerases can act as catalysts of genetic recombination. Champoux envisioned a recombinogenic pathway whereby topoisomerase I would form a covalent adduct with one strand of the DNA duplex and then religate the bound DNA to a heterologous acceptor strand (8). Purified type I topoisomerases catalyze such strand transfer reactions in vitro (9–11) and the vaccinia virus enzyme can do so when expressed in a heterologous system in vivo (12,13).
The catalytic repertoire of vaccinia topoisomerase embraces a variety of recombinogenic DNA strand transfer reactions. Strand transfers involving linear CCCTT-containing DNA substrates have been analyzed in detail (9,10,14). For example, when the scissile bond is situated near the 3′-end of the DNA duplex, covalent adduct formation is accompanied by spontaneous dissociation of the downstream portion of the cleaved strand. The resulting topoisomerase–DNA complex can religate intramolecularly when the 5′-OH single-stranded tail of the non-cleaved strand attacks the 3′-phosphotyrosyl bond. The reaction product is a hairpin DNA loop. The topoisomerase–DNA complex catalyzes intermolecular religation when an exogenous 5′-OH acceptor strand can base pair with the single-strand tail of the activated ‘donor’ complex. The topoisomerase has great power of discrimination between complementary and non-complementary acceptor termini. A different type of end-joining reaction occurs when the scissile phosphate in the CCCTT↓ strand is situated across from a nick in the complementary strand. In this instance, cleavage opposite the nick generates a double-strand break with topoisomerase bound covalently at a blunt end. This DNA can be religated by topoisomerase to a heterologous duplex DNA with blunt 5′-OH termini regardless of the sequence of the acceptor molecule. The sticky end and blunt end intermolecular strand transfer reactions serve as in vitro models for topoisomerase-mediated illegitimate recombination events.
A key insight into the potential recombinogenic role of topoisomerase during vaccinia replication was the finding that vaccinia topoisomerase catalyzes the resolution of a four-way Holliday junction (15). Holliday junctions are intermediates in general homologous recombination and in the site-specific DNA rearrangements catalyzed by tyrosine recombinases. The resolvase activity of topoisomerase was demonstrated using a Holliday structure composed of four synthetic DNA strands of 59, 66, 75 and 68 nt (Fig. 1A). This four-way junction contained 10 complementary bases that permitted branch migration across the 10 bp homologous core (Fig. 1B). The CCCTT cleavage sites for topoisomerase were situated on strands 2 and 4 within the core. Successful and efficient resolution (up to 80% of input DNA resolved) was evinced by conversion of the 5′-32P-labeled junctions into linear duplex products of 59 and 75 bp (Fig. 1A). Junctions labeled on strands 1 or 2 were resolved to 59 bp duplexes, whereas junctions labeled on strands 3 or 4 were converted to 75 bp duplexes (15). This result showed that topoisomerase-catalyzed resolution occurred via action of the enzyme on the CCCTT-containing strands. Sequencing of the 59mer and 75mer recombinant strands confirmed that topoisomerase resolved the junctions by strand cleavage and conservative strand transfer within the CCCTT-containing 10 bp core (15).
Figure 1.

Synthetic Holliday junction substrates with varying branch mobility. (A) The prototype synthetic Holliday junction (junction A) resolved by vaccinia topoisomerase is shown. For convenience of illustration, the junction is depicted in an anti-parallel stacked conformation with the CCCTT target sites in the non-crossover strands (strands 2 and 4) and the scissile bonds (denoted by arrows) situated at the crux. The cleavage sites are located 27 nt from the 5′-end of strand 2 and 36 nt from the 5′-end of strand 4. Resolution of junction A by vaccinia topoisomerase produces two linear duplexes of 59 and 75 bp as shown. (B) The 10 bp mobile core region of junction A is depicted in a square planar conformation with the topoisomerase cleavage sites at the crossover point. The sequences of strands 2 and 3 were altered as shown to reduce junction mobility to 8 (junction B) or 6 bp (junction C). The segments through which the junctions can branch migrate are highlighted in shaded boxes. The T↓ nucleotide that becomes covalently attached to the topoisomerase is defined as position +1; the nucleotides 3′ of the scissile phosphodiester are designated –1, –2, –3, –4, etc.
The aim of the present study was to assess the influence of branch mobility and cleavage site spacing in topoisomerase-catalyzed resolution of Holliday junctions. We find that resolution depends on a spacer DNA segment 3′ of the CCCTT cleavage sites. These results are consistent with a role for topoisomerase in forming the hairpin telomeres of the vaccinia virus genome via reciprocal strand transfer on concatemeric replication intermediates that have been extruded into a mobile cruciform.
RESULTS
Branch mobility in junction resolution
We constructed a new synthetic Holliday structure with asymmetric arms that was designed to limit branch migration to the 6 nt region (GCCCTT) 5′ of the scissile phosphate (Fig. 1B, junction C). This molecule differs from the Holliday junction with a 10 bp mobile core GCCCTTATCC (Fig. 1B, junction A) only at the –1 to –4 nucleotide positions on strands 2 and 3. Junctions A and C that had been 5′-radiolabeled on strand 2 were reacted with vaccinia topoisomerase, then treated with proteinase K. The products were analyzed by electrophoresis through a native polyacrylamide gel (Fig. 2). We found that topoisomerase was incapable of resolving the 6 bp mobile junction into linear DNA over a range of input enzyme concentrations from 20 to 200 nM. Topoisomerase readily resolved the 10 bp mobile junction A to linear products under the same conditions (Fig. 2). Optimal resolution of the 10 bp mobile junction (to an extent of 75% of the total labeled DNA) was achieved at 100 nM topoisomerase, i.e. at a 10:1 ratio of enzyme to junction molecules or 5:1 enzyme to cleavage sites at the junction core.
Figure 2.
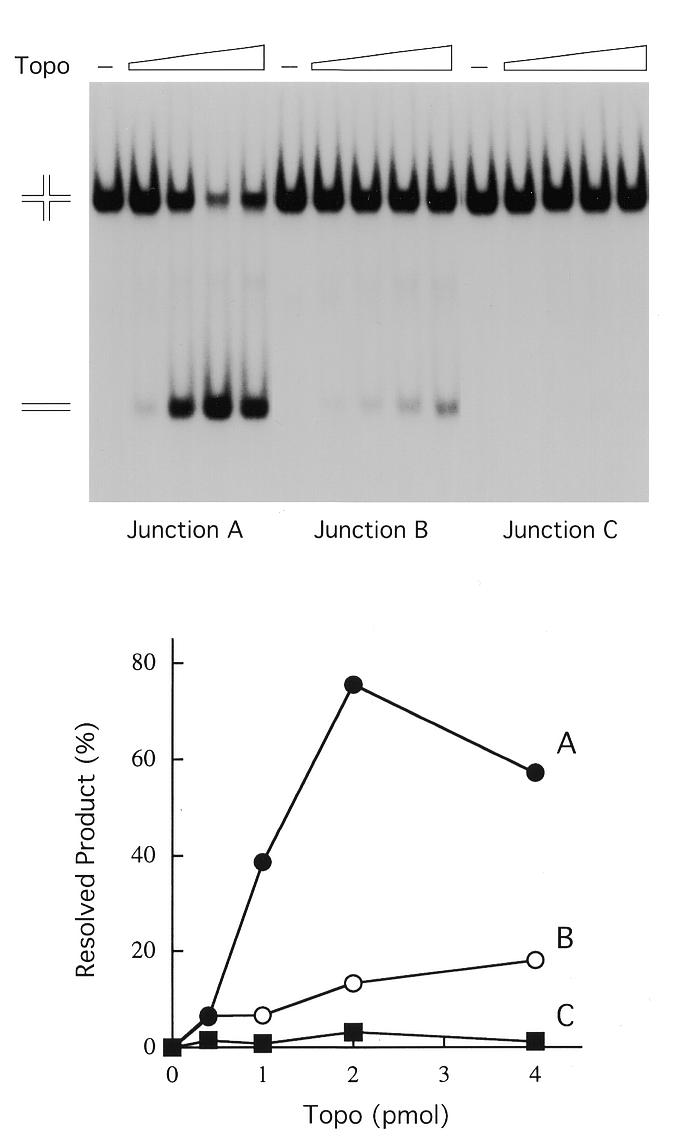
Holliday junction resolution by vaccinia topoisomerase. The procedures for 5′-radiolabeling of individual strands, strand annealing and purification of the Holliday junction by preparative gel electrophoresis were described previously (15). Reaction mixtures (20 µl) containing 50 mM Tris–HCl, pH 8.0, 1.5 mM MgCl2, 10 nM junction A, B or C that had been 5′-labeled on strand 2 and either 0, 20, 50, 100 or 200 nM purified recombinant vaccinia topoisomerase were incubated at 37°C for 15 min. The reactions were quenched by adjusting the mixtures to 0.2% SDS and 0.5 mg/ml proteinase K. After proteolysis at 37°C for 1 h, the DNA products were extracted with phenol and then phenol:chloroform. The aqueous phase was removed and adjusted to 5% glycerol, then the samples were electrophoresed through a 4% polyacrylamide gel in 0.25× TBE (22 mM Tris–borate, 0.6 mM EDTA) at 80 V for 3 h. An autoradiograph of the dried gel is shown in the top panel. The positions of the radiolabeled synthetic Holliday junction and the linear resolution product are indicated at the left. The extents of junction resolution [linear/(linear + junction)] at each level of input topoisomerase were determined by scanning the gel with a phosphorimager and are plotted in the bottom panel.
We then constructed another Holliday junction (Fig. 1B, junction B) with an 8 bp mobile core that was capable of 2 nt of branch migration 3′ of the scissile phosphate. The 8 bp mobile junction was resolved by topoisomerase, albeit less efficiently than the 10 bp mobile junction (Fig. 2). These experiments suggested that a threshold level of branch mobility facilitates junction resolution by vaccinia topoisomerase. Explanations for this effect are considered below.
Binding of vaccinia topoisomerase to junctions A, B and C was examined using a non-denaturing gel mobility shift assay (Fig. 3). The binding reactions contained 10 nM junction DNA 5′-labeled on strand 2. Increasing concentrations of topoisomerase resulted in the formation of protein–DNA complexes of retarded mobility. The shifted species seen at 10–20 nM enzyme appeared to be a closely spaced doublet that may represent junction molecules bound by a single topoisomerase molecule at one or the other CCCTT site at the junction. At 50 nM enzyme, the junctions were bound quantitatively and a new, more slowly migrating discrete complex was detected on junction A. This species is proposed to contain two molecules of topoisomerase bound at the opposing CCCCT sites of the junction (15). The discrete two protomer shifted species was less abundant in the binding reaction with junction B and even scarcer in the reaction with junction C (Fig. 3). The more diffuse higher order complexes seen on all three junctions at 100 nM topoisomerase likely contained additional topoisomerase protomers bound to the DNA arms. Prior studies showed that vaccinia topoisomerase binds non-covalently at non-specific sites on duplex DNA with ∼10-fold lower affinity than it binds non-covalently to CCCTT-containing DNA (16).
Figure 3.
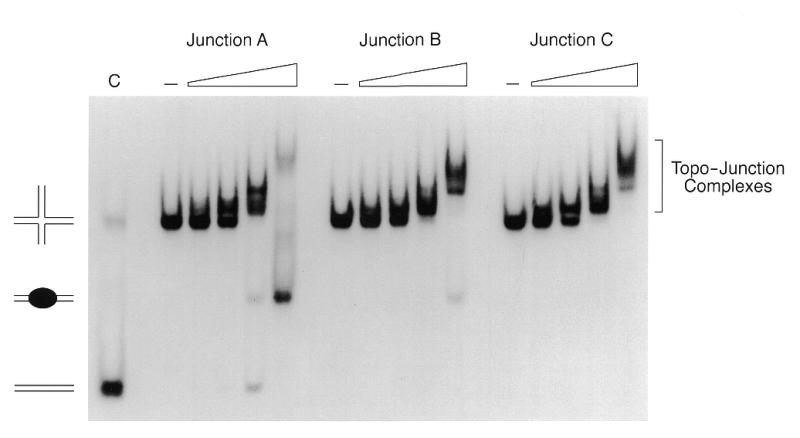
Binding of topoisomerase to the Holliday junctions. Reaction mixtures (20 µl) contained 50 mM Tris–HCl, pH 8.0, 1.5 mM MgCl2, 10 nM junction A, B or C (strand 2 labeled) and increasing concentrations of topoisomerase (10, 20, 50 and 100 nM from left to right in each titration series). Control reactions (–) lacked enzyme. After incubation at 37°C for 15 min, glycerol was added to 5% and the samples were electrophoresed through a 4% polyacrylamide gel in 0.25× TBE at 80 V for 4 h. The gel was dried and subjected to autoradiography. The deproteinized linear duplex junction A resolution product is shown in lane C. The positions of the input junction, the free duplex resolution product and the enzyme-bound resolution product are indicated to the left of the panel. Complexes of topoisomerase bound to the Holliday junction are denoted to the right.
In the binding reactions containing the 10 bp mobile junction A, we noted the appearance at 50–100 nM enzyme of two labeled resolution products that migrated ahead of the input DNA substrate. These corresponded to the free 59 nt linear duplex and shifted 59mer duplex containing bound topoisomerase (Fig. 3). The instructive findings were: (i) that the binding of topoisomerase to 6 bp mobile junction C resulted in no liberation of more rapidly migrating resolution products; (ii) that binding of enzyme to the 8 bp mobile junction B released a much lower amount of resolved product compared to junction A. Taken together with the findings regarding the relative abundance of the two protomer topoisomerase–junction complex, these results suggested that the impaired resolution of junctions with more restricted branch mobility might have been caused by steric hindrance of topoisomerase binding at both cleavage sites on junction C.
Why is junction mobility required for resolution?
Failure to resolve the junction can occur for one of the following reasons: (i) the junction adopts a conformation that cannot be cleaved on either CCCTT strand; (ii) the junction can be cleaved on one of the scissile strands, but not on the other. Note that both CCCTT strands must be cleaved at the same time in order to release linear products. We discriminated between these scenarios by assessing cleavage of the individual strands of the resolved and non-resolved junction molecules. This entailed the preparation of junctions uniquely 5′-labeled on the CCCTT strands (either strand 2 or 4). After reaction with topoisomerase, the proteinase K-digested DNA products were analyzed by denaturing polyacrylamide gel electrophoresis (Fig. 4). Reaction of topoisomerase with the 10 bp mobile junction A resulted in conversion of end-labeled strand 2, originally 66 nt, into a product of 59 nt. Strand 4 was converted from a 68mer to a 75mer (Fig. 4). Additional reaction products derived from strands 2 and 4 were linked covalently to topoisomerase. The size of these species (denoted by Cl in Fig. 4) was consistent with cleavage of strands 2 and 4 at the CCCTT sites; heterogeneity was caused by a variable number of residual amino acids linked to the 3′-phosphate of the cleaved strand after digestion with proteinase K. Religated 59mer and 75mer recombinant strands predominated over the covalent intermediates at the higher concentration of input topoisomerase in this experiment, which represented a 10:1 molar ratio of enzyme to DNA.
Figure 4.
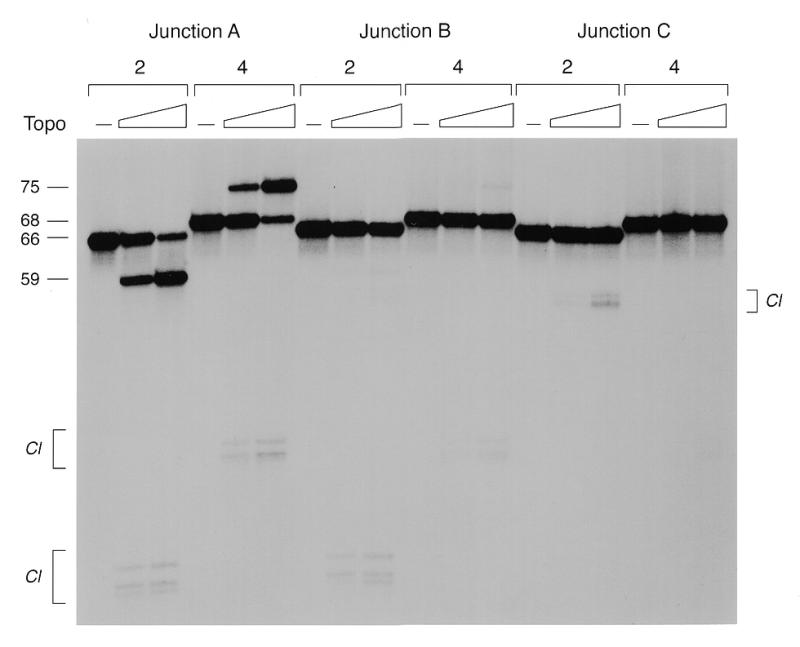
Analysis of the reaction products by denaturing gel electrophoresis. The protease-digested products of the resolution reactions with 10 nM junctions A, B or C and 0, 50 or 100 nM topoisomerase were ethanol precipitated, resuspended in formamide, heat denatured and then electrophoresed through a 12% polyacrylamide gel containing 7 M urea in TBE (90 mM Tris–borate, 25 mM EDTA). An autoradiograph of the gel is shown. The numbers above the bracketed lanes indicate the strand that was 5′-labeled in the junction substrate. The positions and chain lengths of the component scissile strands 2 (66mer) and 4 (68mer) and their respective resolved product strands (59mer and 75mer) are indicated to the left of the panel. Labeled DNA–peptide adducts (Cl) generated by proteolysis of the covalent intermediates are indicated by brackets.
Reaction of topoisomerase with the 8 bp mobile junction B resulted in the formation of covalent adducts on scissile strands 2 and 4 that migrated identically to those formed on strands 2 and 4 of junction A (Fig. 4). However, with junction B there was only trace accumulation of the recombinant 59mer and 75mer strands diagnostic of junction resolution. This result suggested that: (i) the limitation of branch migration to 2 nt 3′ of the scissile phosphate instead of 4 nt did not block cleavage reaction chemistry on the individual strands; (ii) the impediment to resolution on junction B reflected failure to cleave both strands at the same time, e.g. because covalent attachment of topoisomerase at one CCCTT site at the junction hindered cleavage of the opposing site by a second enzyme protomer.
Reaction of topoisomerase with the 6 bp mobile junction C yielded no detectable 59mer or 75mer strand transfer products (Fig. 4), consistent with the conclusion that junction C was not resolvable by the topoisomerase. Whereas low levels of covalent adduct formation at the CCCTT site within the junction was detected on strand 4, we did not observe any cleavage of the opposing CCCTT site on strand 2. Rather, we noted the appearance of a novel cluster of radiolabeled DNA–peptide adducts on strand 2 that migrated at ~55 nt, which arose via transesterification to a cryptic pentapyrimidine cleavage site 5′-TCCTC located distally (at nucleotide 53 of strand 2) in one of the non-mobile arms of the junction (Fig. 1). The size of the DNA–peptide adduct on strand 2 of junction C was consistent with that of the predicted 53mer DNA plus the residual peptide. Thus, limiting junction mobility to the 6 nt 5′ of the scissile phosphate blocked resolution and completely suppressed cleavage of one of the scissile strands, leading to secondary cleavage at a cryptic site in the arm of the four-way DNA structure. We again presume that cleavage at one site in strand 4 was incompatible with simultaneous cleavage at the opposing site in strand 2 because of steric hindrance.
Topoisomerase cleaves its CCCTT target site within an immobile Holliday junction
In principle, the dyad symmetry of the CCCTT element and flanking nucleotides in junction A permits the crossover point to migrate across all 10 of the positions of the core. The location of the scissile phosphates relative to the crossover point can therefore fluctuate between the extreme cases in which the cleavage sites are extruded into the vertical arms of the junction or else retracted into the horizontal arms of the junction (Fig. 5). The capacity for extrusion of the CCCTT sites into the vertical arms is no different for junction C, which cannot be resolved by topoisomerase, than for junction A, which is efficiently resolved. On the other hand, junction C is unable to retract the scissile phosphate past the point at which the TpN dinucleotide is located precisely at the crossover. Is vaccinia topoisomerase capable of cleaving the CCCTT site when it is located at the crux?
Figure 5.

Retracted and extruded configurations of Holliday junction A. See text for discussion.
To answer this question, we constructed an immobile junction containing only one 5′-CCCTT sequence and no symmetry in the vicinity of the crossover (Fig. 6). The substrate was designed so as to place the scissile phosphate at the crossover. The substrate, which was 5′-labeled on the 66mer CCCTT-containing strand, was reacted with topoisomerase and the products were digested with proteinase K and then analyzed by denaturing PAGE. A cluster of labeled species was detected at the size expected for transesterification to the CCCTT site to yield a 27mer DNA linked to one or more amino acids (Fig. 5). About 30% of the input junction was cleaved at a 2:1 molar ratio of topoisomerase to DNA. Note that transesterification on the immobile junction is an equilibrium between the forward cleavage and reverse religation reactions, just as it is on linear DNA. The extent of cleavage declined at higher protein concentrations, presumably because non-specific binding of topoisomerase to the arms either distorted the geometry of the junction or occluded the specific target sequence. We had shown previously that vaccinia topoisomerase cleaves kinked DNAs containing an extrahelical nucleotide inserted on either strand immediately 3′ of the scissile phosphate (9). The present finding that topoisomerase also transesterifies at the crossover point of a fixed Holliday junction underscores the theme that the enzyme tolerates DNA distortions distal to the cleavage site.
Figure 6.
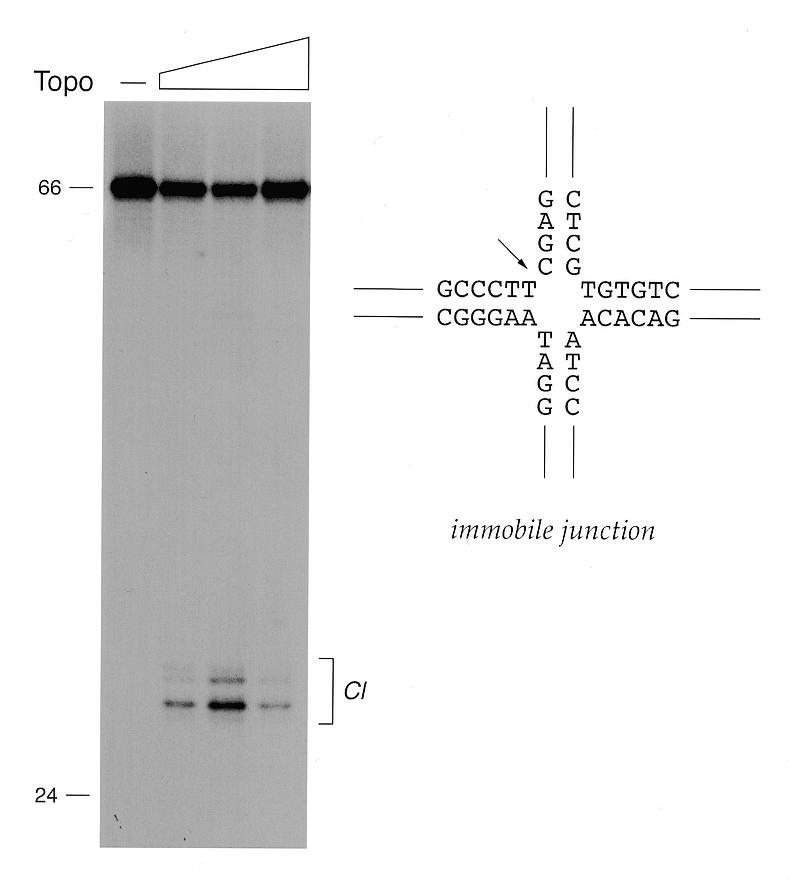
Cleavage at the crux of an immobile Holliday junction. The sequence of the immobile junction in the vicinity of the crossover is depicted in a square planar conformation. The topoisomerase cleavage site is indicated by an arrow. Reaction mixtures containing 5 nM immobile junction (5′-labeled on strand 2) and 0, 5, 10 or 25 nM topoisomerase were incubated at 37°C for 15 min. The protease-digested DNA products were analyzed by denaturing gel electrophoresis as described in the legend to Figure 4. An autoradiograph of a gel is shown. The positions and chain lengths of the input 66mer strand 2 (66mer) and a 5′-labeled 24mer marker strand are indicated to the left. Labeled DNA–peptide adducts (Cl) are indicated by the bracket to the right.
DISCUSSION
Here we have shown that modifications to the mobile core of junction A that progressively limit branch migration diminish, and then abolish, the resolvase activity of vaccinia topoisomerase. The implication is that topoisomerase is unable to simultaneously transesterify to two CCCTT sites at the junction unless they are retracted into the horizontal arms and the scissile phosphates are separated by a spacer DNA (Fig. 5A). In other words, the topoisomerase can resolve the junction when the cleavage sites are in a head-to-head configuration relative to the crossover point (i.e. GCCCTT↓ATCC⊥GGAT↑AAGGGC) but not when they are back-to-back (N↑AAGGGC⊥GCCCTT↓N). The optimal length of the spacer appears to be ∼8 bp, as in junction A. The separation of the scissile phosphates by 4 bp in junction B significantly reduced the efficiency of resolution.
The likely explanation for the failure to resolve the junction with CCCTT in the back-to-back orientation is that binding and cleavage by the first topoisomerase protomer precludes cleavage by a second enzyme. Steric hindrance with binding to the second CCCTT site is probable, given that exonuclease III footprinting studies show tight interaction between covalently bound topoisomerase and a 7–9 bp duplex segment upstream of the scissile phosphate (17). Partial occlusion of the second binding site would ensue when, as in the junction substrates used here, the junction can extrude to the point that the scissile phosphates are separated by only 12 bp (Fig. 5B). We do not exclude the possibility that expanding the limits of branch mobility on the 5′-side the CCCTT sites might overcome steric constraints to simultaneous cleavage and, perhaps, to resolution. However, resolution in that instance would mandate that the crossover point be able to branch migrate through the covalent topoisomerase–DNA complex on the CCCTT side of the cleavage site. Available evidence indicates that covalently bound topoisomerase does not readily release its grip on the duplex segment that includes CCCTT.
In contrast, there is abundant evidence from studies of ‘suicide’ cleavage reactions that vaccinia topoisomerase does not hold on tightly to the DNA 3′ of the scissile phosphate after the covalent intermediate is formed (9,10,14). Therefore, it is plausible that when two topoisomerases cleave the CCCTT sites in a head-to-head orientation in junction A, the covalently bound proteins either (i) release the original 5′-OH leaving strand and take up the other 5′-OH strand across the junction without branch migration or (ii) that the geometry of the complex is flexible enough 3′ of the cleavage sites to permit further branch migration of the scissile phosphates towards the crux.
Efficient resolution of model recombination intermediates by vaccinia topoisomerase suggests a role for the enzyme in the formation of the hairpin telomeres of the vaccinia virus genome. The vaccinia telomeres are 104 nt incompletely base paired hairpin structures that exist in two forms (‘flip’ and ‘flop’) that are inverted and complementary (18). These hairpins link the two strands of the 192 kb genome into one uninterrupted polynucleotide chain. The telomere structure is formed by resolution of concatameric DNA replication intermediates into unit length DNA molecules. The concatamer junctions, which are flanked by large inverted repeats, are converted to mature telomeres by a conservative DNA strand exchange reaction (19–21). Within the inverted repeats, just next to the telomere, are a series of tandemly repeated 69 or 70 bp elements, each of which contains multiple copies of the pentapyrimidine topoisomerase cleavage motif. A simple model for telomere formation invokes extrusion of a cruciform structure with the hairpin telomere at the apex (21). Topoisomerase is an attractive candidate for resolving the cruciform (which is formally equivalent to a mobile Holliday junction) by cleavage and reciprocal strand transfer at the numerous pentapyrimidine sites clustered symmetrically with respect to the apex. This is not to say that the topoisomerase by itself is responsible for telomere resolution during viral infection. Telomere formation requires a vaccinia late promoter element symmetrically flanking the concatamer junction (22,23). A plausible model is that transcription across the concatamer junction (which contains no protein coding potential) serves to drive extrusion of the cruciform from the linear DNA.
Palaniyar et al. (24) recently reported that Shope fibroma virus (SFV) topoisomerase catalyzed the resolution of a supercoiled plasmid DNA containing an extruded cruciform composed of the 1.5 kb concatamer junction from SFV replication intermediates. Included among the reaction products were linear DNAs with hairpin ends. The SFV topoisomerase-catalyzed plasmid resolution reaction was very slow and required large amounts of enzyme. Still, its demonstration in vitro was an important step in solidifying the case for topoisomerase as a candidate telomere resolvase. A key inference from the characterization of the plasmid resolution reaction was a requirement for branch migration by the extruded Holliday junction. Palaniyar et al. (24) suggested that resolution could occur when a junction migrates away from the apex and encounters two topoisomerases bound symmetrically at cleavage sites on the junction arms. This is effectively the scenario we have modeled here, whereby successful Holliday resolution by topoisomerase occurs when the crossover point is located 3′ of the cleavage sites. The resolution reaction with synthetic Holliday junctions is much faster than plasmid cruciform resolution, it requires much less enzyme and the sites of transesterification are clearly defined. Hence, synthetic junctions can be used for further detailed analysis of the conformational states of the DNA and the topoisomerase protomers during the strand transfer reactions.
REFERENCES
- 1.Shuman S. (1998) Biochim. Biophys. Acta, 1400, 321–337. [DOI] [PubMed] [Google Scholar]
- 2.Shuman S. and Prescott,J. (1990) J. Biol. Chem., 265, 17826–17836. [PubMed] [Google Scholar]
- 3.Cheng C., Kussie,P., Pavletich,N. and Shuman,S. (1998) Cell, 92, 841–850. [DOI] [PubMed] [Google Scholar]
- 4.Redinbo M.R., Stewart,L., Kuhn,P., Champoux,J.J. and Hol,W.G.J. (1998) Science, 279, 1504–1513. [DOI] [PubMed] [Google Scholar]
- 5.Redinbo M.R., Champoux,J.J. and Hol,W.G.J. (1999) Curr. Opin. Struct. Biol., 9, 29–36. [DOI] [PubMed] [Google Scholar]
- 6.Guo F., Gopaul,D.N. and Van Duyne,G.D. (1997) Nature, 389, 40–46. [DOI] [PubMed] [Google Scholar]
- 7.Yang W. and Mizuuchi,K. (1997) Structure, 5, 1401–1406. [DOI] [PubMed] [Google Scholar]
- 8.Been M.D. and Champoux,J.J. (1981) Proc. Natl Acad. Sci. USA, 78, 2883–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shuman S. (1992) J. Biol. Chem., 267, 8620–8627. [PubMed] [Google Scholar]
- 10.Shuman S. (1992) J. Biol. Chem., 267, 16755–16758. [PubMed] [Google Scholar]
- 11.Christiansen K. and Westergaard,O. (1994) J. Biol. Chem., 269, 721–729. [PubMed] [Google Scholar]
- 12.Shuman S. (1989) Proc. Natl Acad. Sci. USA, 86, 3489–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shuman S. (1991) Proc. Natl Acad. Sci. USA, 88, 10104–10108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sekiguchi J., Cheng,C. and Shuman,S. (1997) J. Biol. Chem., 272, 15721–15728. [DOI] [PubMed] [Google Scholar]
- 15.Sekiguchi J., Seeman,N.C. and Shuman,S. (1996) Proc. Natl Acad. Sci. USA, 93, 785–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sekiguchi J. and Shuman,S. (1994) Nucleic Acids Res., 22, 5360–5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng C. and Shuman,S. (1999) Biochemistry, 38, 16599–16612. [DOI] [PubMed] [Google Scholar]
- 18.Baroudy B.M., Venkatesan,S. and Moss,B. (1982) Cell, 28, 315–324. [DOI] [PubMed] [Google Scholar]
- 19.Merchlinsky M. and Moss,B. (1986) Cell, 45, 879–884. [DOI] [PubMed] [Google Scholar]
- 20.DeLange A.M., Reddy,M., Scraba,D., Upton,C. and McFadden,G. (1986) J. Virol., 59, 249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Merchlinsky M. (1990) J. Virol., 64, 3437–3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Merchlinsky M. and Moss,B. (1989) J. Virol., 63, 4354–4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu F. and Pickup,D.J. (1991) Virology, 181, 716–720. [DOI] [PubMed] [Google Scholar]
- 24.Palaniyar N., Gerasimopoulos,E. and Evans,D.H. (1999) J. Mol. Biol., 287, 9–20. [DOI] [PubMed] [Google Scholar]