

Abstract
目的
:探索肝细胞癌模型鼠肠道微生态特征。
方法
:将2周龄C57BL/6雄鼠分为正常对照组和肝癌模型组。肝癌模型组在出生后2周给予单次腹腔注射二乙基亚硝胺(DEN),其中存活小鼠自4周龄起腹腔注射1,4-双[2-(3,5-二氯吡啶氧)]苯,每2周一次,共8次。分别于小鼠出生后第10、18和32周随机处死部分实验动物,取肝脏进行组织病理学检查。其中,在小鼠出生后第32周处死动物前无菌条件下收集两组粪便标本,并进行V3~V4高可变区16S rRNA基因组测序,进而对菌群多样性、物种丰度差异、菌群相关性、表型预测以及功能预测进行分析。
结果
:α多样性分析结果显示Good’s coverage均已达到最大值1.00,且正常对照组与肝癌模型组肠道菌群Observed features、Chao1指数、Shannon指数和Simpson指数差异均有统计学意义(均 P<0.05)。β多样性分析结果显示基于加权或未加权Unifrac距离的主坐标PCoA均 R>0,标本的组内差异小于组间差异;两组标本的分离趋势明显( P<0.05)。拟杆菌门、厚壁菌门、放线菌门及髌骨菌门等菌群为正常对照组与肝癌模型组门水平的优势菌群,但肝癌模型组较正常对照组拟杆菌门丰度显著减少( P<0.01),而髌骨菌门丰度显著增加( P<0.05)。正常对照组属水平的优势菌群主要为鼠杆菌科未分类属、副鼠杆菌属、鼠杆菌属、毛螺旋菌科NK4A136属、欧陆森氏菌属等,而肝癌模型组为艾克曼菌属、杜氏杆菌属、鼠杆菌科未分类属、毛螺旋菌科NK4A136属、红椿杆菌科UCG-002属等。两组属水平菌群相对丰度差异有统计学意义的有30个菌属(均 P<0.05)。两组小鼠肠道菌群LEfSe分析共发现14个多级别差异物种(均 P<0.05,LDA评分超过4.0),主要富集在拟杆菌门。其中正常对照组为拟杆菌门、拟杆菌纲、拟杆菌目、鼠杆菌科等10个差异菌群富集,肝癌模型组为杜氏杆菌属、消化链球菌属等4个差异菌群富集。正常对照组肠道优势菌属之间既有正相关关系又有负相关关系(|rho|>0.5, P<0.05),肝癌模型组肠道优势菌属的相关性网络复杂程度比正常对照组降低,所有优势菌群的相互作用均呈较强的正相关。与正常对照组比较,肝癌模型组肠道菌群革兰阳性菌及移动元件菌基因相对丰度均显著上调(均 P<0.05),而革兰阴性菌( P<0.05)及潜在致病性菌基因( P<0.05)相对丰度均显著下调。两组肠道菌群的基因代谢通路差异显著,其中正常对照组富集在能量代谢、细胞分裂及核苷酸代谢等18个代谢通路,而肝癌模型组富集在能量代谢、氨基酸代谢及碳水化合物代谢等12个代谢通路(均 P<0.005)。
结论
:DEN诱导原发性肝癌模型小鼠肠道菌群数减少,菌群构成、菌群相关性、表型和功能均发生改变,其中门水平的拟杆菌门以及属水平的鼠杆菌科未分类属、鼠杆菌属、消化链球菌属、杜氏杆菌属等菌群可能与DEN诱导肝细胞癌的发生有关。
Abstract
Objective
: To explore the characteristics of intestinal microecology in hepatocellular carcinoma (HCC) model mice.
Methods
: C57BL/6 male mice aged 2 weeks were divided into normal control group and HCC model group. Mice in HCC model group were exposed to a single intraperitoneal injection of diethylnitrosamine (DEN) 2 weeks after birth; the surviving mice were intraperitoneally injected with 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene (TCPOBOP), once every 2 weeks for 8 times starting from the 4 th week after birth. Mice in each group were randomly selected and sacrificed at 10 th, 18 th and 32 nd weeks after birth, respectively, the liver tissue samples were obtained for histopathological examination. At the 32 nd week, all mice in both groups were sacrificed and the feces samples were collected under sterile conditions right before the sacrifice. The feces samples were sequenced for the V3-V4 hypervariable regions of the 16S rRNA gene, and the species abundance, flora diversity and phenotype, as well as flora correlation and functional prediction were analyzed.
Results
: Alpha diversity analysis showed that all Good’s coverage reached the maximum value of 1.00, and the differences in the Observed features, Chao1 index, Shannon index and Simpson index of the intestinal flora of mice between normal control group and HCC model group were all statistically significant (all P<0.05). Beta diversity analysis showed that PCoA based on weighted or unweighted Unifrac distances all yielded R>0, confirming that the intra-group differences of the samples were less than the inter-group differences; the trend of separation between the two groups was significant ( P<0.05). Bacteroidetes, Firmicutes, Actinobacteria and Patescibacteria were the dominant taxa at the phylum level in both normal control group and HCC model group. However, compared with normal control group, the abundance of Bacteroidetes in HCC model group was significantly decreased ( P<0.01), while the abundance of Patescibacteria was significantly increased ( P<0.05). Moreover, the dominant taxa at the genus level in normal control group mainly included Muribaculaceae_unclassified, Paramuribaculum, Muribaculum, Lachnospiraceae_NK4A 136 group, Olsenella. The dominant taxa at the genus level in HCC model group mainly included Akkermansia, Dubosiella, Muribaculaceae_unclassified, Lachnospiraceae_NK4A 136 group, Coriobacteriaceae_UCG-002. There were 30 genera with statistically significant differences in relative abundance at the genus level between the two groups (all P<0.05). LEfSe analysis of the intestinal flora of mice in the two groups revealed a total of 14 multi-level differential taxa (all P<0.05, LDA score>4.0), which were mainly enriched in Bacteroidetes. The enrichment of 10 differential taxa including Bacteroidetes, Bacteroidia, Bacteroidales, Muribaculaceae, etc. were found in normal control group, and the enrichment of 4 differential taxa including Dubosiella, Peptostreptococus, etc. were found in HCC model group. There were both positive and negative correlations between the dominant intestinal genera in normal control group (|rho|>0.5, P<0.05), while the correlations of the dominant intestinal genera in HCC model group, being less complex than that in normal control group, were all positive. The relative abundance of gram positive and mobile element containing in the intestinal flora of mice in HCC model group was significantly up-regulated compared with normal control group (both P<0.05), while that of gram negative ( P<0.05) and pathogenic potential ( P<0.05) was significantly down-regulated. The metabolic pathways of the intestinal flora in the two groups were significantly different. For instance, 18 metabolic pathways were enriched in normal control group (all P<0.005), including those related to energy metabolism, cell division, nucleotide metabolism, etc., while 12 metabolic pathways were enriched in HCC model group (all P<0.005), including those related to energy metabolism, amino acid metabolism, carbohydrate metabolism, etc.
Conclusions
: The amount of intestinal flora in DEN-induced primary HCC model mice decreased, and the composition, correlation, phenotype and function of the intestinal flora in mice were significantly altered. Bacteroidetes at the phylum level, as well as several microbial taxa at the genus level such as Muribaculaceae_unclassified, Muribaculum, Peptostreptococus and Dubosiella could be closely associated with DEN-induced primary HCC in mice.
Keywords: Hepatocellular carcinoma, Intestinal flora, Species diversity, Microbial community structure, 16S rRNA sequencing, Diethylnitrosamine, C57BL/6 mouse
二乙基亚硝胺(diethylnitrosamine,DEN);1,4-双[2-(3,5-二氯吡啶氧)]苯{1,4-bis[2-(3,5-dichloropyridyloxy)]benzene, TCPOBOP};苏木精-伊红染色(hematoxylin and eosin staining,HE染色);聚合酶链反应(polymerase chain reaction,PCR);线性判别分析(linear discriminant analysis,LDA);LDA效应大小(LDA effect size,LEfSe);受试者操作特征曲线(receiver operating characteristic curve,ROC曲线);宏基因组谱的统计分析(statistical analysis of metagenomic profiles,STAMP);曲线下面积(area under the curve,AUC);转化生长因子(transforming growth factor,TGF);乙型肝炎病毒(hepatitis B virus,HBV);
原发性肝癌在癌症死亡原因中的排名从2018年的第四位上升至2020年的第三位,其中肝细胞癌(以下简称肝癌)占原发性肝癌的75%~ 85% [1] 。 导致肝癌患者高病死率的主要原因包括早期诊断困难、术后易复发、治疗方法有限且易耐药等。因此亟须寻找肝癌发生相关的潜在生物标志物及治疗靶点,以提高肝癌患者的存活率。
近年来,越来越多的临床前及临床研究提示肠道微生态与肝癌的发生发展及疗效和预后均密切相关 [ 2- 9] 。肝脏和肠道在解剖结构与功能上关系密切,两者通过“肠肝轴”或“肠道-微生物群-肝脏轴”密切联系且相互作用,在维护机体稳态方面发挥重要作用 [ 2, 10] 。目前关于“肝癌-肠道微生态”的临床报道多为横截面研究 [11] ,缺少与疾病发生发展相关的动态分析。临床研究存在标本收集难度高、个体差异大、干扰因素多、随访难度大以及疾病诊断前具体情况追溯难等局限性,动物模型稳定性好、可长期乃至全程观察疾病的发生发展,且标本收集种类多且操作相对简便、实验干扰因素少等 [12] 。两阶段DEN诱导肝癌法,即基因毒性化合物DEN结合非基因毒性化合物如苯巴比妥类TCPOBOP,能够较好地模拟人类肝癌的遗传、免疫和环境特征,包括DNA损伤、炎症反应和纤维化等肿瘤微环境,因此适合用于肝癌的临床前研究 [ 13- 14] 。本研究通过两阶段DEN诱导肝癌法建立小鼠肝癌模型,对模型鼠的粪便标本进行肠道菌群16S rRNA测序,结合生物信息学及统计学分析,了解肝癌模型中肠道菌群分布特点及差异,探索肠道菌群在肝癌发生、发展中的潜在作用,为后续筛选潜在的生物标志物及治疗靶点奠定研究基础。
材料与方法
主要试剂与仪器
DEN和TCPOBOP为美国Sigma公司产品; 4%多聚甲醛为北京白鲨易生物科技有限公司产品;无水乙醇为上海麦克林生化科技有限公司产品;二甲苯为上海沪试实验室器材股份有限公司产品;石蜡为国药集团化学试剂有限公司产品;苏木素、伊红和Qubit为美国Thermo Fisher Scientific公司产品;0.5%盐酸乙醇分化标准溶液为上海化科实验器材有限公司产品;氨水为上海阿拉丁生化科技股份有限公司产品;中性树胶为德国Leica公司产品;Phusion Hot start flex 2× Master Mix为美国NEB公司产品;E.Z.N.A粪便DNA试剂盒为美国Omega公司产品;AMPure XT珠为美国Beckman Coulter公司产品;Illumina文库定量试剂盒为美国KAPA Biosystems公司产品;琼脂糖为德国BioFroxx公司产品;冻存管为美国Corning公司产品;粘附载玻片为江苏世泰实验器材有限公司产品;离心管为美国Axygen公司产品;注射器为浙江龙德医药有限公司产品。
超低温冰箱和Nanodrop 2000分光光度计为美国Thermo Fisher Scientific公司产品;数字切片扫描仪VS200为日本Olympus公司产品;STP120脱水机、AP280-2包埋机和HM335E切片机为上海倍曼生物科技有限公司产品;Agilent 2100生物分析仪为美国Agilent Technologies公司产品;NovaSeq 6000测序仪为美国Illumina公司产品。
制备肝癌小鼠模型
本研究通过浙江中医药大学实验动物管理与伦理委员会审查(IACUC-20190318-06)。健康C57BL/6孕鼠由浙江中医药大学动物实验研究中心[实验动物使用许可号:SYXK(浙)2018-0012]从上海斯莱克公司[实验动物生产许可号:SCXK(浙)2017-0005]购入后,在无特定病原体环境下标准饲养并等待生产。新生鼠标准饲养2周后取 38只雄鼠用于实验(正常对照组17只及肝癌模型组21只)。肝癌模型组除与正常对照组同样标准饲养外,2周龄时单次腹腔注射DEN (20 mg/kg), 并自4周龄起腹腔注射苯巴比妥类TCPOBOP (3 mg/kg, 每2周一次,共8次)。分别于小鼠出生后第10、18、32周,两组各随机处死部分实验动物(正常对照组依次处死6、6、5只小鼠,肝癌模型组依次处死6、6、9只小鼠),分离和收集肝脏并进行大体形态及病理学检查。上述流程见 图1。
图1 .

DEN诱导小鼠肝细胞癌的建模流程示意图
TCPOBOP:1,4-双[2-(3,5-二氯吡啶氧)]苯.
称量小鼠体重和肝重并计算肝体比
如1.2所述分批处死实验动物前,称量每只小鼠的体重,分离肝脏并立即拍照和称量肝脏重量,计算肝体比(肝体比=肝重/体重),以评估DEN等毒性物质暴露引起的小鼠整体及肝脏损伤等反应。
HE染色观察小鼠肝组织病理学变化
按照动物伦理规范处死小鼠后,解剖并分离肝脏,用磷酸盐缓冲液清洗干净后切成小块,立即放入4%多聚甲醛进行固定。固定 24 h后将肝脏进行脱水,并按常规石蜡包埋及切片流程制备成 3~5 μm石蜡切片。对石蜡切片进行HE染色,以中性树胶封片后,用数字切片扫描仪VS200对切片进行多点聚焦扫描成像并进行分析。
16S rRNA测序检测小鼠粪便细菌DNA序列
粪便标本的收集及保存
在小鼠出生后第32周处死动物前,采用无菌操作收集每只小鼠的粪便标本(正常对照组5只小鼠,肝癌模型组 9只小鼠),置于无菌冻存管中,干冰转运并存储于–80 ℃冰箱,用于16S rRNA测序。
基因组DNA提取
使用E.Z.N.A粪便DNA试剂盒提取粪便标本中的细菌DNA,总DNA使用 50 μL洗脱缓冲液洗脱,并储存于 –80 ℃用于PCR检测。提取的DNA采用1%琼脂糖凝胶电泳质检,并用分光光度计定量。
PCR引物设计
用PCR的方法扩增16S rRNA基因V3~V4高变区,805R (5´-GACTACHVGGGTATCTAATCC-3´)与 341F(5´-CCTACGGGGNGGCWGCAG-3´)为其通用引物。每个样品和测序通用引物的5´端加上适合测序的标签序列。
PCR扩增和产物纯化定量
在总体积为 25 μL的反应混合物中进行PCR扩增,其中含 25 ng模板DNA、 12.5 μL PCR预混合液、前后引物各 2.5 μL, 最后用超纯水调整体积。扩增原核16S 片段的PCR条件包括: ① 98 ℃初始变性30 s; ② 98 ℃变性10 s;③ 54 ℃退火 30 s;④ 72 ℃延伸45 s;⑤ 72 ℃延长 10 min。 步骤②~④循环35次。PCR产物经2%琼脂糖凝胶电泳确证后,通过AMPure XT珠纯化,并通过Qubit定量。
16S rRNA测序
扩增子池用于测序,使用Illumina文库定量试剂盒、Agilent 2100生物分析仪评估扩增子文库是否合格(文库浓度大于 2 nmol/L为合格)。经稀释、混合、变性等操作处理后的合格上机测序文库在NovaSeq 6000测序仪上进行 2×250 bp的双端测序以获得原始序列。原始序列均已上传至Sequence Read Archive(SRA)数据库(项目ID:PRJNA827644)。
生物信息学方法分析测序数据
16S rRNA测序数据初步分析
将配对端序列根据标本独特的条形码分配给每个标本,并将建库引入的条形码和引物序列去除,然后用FLASH软件合并匹配端读取。使用QIIME2对原始数据进行高质量过滤,获得纯净数据 [15] 。采用Vsearch 2.3.4软件筛选嵌合序列。采用DADA2对序列进行解调得到特征表和特征序列 [16] ,并根据SILVA(release 132)分类器对特征丰度进行归一化。通过BLAST比对特征序列,并用SILVA数据库对其进行注释。
菌群多样性分析
α多样性分析用于评估单个标本所含有微生物物种的多样性/丰富度以及每种微生物所占比例/均匀度,包括由QIIME2软件计算得到的Chao1指数、Observed features、Good’s coverage、Shannon指数及Simpson指数共5个指标:Good’s coverage反映标本的测序深度;Observed features和Chao1指数反映标本中物种的丰富度;Shannon指数和Simpson指数反映物种的丰富度和均匀度。β多样性分析采用基于距离矩阵(欧式距离以外的其他距离)的PCoA来评估标本间菌群组成与分布差异 [17] ,该指标由QIIME2软件计算并用R3.5.2软件绘制图片。
菌群组成结构及差异菌群分析
基于标本物种相对丰度表,采用曼-惠特尼 U检验对两组标本之间物种差异进行比较, P<0.05为差异有统计学意义的菌群。
菌群多级物种线性判别分析
采用LEfSe方法进行正常对照组与肝癌模型组的多级物种差异分析,以筛选生物标志物 [18] ,并使用ROC曲线分析评估其作为微生物标志物的潜在诊断能力。
菌群相关性分析
采用SparCC法分析属水平丰度前三十的物种相对丰度,获得两两优势属之间的相关性,并将其中|rho|>0.5、 P<0.05的优势属绘制关联网络图。
菌群表型预测及分析
利用Bugbase数据库 [19] 对小鼠肠道菌群的表型进行预测及分析。菌群表型预测及分析包括革兰阳性、革兰阴性、生物膜形成、潜在致病性、移动元件含量、氧利用及氧化胁迫耐受共七方面特征,其中氧利用又包括需氧、厌氧及兼性厌氧。
菌群基因功能预测及分析
使用最新版本PICRUSt2进行16S rRNA基因数据功能预测 [20] ,参考同源基因簇数据库对菌群基因功能注释的结果并结合STAMP进行差异分析 [21] ,得到组间显著差异的菌群基因功能。
统计学方法
采用SPSS 25.0和R 3.5.2软件进行统计分析。非正态分布的计量资料以中位数(上下四分位数)[ M( Q 1, Q 3)]表示,采用曼-惠特尼 U检验进行组间比较;正态分布的计量资料以均数±标准差( )表示,组间比较使用独立样本 t检验方法。 P<0.05表示差异具有统计学意义。
结果
两组肝脏大体形态及组织病理学动态变化
正常对照组各时间点肝脏形态正常、表面光滑、边缘锐利,呈正常的红褐色。肝癌模型组 10、18周龄时肝脏大体形态与正常对照组相似,但32周龄时肝脏大体形态与正常对照组显著不同,肉眼可见肝脏表面出现多个散在、圆形或椭圆形、大小不等的癌结节。组织病理学检查结果可见,正常对照组在三个时间点下肝脏细胞形态均正常,排列整齐有序,无脂肪变性或异型增生;肝癌模型组10周龄时可见肝脏炎症细胞浸润、细胞肿胀、脂肪变性等,18周龄时肝脏内可见细胞内玻璃样变、脂肪变性以及细胞间的胶原纤维沉积等表现,32周龄时肝脏内可见癌变细胞体积增加,胞浆丰富呈嗜酸性,细胞核异型性明显,核仁增大并可见病理性核分裂像,且放大镜下视野范围还可观察到典型的肝纤维化病变,见 图2。病理学检查结果提示,实验终点时肝癌模型组全部造模成功,而正常对照组肝脏形态和组织学无改变。
图2 .

正常对照组与肝癌模型组10、18和32周龄时肝脏大体和组织病理学变化
大体形态学观察可见,正常对照组在三个时间点下肝脏均形态正常、表面光滑、边缘锐利,呈正常的红褐色;肝癌模型组10、18周龄时肝脏大体形态与正常对照组相似,但32周龄时肉眼可见肝脏表面出现多个散在、圆形或椭圆形、大小不等的癌结节. 病理学检查可见正常对照组在三个时间点肝脏细胞形态正常,排列整齐有序,无脂肪变性或异型增生;肝癌模型组10周龄时肝脏可见炎症细胞浸润、细胞肿胀、脂肪变性等,18周龄肝脏可见细胞内玻璃样变、脂肪变性以及细胞间的胶原纤维沉积等表现,32周龄时肝脏可见癌变细胞体积增加,胞浆丰富呈嗜酸性,细胞核异型性明显,核仁增大并可见病理性核分裂像以及典型的肝纤维化和肝硬化病变.
两组体重、肝重及肝体比动态变化
两组小鼠10、18、32周龄时肝癌模型组平均体重均低于正常对照组。10周龄时肝癌模型组平均肝重大于正常对照组,但两组肝重在后续18、32 周龄时无明显差异。10、18、32周龄时肝癌模型组的肝体比均明显高于正常对照组,见 表1。肝癌模型组大体特征变化证实成功构建小鼠肝癌模型。
表1 正常对照组与肝癌模型组10、18和32周龄时体重、肝重及肝体比比较
Table1 Comparison of body weight, liver weight and liver/body weight ratio between normal control group and hepatocellular carcinoma model group at the 10 th, 18 th and 32 nd week after birth
( )
|
组 别 |
体重(g) |
肝重(g) |
肝体比 |
||||||||
|
第10周 |
第18周 |
第32周 |
第10周 |
第18周 |
第32周 |
第10周 |
第18周 |
第32周 |
|||
|
正常对照组 |
28.83± 1.98 |
32.25± 4.98 |
33.88± 2.39 |
1.17± 0.09 |
1.73± 0.23 |
1.29± 0.18 |
0.041±0.002 |
0.054± 0.004 |
0.038± 0.004 |
||
|
肝癌模型组 |
21.60± 0.90 ** |
26.97± 1.62 * |
30.03± 0.99 ** |
1.41± 0.12 ** |
1.60± 0.11 |
1.44± 0.23 |
0.065± 0.004 ** |
0.059± 0.002 * |
0.048± 0.007 * |
||
|
t值 |
8.150 |
2.470 |
3.437 |
–3.992 |
1.298 |
–1.229 |
–13.818 |
–2.918 |
–2.841 |
||
|
P值 |
<0.01 |
<0.05 |
<0.01 |
<0.01 |
>0.05 |
>0.05 |
<0.01 |
<0.05 |
<0.05 |
与正常对照组比较, * P<0.05, ** P<0.01. 第10、18周, n=6;第32周,正常对照组 n=5,肝癌模型组 n=9.
两组肠道菌群多样性分析
两组标本内菌群多样性
α多样性分析结果显示,Good’s coverage均已达到最大值1.00( 附表1),即测序深度基本覆盖标本中所有的物种。正常对照组与肝癌模型组肠道菌群Observed features分别为845.00±89.82和542.56±163.84( P<0.01),Chao1指数分别为855.01±99.68和 550.14±167.85( P<0.01),Shannon指数分别为7.68(7.42,7.76)和5.74(5.66,6.68)( P<0.05),Simpson指数分别为0.99(0.98,0.99)和0.94(0.89,0.97)( P<0.05),差异均有统计学意义。提示肝癌模型组肠道菌群丰富度和均匀度均显著低于正常对照组。
两组标本间菌群多样性
β多样性分析结果 显示,未加权Unifrac距离的PCoA分析结果为 R=0.367, P=0.007;加权Unifrac距离的PCoA分析结果为 R=0.300, P=0.029,见 图3。基于加权/未加权Unifrac距离的主坐标PCoA分析均 R>0,证实标本的组内差异小于组间差异;两组标本的分离趋势明显( P<0.05),证实正常对照组与肝癌模型组肠道菌群结构和组成存在显著差异。
图3 .

正常对照组与肝癌模型组肠道微生物β多样性分析结果
A:非加权的UniFrac PCoA图;B:加权的UniFrac PCoA图. 图中每个点代表单个标本. 正常对照组 n=5,肝癌模型组 n=9.
肠道菌群构成及差异菌群分析
肠道菌群门水平构成及差异
两组门水平优势菌群的占比如 图4。其中,正常对照组拟杆菌门占64.434%(40.656%,69.149%),厚壁菌门占24.591%(17.764%,37.500%),放线菌门占5.312%(3.754%,8.050%),变形菌门占4.762%(4.462%,5.874%);肝癌模型组厚壁菌门占43.738%(30.250%,65.403%),疣微菌门占19.464%(0.113%,30.489%),拟杆菌门占17.590%(12.915%,31.442%),放线菌门占4.998%(4.403%,7.348%),变形菌门占2.544%(2.059%,11.832%)。与正常对照组比较,肝癌模型组拟杆菌门、未分类菌门丰度显著减少(均 P<0.05),髌骨菌门丰度显著增加( P<0.05),见 表2。以上结果提示拟杆菌门、未分类菌门和髌骨菌门可能与肝癌的发生及进展相关。
图4 .
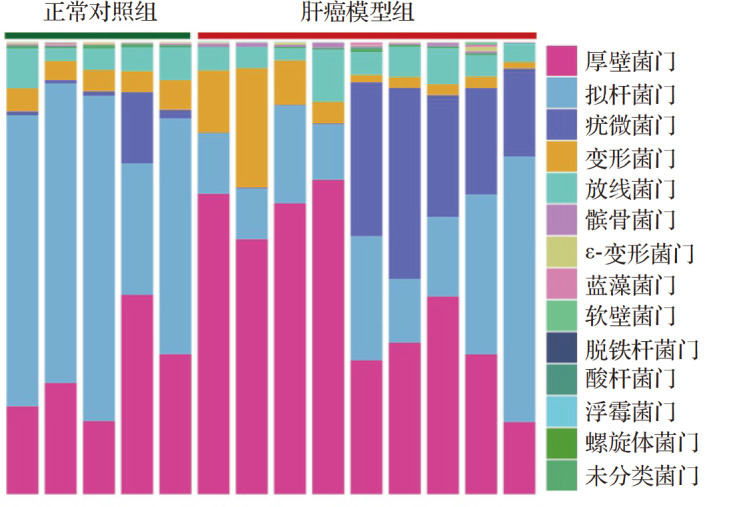
正常对照组与肝癌模型组肠道菌群门水平构成
柱子的长短表示菌群的相对丰度.
表2 正常对照组与肝癌模型组门水平肠道细菌优势菌群相对丰度比较
Table2 Comparison of the relative abundance of predominant intestinal flora at the phylum level between normal control group and hepatocellular carcinoma model group
[ M( Q 1, Q 3), %]
|
组 别 |
n |
厚壁菌门 |
拟杆菌门 |
疣微菌门 |
变形菌门 |
放线菌门 |
髌骨菌门 |
ε-变形菌门 |
|
正常对照组 |
5 |
24.591(17.764, 37.500) |
64.434(40.656, 69.149) |
0.993(0.764, 8.815) |
4.762(4.462, 5.874) |
5.312(3.754, 8.050) |
0.281(0.131, 0.308) |
0.207(0.112, 0.303) |
|
肝癌模型组 |
9 |
43.738(30.250, 65.403) |
17.590(12.915, 31.442) |
19.464(0.113, 30.489) |
2.544(2.059, 11.832) |
4.998(4.403, 7.348) |
0.416(0.321, 0.748) |
0.131(0.038, 0.287) |
|
Z值 |
— |
–1.533 |
–2.600 |
–0.333 |
–0.600 |
–0.200 |
–2.200 |
–0.867 |
|
P值 |
— |
>0.05 |
<0.01 |
>0.05 |
>0.05 |
>0.05 |
<0.05 |
>0.05 |
|
组 别 |
n |
蓝藻菌门 |
软壁菌门 |
脱铁杆菌门 |
酸杆菌门 |
浮霉菌门 |
螺旋体菌门 |
未分类菌门 |
|
正常对照组 |
5 |
0.068(0.046, 0.191) |
0.054(0.045, 0.113) |
0.000(0.000, 0.007) |
0.000(0.000, 0.000) |
0.000(0.000, 0.000) |
0.000(0.000, 0.000) |
0.650(0.502, 0.779) |
|
肝癌模型组 |
9 |
0.010(0.005, 0.304) |
0.048(0.025, 0.125) |
0.006(0.000, 0.023) |
0.000(0.000, 0.002) |
0.000(0.000, 0.000) |
0.000(0.000, 0.000) |
0.361(0.065, 0.469) |
|
Z值 |
— |
–1.133 |
–0.467 |
–1.068 |
–1.094 |
–0.745 |
–0.745 |
–2.333 |
|
P值 |
— |
>0.05 |
>0.05 |
>0.05 |
>0.05 |
>0.05 |
>0.05 |
<0.05 |
—:无相关数据.
肠道菌群属水平构成及差异
共检测到251个菌属,属水平相对丰度前三十位肠道菌群组成及分布见 图5。正常对照组肠道菌群中,鼠杆菌科未分类属占29.474%(23.071%,38.218%),副鼠杆菌属占10.046%(4.684%,11.420%),鼠杆菌属占5.836%(4.598%,7.920%),毛螺旋菌科NK4A136属占2.961%(2.574%,5.927%),欧陆森氏菌属占2.931%(0.473%,4.601%),另枝菌属占2.804%(1.861%,5.118%),杜氏杆菌属占1.989%(1.607%,11.636%),臭气杆菌属占1.345% (1.120%,2.414%),红椿杆菌科UCG-002属占0.714%(0.391%,2.397%);肝癌模型组肠道菌群中,艾克曼菌属占19.464%(0.113%,30.489%),杜氏杆菌属占13.592%(8.877%,23.362%),鼠杆菌科未分类属占9.864%(8.893%,22.861%),毛螺旋菌科NK4A136属占4.730% (1.803%,11.949%),红椿杆菌科UCG-002属占2.169%(1.748%,4.268%),瘤胃菌科UCG-014属占1.905%(1.072%,3.470%),鼠杆菌属占1.806%(1.262%,4.266%),另枝菌属占0.575%(0.162%,1.183%),臭气杆菌属占0.493%(0.394%,0.936%)。基于各菌属相对丰度水平进行曼-惠特尼 U检验分析,筛选出正常对照组与肝癌模型组之间共30个属水平菌群存在差异( P<0.05)。其中,肝癌模型组较正常对照组显著下调的菌属 24个,包括瘤胃梭菌属、颤杆菌属、瘤胃菌科UCG-010属、瘤胃球菌属、臭气杆菌属及瘤胃菌科未分类属等;肝癌模型组较正常对照组显著上调的菌属6个,包括异杆菌属、消化链球菌属、红椿杆菌科UCG-002属、杜氏杆菌属、低嗜盐细菌属以及候选单胞生糖菌属 ( 表3)。 综合分析差异菌群发现,肝癌模型组中大多数差异菌群的丰度均显著低于正常对照组,只有少数差异菌属丰度增加。上述结果提示,肝癌发生过程中,大部分肠道菌属的生长被抑制,少数菌属的生长被促进,导致肠道菌群多样性下降。
图5 .

正常对照组与肝癌模型组肠道菌群属水平构成
柱子的长短表示菌群的相对丰度.
表3 正常对照组与肝癌模型组相对丰度差异有统计学意义属水平肠道菌群
Table3 Genus-level intestinal flora with statistical significance in the relative abundance of intestinal flora between normal control group and hepatocellular carcinoma model group
[ M( Q 1, Q 3), %]
|
组 别 |
n |
异杆菌属 |
消化链球菌属 |
红椿杆菌科 UCG-002属 |
杜氏杆菌属 |
低嗜盐细菌属 |
候选单胞 生糖菌属 |
|
正常对照组 |
5 |
0.000(0.000, 0.006) |
0.000(0.000, 0.000) |
0.714(0.391, 2.397) |
1.989(1.607, 11.636) |
0.000(0.000, 0.000) |
0.281(0.131, 0.308) |
|
肝癌模型组 |
9 |
0.135(0.034, 0.217) |
0.011(0.005, 0.018) |
2.169(1.748, 4.268) |
13.592(8.877, 23.362) |
0.010(0.000, 0.032) |
0.406(0.321, 0.748) |
|
Z值 |
— |
–2.562 |
–2.492 |
–2.200 |
–2.067 |
–2.215 |
–2.200 |
|
P值 |
— |
<0.01 |
<0.05 |
<0.05 |
<0.05 |
<0.05 |
<0.05 |
|
组 别 |
n |
瘤胃梭菌属 |
颤杆菌属 |
瘤胃菌科 UCG-010属 |
瘤胃球菌属 |
臭气杆菌属 |
瘤胃菌科 未分类属 |
|
正常对照组 |
5 |
0.177(0.160, 0.289) |
0.270(0.169, 0.444) |
0.303(0.140, 0.405) |
0.032(0.008, 0.043) |
1.345(1.120, 2.414) |
0.305(0.165, 0.437) |
|
肝癌模型组 |
9 |
0.033(0.012, 0.063) |
0.034(0.004, 0.107) |
0.052(0.012, 0.058) |
0.000(0.000, 0.000) |
0.493(0.394, 0.936) |
0.072(0.051, 0.238) |
|
Z值 |
— |
–2.333 |
–2.202 |
–3.000 |
–2.719 |
–2.200 |
–2.067 |
|
P值 |
— |
<0.05 |
<0.05 |
<0.01 |
<0.05 |
<0.05 |
<0.05 |
|
组 别 |
n |
瘤胃菌科 UCG-005属 |
普雷沃菌科 NK3B31属 |
埃格特菌属 |
邓氏杆菌属 |
丹毒丝菌科 未分类属 |
瘤胃菌科 UCG-007属 |
|
正常对照组 |
5 |
0.017(0.013, 0.020) |
0.148(0.050, 0.274) |
0.148(0.076, 0.285) |
0.163(0.058, 0.191) |
0.150(0.061, 0.230) |
0.028(0.011, 0.033) |
|
肝癌模型组 |
9 |
0.000(0.000, 0.007) |
0.000(0.000, 0.000) |
0.053(0.032, 0.086) |
0.000(0.000, 0.000) |
0.025(0.000, 0.046) |
0.000(0.000, 0.000) |
|
Z值 |
— |
–2.386 |
–3.006 |
–2.200 |
–3.496 |
–2.494 |
–3.496 |
|
P值 |
— |
<0.05 |
<0.05 |
<0.05 |
<0.01 |
<0.05 |
<0.01 |
|
组 别 |
n |
DTU014_ unclassified属 |
拟杆菌目 未分类属 |
贪噬菌属 |
另枝菌属 |
Candidatus_ Stoquefichus属 |
气单胞菌属 |
|
正常对照组 |
5 |
0.042(0.014, 0.058) |
0.247(0.079, 0.303) |
0.007(0.003, 0.012) |
2.804(1.861, 5.118) |
0.048(0.017, 0.094) |
0.028(0.021, 0.043) |
|
肝癌模型组 |
9 |
0.000(0.000, 0.000) |
0.011(0.005, 0.057) |
0.000(0.000, 0.000) |
0.574(0.162, 1.183) |
0.000(0.000, 0.000) |
0.000(0.000, 0.019) |
|
Z值 |
— |
–2.719 |
–2.603 |
–3.006 |
–2.867 |
–2.719 |
–2.567 |
|
P值 |
— |
<0.05 |
<0.01 |
<0.05 |
<0.01 |
<0.05 |
<0.05 |
|
组 别 |
n |
Millionella属 |
乳头杆菌属 |
鼠杆菌属 |
普雷沃菌科 UCG-001属 |
鼠杆菌科 未分类属 |
未分类属 |
|
正常对照组 |
5 |
0.042(0.032, 0.091) |
0.020(0.010, 0.069) |
5.836(4.598, 7.920) |
0.368(0.178, 0.502) |
29.474(23.071, 38.218) |
0.650(0.502, 0.779) |
|
肝癌模型组 |
9 |
0.000(0.000, 0.000) |
0.000(0.000, 0.012) |
1.806(1.262, 4.266) |
0.024(0.000, 0.123) |
9.864(8.893, 22.861) |
0.361(0.065, 0.470) |
|
Z值 |
— |
–3.006 |
–2.349 |
–2.067 |
–2.612 |
–2.333 |
–2.333 |
|
P值 |
— |
<0.05 |
<0.05 |
<0.05 |
<0.01 |
<0.05 |
<0.05 |
—:无相关数据.
肠道菌群多级物种差异判别
LEfSe分析结果显示,两组肠道菌群中发现14个差异微生物类群(均 P<0.05,LDA评分超过4.0),主要富集在拟杆菌门( 图6和 附表2)。其中,正常对照组中发现10个差异微生物类群,分别为拟杆菌门、拟杆菌纲、拟杆菌目、鼠杆菌科、鼠杆菌科未分类属、鼠杆菌科未分类种、鼠杆菌属、理研菌科、另枝菌属及鼠杆菌属 sp.种;肝癌模型组中发现4个差异微生物类群,分别为未培养的消化链球菌属 sp.种、消化链球菌属、杜氏杆菌属及杜氏杆菌属未分类种。ROC曲线分析结果显示,拟杆菌门、拟杆菌纲、拟杆菌目、理研菌科、另枝菌属诊断肝癌的AUC均大于0.9,其余物种的AUC值也均大于0.8( 表4)。结果提示,上述拟杆菌门等10个差异菌群可能在维持小鼠健康肠道微生态中发挥了重要作用,而消化链球菌属、杜氏杆菌属等四个差异菌群可能在模型鼠肝癌发生中发挥了重要作用。
图6 .

正常对照组与肝癌模型组肠道菌群LEfSe分析结果
A:肠道菌群进化分支图,显示与正常对照组或肝癌模型组相关微生物群的系统发育分布,其中由内至外辐射的圆圈代表了由界(单个圆圈)至属(或种)的分类级别,小圆圈直径大小与相对丰度大小呈正比;黄色点为正常对照组与肝癌模型组之间无显著差异的微生物类群,绿色点为在正常对照组中起重要作用的微生物类群,红色点为在肝癌模型组中起重要作用的微生物类群. B:线性判别分析分数直方图. 正常对照组 n=5,肝癌模型组 n=9.
表4 差异肠道菌群诊断肝癌的ROC曲线分析结果
Table4 ROC curve analysis of differential intestinal flora for the diagnosis of primary hepatocellular carcinoma
|
菌群类别 |
诊断标准(%) |
AUC |
敏感度 |
特异度 |
P 值 |
|
拟杆菌门 |
≤28.297 |
0.933 |
1.000 |
0.778 |
0.009 |
|
拟杆菌纲 |
≤28.292 |
0.933 |
1.000 |
0.778 |
0.009 |
|
拟杆菌目 |
≤28.292 |
0.933 |
1.000 |
0.778 |
0.009 |
|
理研菌科 |
≤1.657 |
0.911 |
1.000 |
0.778 |
0.014 |
|
鼠杆菌科 |
≤37.235 |
0.889 |
0.800 |
0.889 |
0.020 |
|
另枝菌属 |
≤1.524 |
0.978 |
1.000 |
0.889 |
0.004 |
|
消化链球菌属 |
≥0.005 |
0.889 |
0.778 |
1.000 |
0.020 |
|
鼠杆菌科未分类属 |
≤27.696 |
0.889 |
0.800 |
0.889 |
0.020 |
|
杜氏杆菌属 |
≥4.704 |
0.844 |
1.000 |
0.600 |
0.039 |
|
鼠杆菌属 |
≤3.825 |
0.844 |
1.000 |
0.778 |
0.039 |
|
未培养的消化链球菌属sp.种 |
≥0.005 |
0.889 |
0.778 |
1.000 |
0.020 |
|
鼠杆菌科未分类种 |
≤27.696 |
0.889 |
0.800 |
0.889 |
0.020 |
|
鼠杆菌属sp.种 |
≤3.074 |
0.889 |
1.000 |
0.889 |
0.020 |
|
杜氏杆菌属未分类种 |
≥4.704 |
0.844 |
1.000 |
0.600 |
0.039 |
ROC:受试者操作特征;AUC:曲线下面积.
肠道菌群优势属关联性分析
采用SparCC法分别分析正常对照组与肝癌模型组属水平丰度前三十位菌群的相对丰度,获得两两优势属之间的相关性,并基于其中|rho|>0.5, P<0.05的优势属构建关联网络( 图7)。结果显示,与正常对照组比较,肝癌模型组肠道优势菌属相关性网络的复杂程度降低,所有优势菌群的相互作用均呈较强的正相关,如红椿杆菌科UCG-002属与杜氏杆菌属、艾克曼菌属与鼠杆菌科未分类属、艾克曼菌属与鼠杆菌属、艾克曼菌属与欧陆森氏菌属、鼠杆菌科未分类属与鼠杆菌属、梭菌目未分类属与毛螺旋菌科NK4A136属、伯克霍尔德菌属与苏黎世杆菌属。然而,正常对照组肠道优势菌属之间既有正相关关系,如鼠杆菌属与副鼠杆菌属、欧陆森氏菌属与副鼠杆菌属、杜氏杆菌属与木质真菌属、红椿杆菌科UCG-002属与杜氏杆菌属、鼠杆菌属与欧陆森氏菌属、艾克曼菌属与毛螺旋菌科NK4A136属、艾克曼菌属与木质真菌属、艾克曼菌属与红椿杆菌科UCG-002属、拟杆菌属与瘤胃菌科UCG-014属、红椿杆菌科UCG-002属与木质真菌属,又有负相关关系,如艾克曼菌属与副鼠杆菌属、红椿杆菌科UCG-002属与副鼠杆菌属、杜氏杆菌属与瘤胃菌科UCG-014属。上述结果提示肝癌模型组大部分肠道优势菌属之间原有的共生共存与竞争抑制同时存在的关系变成了以共生共存为主的新关系。
图7 .

正常对照组和肝癌模型组丰度前三十位细菌属的相关性网络
绿色节点为正常对照组,红色节点为肝癌模型组;节点大小代表中心度,中心度越大,相关对象个数越多;节点 之间的连接表明两个属之间存在相关性,相关性系数 |rho|>0.5, P< 0.05;线条粗细代表相关性强弱,线条越粗表明相关性越强,而线条越细表明相关性越弱;实线表明正相关,虚线表明负相关.
肠道菌群表型预测及分析
与正常对照组比较,肝癌模型组肠道菌群革兰阳性菌及移动元件菌基因相对丰度均显著上调(均 P<0.05),而革兰阴性菌及潜在致病性菌基因相对丰度均显著下调(均 P<0.05)。此外,两组肠道菌群在生物膜形成、氧利用及氧化胁迫耐受三方面表型基因的相对丰度差异无统计学意义 ( 图8 和 附表3)。上述结果说明,肝癌模型组肠道菌群表型发生显著改变,肠道微生态失调。
图8 .

正常对照组和肝癌模型组各类表型肠道菌群的表达丰度比较
组间比较, * P<0.05.
肠道菌群基因功能预测及分析
两组肠道菌群共有462条基因代谢通路差异有统计学意义(均 P<0.05),其中 P<0.005的差异基因代谢通路有30条( 图9和 附表4)。正常对照组表现为能量代谢、细胞分裂、核苷酸代谢、翻译及转录等18条相关基因代谢通路的富集,而肝癌模型组则表现为能量代谢、氨基酸代谢、碳水化合物代谢、翻译及翻译后修饰等12条相关基因代谢通路的富集,表明肝癌模型组肠道菌群大量基因代谢通路的相对丰度发生了显著变化。
图9 .

菌群基因功能预测与代谢通路差异分析
根据同源基因簇数据库功能注释结果筛选出差异有统计学意义的功能, 横柱状图代表富集在该代谢通路的丰度分别在正常对照组与肝癌模型组标本中所有代谢通路的百分比.
讨论
大量研究证实,肠道菌群与帕金森综合征、高血压、糖尿病及癌症等多种人类疾病关系密切,肠道菌群甚至因此被称为“被遗忘的器官” [ 22- 23] 。越来越多的证据表明肺癌、乳腺癌、肝癌、胰腺癌等多种癌变中均存在肠道菌群的改变 [ 8- 9] 。由于癌症具有器官特异性、高度的复杂性及异质性,特定癌症与肠道微生态相互作用的具体模式、特点及机制亟须进一步深入研究。然而,由于患者的个体差异以及入组人群所在地区、饮食习惯、样本量大小等因素的干扰,关于肝癌患者肠道微生态特征的临床研究报道间存在较大差异。如Ni等 [24] 认为肝癌患者的粪便微生物群中变形菌门显著增加;汤莉等 [25] 报道肝癌患者肠道中双歧杆菌、乳杆菌和拟杆菌丰度显著降低,而肠杆菌和酵母菌丰度显著增高;郑微等 [26] 则认为肝癌患者肠道菌群中副血链球菌、唾液链球菌、变形链球菌、嗜热链球菌、副流感嗜血杆菌、韦荣球菌及殊异韦荣菌的丰度显著增加,而艾克曼菌、嗜黏蛋白艾克曼菌、普雷沃菌及另枝菌的丰度显著降低;孙亮等 [27] 发现肝癌患者肠道菌群中拟杆菌属/拟杆菌门、大肠杆菌螺旋杆菌属/变形菌门所占比例显著升高,而双歧杆菌属/放线菌门、梭菌属/厚壁菌门所占比例显著降低。
构建和利用具有多重优势的肝癌动物模型,有望推动肝癌-肠道微生态相关研究进程 [8] 。目前基于肝癌动物模型探索肠道微生态变化仅见2018年甄宏德等 [28] 报道,研究者基于肝癌H-ras12V转基因小鼠模型发现巴恩斯氏菌属、拟普雷沃菌属、拟杆菌属等菌群可能与肝癌的发生存在密切联系。但由于其转基因动物模型缺乏肝癌常见的肝纤维化或肝硬化的病变过程,导致相关研究结果的临床意义较为局限。尽管HBV感染是目前我国肝癌患者的主要病因,但由于HBV可感染的宿主谱极其有限,HBV感染或其过表达无法在非灵长类动物模型中模拟人类肝炎及后续肝纤维化、肝癌的病理学变化 [29] 。长期以来,两阶段DEN诱导的小鼠肝癌模型被公认为是最经典和最接近临床肝癌发生发展过程的动物模型。鉴于此,本研究成功构建了两阶段DEN诱导肝癌小鼠动物模型,其病理过程与人类肝癌相似 [13] ,且该肝癌造模过程中小鼠体重、肝重的动态变化与肝癌的病理学变化过程相吻合 [ 30- 31] 。在此动物模型基础上,本研究开展了肝癌模型肠道菌群16S rRNA测序及生物信息学分析。首先,采用α多样性分析与β多样性分析来评估肠道微生态整体多样性及群落结构分布。结果显示,所有标本Good’s coverage均已达到最大值1.00,说明测序深度基本覆盖标本中所有的菌群;而肝癌模型组肠道菌群Observed features、Chao1指数、Shannon指数、Simpson指数均显著低于正常对照组,说明肝癌模型组肠道菌群的丰度和均匀度均逊于正常对照组。进一步采用基于距离矩阵的PCoA分析 [17] ,发现正常对照组与肝癌模型组标本的菌群构成及分布存在显著差异。上述α多样性和β多样性分析结果证实肝癌小鼠肠道菌群多样性减小、群落分布均匀度降低,肠道微生态失调。
本研究数据显示,正常对照组肠道属水平优势菌物种包括鼠杆菌科未分类属、毛螺菌属NK4A136、另枝菌属、拟普雷沃菌属、拟杆菌属、鼠杆菌属和乳杆菌属,与黄树武等 [32] 报道的结果基本一致。进一步分析数据显示,肠道门水平菌群变化主要为拟杆菌门占比明显下降而髌骨菌门占比显著增加;属水平的变化则主要为杜氏杆菌属、红椿杆菌科UCG-002属及候选单胞生糖菌属占比显著增加,而鼠杆菌科未分类属、鼠杆菌属、另枝菌属、臭气杆菌属及拟普雷沃菌属占比显著减少。为筛选出同时具有统计学意义和生物相关性的生物标志物,采用综合了肠道菌群统计学差异及其对分组结果的影响力大小的LEfSe分析,结果发现拟杆菌门、拟杆菌纲、拟杆菌目、鼠杆菌科、鼠杆菌科未分类属、鼠杆菌科未分类种、鼠杆菌属、理研菌科、另枝菌属及鼠杆菌属 sp.种共10个差异菌群可能在维持小鼠健康肠道微生态中发挥了重要作用,而未培养的消化链球菌属 sp.种、消化链球菌属、杜氏杆菌属及杜氏杆菌属未分类种属共4个差异菌群可能在肝癌发生中发挥重要作用。其中,肝癌模型组拟杆菌门、拟杆菌目未分类属、另枝菌属丰度均显著减少,消化链球菌属丰度显著增加,厚壁菌门变化不明显,这与肝癌患者肠道微生态的部分变化特征一致 [ 24- 26] ,证实DEN诱导肝癌模型可以较好模拟肝癌患者肠道菌群的特点。
将两组肠道属水平优势菌群的相关性可视化后,发现正常对照组物种相关性网络中关联性强/中心度大的节点占据了大部分位置,且同时存在共生共存与竞争抑制的生态关系。然而,与正常对照组比较,仅有红椿杆菌科UCG-002属与杜氏杆菌属的正相关关系在肝癌模型组中继续存在,其余诸多优势菌属的相关性均发生改变,且肝癌模型组优势物种之间主要是共生共存的生态关系,缺乏正常对照组相互制约、相互平衡的自然状态。这些结果表明,肝癌模型组肠道微生态系统失调,其环境更倾向于支持某些特定或同类菌群的发展,导致原有的优势菌属之间失去了共生共存与竞争抑制的平衡关系,代之以共生共存关系为主的单一新生态系统。这与健康成年小鼠体内肠道菌群具有丰富的多样性并以优势菌属间互利共存与竞争抑制相结合的菌群稳态为特征 [ 33- 34] ,以及临床研究中观察到肝癌患者肠道微生物之间表现出较健康人群更单一的物种关联网络、细菌间相互作用多为正相关或极少出现负相关的结果一致 [35] 。
为了解不同的菌群表型与肝癌的关系,本研究进一步对菌群表型即细菌特定的物理外观或成分特征进行预测分析。与正常对照组比较,肝癌模型组肠道菌群表型中革兰阳性菌基因相对丰度显著上调、革兰阴性菌基因相对丰度显著下调,这与肝癌模型组肠道菌群在门水平的主要变化特征为厚壁菌门(革兰阳性菌)占比增加,而拟杆菌门(革兰阴性菌)占比下降的结果相一致。菌群的另一重要表型特征为可移动基因元件,包括自转移广宿主质粒、转座子、整合子、插入序列共同区等 [36] 。研究证实,可移动基因元件能通过随机插入癌症相关基因或诱导易位促进癌症的发生与进展 [37] 。此外,可移动基因元件的水平转移不仅可以促使微生物通过整合外源性基因而实现基因组转化或进化,还可在细菌适应特定环境压力过程中发挥重要作用 [36] 。较多研究表明,可移动基因元件是细菌获得、发展和传播耐药性的关键机制 [38] 。长期以来,科学界认为微生物在癌症中的作用主要归因于慢性炎症或毒素诱导的突变,而细菌的DNA或可移动基因元件不能整合到人类基因组中。而近年研究发现,细菌插入突变可能发生在癌细胞中,因此可移动基因元件介导的生物过程为探索新的生物修复途径和新的疾病治疗策略提供了独特思路 [37] 。本研究数据显示肝癌模型组肠道菌群可移动基因元件较正常对照组显著上调,符合该组小鼠为适应化学毒物暴露及癌变等环境刺激/压力而发生转化或进化的预期。肠道菌群基因组中的可移动基因元件与肝癌发生发展过程中的菌群进化以及在菌群-宿主互作中发挥作用值得进一步深入研究。
综上,本研究通过16S rRNA测序及生物信息学分析揭示了DEN诱导肝癌小鼠肠道菌群的特征及变化,为阐明肝癌与肠道微生物的互作及具体机制提供新的实验依据。值得关注的是,肠道微生态特征不仅与肝癌的发生有关,也与肝癌进展、患者治疗反应及预后存在密切联系 [ 2, 5, 8, 24, 39- 43] 。因此,本研究的发现为筛选肝癌新型生物标志物,以及研制微生物制剂用于肝癌治疗等提供了基础。
Supporting information
Acknowledgments
浙江中医药大学动物实验研究中心、浙江省人民医院肿瘤生物学与创新治疗实验室在动物实验实施过程中给予支持
COMPETING INTERESTS
所有作者均声明不存在利益冲突
Funding Statement
国家自然科学基金(21722405,22075243); 浙江大学“百人计划”研究员启动基金
References
- 1.SUNG H, FERLAY J, SIEGEL R L, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries[J] CA Cancer J Clin. . 2021;71(3):209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.SCHWABE R F, GRETEN T F. Gut microbiome in HCC——mechanisms, diagnosis and therapy[J] J Hepatol. . 2020;72(2):230–238. doi: 10.1016/j.jhep.2019.08.016. [DOI] [PubMed] [Google Scholar]
- 3.YOSHIMOTO S, LOO T M, ATARASHI K, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome[J] Nature. . 2013;499(7456):97–101. doi: 10.1038/nature12347. [DOI] [PubMed] [Google Scholar]
- 4.QIN N, YANG F, LI A, et al. Alterations of the human gut microbiome in liver cirrhosis[J] Nature. . 2014;513(7516):59–64. doi: 10.1038/nature13568. [DOI] [PubMed] [Google Scholar]
- 5.MA C, HAN M, HEINRICH B, et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells[J/OL] Science. . 2018;360(6391):eaan5931. doi: 10.1126/science.aan5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.BEHARY J, AMORIM N, JIANG X T, et al. Gut microbiota impact on the peripheral immune response in non-alcoholic fatty liver disease related hepatocellular carcinoma[J] Nat Commun. . 2021;12(1):187. doi: 10.1038/s41467-020-20422-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.SEPICH-POORE G D, ZITVOGEL L, STRAUSSMAN R, et al. The microbiome and human cancer[J/OL] Science. . 2021;371(6536):eabc4552. doi: 10.1126/science.abc4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.YU L X, SCHWABE R F. The gut microbiome and liver cancer: mechanisms and clinical translation[J] Nat Rev Gastroenterol Hepatol. . 2017;14(9):527–539. doi: 10.1038/nrgastro.2017.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.ZHOU A, TANG L, ZENG S, et al. Gut microbiota: a new piece in understanding hepatocarcinogenesis[J] Cancer Lett. . 2020;474:15–22. doi: 10.1016/j.canlet.2020.01.002. [DOI] [PubMed] [Google Scholar]
- 10.NAKANISHI T, FUKUI H, WANG X, et al. Effect of a high-fat diet on the small-intestinal environment and mucosal integrity in the gut-liver axis[J] Cells. . 2021;10(11):3168. doi: 10.3390/cells10113168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.黄佳文, 朱 娟, 黄慧瑶, 等. 中国肝癌及相关疾病状态健康效用值及失能权重研究的系统评价[J]. 中国循证医学杂志, 2018, 18(5): 410-417 ; HUANG Jiawen, ZHU Juan, HUANG Huiyao, et al. Health utility scores and disability weights of liver cancer and related diseases in China: a systematic review[J]. Chinese Journal of Evidence-based Medicine, 2018, 18(5): 410-417. (in Chinese)
- 12.刘 嘉, 申恒巧, 顾红梅, 等. 肝癌动物模型的构建及其应用现状[J]. 肿瘤药学, 2013, 3(1): 13-16 ; LIU Jia, SHEN Hengqiao, GU Hongmei, et al. Establishment of the animal models of hepatic carcinoma and their applications[J]. Anti-tumor Pharmacy, 2013, 3(1): 13-16. (in Chinese)
- 13.GU C Y, LEE T K W. Preclinical mouse models of hepatocellular carcinoma: an overview and update[J] Exp Cell Res. . 2022;412(2):113042. doi: 10.1016/j.yexcr.2022.113042. [DOI] [PubMed] [Google Scholar]
- 14.LI Z, TUTEJA G, SCHUG J, et al. Foxa1 and Foxa2 are essential for sexual dimorphism in liver cancer[J] Cell. . 2012;148(1-2):72–83. doi: 10.1016/j.cell.2011.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.BOLYEN E, RIDEOUT J R, DILLON M R, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2[J] Nat Biotechnol. . 2019;37(8):852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.CALLAHAN B J, MCMURDIE P J, ROSEN M J, et al. DADA2: high-resolution sample inference from Illumina amplicon data[J] Nat Methods. . 2016;13(7):581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.GOWER J C. Some distance properties of latent root and vector methods used in multivariate analysis[J] Biometrika. . 1966;53(3-4):325–338. doi: 10.1093/biomet/53.3-4.325. [DOI] [Google Scholar]
- 18.SEGATA N, IZARD J, WALDRON L, et al. Metagenomic biomarker discovery and explanation[J] Genome Biol. . 2011;12(6):R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.WARD T, LARSON J, MEULEMANS J, et al. BugBase predicts organism-level microbiome phenotypes[J]. bioRxiv, 2017. DOI: 10.1101/133462
- 20.DOUGLAS G M, MAFFEI V J, ZANEVELD J, et al. PICRUSt2: an improved and extensible approach for metagenome inference[J]. bioRxiv, 2019. DOI: 10.1101/672295
- 21.PARKS D H, TYSON G W, HUGENHOLTZ P, et al. STAMP: statistical analysis of taxonomic and functional profiles[J] Bioinformatics. . 2014;30(21):3123–3124. doi: 10.1093/bioinformatics/btu494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DIETERT R, DIETERT J. The microbiome and sustainable healthcare[J] Healthcare. . 2015;3(1):100–129. doi: 10.3390/healthcare3010100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.CHEN Y, ZHOU J, WANG L. Role and mechanism of gut microbiota in human disease[J] Front Cell Infect Microbiol. . 2021;11:625913. doi: 10.3389/fcimb.2021.625913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.NI J, HUANG R, ZHOU H, et al. Analysis of the relationship between the degree of dysbiosis in gut microbiota and prognosis at different stages of primary hepatocellular carcinoma[J] Front Microbiol. . 2019;10:1458. doi: 10.3389/fmicb.2019.01458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.汤 莉, 吴伶莉. 原发性肝癌患者肠道菌群变化与内毒素和炎性因子水平的关系[J]. 中国微生态学杂志, 2021, 33(10): 1162-1165 ; TANG Li, WU Lingli. Correlation between intestinal flora changes and levels of endotoxin and inflammatory factors in patients with primary liver cance[J]. Chinese Journal of Microecology, 2021, 33(10): 1162-1165. (in Chinese)
- 26.郑 微, 赵 鹏, 张永宏, 等. 宏基因组测序技术分析原发性肝癌患者肠道菌群特征[J/CD]. 中华实验和临床感染病杂志(电子版), 2021, 15(3): 149-157 ; ZHENG Wei, ZHAO Peng, ZHANG Yonghong, et al. Metagenomic analysis on characteristics of intestinal flora of patients with primary liver cancer[J/CD]. Chinese Journal of Liver Diseases (Electronic), 2021, 15(3): 149-157. (in Chinese)
- 27.孙 亮, 周 旋, 刘桂治, 等. 肝癌患者肠道微生态结构变化的特点分析[J]. 肝脏, 2020, 25(3): 267-269 ; SUN Liang, ZHOU Xuan, LIU Guizhi, et al. Characteristics of intestinal microecological structure changes in patients with hepatocellular carcinoma[J]. Chinese Hepatology, 2020, 25(3): 267-269. (in Chinese)
- 28.甄宏德, 王爱国, 钱 祥, 等. 原发性肝癌H-ras12V小鼠肠道微生态的初步研究[J]. 中国微生态学杂志, 2018, 30(2): 132-136 ; ZHEN Hongde, WANG Aiguo, QIAN Xiang, et al. A preliminary study on intestinal microflora of mice with primary liver cancer[J]. Chinese Journal of Microecology, 2018, 30(2): 132-136. (in Chinese)
- 29.DU Y, BROERING R, LI X, et al. In vivo mouse models for hepatitis B virus infection and their application[J] . Front Immunol. . 2021;12:766534. doi: 10.3389/fimmu.2021.766534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.FERENCI P, FRIED M, LABRECQUE D, et al. Hepatocellular carcinoma (HCC)[J] J Clin Gastroenterol. . 2010;44(4):239–245. doi: 10.1097/MCG.0b013e3181d46ef2. [DOI] [PubMed] [Google Scholar]
- 31.张 鹏. 肝炎肝硬化和酒精性肝硬化的B超影像差异研究[J]. 系统医学, 2017, 2(17): 86-90 ; ZHANG Peng. Research on difference in ultrasound B images of hepatitis liver cirrhosis and alcoholic liver cirrhosis[J]. Systems Medicine, 2017, 2(17): 86-90. (in Chinese)
- 32.黄树武, 闵凡贵, 王 静, 等. 常用小鼠、大鼠肠道菌群比较研究[J]. 中国实验动物学报, 2021, 29(6): 777-784 ; HUANG Shuwu, MIN Fangui, WANG Jing, et al. Comparative study of intestinal flora in common mice and rats[J]. Acta Laboratorium Animalis Scientia Sinica, 2021, 29(6): 777-784. (in Chinese)
- 33.BENSON A K, KELLY S A, LEGGE R, et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors[J] Proc Natl Acad Sci U S A. . 2010;107(44):18933–18938. doi: 10.1073/pnas.1007028107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.谭 振, 翟丽维, 陈少康, 等. 肠道微生物与宿主遗传背景互作关系的研究进展[J]. 中国畜牧杂志, 2016, 52(5): 84-88 ; TAN Zhen, ZHAI Liwei, CHEN Shaokang, et al. Research progress of intestinal microbes interactions with the host genetic background[J]. Chinese Journal of Animal Science, 2016, 52(5): 84-88. (in Chinese)
- 35.LIU Q, LI F, ZHUANG Y, et al. Alteration in gut microbiota associated with hepatitis B and non-hepatitis virus related hepatocellular carcinoma[J] Gut Pathog. . 2019;11(1):1–3. doi: 10.1186/s13099-018-0281-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.VALE F F, LEHOURS P, YAMAOKA Y. Editorial: the role of mobile genetic elements in bacterial evolution and their adaptability[J] Front Microbiol. . 2022;13:849667. doi: 10.3389/fmicb.2022.849667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.ROBINSON K M, DUNNING HOTOPP J C. Mobile elements and viral integrations prompt considerations for bacterial DNA integration as a novel carcinogen[J] Cancer Lett. . 2014;352(2):137–144. doi: 10.1016/j.canlet.2014.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.BROADERS E, GAHAN C G M, MARCHESI J R. Mobile genetic elements of the human gastrointestinal tract[J] Gut Microbes. . 2013;4(4):271–280. doi: 10.4161/gmic.24627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.DAPITO D H, MENCIN A, GWAK G Y, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4[J] Cancer Cell. . 2012;21(4):504–516. doi: 10.1016/j.ccr.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.李洪波, 匡 黎. 肠道共生菌及代谢产物与肝癌进展的相关性分析[J]. 中西医结合肝病杂志, 2021, 31(10): 897-900 ; LI Hongbo, KUANG Li. A correlation analysis of intestinal commensal bacteria and metabolites with liver cancer progression[J]. Chinese Journal of Integrated Traditional and Western Medicine on Liver Diseases, 2021, 31(10): 897-900. (in Chinese)
- 41.SHANG-GUAN K, WANG M, HTWE N M P S, et al. Lipopolysaccharides trigger two successive bursts of reactive oxygen species at distinct cellular locations[J] Plant Physiol. . 2018;176(3):2543–2556. doi: 10.1104/pp.17.01637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.HARTMANN P, HAIMERL M, MAZAGOVA M, et al. Toll-like receptor 2-mediated intestinal injury and enteric tumor necrosis factor receptor I contribute to liver fibrosis in mice[J] Gastroenterology. . 2012;143(5):1330–1340.e1. doi: 10.1053/j.gastro.2012.07.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.ZHENG Y, WANG T, TU X, et al. Gut microbiome affects the response to anti-PD-1 immunotherapy in patients with hepatocellular carcinoma[J] J Immunother Cancer. . 2019;7(1):193. doi: 10.1186/s40425-019-0650-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.