


Abstract
Individuals with mutations in the WRN gene suffer from Werner syndrome, a disease with early onset of many characteristics of normal aging. The WRN protein (WRNp) functions in DNA metabolism, as the purified polypeptide has both 3′→5′ helicase and 3′→5′ exonuclease activities. In this study, we have further characterized WRNp exonuclease activity by examining its ability to degrade double-stranded DNA substrates containing abnormal and damaged nucleotides. In addition, we directly compared the 3′→5′ WRNp exonuclease activity with that of exonuclease III and the Klenow fragment of DNA polymerase I. Our results indicate that the presence of certain abnormal bases (such as uracil and hypoxanthine) does not inhibit the exonuclease activity of WRNp, exonuclease III or Klenow, whereas other DNA modifications, including apurinic sites, 8-oxoguanine, 8-oxoadenine and cholesterol adducts, inhibit or block WRNp. The ability of damaged nucleotides to inhibit exonucleolytic digestion differs significantly between WRNp, exonuclease III and Klenow, indicating that each exonuclease has a distinct mechanism of action. In addition, normal and modified DNA substrates are degraded similarly by full-length WRNp and an N-terminal fragment of WRNp, indicating that the specificity for this activity lies mostly within this region. The biochemical and physiological significance of these results is discussed.
INTRODUCTION
Werner syndrome (WS) is a human autosomal recessive disease characterized by signs of premature aging (reviewed in 1,2). Although not all characteristics of normal aging are accelerated in WS, affected individuals do display early graying and loss of hair, wrinkling and ulceration of the skin, atherosclerosis, osteoporosis and cataracts. WS patients also have an increased frequency of malignancies, diabetes mellitus type II and hypertension. Premature aging characteristics begin to appear during adolescence and WS individuals die (usually of cardiovascular disease or cancer) almost always before age 50. Thus, WS has been used increasingly as a model system to study human aging.
The premature aging phenotype of WS is caused by mutations in a single gene, known as WRN (3). The amino acid sequence of the WRN gene contains seven sequence motifs in its central region (see Fig. 1) with homology to the RecQ family of helicases that unwind DNA–DNA and DNA–RNA duplexes (3). The RecQ helicases have been implicated in many important nucleic acid metabolism pathways, including transcription, replication, chromosome segregation, recombination and repair (4–6). The human members of the RecQ family include the WRNp, BLM, RECQL, RECQ4 and RECQ5 proteins (3,7–10). Several investigators (11–13) have shown that WRNp, like other biochemically characterized RecQ homologs, has an associated helicase activity with 3′→5′ directionality. Unlike other known members of the RecQ family, WRNp contains sequence blocks near its N-terminus that have homology to a family of 3′→5′ exonucleases (14). The inherent ability of 3′→5′ exonucleases to remove nucleotides from the 3′-termini of DNA strands and, in the process, produce a substrate amenable to polymerization makes them virtually indispensable in DNA replication, repair and recombination pathways. Purified WRNp has recently been shown to have 3′→5′ exonuclease activity on double-stranded DNA with recessed 3′-ends (15,16). Notably, a point mutation at a conserved amino acid within the exonuclease domain of WRNp completely abolishes this 3′→5′ exonuclease activity (15). Despite the preliminary characterization of the enzymatic functions of WRNp, the precise cellular roles of both the helicase and exonuclease activities of WRNp remain unknown.
Figure 1.

Structure–function map of wild-type and mutant WRN proteins. Important amino acid sequence regions of wild-type (WT) WRNp, WRN-X, WRN-K and WRN-N are noted with blocks as specified in the legend (at top). The three exonuclease (gray) and seven helicase (black) sequence motifs are labeled with Roman numerals above the map for wild-type WRNp. Asterisks mark the approximate positions of point mutations in the WRN-X (Glu84→Ala, E84A) and WRN-K (Lys577→Met, K577M) proteins. The WRN-N mutant is truncated containing only the exonuclease sequence motifs.
Cells from WS patients have been the subject of a number of studies. Primary fibroblasts from WS patients exhibit premature cellular senescence (17,18), probably due to accelerated loss of telomeric sequences (19,20). When compared to normal cells, the most established biochemical property of WS cells is elevated genomic instability, typified by increased levels of DNA deletions, insertions and translocations (21,22). Thus, WRNp appears to be involved in maintenance of genomic integrity, apparently through a crucial function in DNA metabolism. Cells with mutations in WRN show sensitivity to some DNA damaging agents, including 4-nitroquinoline-1-oxide (4NQO) (23) and topoisomerase I inhibitors (24), and the 4NQO sensitivity of a WS cell can be complemented by fusion with a wild-type cell (25). Notably, treatment of cells with 4NQO causes the formation of several bulky quinoline adducts as well as apurinic/apyrimidinic sites and 8-oxoguanine (8-oxoG) lesions in DNA (26). These studies suggest a possible role for WRNp in DNA damage processing. Several other premature aging and cancer-prone hereditary diseases, such as Cockayne syndrome, xeroderma pigmentosum and Bloom syndrome, are characterized by genomic instability due to defined defects in DNA repair pathways (27).
To further characterize the enzymatic properties of WRNp and assess whether it might function in DNA damage processing, we investigated the effect of abnormal or damaged nucleotides in double-stranded DNA on the 3′→5′ exonuclease activity of WRNp. We also directly compared the exonuclease activity of WRNp with that of two well-studied 3′→5′ exonucleases from Escherichia coli, the Klenow fragment of DNA polymerase I (Klenow) and exonuclease III (exo III). Our results indicate that exo III, Klenow and WRNp have significantly different molecular mechanisms for the 3′→5′ digestion of normal and modified double-stranded DNA. In particular, the exonucleolytic activity of WRNp is greatly inhibited by the presence of minor oxidative damage or bulky lesions in DNA. Moreover, our results also demonstrate that the specificity for exonuclease activity lies predominantly but not completely within the N-terminal 368 amino acids of WRNp. The observed inhibition of 3′→5′ exonuclease activity by perturbations in double-stranded DNA suggests that WRNp could potentially be involved in sensing but probably not removing damaged nucleotides as part of its function in DNA metabolism.
MATERIALS AND METHODS
Enzymes
Exo III and Klenow were purchased from Boehringer Mannheim (Indianapolis, IN) and T4 polynucleotide kinase was from New England Biolabs (Beverly, MA). Escherichia coli formamidopyrimidine glycosylase (Fpg) and human apurinic/apyrimidinic endonuclease (APE) were gifts from A. Grollman (State University of New York, Stony Brook, NY) and S. Wilson (NIEHS, Research Triangle Park, NC), respectively.
Recombinant wild-type and mutant WRN proteins were overproduced and purified using a baculovirus/insect cell system. Figure 1 maps the functional regions of WRNp and the locations of amino acid sequence alterations present in each mutant protein used in this study. Matthew Gray (University of Washington, Seattle, WA) kindly provided baculovirus containing cDNA sequences coding either for a full-length WRNp with a methionine substitution at an absolutely conserved lysine residue (K577M) in motif I of the helicase domain (WRN-K) or only for the exonuclease portion (N-terminal 368 amino acids) of WRNp (WRN-N). Baculovirus containing a point mutation (E84A) in the exonuclease domain of WRNp (WRN-X) was a gift of Shurong Huang and Judy Campisi (University of California, Berkeley, CA). All WRN baculoviral constructs contained an N-terminal hexahistidine tag to facilitate purification by Ni2+ affinity chromatography. After infection of Sf9 insect cells with the appropriate baculovirus and overproduction of the corresponding recombinant protein, full-length wild-type WRNp, WRN-K and WRN-X were purified by a combination of DEAE–Sepharose (Amersham Pharmacia, Piscataway, NJ), Q-Sepharose (Pharmacia) and Ni–NTA (Gibco BRL, Rockville, MD) chromatography as described previously (28). The WRN-N polypeptide was more highly overproduced and more soluble in Sf9 insect cells than full-length WRNp (Orren, unpublished results), thus WRN-N purified only by Ni–NTA chromatography was suitable for use.
DNA substrates
Single-stranded DNA oligomers (21mer, 32mer and 43mer) containing only normal nucleotides were obtained from Gibco BRL. 32mers containing unusual or modified [uracil, hypoxanthine, ethenoadenine, 8-oxoadenine (8-oxoA), 8-oxoG, an apurinic site or cholesterol] nucleotides were purchased from Midland (Midland, TX). The sequences of the various oligomers and the positions of abnormal and damaged bases are shown in Table 1. Individual 32mers and the 21mer (7 pmol each) were 5′-labeled with [γ-32P]ATP (60 µCi, 3000 Ci/mmol) and polynucleotide kinase (10 U) using standard conditions. For construction of a double-stranded DNA substrate with one blunt end and one 3′-recessed (5′-overhang) end, labeled 32mers (undamaged, abnormal or damaged) were mixed with a 2-fold excess of unlabeled 43mer, heated together at 90°C for 5 min, then cooled slowly to 25°C. A substrate with both 3′- and 5′-overhangs was prepared similarly using the labeled 21mer and the unlabeled 43mer. Annealed double-stranded substrates were then separated from unannealed and excess single-stranded oligomers by non-denaturing 12% PAGE. Intact double-stranded DNA substrates were recovered using a Qiaex II gel extraction kit (Qiagen, Valencia, CA) and stored at 4°C.
Table 1. DNA oligomers.

The nucleotide modifications are underlined and depicted as follows: U, uracil; H, hypoxanthine; G°, 8-oxoG; A°, 8-oxoA; E, ethenoadenine; X, cholesterol; ^, apurinic site.
The unmodified 43mer to which the 21mer and all 32mers were annealed is listed in 3′→5′ orientation at the bottom, aligned with the oligomer above with regard to base pairing structure.
Exonuclease assays
WRNp exonuclease assays were carried out in buffer containing 40 mM Tris (pH 8.0), 4 mM MgCl2, 1 mM ATP, 0.1 mg/ml bovine serum albumin (BSA) and 5 mM dithiothreitol (DTT). Klenow exonuclease assays were carried out in 50 mM Tris (pH 7.4), 10 mM MgCl2 and 1 mM DTT. Exonuclease III reactions were carried out in 67 mM Tris (pH 7.4), 0.66 mM MgCl2 and 1 mM β-mercaptoethanol. Undamaged, abnormal and damaged DNA substrates (3 fmol) were incubated with Klenow (2 U), exonuclease III (1 U), WRNp (120 fmol), WRN-X (120 fmol), WRN-K (120 fmol) or WRN-N (120 fmol) for 1 h at 37°C and the reactions were stopped by addition of an equal volume of formamide loading buffer (80% formamide, 0.5× TBE, 0.1% xylene cyanol and 0.1% bromophenol blue). The digestion products of these reactions were separated on denaturing 15% polyacrylamide gels and visualized using a phosphorimager (Molecular Dynamics, Sunnyvale, CA). Quantitative comparison of individual lanes was accomplished using ImageQuant software. Total radioactivity in individual lanes containing the same substrate was comparable. Apparent differences between lanes are due to the digestion of substrates to mononucleotides and very short oligonucleotides that are not displayed in the experimental results (Figs 2–4).
Figure 2.
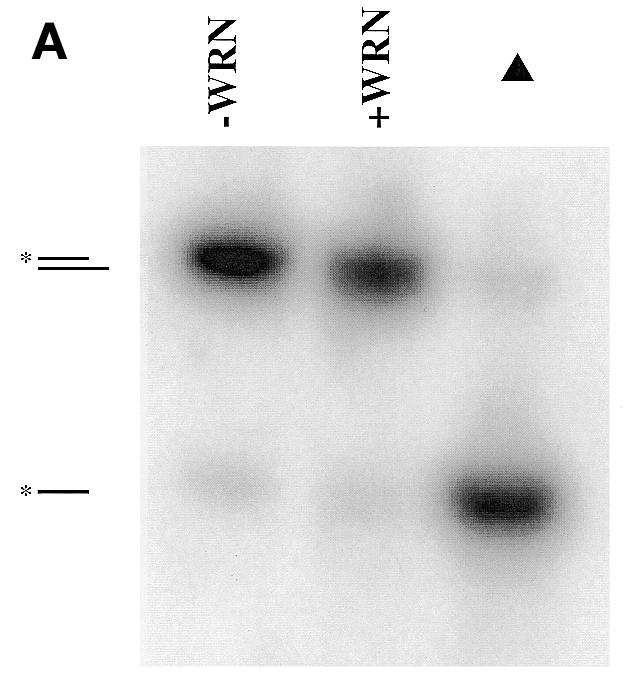

Activities of WRNp on an unmodified, 5′-overhang DNA substrate. (A) Assay of WRNp helicase activity on the 5′-overhang DNA substrate. The unmodified substrate was incubated with or without WRNp (150 fmol) for 1 h at 37°C and the DNA products were analyzed on a non-denaturing 12% polyacrylamide gel. Under these non-denaturing electrophoretic conditions, migration of double-stranded DNA species is dictated by the length of the unlabeled strand (data not shown) and thus WRNp exonuclease degradation products cannot be resolved by this method. As a marker for the migration of the labeled strand, the DNA substrate was loaded onto the gel immediately following heat denaturation (denoted by the triangle). The positions of the intact double-stranded substrate and the labeled single-stranded 32mer are denoted to the left. (B) Assay of exo III, Klenow and WRNp 3′→5′ exonuclease activities on the unmodified, 5′-overhang DNA substrate. The unmodified DNA substrate (depicted at the top) was incubated without enzyme (–enzyme) or with exo III (1 U), Klenow (2 U) or WRNp (120 fmol) at 37°C for 1 h. After heating at 90°C, the single-stranded DNA products were separated on a 15% denaturing polyacrylamide gel and visualized by phosphorimaging. The identities of the 3′-end nucleotide for each single-stranded degradation product are denoted to the right.
Figure 4.

The 3′→5′ exonuclease activities of WRNp, exo III and Klenow on substrates containing 8-oxoG, 8-oxoA, an apurinic site or a cholesterol adduct. The DNA substrates were as depicted in Figure 2B, except that 8-oxoG, 8-oxoA, an apurinic site or cholesterol was substituted for a normal nucleotide at a defined position. The sequence and base pairing structure near the 5′-overhang end of each substrate is shown at the top. The hydroxyl groups at position 8 of guanine and adenine are denoted by ° above the modified nucleotide and the cholesterol moiety is indicated by X. For each substrate, the abnormal base pair is capitalized and in bold and the modified nucleotide is underlined, with the exception of the apurinic site, which is indicated by ^. Exonuclease reactions and subsequent analysis were carried out as in Figure 2B. For each substrate, the position of the degradation product having the modified nucleotide at the 3′-terminus of the labeled strand is denoted with arrows.
Endonuclease and helicase assays
The presence and location of 8-oxoG and 8-oxoA in double-stranded substrates was tested using Fpg; apurinic sites were examined likewise using APE. Reactions containing 8-oxoG- or 8-oxoA-modified substrate (0.75 fmol) and Fpg (10–50 ng) were incubated for 1 h at 37°C in buffer containing 20 mM HEPES (pH 7.4), 50 mM KCl, 5 mM EDTA, 5 mM DTT, 0.1 mg/ml BSA and 5% glycerol. Reactions containing double-stranded DNA substrate (3 fmol) with an apurinic site and APE (25 ng) were incubated for 1 h at 37°C in buffer containing 40 mM HEPES (pH 7.8), 5 mM MgCl2, 0.1 mM EDTA and 1 mM DTT. All endonuclease reaction products were analyzed using denaturing PAGE and phosphorimaging as described above.
Helicase reactions with unmodified 32mer/43mer and 21mer/43mer DNA substrates were carried out for 1 h at 37°C in the same buffer used for WRNp exonuclease reactions. The reactions were terminated by addition of 0.5 vol helicase loading dye containing 50 mM EDTA, 0.9% SDS, 0.1% bromophenol blue, 0.1% xylene cyanol and 40% glycerol. The DNA products were resolved on non-denaturing 12% polyacrylamide gels and radiolabeled products were visualized by phosphorimaging.
RESULTS
Activities of WRNp, Klenow and exo III on undamaged substrates
WRNp has been previously shown to have both 3′→5′ helicase and 3′→5′ exonuclease activities (15,16,29,30). Another report (31) suggests that WRNp has 5′→3′ exonuclease activity. Using several different substrates, we consistently observe a 3′→5′ exonuclease activity associated with WRNp, and a recombinant WRN protein with a point mutation at a conserved residue in the exonuclease domain (WRN-X) lacks this 3′→5′ exonuclease activity (15,32). Although not required, ATP significantly stimulates the exonuclease activity of WRNp (16) by an unknown mechanism. Our experiments included ATP in WRNp-containing reactions to promote optimal exonuclease activity. The WRNp helicase activity prefers a single-stranded 3′-overhang (recessed 5′-end) and does not detectably unwind DNA substrates with blunt ends. In contrast, the 3′→5′ exonuclease activity requires a single-stranded 5′-overhang (recessed 3′-end) and does not significantly degrade single-stranded DNA or double-stranded DNA with blunt ends (16).
To minimize WRNp helicase activity and limit 3′→5′ exonuclease activity to one (labeled) strand, we designed a DNA substrate with one blunt end and an 11 nt 5′-overhang on the unlabeled strand (Table 1 and Fig. 2B, top). As expected, this substrate was not detectably unwound by the helicase activity of WRNp (Fig. 2A). However, a related substrate containing a 3′-overhang as well as a 5′-overhang (a labeled 21mer annealed to the same 42mer) was unwound by WRNp (data not shown), attesting to the ability of our WRNp preparations to carry out DNA unwinding reactions. Although not a helicase substrate for WRNp, the blunt end/5′-overhang substrate was readily acted on by the exonuclease activity of WRNp, resulting in stepwise, single nucleotide shortening of the labeled strand with the expected 3′→5′ directionality (Fig. 2B). The extent of digestion from the 3′-end of this substrate is directly related to the WRNp concentration (data not shown). This unmodified substrate was also subject to the 3′→5′ exonuclease activity of either Klenow or exo III and the concentration of both enzymes was adjusted to achieve almost the same extent of digestion as WRNp (Fig. 2B). Importantly, WRNp, Klenow and exo III have subtle but significant differences in digestion patterns, indicating possibly different substrate recognition parameters. For example, in comparison to exo III and Klenow, WRNp appears to have difficulty removing the second deoxyadenylate residue from the 3′-end. The differences between WRNp and Klenow are particularly noteworthy, considering that both share sequence homology to the RNase D family of nucleases (14).
Abnormal and damaged DNA substrates
As WRNp has been postulated to have a role in maintaining genomic stability, we wanted to determine whether the 3′→5′ exonuclease activity of WRNp could act on double-stranded DNA containing modified nucleotides. The 32mer/43mer substrate was used, except that a modified (uracil, hypoxanthine, ethenoadenine, 8-oxoA, 8-oxoG, apurinic site or cholesterol adduct) nucleotide was introduced within the double-stranded region at a specific position between 4 and 9 nt from the end of the recessed 3′-strand (Table 1). Modifications were chosen to represent a wide variety of structural perturbations in DNA, from helix-distorting lesions (cholesterol and apurinic sites) to minor base modifications (8-oxoA, 8-oxoG and ethenoadenine) and unusual bases (uracil and hypoxanthine) that result in marginal or no disruption of base pairing. Cholesterol-containing substrates have been used regularly in nucleotide excision repair assays because this lesion creates a major distortion of the DNA structure (33), although it should be noted that the cholesterol moiety is not adducted to the nucleotide but instead replaces the entire guanine deoxynucleotide in that particular substrate. All other substitutions were made while maintaining the relevant base pairing structure of double-stranded DNA, i.e. uracil opposite adenine, hypoxanthine opposite thymine, ethenoadenine opposite thymine, 8-oxoG opposite cytosine, 8-oxoA opposite thymine and the apurinic site opposite cytosine. Notably, apurinic sites and 8-oxoG lesions are formed after treatment of cells with 4NQO (26), an agent to which WS cells are hypersensitive (23,25). The presence and location of the apurinic site and 8-oxoG modifications were confirmed by quantitative incision with APE or Fpg, respectively (data not shown). Our DNA substrate containing 8-oxoA was not detectably incised by Fpg.
Comparison of exonuclease activity on substrates containing uracil, hypoxanthine and ethenoadenine
The 3′→5′ exonuclease activities of exo III, Klenow and WRNp on substrates containing uracil, hypoxanthine or ethenoadenine are directly compared in Figure 3. The positions of these base substitutions are depicted in the sequence above each panel (underlined) and in the degradation pattern to the right of each panel (with arrows). Replacement of normal nucleotides with uracil, hypoxanthine or ethenoadenine resulted in little or no change in the ability of WRNp to digest through the region of sequence containing the modification. The substitution of hypoxanthine for adenine actually appears to enhance the ability of WRNp to digest this sequence, as judged by the loss of the pause site (at that position) that is present in both the normal and the uracil substrate digestion patterns. Similarly to WRNp, degradation of substrate by Klenow was also not significantly affected by uracil, hypoxanthine or ethenoadenine nucleotides. In contrast, the ethenoadenine nucleotide appears to significantly inhibit the activity of exo III, even though the presence of uracil or hypoxanthine had little or no effect. Intriguingly, the inhibition of exo III activity by ethenoadenine occurs up to and beyond the position of the modified nucleotide (Fig. 3, right, indicated by arrow).
Figure 3.

The 3′→5′ exonuclease activities of WRNp, exo III and Klenow on substrates containing uracil, hypoxanthine or ethenoadenine. The DNA substrates were as depicted in Figure 2B, except that uracil, hypoxanthine or ethenoadenine (ethenoA) was substituted for a normal nucleotide at a defined position. The sequence and base pairing structure near the 5′-overhang end of each substrate is shown at the top. For each substrate, the abnormal base pair is capitalized and in bold and the modified nucleotide is underlined, with U, H and E representing uracil, hypoxanthine and ethenoadenine, respectively. Exonuclease reactions and analysis of DNA products were carried out as described in Figure 2B. For each substrate, the position of the degradation product having the modified nucleotide at the 3′-terminus of the labeled strand is denoted with arrows.
Comparison of exonuclease activity on substrates containing 8-oxo-guanine, 8-oxo-adenine, an apurinic site or a cholesterol adduct
The 3′→5′ exonuclease activities of exo III, Klenow and WRNp on substrates containing 8-oxoG, 8-oxoA, an apurinic site or a cholesterol adduct are directly compared in Figure 4. Again, the positions of the modified nucleotides in the sequence and the digestion pattern are indicated as described above. Importantly, the removal of the first guanine deoxynucleotide from the 3′-end confirms the ability of each enzyme to initiate exonucleolytic digestion on all substrates containing damaged nucleotides. To facilitate comparison of the WRNp exonuclease activity on different substrates, we calculated the percentage of the total radioactivity in each lane associated with the 10 longest oligonucleotide products after digestion of undamaged and uracil-, 8-oxoA-, 8-oxoG-, cholesterol- and apurinic site-containing substrates with WRNp (Fig. 5). This 3-dimensional representation is particularly helpful in emphasizing, for each damaged substrate, the point at which WRNp exonuclease is inhibited (tallest shape) with respect to the position of the damaged nucleotide (denoted by asterisk). It is clear from this analysis that the undamaged and uracil-containing substrate are degraded almost identically by WRNp. In stark contrast to our results with substrates containing uracil, hypoxanthine or ethenoadenine, the exonuclease activity of WRNp is markedly inhibited by the presence of 8-oxoG, 8-oxoA, an apurinic site or a cholesterol adduct (Figs 4 and 5). The substrates containing 8-oxoG or an apurinic site appear to completely block the ability of WRNp to digest through the region containing the damaged nucleotide, while those containing 8-oxoA and cholesterol allow only a marginal amount of degradation past the damaged nucleotide (Figs 4 and 5). Increasing the WRNp concentration increased the extent of digestion 3′ to the 8-oxoG nucleotide but did not overcome this blockage (data not shown), suggesting both a kinetic and steric effect of at least this modification on exonuclease activity. In the case of the 8-oxoA substrate, the slightly degraded state of the substrate alone without added WRNp may account for some if not most of the radioactivity detected beyond the position of the modified nucleotide (compare the –enzyme lane with that of WRNp, Fig. 4). For substrates containing 8-oxoG, 8-oxoA, cholesterol or an apurinic site, inhibition of WRNp exonucleolytic digestion is apparent at nucleotide positions 3′ to the modified nucleotide. This suggests either that the modified nucleotide locally disrupts DNA structure 3′ to the modification or that WRNp can sense abnormal DNA structures at short distances internal to the 3′-end of the DNA.
Figure 5.

Comparison of WRNp 3′→5′ exonuclease activities on undamaged and modified DNA substrates. WRNp exonuclease degradation patterns on undamaged and modified DNA substrates (data from Figs 2–4) are quantitatively compared. Phosphorimager analysis was used to quantitate the amount of radioactivity associated with each band with respect to the total radioactivity present in all degradation products in an individual exonuclease reaction (single lane of a gel). The percentage of the total radioactivity contained in the first 10 degradation products nearest the 3′-end of the labeled strand of each substrate is then plotted in 3-dimensional bar graph form. Note that, except for the undamaged and uracil-containing substrates, virtually all of the total radioactivity is found in DNA products degraded 10 nt or less. The identities of the 3′-end nucleotide for each single-stranded degradation product are labeled on the x-axis and the positions of the modifications in each respective substrate (defined on the right axis) are denoted with asterisks.
The action of Klenow on these same substrates is significantly different than that of WRNp. Klenow can digest through regions of DNA containing an apurinic site, an 8-oxoG lesion or a cholesterol moiety, although its exonuclease activity is inhibited somewhat by 8-oxoG and cholesterol, as judged by the detection of prominent pause sites 3′ to these modifications (Fig. 4). Like WRNp, the exonuclease activity of Klenow is blocked by the presence of 8-oxoA. However, Klenow is able to remove nucleotides up to the 8-oxoA:T base pair itself, while WRNp appears to stop primarily 1 nt 3′ to 8-oxoA. Additional differences in the specificities of 3′→5′ exonucleases are demonstrated by the activity of exo III on DNA substrates containing 8-oxoG, 8-oxoA or cholesterol. The apurinic site itself is a substrate for the apurinic/apyrimidinic endonuclease activity of exo III, as shown by the prominent band at that position (Fig. 4), therefore, it is not possible to accurately assess the effect of apurinic sites on the exonuclease activity of exo III. Like WRNp and Klenow, exo III is inhibited but not completely blocked by the presence of cholesterol. However, in contrast to WRNp and Klenow, exo III has little problem digesting through regions containing 8-oxoG or 8-oxoA modifications. Comparison of the activities of exo III on unmodified and modified substrates (Figs 2B and 4) indicates the presence of a pause site caused by 8-oxoG. Nevertheless, both exo III and Klenow appear to be much more robust 3′→5′ exonucleases than WRNp.
Exonuclease activities of mutant WRN proteins
The homology to the RNase D family of 3′→5′ exonucleases lies within three discrete sequence blocks in the N-terminal 335 amino acids of WRNp (14; see also Fig. 1). The remainder of WRNp is comprised of a central region with seven motifs indicative of DNA-dependent ATPase/helicase activity (Fig. 1) and a C-terminal region with an, as yet, unknown function. Although the exonuclease homology is wholly contained in the N-terminus of WRNp, it is possible that the remainder of the protein is necessary for or could influence exonuclease activity. In this regard, the potential exonuclease activities of two mutant WRN proteins were investigated. The WRN-K mutant is a full-length protein that contains a single amino acid change (K577M) at an absolutely conserved lysine residue in motif I of the helicase domain (Fig. 1). This mutation completely eliminates the ATPase and helicase activities of WRNp (11,13). The WRN-N mutant is a truncated protein that contains only the N-terminal 368 amino acids of WRNp (Fig. 1). Thus, it contains all of the exonuclease homology but lacks the remainder of the protein, including the entire helicase domain. The exonuclease activities of these two mutant proteins on unmodified and modified DNA substrates were directly compared to that of the full-length wild-type WRNp (Fig. 6). Both WRN-N and WRN-K have significant 3′→5′ exonuclease activity on the unmodified substrate and the activity of WRN-K on substrates containing 8-oxoA, ethenoadenine or an apurinic site appears to closely mimic that of wild-type WRNp. Subtle differences in the digestion patterns of wild-type WRNp and WRN-K are probably due to a slightly lower activity of the WRN-K preparation. Although the WRN-N mutant and wild-type WRNp have nearly identical activity on unmodified and 8-oxoA substrates, their digestion patterns on substrates containing an apurinic site or ethenoadenine are somewhat different. There is little or no degradation of the substrate containing an apurinic site by WRN-N, while both WRNp and WRN-K are able to partially remove the first two nucleotides before being blocked by the presence of the lesion. The exonuclease activity of WRN-N also appears to be significantly inhibited by the presence of ethenoadenine, as evidenced by the appearance of a major pause site just 3′ to the modified nucleotide. These results indicate that the N-terminal portion of WRNp forms an independent, functional exonuclease domain that is subtly influenced by the remainder of the protein. This influence on specificity is seemingly independent of any requirement for ATP hydrolysis or DNA helicase activity.
Figure 6.

Comparison of the 3′→5′ exonuclease activities of wild-type and mutant WRN proteins. Unmodified DNA substrate (undamaged) or substrate containing ethenoadenine (ethenoA), 8-oxoA or an apurinic site was incubated without enzyme (–enzyme) or with wild-type WRNp (WRN-WT), WRN-N or WRN-K (120 fmol each) at 37°C for 1 h. The labeled DNA products for each individual reaction were analyzed and visualized as described in Figure 2B.
DISCUSSION
The lack of functional WRNp causes elevated chromosomal instability and premature cellular senescence and eventually results in the premature aging phenotype of WS. The primary amino acid sequence of WRNp revealed a high degree of homology to known DNA helicases (of the RecQ family) and exonucleases in the central and N-terminal portions of the protein, respectively (3,14). Like other RecQ family members characterized thus far, WRNp has DNA-dependent ATPase/helicase activity with a 3′→5′ directionality (11–13,28). The N-terminal region (amino acids 1–335) of WRNp is homologous to the RNase D family of nucleases that includes the 3′→5′ exonuclease domains of DNA polymerase I (from E.coli, Haemophilis influenzae, Rhizobium leguminosarum and Synechocystis sp.) (14). Recently, WRNp has been shown to have exonuclease activity with 3′→5′ directionality (15,16), although another study reports a 5′→3′ directionality (31). Our results are consistent with the notion that WRNp has only 3′→5′ exonuclease activity.
The results of our studies significantly extend the characterization of the basic enzymatic activities of WRNp. The inability of WRNp to unwind a DNA substrate with one blunt end and one single-stranded 5′-overhang (recessed 3′-end) confirms the requirement for a single-stranded 3′-region for helicase activity. Using this helicase-resistant substrate, our experiments clearly show that WRNp catalyzes the stepwise 3′→5′ exonucleolytic degradation of the double-stranded region of this substrate only from the recessed 3′-end. Exonuclease activity was also examined in two WRNp mutants of interest: (i) a full-length protein (WRN-K) containing a single amino acid substitution (K577M) at a conserved lysine residue in helicase motif I; (ii) a deletion mutant (WRN-N) containing only the N-terminal 368 amino acids. The mutation in WRN-K completely eliminates ATPase and helicase activity (11,13,28) but retains 3′→5′ exonuclease activity. Similar results on different DNA substrates have been reported (15,29). Our results show that the patterns of exonucleolytic digestion by WRN-K on normal and modified DNA substrates closely resemble those of wild-type WRNp, indicating that the lack of ATP hydrolysis and DNA unwinding activities have little effect on the specificity of exonuclease activity. The WRN-N deletion mutant also possesses 3′→5′ exonuclease activity that is very similar to that of full-length WRNp on unmodified and 8-oxoA-containing substrates. However, in comparison to full-length WRNp, the activity of WRN-N is slightly altered on substrates containing an apurinic site or ethenoadenine. Although other investigators have also shown that N-terminal fragments of WRNp exhibit 3′→5′ exonuclease activity (15,29), this study is the first to demonstrate distinct differences in activity or specificity between truncated N-terminal fragments and the full-length wild-type WRNp. These results indicate that, similar to the exonuclease domain of DNA polymerase I of E.coli (34), the N-terminal portion of WRNp folds into a domain that can function independently as a 3′→5′ exonuclease, although other regions of WRNp do have at least a minor influence on exonuclease activity and/or specificity. This influence could occur through contact of other parts of WRNp with either the exonuclease domain or the DNA substrate. In a similar manner, other proteins might also affect the specificity and activity of the WRNp exonuclease.
The studies presented here directly compare the 3′→5′ exonuclease activities of WRNp, exo III and Klenow fragment on various DNA substrates. Our results clearly show that the exonucleolytic activities of wild-type WRNp, exo III and Klenow fragment are significantly different from one another on both unmodified substrates and substrates containing non-Watson–Crick nucleotide pairs. The differences between WRNp and Klenow are striking despite the shared sequence homology in their exonuclease domains. On unmodified substrate, differences in digestion patterns/pause sites caused by digestion with WRNp, exo III and Klenow are clearly evident. The exonucleolytic activity of Klenow is only inhibited by either 8-oxoA or a cholesterol adduct, while exo III is significantly inhibited only by ethenoadenine or cholesterol. WRNp, being blocked by 8-oxoA, 8-oxoG, apurinic sites or a cholesterol moiety, appears to be generally more sensitive to the presence of modifications than either Klenow fragment or exo III. This could be due to the individual mechanisms by which WRNp, exo III and Klenow catalyze removal of mononucleotides. In this regard, the ability of Klenow to act on single-stranded DNA substrates and the ability of exo III to act on double-stranded DNA with mismatched 3′-termini of up to 3 nt (35) may allow them to degrade through regions containing certain damaged nucleotides that destabilize DNA ends. In contrast, WRNp does not degrade single-stranded DNA and has weak exonucleolytic activity on duplex DNA substrates with two mismatched nucleotides at the 3′-terminus, although a single mismatched nucleotide at the 3′-end is an excellent substrate for WRNp (16). The fact that WRNp exonuclease activity is inhibited several nucleotides 3′ to the modification suggests that either the protein can sense abnormalities in DNA structure at short distances away from the active site or that the modification causes localized destabilization of the DNA. Our result showing the specific inhibition of WRNp exonuclease activity at several positions 3′ to a reasonably stable 8-oxoG:C base pair suggests that sensing of DNA perturbations at short distances 5′ to the active site may contribute to a lowering of WRNp exonuclease activity.
Although the precise biochemical role of WRNp is yet to be determined, one could envision a role in DNA damage processing due to the specific properties of the WRNp 3′→5′ exonuclease activity. Our finding that this activity is uniquely inhibited or blocked by certain types of DNA lesions suggests that WRNp could be involved in sensing but not removing those types of DNA damage. Another study has demonstrated that WRNp has no specific binding affinity for several types of DNA lesions within the context of double-stranded DNA (28). WRNp can, however, hydrolyze single base pair mismatches at a recessed 3′-end (13), suggesting a possible role in proofreading during polymerization, an idea linked to the co-purification of WRNp with a replication complex (24) and the putative functional interaction between WRNp and human polymerase δ (36). The exonuclease activity of WRNp could also potentially be involved in processing the 3′-ends of duplex DNA molecules during strand break repair and/or recombination processes, a notion that is strengthened by our very recent finding (32) that the Ku heterodimer physically interacts with WRNp and stimulates its exonuclease activity. Undoubtedly, the physiological role of WRNp must involve its helicase activity as well. However, it remains to be demonstrated if and how the exonuclease and helicase activities of WRNp would act coordinately in any of these processes.
The blockage of WRNp exonuclease activity by different types of DNA modifications may be a useful experimental tool either to examine DNA structure or to pinpoint sites of DNA damage. The exonuclease activity of T4 DNA polymerase has previously been exploited for the purpose of detecting bulky types of DNA damage such as cisplatin adducts (37). As it appears to be very sensitive to perturbations in DNA, WRNp could be used for the detection and hybridization mapping of minor base modifications that do not block other 3′→5′ exonucleases. In particular, the exonuclease activity of WRNp is blocked by 8-oxoA and 8-oxoG, both having hydroxyl groups at the C8 position in the major groove of DNA. In contrast, the exonuclease activity of T4 DNA polymerase is not blocked even by the more bulky aminofluorene and aminoquinoline-1-oxide adducts covalently attached to the C8 position of guanine (38,39). The WRN-N truncated protein may be particularly well suited for detection of damage, as it has similar specificity yet is more highly overproduced and more soluble than full-length WRNp (Orren, unpublished observations) and also lacks helicase activity. However, more DNA lesions in multiple sequence contexts need to be tested for their effect on WRNp exonuclease before its utility as a tool for probing DNA structure can be truly ascertained.
The RecQ helicases are likely involved in multiple and perhaps overlapping pathways that function to maintain genomic stability. However, the homology between the human RecQ family members lies almost exclusively within the helicase domain and significant N-terminal and C-terminal portions of each RecQ member are dissimilar. Unlike the other human RecQ family members, the N-terminal 335 amino acids of WRNp shows homology to a family of 3′→5′ exonucleases (14). A loss of function of WRNp results in WS, a condition that is phenotypically different from both Bloom and Rothmund–Thomson syndromes, diseases that are the result of loss of function of the RecQ family members BLM and RecQ4, respectively. This indicates that WRNp has a unique function in DNA metabolism and that the loss of either helicase or exonuclease activity associated with WRNp could contribute to the premature aging phenotype of WS.
Acknowledgments
ACKNOWLEDGEMENTS
The authors would like to thank Jason Piotrowski for technical assistance, the Danish Center for Molecular Gerontology for helpful interactions and Dr Robert M. Brosh Jr for critical reading of the manuscript.
REFERENCES
- 1.Martin G.M. (1996) Exp. Gerontol., 31, 49–59. [DOI] [PubMed] [Google Scholar]
- 2.Goto M. (1997) Mech. Ageing Dev., 98, 239–254. [DOI] [PubMed] [Google Scholar]
- 3.Yu C.-E., Oshima,J., Fu,Y.-H., Wijsman,E.M., Hisama,F., Alisch,R., Matthews,S., Nakura,J., Miki,T., Ouais,S., Martin,G.M., Mulligan,J. and Schellenberg,G.D. (1996) Science, 272, 258–262. [DOI] [PubMed] [Google Scholar]
- 4.Lee S.-K., Johnson,R.E., Yu,S.-L., Prakash,L. and Prakash,S. (1999) Science, 286, 2339–2342. [DOI] [PubMed] [Google Scholar]
- 5.Chakraverty R.K. and Hickson,I.D. (1999) Bioessays, 21, 286–294. [DOI] [PubMed] [Google Scholar]
- 6.Shen J.-C. and Loeb,L.A. (2000) Trends Genet., 16, 213–220. [DOI] [PubMed] [Google Scholar]
- 7.Puranam K.L. and Blackshear,P.J. (1994) J. Biol. Chem., 269, 29838–29845. [PubMed] [Google Scholar]
- 8.Ellis N.A., Groden,J., Ye,T.-Z., Straughen,J., Lennon,D.J., Ciocci,S., Proytcheva,M. and German,J. (1995) Cell, 83, 655–666. [DOI] [PubMed] [Google Scholar]
- 9.Kitao S., Ohsugi,I., Ichikawa,K., Goto,M., Furuichi,Y. and Shimamoto,A. (1998) Genomics, 54, 443–452. [DOI] [PubMed] [Google Scholar]
- 10.Kitao S., Shimamoto,A., Goto,M., Miller,R.W., Smithson,W.A., Lindor,N.M. and Furuichi,Y. (1999) Nature Genet., 22, 82–84. [DOI] [PubMed] [Google Scholar]
- 11.Gray M.D., Shen,J.-C., Kamath-Loeb,A.S., Blank,A., Sopher,B.L., Martin,G.M., Oshima,J. and Loeb,L.A. (1997) Nature Genet., 17, 100–103. [DOI] [PubMed] [Google Scholar]
- 12.Suzuki N., Shimamoto,A., Imamura,O., Kuromitsu,J., Kitao,S., Goto,M. and Furuichi,Y. (1997) Nucleic Acids Res., 25, 2973–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brosh R.M. Jr, Orren,D.K., Nehlin,J.O., Ravn,P.H., Kenny,M.K., Machwe,A. and Bohr,V.A. (1999) J. Biol. Chem., 274, 18341–18350. [DOI] [PubMed] [Google Scholar]
- 14.Mian I.S. (1997) Nucleic Acids Res., 25, 3187–3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang S., Li,B., Gray,M.D., Oshima,J., Mian,I.S. and Campisi,J. (1998) Nature Genet., 20, 114–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamath-Loeb A.S., Shen,J.C., Loeb,L.A. and Fry,M. (1998) J. Biol. Chem., 273, 34145–34150. [DOI] [PubMed] [Google Scholar]
- 17.Martin G.M., Sprague,C.A. and Epstein,C.J. (1970) Lab. Invest., 23, 86–92. [PubMed] [Google Scholar]
- 18.Saito H. and Moses,R.E. (1991) Exp. Cell Res., 192, 373–379. [DOI] [PubMed] [Google Scholar]
- 19.Schulz V.P., Zakian,V.A., Ogburn,C.E., McKay,J., Jarzebowicz,A.A., Edland,S.D. and Martin,G.M. (1996) Hum. Genet., 97, 750–754. [DOI] [PubMed] [Google Scholar]
- 20.Tahara H., Tokutake,Y., Maeda,S., Kataoka,H., Watanabe,T., Satoh,M., Matsumoto,T., Sugawara,M., Ide,T., Goto,M., Furuichi,Y. and Sugimoto,M. (1997) Oncogene, 15, 1913–1920. [DOI] [PubMed] [Google Scholar]
- 21.Fukuchi K.-I., Martin,G.M. and Monnat,R.J.,Jr (1989) Proc. Natl Acad. Sci. USA, 86, 5893–5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stefanini M., Scappaticci,S., Lagomarsini,P., Borroni,G., Berardesca,E. and Nuzzo,F. (1989) Mutat. Res., 219, 179–185. [DOI] [PubMed] [Google Scholar]
- 23.Ogburn C.E., Oshima,J., Poot,M., Chen,R., Hunt,K.E., Gollahon,K.A., Rabinovitch,P.S. and Martin,G.M. (1997) Hum. Genet., 101, 121–125. [DOI] [PubMed] [Google Scholar]
- 24.Lebel M. and Leder,P. (1998) Proc. Natl Acad. Sci. USA, 95, 13097–13102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prince P.R., Ogburn,C.E., Moser,M.J., Emond,M.J., Martin,G.M. and Monnat,R.J.,Jr (1999) Hum. Genet., 105, 132–138. [DOI] [PubMed] [Google Scholar]
- 26.Menichini P., Fronza,G., Tornaletti,S., Galiegue-Zouitina,S., Bailleul,B., Loucheux-Lefebvre,M.-H., Abbondandolo,A. and Pedrini,A.M. (1989) Carcinogenesis, 10, 1589–1593. [DOI] [PubMed] [Google Scholar]
- 27.Bohr V.A., Dianov,G., Balajee,A., May,A. and Orren,D.K. (1998) J. Invest. Dermatol. Symp. Proc., 3, 11–13. [PubMed] [Google Scholar]
- 28.Orren D.K., Brosh,R.M.,Jr, Nehlin,J.O., Machwe,A., Gray,M.D. and Bohr,V.A. (1999) Nucleic Acids Res., 27, 3557–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shen J.-C., Gray,M.D., Oshima,J. and Loeb,L.A. (1998) Nucleic Acids Res., 26, 2879–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen J.-C., Gray,M.D., Oshima,J., Kamath-Loeb,A.S., Fry,M. and Loeb,L.A. (1998) J. Biol. Chem., 273, 34139–34144. [DOI] [PubMed] [Google Scholar]
- 31.Suzuki N., Shiratori,M., Goto,M. and Furuichi,Y. (1999) Nucleic Acids Res., 27, 2361–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cooper,M.C., Machwe,A., Orren,D.K., Brosh,R.M., Ramsden,D. and Bohr,V.A. (2000) Genes Dev., 14, 907–912. [PMC free article] [PubMed] [Google Scholar]
- 33.Matsunaga T., Mu,D., Park,C.-H., Reardon,J.T. and Sancar,A. (1995) J. Biol. Chem., 270, 20862–20869. [DOI] [PubMed] [Google Scholar]
- 34.Ollis D.L., Brick,P., Hamlin,R., Xuong,N.G. and Steitz,T.A. (1985) Nature, 313, 762–766. [DOI] [PubMed] [Google Scholar]
- 35.Brutlag D. and Kornberg,A. (1972) J. Biol. Chem., 247, 241–248. [PubMed] [Google Scholar]
- 36.Kamath-Loeb A.S., Johansson,E., Burgers,P.M.J. and Loeb,L.A. (2000) Proc. Natl Acad. Sci. USA, 97, 4603–4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rampino N.J. and Bohr,V.A. (1994) Proc. Natl Acad. Sci. USA, 91, 10977–10981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bichara M. and Fuchs,R.P.P. (1985) J. Mol. Biol., 183, 341–351. [DOI] [PubMed] [Google Scholar]
- 39.Panigrahi G.B. and Walker,I.G. (1990) Biochemistry, 29, 2122–2126. [DOI] [PubMed] [Google Scholar]