

Abstract
INTRODUCTION.
APOE genotype is a driver of cognitive decline and dementia. We used causal mediation methods to characterize pathways linking APOE genotype to late-life cognition through Alzheimer’s disease (AD) and non-AD neuropathologies.
METHODS.
We analyzed autopsy data from 1,671 individuals from the ROS/MAP/MARS studies with cognitive assessment within 5 years of death and autopsy measures of AD (amyloid-β, neurofibrillary tangles), vascular (athero/arteriolo-sclerosis, micro/macro-infarcts) and non-AD neurodegenerative neuropathologies (TDP-43, Lewy Bodies, amyloid angiopathy, hippocampal sclerosis).
RESULTS.
The detrimental effect of APOE-ε4 on cognition was mediated by summary measures of AD and non-AD neurodegenerative neuropathologies but not vascular neuropathologies; effects were strongest in individuals with dementia. The protective effect of APOE-ε2 was partly mediated by AD neuropathology and stronger in women than men.
DISCUSSION.
APOE genotype influences cognition and dementia through multiple neuropathological pathways, with implications for different therapeutic strategies targeting people at increased risk for dementia.
Keywords: APOE, neuropathology, causal mediation, amyloid, tau, multiple pathologies, dementia, aging, cognition
1. Background
The apolipoprotein (APOE) genotype is the strongest known genetic risk factor for dementia [1]. Compared to the APOE-ε3/ε3 genotype, the APOE-ε4 allele is associated with more severe cognitive decline and higher dementia risk, while APOE-ε2 has the opposite effect [2–5]. New insights into mechanisms underlying these effects could enhance our understanding of disease pathogenesis and aid the development of strategies to prevent or delay disease [6].
Prior mediation analyses have shown that effects of the APOE-ε2 and -ε4 alleles on cognition are partially mediated through their effects on Alzheimer’s disease (AD) neuropathology, including amyloid-β and tau [7–10]. However, effects of the APOE genotype extend beyond its impact on AD neuropathology. The APOE protein has a central role in lipid metabolism, and APOE genotype is associated with cardiovascular disease and cardiovascular disease risks, including stroke, atherosclerosis, white matter hyperintensities, and myocardial infarction, as well as levels of oxidative stress and inflammation [11–17]. One prior study found TDP-43 significantly mediated the association between the APOE-ε4 allele and hippocampal sclerosis and was associated with faster cognitive decline [18]. Another study found that cholesterol partially mediated the association between the APOE-ε2 allele and verbal memory [19]. However, other efforts to establish non-AD mediators of the association between the either the APOE-ε2 or -ε4 allele and cognition or dementia have yielded null findings [10,20–22].
This study used data from the Religious Orders Study, Memory and Aging Project, and Minority Aging Research Study (ROS/MAP/MARS) to advance and extend prior research on mediators of the effect of both the APOE-ε2 and ε4 alleles on cognition. We used causal mediation methods, which have distinct advantages over traditional mediation approaches, including allowing for interaction between exposures (i.e. APOE genotype) and mediators (i.e. neuropathologies) [23]. Although evidence suggests that APOE genotype interacts with AD neuropathologies, few prior studies used these methods [24–26]. Additionally, we assessed a range of different measures of AD neuropathology to better understand which components have the strongest mediating effects. Because APOE has a wide range of biological effects, we also assessed the role of vascular (e.g. atherosclerosis, microinfarcts) and non-AD neurodegenerative (e.g. TDP-43, cerebral amyloid angiopathy) pathologies. Finally, we evaluated differences in mediating pathways by sex and clinical dementia status, as existing evidence suggests both factors moderate associations between APOE genotype and both cognition and neuropathology [27–31].
2. Method
2.1. Study sample
We used data from three ongoing cohort studies conducted through the Rush Alzheimer’s Disease Center. The Religious Orders Study (ROS) has recruited Catholic nuns and priests in the US since 1994, the Memory and Aging Project (MAP) has recruited older adults from northeastern Illinois since 1997, and the Minority Aging Research Study (MARS) has recruited older African Americans from northeastern Illinois since 2004. All participants gave informed consent and each study was approved by the Rush University Medical Center Institutional Review Board.
Of the 2,330 decedents in the three studies, there were 1,834 participants with autopsy data (79%). We excluded those with missing data on APOE genotype (N=61), those with an ε2/ε4 genotype (N=38), those with missing data on global cognition (N=5), and those with greater than 5 years between their last cognitive assessment and death (N=53). We additionally excluded those who identified as a race other than White/Black, as this group was too small to appropriately characterize (N=6). For analyses with each mediator, we excluded individuals with missing data on the mediator of interest (min N=0; max N=189) (Appendix figure S1).
2.2. APOE Genotyping
APOE genotype was determined by sequencing codons 112 and 158 of the APOE gene in DNA extracted from peripheral blood or frozen postmortem brain tissue. APOE-ε2 or -ε4 carriers were defined as individuals with either 1 or 2 copies of the ε2 or ε4 allele, respectively; hetero- and homozygotes were grouped together. APOE-ε2/4 carriers were excluded from analyses.
2.3. Measures of Alzheimer’s disease (AD)
We used pathologic diagnosis of AD using the National Institute on Aging (NIA) Reagan criteria (0–3 point scale) as our primary measure of AD neuropathology [32]. NIA-Reagan criteria are based on Braak stage (summarizing neurofibrillary tangle pathology) and CERAD neuritic plaque score.
In addition to NIA-Reagan score, Braak score, and CERAD score, we examined four continuous measures of AD neuropathology. Amyloid-β neuritic plaques, diffuse plaques, and neurofibrillary tangles were visualized in five cortical areas using Bielschowsky silver staining; scaled scores were averaged across regions. A continuous measure of global AD neuropathology was computed as the average of these three summary scores (details in Appendix). Because continuous variables were right-skewed, we used square root transformed measures [33].
2.4. Non-AD neuropathological measures and summary scores
We considered 8 non-AD neuropathologies: arteriolosclerosis, atherosclerosis, cerebral amyloid angiopathy (CAA), Lewy body pathology, hippocampal sclerosis, macroinfarcts, microinfarcts, and TDP-43. To limit the number of primary analyses, we created two composites (vascular [arteriolosclerosis, atherosclerosis, macro- and microinfarcts] vs. neurodegenerative [CAA, Lewy bodies, hippocampal sclerosis, TDP-43]) based on parallel analysis and principal components analysis (see Appendix). Although CAA could be conceptualized as a vascular pathology, based on our data driven analysis and prior investigations into genetic and non-genetic risks for CAA, we categorized CAA as a neurodegenerative pathology [34,35]. Indicators were scaled and summed such that each indicator contributed equally; z-scores were computed so results would be interpretable in terms of standard deviation units.
2.6. Measurement of Cognition
We used global cognition proximate to death as our primary outcome. Global cognition was computed as the average z-score of 19 cognitive tests, covering a range of cognitive domains, including memory, executive functioning, and visuospatial ability (details in Appendix) [36]. We averaged cognitive scores from the last three annual visits before death to provide a more stable measure of cognition at the end of life. Where there were fewer than three visits with cognitive test scores available before death, we used scores from either 2 visits (N=78) or 1 visit (N=88). An ANOVA test for differences in cognitive z-score for those with 2 or 1 visit compared to 3 visits revealed no significant differences (p=0.34).
2.5. Other key variables
Age at death, self-reported sex, self-reported race (Black vs. White), self-reported years of education, and the interval between death and the last cognitive visit were used as covariates in statistical models. Clinical dementia status at death was determined by adjudication procedures based on clinical data and was blinded to autopsy findings [36].
2.6. Causal mediation methods
We used causal mediation methods to assess whether AD and non-AD neuropathologies mediated associations between APOE genotype and cognition [37,38]. We estimated natural direct and indirect effects using a counterfactual framework. Within this framework, we quantified comparisons in expected cognitive levels of participants in three hypothetical situations: (1) the scenario in which all participants had the exposure of interest (either the ε2 or ε4 allele) and the mediator value they would naturally be expected to have given their exposure, (2) the scenario in which all participants had the reference exposure (ε3/ε3 genotype) and the mediator value they would naturally be expected to have given this reference exposure, and (3) the cross-world scenario in which all participants had the exposure of interest (either the ε2 or ε4 allele) and the mediator value they would be expected to have if they had instead had the reference exposure. The natural indirect effect contrasts scenarios (1) and (3); only the mediator value changes while exposure level is held constant. The natural direct effect contrasts scenarios (2) and (3); only the exposure changes while the mediator level is held constant. We estimated contrasts using regression modeling to predict counterfactual outcomes for all participants and subtracting these outcomes to quantify effects on the additive scale (details in the Appendix). We used bootstrapping with 1,000 replications to calculate 95% Confidence Intervals.
2.7. Analytic plan
We first characterized distributions of demographic characteristics, cognitive scores, and neuropathologic features by APOE status. We made select comparisons using chi-squared test for categorical variables and t-tests for continuous variables. Mediation analyses had five steps (Figure 1). We assessed whether NIA Reagan Score mediated the association between APOE genotype (ε2 or ε4 carriers compared to ε3/ε3 genotype) and cognition proximal to death. Then, we contrasted mediation results across different measures of AD neuropathology: NIA Reagan score, CERAD score, Braak stage, and continuous measures of global AD neuropathology density (neuritic and diffuse plaques, and neurofibrillary tangles). Next, we assessed whether summary measures of vascular and non-AD neurodegenerative pathologies mediated associations between APOE genotype and cognition. If the summary measure was a statistically significant mediator, we evaluated the component parts and repeated mediation analyses after controlling for NIA Reagan Score to understand if the observed mediation was independent of AD neuropathology. We then repeated mediation analyses for our primary AD and any significant non-AD neuropathological measures after stratifying by sex to understand whether processes differ between men and women. Last, we repeated analyses after stratifying by clinical dementia status at death to further probe whether effects where driven by those with clinical dementia.
Figure 1.
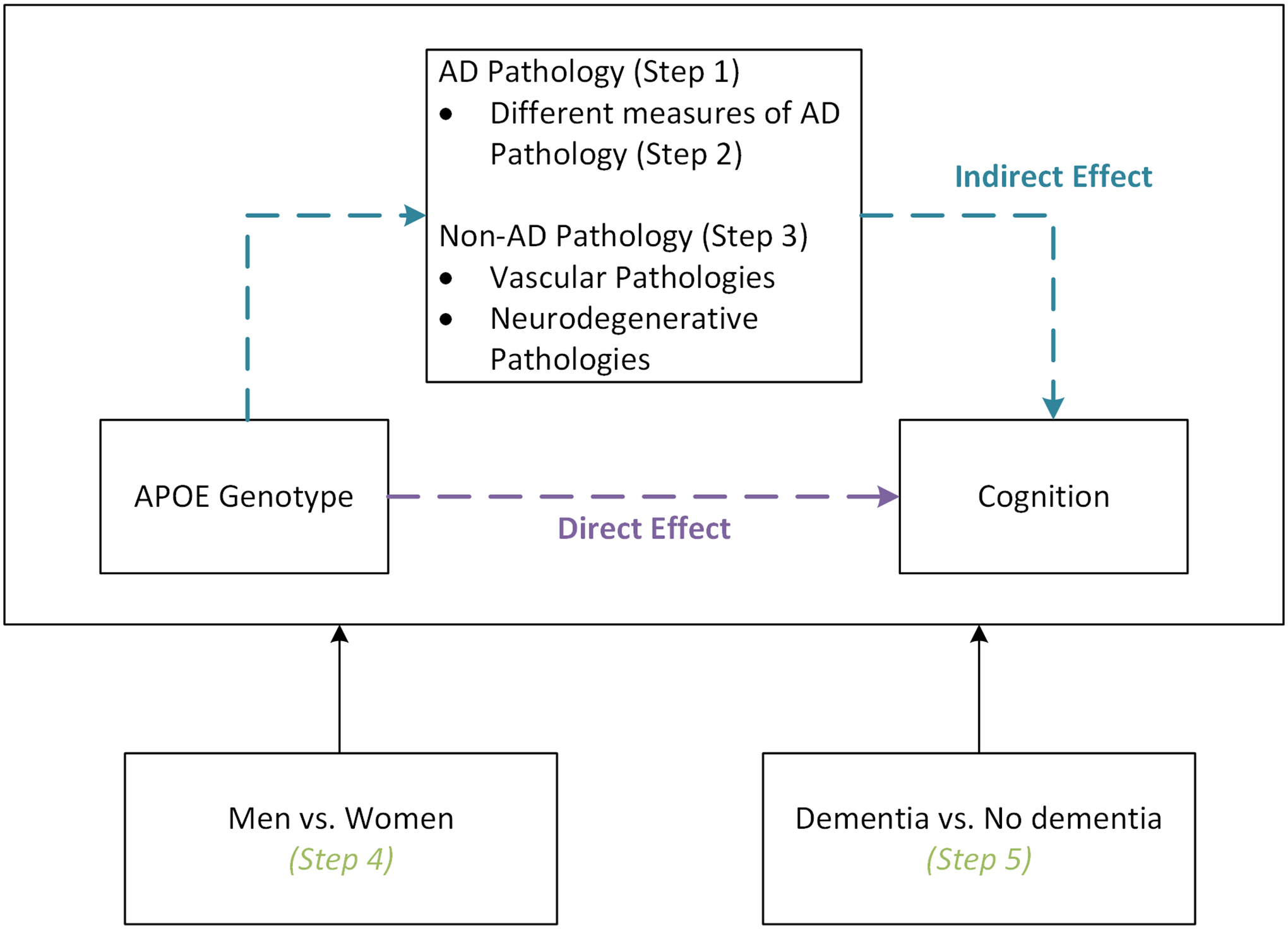
Conceptual framework outlining the analytic steps included in causal mediation analyses. In the first step, we assessed whether AD pathology mediated the association between APOE genotype and late-life cognition. We then characterized variation in the observed associations across different measures of AD pathology. Third, we assessed whether vascular or neurodegenerative non-AD pathology mediated the association of interest. Finally, we explored whether observed mediating effects varied by sex or clinical dementia status.
3. Results
3.1. Sample characteristics
Of 1,671 total participants, 399 were APOE-ε4 carriers (28 homozygotes) and 232 were APOE-ε2 carriers (9 homozygotes) (Table 1). Relative to the APOE-ε3/ε3 genotype, APOE-ε4 carriers were younger at death (p<0.001), had a longer interval between their last visit and death (p=0.027), had more years of education (p=0.008), and were more likely to be Black (p=0.034). APOE- ε2 carriers had fewer years of education than those with the APOE-ε3/ε3 genotype (p=0.040); differences on other demographic variables were not statistically significant.
Table 1.
Sample characteristics, cognitive outcomes and select neuropathological measures by APOE genotype in the combined Religious Orders Study/Memory and Aging Project/Minority Aging Research Project sample (N=1671)
| APOE E2 carriers | APOE E3/E3 | APOE E4 carriers | |
|---|---|---|---|
| Number of Participants, N | 232 | 1040 | 399 |
| Number of homozygotes, N (%) | 9 (3.9) | 1040 (100.0) | 28 (7.0) |
| Age at death in years, mean (SD) | 90.4 (7.0) | 89.5 (6.7) | 88.2 (6.6) |
| Interval between death and last visit in years, mean (SD) | 0.9 (0.8) | 1.0 (0.9) | 1.1 (1.0) |
| Education in years, mean (SD) | 15.7 (3.2) | 16.2 (3.7) | 16.8 (3.6) |
| Females, N (%) | 165 (71.1) | 697 (67.0) | 261 (65.4) |
| White participants, N (%) | 220 (94.8) | 999 (96.1) | 372 (93.2) |
| Cognitive level - global, mean (SD) | −0.4 (0.9) | −0.6 (0.9) | −1.1 (1.1) |
| Clinical dementia at death, N (%) | 81 (34.9) | 424 (40.8) | 246 (61.7) |
| Pathological AD#, N (%) | 107 (46.1) | 620 (59.6) | 327 (82.0) |
| CERAD score | |||
| None, N (%) | 94 (40.5) | 287 (27.6) | 33 (8.3) |
| Mild, N (%) | 26 (11.2) | 88 (8.5) | 22 (5.5) |
| Moderate, N (%) | 77 (33.2) | 378 (36.3) | 138 (34.6) |
| Severe, N (%) | 35 (15.1) | 287 (27.6) | 206 (51.6) |
| Braak stage | |||
| Stage 0, N (%) | 2 (0.9) | 16 (1.5) | 2 (0.5) |
| Stage I-II, N (%) | 47 (20.3) | 186 (17.9) | 41 (10.3) |
| Stage III-IV, N (%) | 161 (69.4) | 600 (57.7) | 182 (45.6) |
| Stage V-VI, N (%) | 22 (9.5) | 238 (22.9) | 174 (43.6) |
| Z-score of vascular pathology, mean (SD) | 0.1 (1.1) | 0.0 (1.0) | 0.0 (1.0) |
| Z-score of non-AD neurodegenerative pathology, mean (SD) | −0.2 (0.8) | −0.2 (0.9) | 0.3 (1.1) |
SD = standard deviation,
based on NIA-Reagan score intermediate or high
3.2. NIA Reagan score as a mediator of the association between APOE genotype and cognition
Compared to those with the APOE-ε3/ε3 genotype, those with the APOE-ε4 genotype had higher NIA Reagan scores (Figure 2 Panel A) and lower levels of global cognition (Figure 2 Panel B), whereas those with the APOE-ε2 genotype had lower NIA Reagan scores and higher global cognition scores (Appendix Table S1). NIA Reagan score significantly mediated both the harmful effect of the APOE-ε4 allele and the protective effect of APOE-ε2 allele on global cognition (Figure 2 Panel C). Those with the APOE-ε4 genotype had cognitive scores that were −0.32 (95% CI −0.18 – −0.51) SD units lower than those with the APOE-ε3/ε3 genotype mediated through changes in NIA Reagan score, whereas we estimated that the APOE-ε2 genotype was associated with a global cognitive score 0.13 (95% CI 0.03 – 0.26) SD greater than those with the APOE-ε3/ε3 due to changes in NIA Reagan score. We also found evidence of significant direct effects of APOE-ε4 and -ε2 alleles on global cognition, indicating that other factors beyond NIA Reagan score contribute to the effect of APOE genotype on cognition (Figure 2 Panel C).
Figure 2.

Associations between APOE status and NIA Reagan Score [A], APOE Status and Global Cognition [B], and the indirect and direct effects of the APOE-ε4 and ε2 alleles on cognition from causal mediation analysis [C]. The indirect effect represents the effect of APOE genotype on cognition through changes in NIA Reagan score; the direct effect represents the effect of APOE genotype on cognition via other pathways. The reference category in causal mediation models was the group of participants with an ε3/ε3 genotype. Categories of None and Low on the NIA Reagan score were collapsed for visualization purposes due to low cell counts in the None category.
3.3. Differences between AD neuropathological measures as mediators
APOE genotype was strongly associated with all AD neuropathology measures examined and all AD neuropathology measures were associated with late-life cognition (Appendix Tables S2 & S3). Mediation findings were generally consistent across the range of AD neuropathology measures available (Appendix Figure S4). There were no clear differences when comparing continuous measures (i.e. continuous neurofibrillary tangles measure) versus summary ordinal variables (i.e. Braak stage). However, compared with other measures, when considering diffuse plaques as a mediator, the indirect (mediational) effect was smaller and the direct effect was larger. For example, the effect of the APOE-ε4 allele on cognition via changes in diffuse plaques was −0.12 SD units (95% CI −0.05 – −0.19) but the effect via changes in CERAD score was −0.54 SD units (95% CI −0.36 – −0.71).
3.4. Mediating role of non-AD pathologies on the effect of APOE genotype on cognition
All non-AD pathologies considered were associated with late-life cognition (Appendix Table S4). However, only the APOE- ε4 allele was associated with non-AD neurodegenerative pathologies and APOE genotype was not associated with vascular pathologies considered (Appendix Table S5). In line with these descriptive findings, the composite measure of vascular pathologies did not mediate the effect of APOE genotype on cognition (Figure 3). However, the summary score of non-AD neurodegenerative pathologies was a significant mediator of the effect of the APOE-ε4 allele on cognition; we estimated that differences in non-AD neurodegenerative pathologies were responsible for those with the APOE-ε4 allele having a global cognitive functioning score that was 0.21 (95% CI 0.15 – 0.28) SD units lower than those with the APOE-ε3/ε3 genotype. In comparison, non-AD neurodegenerative pathology was not a significant mediator of the effect of the APOE-ε2 allele on cognition (indirect effect: 0.01 [95% CI −0.02–0.04]). Individual mediation models evaluating each of the four components of the non-AD neurodegenerative pathology summary score indicated that patterns generally held, although the indirect (mediational) effect of APOE-ε4 was larger for TDP-43 and cerebral amyloid angiopathy compared to hippocampal sclerosis and Lewy body pathology (Appendix Figure S5). The indirect effect of the APOE-ε4 genotype on cognition via the non-AD neurodegenerative pathology summary score remained statistically significant in models controlling for NIA Reagan score, indicating this mediation finding was not attributable to correlations between AD and non-AD neurodegenerative pathologies.
Figure 3.
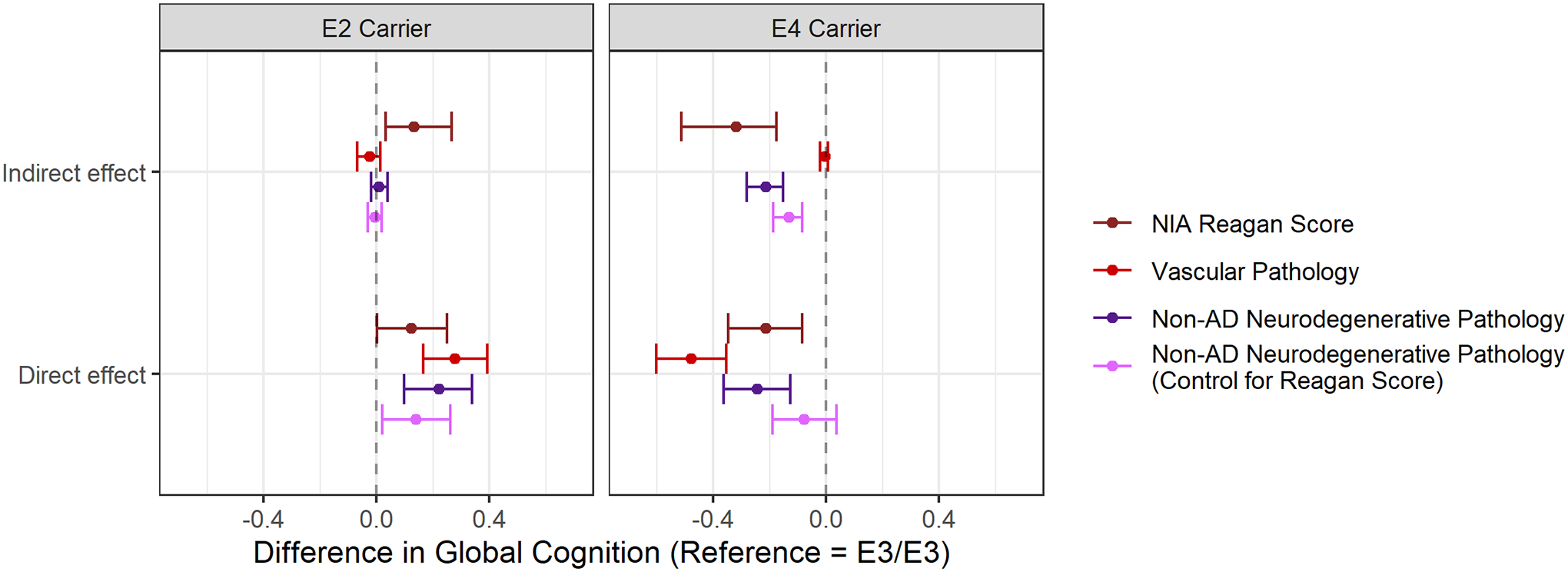
Comparison between NIA Reagan Score and non-AD Pathologies (vascular and non-AD neurodegenerative pathologies) as mediators of the association between APOE genotype and cognitive functioning. Those with the APOE ε3/ε3 genotype are the reference group in all analyses. Indirect effect represents the effect of APOE genotype on cognition through changes in the mediator; direct effect represents the effect of APOE genotype on cognition via other pathways. When controlling for NIA Reagan score in the entire mediation analysis, effects can be interpreted as the effect of APOE genotype on cognition, independent of NIA Reagan score.
3.5. Sex differences in AD and non-AD mediators of APOE genotype on cognition
Women were more likely to have a higher NIA Reagan Score compared to men, although results were not statistically significant (Figure 4 Panel A; Appendix Table S6). Additionally, when considering global cognition as an outcome, there was a significant interaction between sex and the APOE-ε2 allele, such that among women APOE- ε2 carriers had better global cognition compared to those with the APOE-ε3/ε3 genotype; a similar difference was not present in men (Figure 4 Panel C). NIA Reagan score and non-AD neurodegenerative pathologies both significantly mediated the association between the APOE-ε4 allele and cognition in men and women. While not achieving statistical significance, the mediational effect of NIA Reagan Score was larger in women (SD unit difference: −0.41 [95% CI −0.17 – −0.72]) compared to men (SD unit difference: −0.21 [95% CI −0.04 – −0.45]). The effect of the APOE-ε2 allele on cognition via NIA Regan score was statistically significant in women by not men. Additionally, we found strong direct effects of the APOE-ε2 allele on cognition through pathways other than the considered mediators in women but not in men.
Figure 4.

Associations between APOE genotype and NIA Reagan Score [A], APOE genotype and Non-AD pathology [B], APOE genotype and global cognition [C] and the indirect and direct effects of APOE genotype on cognition through NIA Reagan score and non-AD pathologies [D] stratified by sex. The indirect effect represents the effect of APOE genotype on cognition through changes in NIA Reagan score; the direct effect represents the effect of APOE genotype on cognition via other pathways. The reference category in causal mediation models was the group of participants with an ε3/ε3 genotype. Categories of None and Low on the NIA Reagan score were collapsed for visualization purposes due to low cell counts in the None category.
3.6. Difference in AD and non-AD mediators of APOE genotype on cognition by clinical dementia status
Those with dementia at death had higher NIA Reagan scores (Figure 5 Panel A), higher levels of non-AD neurodegenerative pathologies (Figure 5 Panel B), and lower levels of cognitive functioning (Figure 5 Panel C). The APOE-ε4 allele was significantly associated with NIA-Reagan scores and non-AD neurodegenerative pathology in those with and without dementia (Appendix Table S7). Mediation analyses suggested that NIA Reagan score mediated the effect of the APOE-ε4 allele on cognition in those with and without dementia (Figure 5 Panel D). In contrast, non-AD neurodegenerative pathology was a significant mediator of the association between the APOE-ε4 allele in those with dementia but not in those who without clinical dementia at death. The indirect effect of the APOE-ε2 allele on cognition through NIA Reagan score was only observed in those without dementia (Figure 5 Panel D).
Figure 5.

Associations between APOE genotype and NIA Reagan Score [A], APOE genotype and Non-AD pathology [B], APOE genotype and global cognition [C] and the indirect and direct effects of APOE genotype on cognition through NIA Reagan score and non-AD pathologies [D] stratified by clinical dementia status at the time of death. The indirect effect represents the effect of APOE genotype on cognition through changes in NIA Reagan score; the direct effect represents the effect of APOE genotype on cognition via other pathways. The reference category in causal mediation models was the group of participants with an ε3/ε3 genotype. Categories of None and Low on the NIA Reagan score were collapsed for visualization purposes due to low cell counts in the None category.
4. Discussion
Our study showed that both AD and non-AD neurodegenerative pathology mediated associations between APOE genotype and late-life cognition. Although effects of the APOE-ε2 and -ε4 alleles were in opposite directions, discrepancies in mediation results suggest mechanistic pathways may differ. AD neuropathology mediated the effect of the APOE-ε4 allele on cognition in those with and without dementia; however, the protective mediating effect of AD pathology in the association between the APOE-ε2 allele and cognition was driven by those without clinical dementia at death. Non-AD neurodegenerative pathologies were a mediator for the association between APOE-ε4 (but not the ε2 allele) and cognitive function, and this finding was driven by those with clinical dementia. The effect of the APOE-ε4 allele on cognition through non-AD neurodegenerative pathology remained statistically significant even after controlling for AD neuropathology, indicating that the mediating effect of non-AD neurodegenerative pathology is not merely due to its correlation with AD neuropathology. Instead, it suggests the presence of multiple pathways through which the APOE-ε4 allele is associated with cognitive impairment and dementia.
Findings were generally consistent across the majority of AD neuropathology measures considered, regardless of the proteinopathy they reflected (amyloid-β versus tau) or psychometric properties (ordinal scales versus quantitative measures). However, estimates for mediation through diffuse plaques were smaller than other measures, in line with the hypothesis that diffuse plaques are less neurotoxic and play a smaller role in associations between APOE genotype and cognition [39,40]. Estimates of total and mediated effects were somewhat smaller using continuous measures of AD neuropathologies compared to ordinalized forms of the same underlying pathology. In this study, ordinal measures take into account neuropathology location, whereas continuous variables average all brain regions; location of neuropathologies may contain important additional information. Although many researchers argue against categorizing continuous variables, our findings suggest that the ordinal forms of the neuropathology measures examined isolate meaningful variation in constructs while ameliorating the noise imposed by sampling and measurement error inherent in standard neuropathology methods [41,42].
Prior studies have found that vascular neuropathologies such as atherosclerosis, lacunar infarcts, and micro- and macro-infarcts did not significantly mediate the association between APOE genotype and late-life cognition, in line with our findings [10,21,22]. The only vascular pathway that has previously shown consistent links with APOE genotype is CAA [43]; however, we grouped CAA with neurodegenerative pathologies in this study based on a data driven approach (see details in Appendix). Despite these findings, individuals with Alzheimer’s disease commonly have comorbid vascular disease, which can lead to worse cognitive and clinical outcomes [44–47]. In tandem, results suggest that while vascular pathologies play a role in Alzheimer’s disease, the APOE genotype is not a shared mechanistic pathway linking late-life cognition and vascular pathways other than CAA.
However, in our sample, a summary measure of non-AD neurodegenerative pathologies significantly mediated the association between the APOE-ε4 allele and late-life cognition. This finding aligns with prior evidence that TDP-43 is associated with the APOE-ε4 allele and faster cognitive decline [18]. The breakdown of our non-AD neurodegenerative pathology score indicated that of components considered, measures of TDP-43 and cerebral amyloid angiopathy had the largest contributions to effects observed for our summary score. Accumulating evidence supports a role for concomitant Lewy body, cerebral amyloid angiopathy, hippocampal sclerosis, and TDP-43 pathology in cognitive deficits of patients with Alzheimer’s disease [48–52]. Our results suggest that the APOE-ε4 allele represents one potential mechanism explaining some degree of interconnectedness between AD and non-AD neurodegenerative pathologies in late-life cognitive outcomes. In contrast, we did not find that non-AD neurodegenerative pathology mediated the protective effect of APOE-ε2 on late-life cognition. The absence of such a pathway may help explain the smaller protective effect of the ε2 allele in comparison to the harmful effect of the ε4 allele [31,53].
The majority of existing studies suggest that harmful effects of the APOE-ε4 allele are greater in women than men [28,31,54]. However, we found no sex difference in the effect of the APOE-ε4 allele on late-life cognition, in line with a smaller minority of published work [27,55]. Discrepant results could be due to the older age of participants in our sample, as effects of APOE genotype may be weaker at older ages [56]. In comparison, existing literature on sex differences in APOE-ε2 is more sparse and mixed [19,54,57]. In our sample, we found that the APOE-ε2 allele was protective for late-life cognition in women but not men. The effect of APOE-ε2 on cognition was partially mediated by AD neuropathology, but not non-AD neuropathology in women. The direct effect of APOE-ε2 in women suggests that there is some effect of APOE-ε2 in women through pathways other than those considered here; factors related to immune and inflammatory responses or childhood cognitive development could be candidate pathways as there are known sex differences and ties to APOE genotype [58–61].
Our results have implications for drug development. Aducanumaub and the majority of drugs for Alzheimer’s disease in phase III clinical trials, target the removal of amyloid-β as a putative mechanism by which to alter disease pathogenesis [62,63]. Many clinical trials for anti-amyloid therapies showed large decreases in amyloid-β, but had inconclusive or null results for clinical outcomes [64,65]. Our results indicated the effects of the APOE-ε4 allele are mediated not only through AD neuropathology but also through non-AD neurodegenerative pathology, suggesting that the causes of ε4-associated late-life cognitive impairment are multifaceted in those with the APOE-ε4 allele. Therefore, a single approach targeting one molecule (amyloid-β) may not be sufficient to alter outcomes for APOE- ε4 carriers. Personalized, genotype-specific treatments may be needed and combination therapy, pairing an anti-amyloid treatment with drugs targeting other neuropathological processes, could be a promising approach [66,67].
Notable strengths of the present study include the use of a large, well-characterized autopsy sample, the comparison of different measures of AD neuropathologies with largely consistent findings, and the implementation of causal mediation methods. However, some limitations warrant consideration. First, we were unable to establish temporality between the mediator and outcome; in fact, our outcome (late-life cognition) was assessed before neuropathology mediators. However, the processes that result in neuropathologic burden observed at autopsy take decades and theoretical models posit that pathology in the brain leads to cognitive deficits and clinical dementia [68]. Second, our sample is predominantly white and highly educated. Future research should explore whether findings will generalize to more diverse samples, especially given known interactions between APOE genotype and race [69,70]. Finally, differences in sample size between APOE-ε2 and APOE-ε4 carriers (N=232 for APOE-ε2; N=399 for APOE-ε4) led to differences in power and may affect comparisons. However, in addition to differences in statistical significance of estimates, we also observed differences in the magnitude of effect sizes.
In summary, we found that both AD and non-AD neurodegenerative pathology mediated the association between the APOE-ε4 allele and late-life cognition. Our study shows that underlying genotypes can lead to more than one downstream cause of late-life cognitive impairment, and emphasizes the importance of considering both the traditional targets of AD research, amyloid-β and tau, as well as the development of other pathological processes in APOE-ε4 carriers.
Supplementary Material
Highlights.
Both APOE-ε2 and -ε4 effects on late-life cognition are mediated by AD neuropathology
Estimated mediated effects of most measures of AD neuropathology were similar
Non-AD neurodegenerative pathologies mediate the effect of ε4 independently from AD
Non-AD vascular pathologies did not mediate the effect of APOE genotype on cognition
Protective effect of APOE-ε2 on cognition was stronger in women
Research in Context.
Systematic Review.
We used PubMed and Google Scholar to review literature on i) the associations between APOE genotype and autopsy measures of AD and non-AD neuropathologies, and ii) mediation approaches to understand the effect of APOE genotype on cognitive deficits through neuropathological measures.
Interpretation.
Based on a sample of 1,671 participants with neuropathological assessment, we showed that the detrimental effect of APOE-ε4 on late-life cognition is mediated by both AD and non-AD neurodegenerative neuropathologies (TDP-43, cerebral amyloid angiopathy, hippocampal sclerosis, Lewy body pathology), especially in individuals with dementia. The protective effect of APOE-ε2 on cognitive function is partly mediated by AD neuropathology.
Future directions.
These results highlight the multiple pathways linking APOE genotype to cognitive decline and dementia, emphasizing the need for interventions that target multiple neuropathological processes to maximize therapeutic benefits. Investigations in more racially and socially diverse groups are needed to better characterize the impact of APOE genotype.
Acknowledgements:
This manuscript resulted from a conference that was funded in part by a grant from the National Institute on Aging to the University of California, Davis (R13 AG030995, Dan Mungas PI). We thank Dr. M. Maria Glymour (UCSF) for her help and guidance in the analytic aspects of the analyses.
Funding Information:
This work was supported by the National Institutes of Health [grant numbers T32AG000247, K99AG065501, R01AG072475, R01AG059727, UF1NS125513, P30AG066546, P30AG010161/P30AG072975, R01AG015819, R01AG17917, R01AG022018, RF1AG054617], the Harquail Centre for Neuromodulation, the Dr. Sandra Black Centre for Brain Resilience & Recovery and Canadian Institutes of Health Research (173253, 438475), and the Texas Alzheimer’s Research and Care Consortium (2020-58-81-CR).
Footnotes
Declaration of Interests: J.A.S.: Scientific Advisory Board/Consultant: AVID radiopharmaceuticals (subsidiary of Lilly), Alnylam Pharmaceuticals, Apellis Pharmaceuticals, Takeda Pharmaceuticals, National Hockey League. No other authors have conflicts to disclose.
References
- [1].Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PHS, Pericak-Vance MA, Joo SH, et al. Association of apolipoprotein E allele ϵ4 with late‐onset familial and sporadic Alzheimer’s disease. Neurology 1993;43:1467–1467. 10.1212/WNL.43.8.1467. [DOI] [PubMed] [Google Scholar]
- [2].Blair CK, Folsom AR, Knopman DS, Bray MS, Mosley TH, Boerwinkle E, et al. APOE genotype and cognitive decline in a middle-aged cohort. Neurology 2005;64:268–76. 10.1212/01.WNL.0000149643.91367.8A. [DOI] [PubMed] [Google Scholar]
- [3].Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A 1993;90:1977–81. 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Tiraboschi P, Hansen LA, Masliah E, Alford M, Thal LJ, Corey-Bloom J. Impact of APOE genotype on neuropathologic and neurochemical markers of Alzheimer disease. Neurology 2004;62:1977–83. 10.1212/01.WNL.0000128091.92139.0F. [DOI] [PubMed] [Google Scholar]
- [5].Kim J, Basak JM, Holtzman DM. The Role of Apolipoprotein E in Alzheimer’s Disease. Neuron 2009;63:287–303. 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yamazaki Y, Painter MM, Bu G, Kanekiyo T. Apolipoprotein E as a Therapeutic Target in Alzheimer’s disease: A Review of Basic Research and Clinical Evidence. CNS Drugs 2016;30:773–89. 10.1007/s40263-016-0361-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Serrano-Pozo A, Qian J, Monsell SE, Betensky RA, Hyman BT. APOEε2 is associated with milder clinical and pathological Alzheimer’s disease. Ann Neurol 2015;77:917–29. 10.1002/ana.24369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Koffie RM, Hashimoto T, Tai H-C, Kay KR, Serrano-Pozo A, Joyner D, et al. Apolipoprotein E4 effects in Alzheimer’s disease are mediated by synaptotoxic oligomeric amyloid-β. Brain 2012;135:2155–68. 10.1093/brain/aws127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bennett DA, Schneider JA, Wilson RS, Bienias JL, Berry-Kravis E, Arnold SE. Amyloid mediates the association of apolipoprotein E e4 allele to cognitive function in older people. Journal of Neurology, Neurosurgery & Psychiatry 2005;76:1194–9. 10.1136/jnnp.2004.054445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gavett BE, John SE, Gurnani AS, Bussell CA, Saurman JL. The Role of Alzheimer’s and Cerebrovascular Pathology in Mediating the Effects of Age, Race, and Apolipoprotein E Genotype on Dementia Severity in Pathologically-Confirmed Alzheimer’s Disease. J Alzheimers Dis 2016;49:531–45. 10.3233/JAD-150252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mahley RW. Central Nervous System Lipoproteins. Arteriosclerosis, Thrombosis, and Vascular Biology 2016;36:1305–15. 10.1161/ATVBAHA.116.307023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Khan TA, Shah T, Prieto D, Zhang W, Price J, Fowkes GR, et al. Apolipoprotein E genotype, cardiovascular biomarkers and risk of stroke: Systematic review and meta-analysis of 14 015 stroke cases and pooled analysis of primary biomarker data from up to 60 883 individuals. International Journal of Epidemiology 2013;42:475–92. 10.1093/ije/dyt034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Davignon J, Gregg RE, Sing CF. Apolipoprotein E polymorphism and atherosclerosis. Arteriosclerosis: An Official Journal of the American Heart Association, Inc 1988. 10.1161/01.ATV.8.1.1. [DOI] [PubMed] [Google Scholar]
- [14].Luc G, Bard JM, Arveiler D, Evans A, Cambou JP, Bingham A, et al. Impact of apolipoprotein E polymorphism on lipoproteins and risk of myocardial infarction. The ECTIM Study. Arterioscler Thromb 1994;14:1412–9. 10.1161/01.atv.14.9.1412. [DOI] [PubMed] [Google Scholar]
- [15].Ramassamy C, Averill D, Beffert U, Theroux L, Lussier-Cacan S, Cohn JS, et al. Oxidative insults are associated with apolipoprotein E genotype in Alzheimer’s disease brain. Neurobiol Dis 2000;7:23–37. 10.1006/nbdi.1999.0273. [DOI] [PubMed] [Google Scholar]
- [16].Lynch JR, Tang W, Wang H, Vitek MP, Bennett ER, Sullivan PM, et al. APOE Genotype and an ApoE-mimetic Peptide Modify the Systemic and Central Nervous System Inflammatory Response *. Journal of Biological Chemistry 2003;278:48529–33. 10.1074/jbc.M306923200. [DOI] [PubMed] [Google Scholar]
- [17].Brickman AM, Schupf N, Manly JJ, Stern Y, Luchsinger JA, Provenzano FA, et al. APOE ε4 and risk for Alzheimer’s disease: do regionally distributed white matter hyperintensities play a role? Alzheimers Dement 2014;10:619–29. 10.1016/j.jalz.2014.07.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yang H-S, Yu L, White CC, Chibnik LB, Chhatwal JP, Sperling RA, et al. Evaluation of TDP-43 proteinopathy and hippocampal sclerosis in relation to APOE ε4 haplotype status: a community-based cohort study. Lancet Neurol 2018;17:773–81. 10.1016/S1474-4422(18)30251-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lamonja-Vicente N, Dacosta-Aguayo R, López-Olóriz J, Prades-Senovilla L, Roig-Coll F, Castells-Sánchez A, et al. Sex-Specific Protective Effects of APOE ε2 on Cognitive Performance. The Journals of Gerontology: Series A 2021;76:41–9. 10.1093/gerona/glaa247. [DOI] [PubMed] [Google Scholar]
- [20].Loika Y, Feng F, Loiko E, Kulminski AM. Mediation of the APOE associations with Alzheimer’s and coronary heart diseases through body mass index and lipids. GeroScience 2021. 10.1007/s11357-021-00458-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mortimer JA, Snowdon DA, Markesbery WR. The Effect of APOE-ε4 on Dementia is Mediated by Alzheimer Neuropathology. Alzheimer Dis Assoc Disord 2009;23:152–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Li Y, Schneider JA, Bennett DA. Estimation of the mediation effect with a binary mediator. Statistics in Medicine 2007;26:3398–414. 10.1002/sim.2730. [DOI] [PubMed] [Google Scholar]
- [23].VanderWeele TJ. Mediation Analysis: A Practitioner’s Guide. Annual Review of Public Health 2016;37:17–32. 10.1146/annurev-publhealth-032315-021402. [DOI] [PubMed] [Google Scholar]
- [24].Weigand AJ, Thomas KR, Bangen KJ, Eglit GML, Delano-Wood L, Gilbert PE, et al. APOE interacts with tau PET to influence memory independently of amyloid PET in older adults without dementia. Alzheimer’s & Dementia 2021;17:61–9. 10.1002/alz.12173. [DOI] [PubMed] [Google Scholar]
- [25].Mielke MM, Machulda MM, Hagen CE, Christianson TJ, Roberts RO, Knopman DS, et al. Influence of amyloid and APOE on cognitive performance in a late middle-aged cohort. Alzheimer’s & Dementia 2016;12:281–91. 10.1016/j.jalz.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mormino EC, Betensky RA, Hedden T, Schultz AP, Ward A, Huijbers W, et al. Amyloid and APOE ε4 interact to influence short-term decline in preclinical Alzheimer disease. Neurology 2014;82:1760–7. 10.1212/WNL.0000000000000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sundermann EE, Tran M, Maki PM, Bondi MW. Sex differences in the association between apolipoprotein E ε4 allele and Alzheimer’s disease markers. Alzheimers Dement (Amst) 2018;10:438–47. 10.1016/j.dadm.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Makkar SR, Lipnicki DM, Crawford JD, Kochan NA, Castro-Costa E, Lima-Costa MF, et al. APOE ε4 and the Influence of Sex, Age, Vascular Risk Factors, and Ethnicity on Cognitive Decline. The Journals of Gerontology: Series A 2020;75:1863–73. 10.1093/gerona/glaa116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hohman TJ, Dumitrescu L, Barnes LL, Thambisetty M, Beecham G, Kunkle B, et al. Sex-Specific Association of Apolipoprotein E With Cerebrospinal Fluid Levels of Tau. JAMA Neurol 2018;75:989–98. 10.1001/jamaneurol.2018.0821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hayden KM, Zandi PP, West NA, Tschanz JT, Norton MC, Corcoran C, et al. Effects of Family History and Apolipoprotein E ε4 Status on Cognitive Decline in the Absence of Alzheimer Dementia: The Cache County Study. Archives of Neurology 2009;66:1378–83. 10.1001/archneurol.2009.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of Age, Sex, and Ethnicity on the Association Between Apolipoprotein E Genotype and Alzheimer Disease: A Meta-analysis. JAMA 1997;278:1349–56. 10.1001/jama.1997.03550160069041. [DOI] [PubMed] [Google Scholar]
- [32].Consensus Recommendations for the Postmortem Diagnosis of Alzheimer’s Disease. Neurobiology of Aging 1997;18:S1–2. 10.1016/S0197-4580(97)00057-2. [DOI] [PubMed] [Google Scholar]
- [33].Osborne J. Notes on the use of data transformations. Practical Assessment, Research, and Evaluation 2019;8. 10.7275/4vng-5608. [DOI] [Google Scholar]
- [34].Rannikmäe K, Samarasekera N, Martînez-Gonzâlez NA, Salman RA-S, Sudlow CLM. Genetics of cerebral amyloid angiopathy: systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 2013;84:901–8. 10.1136/jnnp-2012-303898. [DOI] [PubMed] [Google Scholar]
- [35].Yamada M Risk Factors for Cerebral Amyloid Angiopathy in the Elderly. Annals of the New York Academy of Sciences 2002;977:37–44. 10.1111/j.1749-6632.2002.tb04797.x. [DOI] [PubMed] [Google Scholar]
- [36].Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, Schneider JA. Religious Orders Study and Rush Memory and Aging Project. J Alzheimers Dis 2018;64:S161–89. 10.3233/JAD-179939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Pearl J. Causal inference in statistics: An overview. Statistics Surveys 2009;3:96–146. [Google Scholar]
- [38].Robins JM, Greenland S. Identifiability and Exchangeability for Direct and Indirect Effects. Epidemiology 1992;3:143–55. [DOI] [PubMed] [Google Scholar]
- [39].Mrak RE. Neuropathology and the Neuroinflammation Idea. Journal of Alzheimer’s Disease 2009;18:473–81. 10.3233/JAD-2009-1158. [DOI] [PubMed] [Google Scholar]
- [40].Malek-Ahmadi M, Perez SE, Chen K, Mufson EJ. Neuritic and Diffuse Plaque Associations with Memory in Non-Cognitively Impaired Elderly. Journal of Alzheimer’s Disease 2016;53:1641–52. 10.3233/JAD-160365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bennette C, Vickers A. Against quantiles: categorization of continuous variables in epidemiologic research, and its discontents. BMC Med Res Methodol 2012;12:21. 10.1186/1471-2288-12-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].van Walraven C, Hart RG. Leave ‘em alone - why continuous variables should be analyzed as such. Neuroepidemiology 2008;30:138–9. 10.1159/000126908. [DOI] [PubMed] [Google Scholar]
- [43].Nelson PT, Pious NM, Jicha GA, Wilcock DM, Fardo DW, Estus S, et al. APOE-ε2 and APOE-ε4 Correlate With Increased Amyloid Accumulation in Cerebral Vasculature. Journal of Neuropathology & Experimental Neurology 2013;72:708–15. 10.1097/NEN.0b013e31829a25b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA 1997;277:813–7. [PubMed] [Google Scholar]
- [45].Nagy Z, Esiri MM, Jobst KA, Morris JH, King EM-F, McDonald B, et al. The Effects of Additional Pathology on the Cognitive Deficit in Alzheimer Disease. Journal of Neuropathology & Experimental Neurology 1997;56:165–70. 10.1097/00005072-199702000-00007. [DOI] [PubMed] [Google Scholar]
- [46].Rabin JS, Schultz AP, Hedden T, Viswanathan A, Marshall GA, Kilpatrick E, et al. Interactive associations of vascular risk and β-amyloid burden with cognitive decline in clinically normal elderly individuals findings from the Harvard Aging Brain Study. JAMA Neurology 2018;75:1124–31. 10.1001/jamaneurol.2018.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Toledo JB, Arnold SE, Raible K, Brettschneider J, Xie SX, Grossman M, et al. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain 2013;136:2697–706. 10.1093/brain/awt188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Meneses A, Koga S, O’Leary J, Dickson DW, Bu G, Zhao N. TDP-43 Pathology in Alzheimer’s Disease. Mol Neurodegener 2021;16:84. 10.1186/s13024-021-00503-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Shih Y-H, Tu L-H, Chang T-Y, Ganesan K, Chang W-W, Chang P-S, et al. TDP-43 interacts with amyloid-β, inhibits fibrillization, and worsens pathology in a model of Alzheimer’s disease. Nat Commun 2020;11:5950. 10.1038/s41467-020-19786-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kotzbauer PT, Trojanowsk JQ, Lee VM. Lewy body pathology in Alzheimer’s disease. J Mol Neurosci 2001;17:225–32. 10.1385/jmn:17:2:225. [DOI] [PubMed] [Google Scholar]
- [51].Nag S, Yu L, Capuano AW, Wilson RS, Leurgans SE, Bennett DA, et al. Hippocampal sclerosis and TDP-43 pathology in aging and Alzheimer disease. Annals of Neurology 2015;77:942–52. 10.1002/ana.24388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Thal DR, Griffin WST, de Vos RAI, Ghebremedhin E. Cerebral amyloid angiopathy and its relationship to Alzheimer’s disease. Acta Neuropathol 2008;115:599–609. 10.1007/s00401-008-0366-2. [DOI] [PubMed] [Google Scholar]
- [53].Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet 1994;7:180–4. 10.1038/ng0694-180. [DOI] [PubMed] [Google Scholar]
- [54].Altmann A, Tian L, Henderson VW, Greicius MD. Sex Modifies the APOE-Related Risk of Developing Alzheimer’s Disease. Ann Neurol 2014;75:563–73. 10.1002/ana.24135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Beydoun MA, Beydoun HA, Kaufman JS, An Y, Resnick SM, O’Brien R, et al. Apolipoprotein E ε4 Allele Interacts with Sex and Cognitive Status to Influence All-Cause and Cause-Specific Mortality in U.S. Older Adults. Journal of the American Geriatrics Society 2013;61:525–34. 10.1111/jgs.12156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Corrada MM, Paganini-Hill A, Berlau DJ, Kawas CH. Apolipoprotein E genotype, dementia, and mortality in the oldest old: The 90+ Study. Alzheimer’s & Dementia 2013;9:12–8. 10.1016/j.jalz.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Neu SC, Pa J, Kukull W, Beekly D, Kuzma A, Gangadharan P, et al. Apolipoprotein E Genotype and Sex Risk Factors for Alzheimer Disease: A Meta-analysis. JAMA Neurol 2017;74:1178–89. 10.1001/jamaneurol.2017.2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol 2016;16:626–38. 10.1038/nri.2016.90. [DOI] [PubMed] [Google Scholar]
- [59].Keene CD, Cudaback E, Li X, Montine KS, Montine TJ. Apolipoprotein E isoforms and regulation of the innate immune response in brain of patients with Alzheimer’s disease. Current Opinion in Neurobiology 2011;21:920–8. 10.1016/j.conb.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Reynolds CA, Smolen A, Corley RP, Munoz E, Friedman NP, Rhee SH, et al. APOE effects on cognition from childhood to adolescence. Neurobiology of Aging 2019;84:239.e1–239.e8. 10.1016/j.neurobiolaging.2019.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Chang L, Douet V, Bloss C, Lee K, Pritchett A, Jernigan TL, et al. Gray matter maturation and cognition in children with different APOE ε genotypes. Neurology 2016;87:585–94. 10.1212/WNL.0000000000002939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K. Alzheimer’s disease drug development pipeline: 2020. Alzheimer’s & Dementia: Translational Research & Clinical Interventions 2020;6:e12050. 10.1002/trc2.12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Knopman DS, Jones DT, Greicius MD. Failure to demonstrate efficacy of aducanumab: An analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimer’s & Dementia 2021;17:696–701. 10.1002/alz.12213. [DOI] [PubMed] [Google Scholar]
- [64].Kwan ATH, Arfaie S, Therriault J, Rosa-Neto P, Gauthier S. Lessons Learnt from the Second Generation of Anti-Amyloid Monoclonal Antibodies Clinical Trials. DEM 2020;49:334–48. 10.1159/000511506. [DOI] [PubMed] [Google Scholar]
- [65].Castello MA, Jeppson JD, Soriano S. Moving beyond anti-amyloid therapy for the prevention and treatment of Alzheimer’s disease. BMC Neurology 2014;14:169. 10.1186/s12883-014-0169-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Delrieu J, Bateman RJ, Touchon J, Sabbagh M, Cummings J. The Future of AD Clinical Trials with the Advent of Anti-Amyloid Therapies: An CTAD Task Force Report. J Prev Alzheimers Dis 2022. 10.14283/jpad.2022.48. [DOI] [PubMed] [Google Scholar]
- [67].Fessel J. Reversing Alzheimer’s disease dementia with clemastine, fingolimod, or rolipram, plus anti-amyloid therapy. Alzheimer’s & Dementia: Translational Research & Clinical Interventions 2022;8:e12242. 10.1002/trc2.12242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Jack CR, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. The Lancet Neurology 2010;9:119–28. 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Beydoun MA, Weiss J, Beydoun HA, Hossain S, Maldonado AI, Shen B, et al. Race, APOE genotypes, and cognitive decline among middle-aged urban adults. Alzheimer’s Research & Therapy 2021;13:120. 10.1186/s13195-021-00855-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Chan ML, Meyer OL, Farias ST, Whitmer RA, Rajan K, Olichney J, et al. APOE Effects on Late Life Cognitive Trajectories in Diverse Racial/Ethnic Groups. Journal of the International Neuropsychological Society 2022:1–10. 10.1017/S1355617722000030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.