


Abstract
We isolated RNA polymerase (RNAP) from Rhizobium leguminosarum, the nitrogen-fixing symbiont of peas and Vicia. Its 91 kDa subunit, which is homologous to σ70 of Escherichia coli RNAP, is necessary for transcription of the regulatory nodD gene, which in the presence of certain flavonoids induces transcription of other nod genes that are needed for the early steps of infection. We also show that negative autoregulation of nodD was achieved through competition between RNAP and NodD for their respective binding sites, which largely overlap. Combined with the result that high concentrations of the flavonoid inducer naringenin relieved the binding affinity of NodD for its target DNA, the way in which the nod genes are activated is discussed.
INTRODUCTION
Rhizobia, symbionts of leguminous plants, are distinguished from other Gram-negative bacteria by their possession of such genes as nodulation (nod, nol and noe) genes and nitrogen fixing (nif and fix) genes. Many of the nod, nol and noe genes are involved in synthesis of the lipochitooligosaccharide Nod factors which induce root hair curling and division of cortical cells in plants, leading to nodules where nitrogen fixation occurs (1–3).
Expression of most nod genes requires the presence of plant-derived inducers, usually flavonoids, and the presence of the regulatory protein NodD (4). Some species, e.g. Rhizobium leguminosarum bv. viciae and R.leguminosarum bv. trifolii, have only one nodD gene, whereas Bradyrhizobium japonicum, Rhizobium sp. NGR234, Rhizobium meliloti and Rhizobium tropici variously harbor two to five copies of nodD (5). Products of different nodD genes may perceive different inducers to fine tune the nodulation process (6). NodD is a DNA-binding protein that belongs to the LysR family of prokaryotic transcriptional regulators (7,8). It binds to conservative sequences (nod boxes) that are upstream of inducible nod operons (9–11). Although the specific binding of most NodDs to the nod box was not affected by flavonoids in vitro, experiments with mutant and chimeric NodDs suggested that the inducer molecules interact directly with NodD protein (12–14). The binding of NodD to its target DNA has been correlated with the inducible expression of nod genes in R.meliloti and Azorhizobium caulindans (15,16). In R.leguminosarum bv. viciae, however, cell-free extracts from strains containing class II NodD mutants, which are defective in their ability to autoregulate but retain their ability to activate nodABC and nodF expression in the presence of flavonoids, appeared not to bind the nod box in vitro (9,12). Therefore, the correlation between binding of NodD to the nod box and its activation and/or repression functions needs to be determined in R.leguminosarum bv. viciae.
Regulation of the nodD gene itself differs in different species. For example, in R.meliloti, both the nodD1 and nodD2 genes are subject to repression by NolR protein (17), while nodD3 and syrM constitute a self-amplifying positive regulatory circuit (18). In B.japonicum, the nodD1 gene is inducible in the presence of flavonoids (19); in contrast, the single nodD gene of R.leguminosarum bv. viciae is negatively autoregulated by its own product both in the presence and absence of flavonoids (20).
Despite the knowledge available on the regulation of nod genes in rhizobia, little is known about the basic transcriptional component RNA polymerase (RNAP). It is known that σ54-dependent RNAPs in rhizobia control, together with the upstream activator protein NifA, transcription of the genes for nitrogen fixation and other cellular functions (21,22), while the primary sigma factors are thought to be responsible for expression of housekeeping genes (23–25). However, the identity of the sigma factor responsible for transcription of nod genes remains unknown. Fisher et al. reported that RNAP containing a σ70-like sigma factor initiated in vitro transcription of the nodD1 gene of R.meliloti (26).
In R.leguminosarum bv. viciae, nodD has been identified as the regulatory gene for all the nod genes and its product represses its own transcription independent of inducers while activating the transcription of the other nod genes in the presence of plant-derived inducers such as naringenin (20,27). Lotz et al. (28) reported on the purification of RNAP from R.leguminosarum, but no further studies were done on this important enzyme.
We have established earlier an in vitro transcription system with RNAP purified from R.leguminosarum (29). In this report, using this system we prove that the 91 kDa subunit of RNAP is homologous to Escherichia coli σ70 and is required for nodD transcription. We also show that negative autoregulation of nodD is achieved via blocking the access of RNAP to the nodD promoter by NodD. Further, we show that high concentrations of the inducer naringenin have effects on the NodD–DNA complex.
MATERIALS AND METHODS
Bacterial strains, plasmids and DNA fragments
Rhizobium leguminosarum 8401 is a streptomycin-resistant strain of R.leguminosarum bv. phaseoli cured of its Sym plasmid (30); R.leguminosarum bv. viciae 248 is a wild-type isolate which contains the Sym plasmid pRL1JI (31); E.coli strain JM109 was used as the source of purified E.coli RNAP.
Plasmid pIJ1518 was consructed from pKT230 with a 1.7 kb BclI fragment containing the nodD gene under the control of the streptomycin resistance gene promoter and is also Kmr (20). pUC18AD was constructed by recloning the PstI–EcoRI insert from vector M13mp8-IJ487 (9) into pUC18 and is Apr. pMP220, a broad host range vector, is Tcr (21).
The DNA restriction fragments used in this study were obtained as follows. Fragment EB, 306 bp in length, was generated by EcoRI and BamHI digestion of pUC18AD; fragment ES, of 312 bp, was generated by EcoRI and SalI digestion of pUC18AD. Both fragments contain the nodA–nodD intergenic DNA, within which two intact overlapping promoters, nodD and nodA, are transcribed in opposite orientations (27,32; Fig. 1A). Fragment PE, obtained by PvuII and EcoRI digestion of pUC18, is 180 bp in length and contains the promoter of the lacZ gene. The ES fragment was radiolabeled for gel retardation and DNase I footprinting as described previously (29).
Figure 1.

Diagram of the nodA–nodD intergenic region. (A) The partial map of pUC18AD that carries the nodA–nodD intergenic DNA. The transcription start site for nodDp1 is numbered +1. The 306 bp EcoRI–BamHI and 312 bp EcoRI–SalI fragments (EB and ES, respectively) were used in this study. The transcription start site of nodA (14) is also numbered. A1, A2 and A3 are inverted repeat sequences (32). (B) The relevant nucleotide sequence of the nodA–nodD intergenic DNA and summary of the footprinting results. Thick and thin lines indicate the ‘footprints’ of RNAP and NodD, respectively. Triangular arrows and barbed arrows denote hypersensitive sites for RNAP and NodD, respectively. The transcription start sites of nodA and nodDp1 are indicated by divergent horizontal arrows. The bold ATG indicates the initiation codon of NodD protein. The canonical nod box is boxed. The double underlines designate the –35/–10 sequences of nodDp1. The mutant nucleotides are also labeled in bold.
Purification and characterization of RNAP
Rhizobium leguminosarum 8401 was grown aerobically in TY medium (33) at 28°C for 36–40 h and harvested by centrifugation. All purification steps were performed at 4°C unless otherwise stated. The cells (10 g wet weight) were resuspended in 50 ml buffer G (10 mM Tris–HCl pH 8.0, 1 mM EDTA, 5% glycerol, 0.24 M NaCl, 10 mM β-mercaptoethanol, 0.4 mg/ml lysozyme and 23 µg/ml phenylmethylsulfonyl fluoride) and sonicated (30 s × 25 times at 80% output on a Ultrasonics W375 sonicator) in a salt/ice bath. The lysate was mixed with ∼1.3 vol of TEGB buffer (10 mM Tris–HCl pH 8.0, 1 mM EDTA, 5% glycerol, 10 mM β-mercaptoethanol) containing 0.2 M NaCl, then centrifuged at 10 000 r.p.m. for 30 min (Sorvall SS-34 rotor). The crude extract was treated with 10% (v/v) polyethylenimine to a final concentration of 0.35%. The pellet obtained after centrifugation (10 000 r.p.m. for 30 min) was washed with TEGB buffer (0.5 M NaCl) in a glass homogenizer and then dissolved in TEGB buffer (1 M NaCl). Ammonium sulfate was added to 33% final saturation (18 g/100 ml) with constant stirring for 2 h and was then centrifuged. The collected supernatant was again precipitated by adding ammonium sulfate to 50% final saturation (10.75 g/100 ml). The precipitate was then resuspended in 45% saturated ammonium sulfate (25.8 g/100 ml) prepared in TEGB buffer, stirred for 2 h and then centrifuged. The precipitate was dissolved in an appropriate volume of TEGB buffer to make a solution with equal conductivity to TEGB buffer (0.2 M NaCl). This solution was loaded onto a pre-equilibrated column of heparin–Sepharose CL6B (1.5 × 10.3 cm; Pharmacia, Sweden), from which it was eluted with a linear 0.2–1.0 M NaCl gradient in TEGB buffer. Fractions containing RNAP as determined by SDS–PAGE and non-specific transcription assays (described below) were pooled, concentrated, dialyzed against TEGB buffer (0.1 M NaCl) containing 50% glycerol, and stored at –20°C. Sometimes minor impurities in the preparation were further removed by passing through a column of DE52 eluted with a linear 0.1–0.5 M NaCl salt gradient. About 3 mg RNAP could be obtained from 10 g cells using this method. RNAPs of R.leguminosarum bv. viciae 248 and E.coli JM109 were also purified using the procedures described above.
RNAP activity was judged by a non-specific transcription assay. The assay mixture contained, in a volume of 10 µl, 40 mM Tris–HCl pH 8.0, 10 mM MgCl2, 0.1 mM DTT, 150 mM KCl, 0.5 mg/ml BSA, 0.15 mM ATP, CTP and GTP, 10 µCi [α-32P]UTP (3000 Ci/mmol, 10 µCi/µl) and 0.15 mg/ml calf thymus DNA as template. RNA synthesis was initiated by addition of 5.0 µl enzyme fraction followed by incubation at 28°C for 30 min and was terminated by spotting the reaction onto DE81 filters. Unincorporated ribonucleotides were removed by washing the filters with 0.3 M sodium phosphate buffer (pH 6.5) three times. Radioactivity was determined in a liquid scintillation counter.
The subunits of RNAPs were separated by 10% SDS–PAGE and electroblotted onto BA-85 membrane (S&S, Germany), then probed with monospecific anti-E.coli σ70 antibody (a gift of Dr A. Ishihama; 34) at 1:1000 dilution. Color development was with a goat anti-rabbit IgG–horseradish peroxidase conjugate and 3,3′-diaminobenzidine substrate system.
Purification of NodD protein
Two forms of wild-type NodD protein of different purities were used in this study. Form I NodD was partially purified as described before (29); this was used for footprinting experiments. In order to remove endogenous DNA contamination from Form I NodD protein, which may affect in vitro transcription, Form II NodD protein was prepared by purification by DNA affinity chromatography as follows. First, a DNA affinity column with the ES fragment as ligand was prepared. About 100 µg of purified ES fragment was 3′-end filled in and labeled with Klenow enzyme (Boehringer Mannheim, Germany) and biotin-14-dATP (Gibco BRL, USA), then ethanol precipitated twice to remove free dATP. The biotin-labeled fragments were coupled to 1.0 ml streptavidin–agarose (Gibco BRL) with an efficiency of coupling of 100%. The prepared DNA affinity column was equilibrated with binding buffer (20 mM Tris–HCl pH 8.0, 0.1 mM EDTA, 0.1 mM DTT, 3% glycerol, 50 µg/ml BSA, 0.1 M KCl, 5 mM CaCl2). Second, ∼5.0 g R.leguminosarum 8401 (pIJ1518) cells were resuspended in 25 ml buffer G and lysed by sonication (30 s × 20 times) in an ice/salt bath. The lysate was cleared by supercentrifugation at 100 000 g for 1 h at 4°C. The supernatant was loaded onto a pre-equilibrated DE-52 column (Whatman, Germany), from which it was eluted with a linear 0.1–1.0 M NaCl gradient in TEGB buffer. Fractions containing NodD protein (monitored by binding activity with gel retardation and SDS–PAGE) were pooled, concentrated and dialyzed against TEGB buffer (0.1 M NaCl). The dialysis sample was then loaded onto the equilibrated DNA affinity column. The column was washed with 10 vol of binding buffer and eluted with high salt buffer (binding buffer containing 0.5 M KCl). The eluent was desalted by dialyzing against TEGB buffer (0.1 M KCl), concentrated, and kept as Form II NodD protein. The purity of Form II NodD was ∼80% as judged by SDS–PAGE but the amount recovered was lower. Repeated preparation was therefore required to make sufficient amounts.
Gel retardation
Gel retardation was performed in a total volume of 10 µl of binding buffer as described before (29). In assaying the RNAP–DNA interaction alone, RNAP (1.0 µg) and labeled DNA fragment (∼2.5 c.p.s./ng) were incubated at 28°C for 15 min and then the reaction mixture was treated with 3.0 µg heparin for 3 min before loading onto a non-denaturing gel.
In detecting the interaction between NodD and DNA, 100 ng calf thymus DNA as competitor was present in the reaction mixture containing NodD (0.3 µg) and the labeled DNA fragment (2.5 c.p.s./ng).
In the titration experiment, RNAP (0–2.0 µg) and NodD (0–1.2 µg) were simultaneously added to labeled DNA and incubated at 28°C for 15 min; no competitor was added to this reaction mixture.
DNase I footprinting
To determine the DNA sequence protected by RNAP or NodD, DNase I footprinting reactions were performed. RNAP (3.0 µg) or Form I NodD (3.0 µg) was incubated with the labeled DNA fragment (50 c.p.s.) for 30 min at 28°C in 30 µl of transcription buffer. Then 2.0 µl of DNase I (2.0 µg/µl; Boehringer Mannheim) were added and digestion allowed to proceed for 30 s. The reaction was terminated by mixing with 8.0 µl of 1.5 M sodium acetate, pH 5.2, containing 20 mM EDTA, 100 µg/ml yeast tRNA and extracting with phenol/chloroform. The DNA in the aqueous phase was precipitated with ethanol and the samples were analyzed on 6% denaturing polyacrylamide gels against an A+G ladder produced by the method of Liu and Hong (35). To detect the effects of naringenin on NodD footprints, naringenin (0.1–10 mM, dissolved in 50% ethanol) was added to the reaction mixture to a final concentration of 0.01–1 mM and incubated with the NodD–DNA complex for 30 min before adding DNase I.
In vitro transcription
The transcription assay was performed in a total volume of 20 µl of transcription buffer (40 mM Tris–HCl pH 8.0, 5 mM MgCl2, 2 mM spermidine, 0.15 M KCl, 0.1 mM EDTA, 0.1 mM DTT, 1 U/µl RNasin, 100 µg/ml BSA). Linear template fragment (ES or EB or PE, 50 ng) was incubated with RNAP (1.0 µg) for 20 min at 28°C, then 10 µl of prewarmed NTP/heparin mixture (0.15 mM ATP, GTP and CTP, 0.015 mM UTP, 200 µg/ml heparin, 10 µCi [α-32P]UTP) was added. After incubation for another 10 min, the reaction was terminated with 30 µl of stop solution (9 M ammonium acetate, 200 µg/ml yeast tRNA, 40 mM EDTA) and precipitated with 100 µl of ethanol. The pellet was dissolved in 5.0 µl of formamide loading buffer and analyzed on a 6% sequencing gel. To detect the effects of NodD protein or antibody on the transcription of nodD, Form II NodD (0–1.2 µg), anti-σ70 (34) or anti-NodD (a gift of Dr H. R. M. Schlaman; 36) was incubated with RNAP and DNA template for 20 min before initiation of transcription.
Primer extension
To determine the transcription start sites of the nodD gene, a primer complementary to ∼21–45 nt downstream of the nodD initiation codon was synthesized and 5′-end-labeled to high specific activity (∼5000 d.p.m./ng) with [γ-32P]ATP and T4 polynucleotide kinase. The sequence of the primer was 5′-gtcgagtgctacaagaaggtttaga. Usually, 15~20 ng primer was incubated with in vitro transcribed RNAs or 30 µg total RNA isolated from R.leguminosarum 8401 (pMP220ADw) in 50 µl of 1× SSC solution at 85°C for 10 min, at 52°C for 5 min and then at 30°C for 1 h. The subsequent steps were carried out as described before (37). In vitro transcribed RNAs were synthesized as follows. An aliquot of 200 ng ES fragment was incubated with ∼10 µg of 8401 RNAP at 28°C for 30 min in 30 µl of transcription buffer containing 5 mM ATP, UTP, CTP and GTP and 200 µg/ml heparin. The reaction was terminated by inactivation of RNAP at 65°C for 5 min. The DNA template was removed by adding RNase-free DNase to 0.1 U/µl and incubating at 37°C for 2 h. After phenol/chloroform extraction, the aqueous phase was precipitated, washed and dried. Total bacterial RNA was extracted by the hot phenol method as described previously (38).
Site-directed mutagenesis
Site-directed mutagenesis of nodA–nodD intergenic DNA was carried out by the method of Kunkel (39). The single-strand template was extracted from phagemid pUCZW, which was constructed by inserting the PstI–EcoRI fragment of pUC18AD into plasmid pUC119. The sequences of the two synthesized oligonucleotides for mutagenesis were 5′-ggcaatcagctatggaa-3′ and 5′-aaattgattggttggatg-3′, corresponding to 15~31 and 31~48 nt upstream of the nodD translation start codon, respectively. The two mutant fragments obtained by digesting pUCZW with EcoRI and PstI and the wild-type fragment ES were recloned into the reporter plasmid pMP220, resulting in pMP220ADm1, pMP220ADm2 and pMP220ADw with the nodD promoter preceding a promoterless lacZ gene. pMP220ADm1, pMP220ADm2 and pMP220ADw were transferred into rhizobia by conjugation in triparental matings as described by Figurski and Helinski (40). β-Galactosidase activity was assayed as described by Rossen et al. (20).
RESULTS
The 91 kDa subunit of R.leguminosarum RNAP is homologous to E.coli σ70
RNAPs were purified from R.leguminosarum 8401 and R.leguminosarum bv. viciae 248 by a modified method combining advantages of several previous protocols (Materials and Methods). Both RNAPs showed nearly the same composition of subunits and their purities were >95% as judged by estimating the Coomassie blue stained band intensities on 10% SDS–PAGE gels (Fig. 2A, lanes 3 and 4). Three major bands, from top to bottom, on the gel had apparent molecular masses of 160, 91 and 45 kDa. The 160 kDa band could be further resolved into two discrete bands if the separating gel was reduced in its concentration (data not shown). The overall banding patterns for the purified RNAPs were similar to those purified from other rhizobia (23,28,41,42). These protein components were found to be equivalent to the β′, β, α and σ subunits of E.coli RNAP (Fig. 2A, lane 2). The 91 kDa subunit was further proved to be the E.coli σ70 homolog, as it reacted with a specific anti-E.coli σ70 antibody as shown by western blotting (Fig. 2B, lanes 1–3).
Figure 2.

(A) SDS–PAGE analysis of RNAPs. Lane 1, protein size markers; lane 2, E.coli RNAP (5.0 µg); lane 3, R.leguminosarum 8401 RNAP (5.0 µg); lane 4, R.leguminosarum bv. viciae 248 RNAP (5.0 µg). The sizes of markers are shown next to the arrows on the left and the subunits of E.coli RNAP are indicated on the right. (B) Western blotting to probe the E.coli σ70 homologous subunit in rhizobial RNAPs. RNAPs from E.coli (lane 1), R.leguminosarum 8401 (lane 2) and R.leguminosarum bv. vicae 248 (lane 3) were immunoreacted with anti-σ70.
Formation of an open complex by RNAP binding to the nodD promoter
The ES fragment carrying the R.leguminosarum bv. viciae nodA–nodD intergenic DNA was incubated with RNAP and the protein–DNA complexes were analyzed by gel retardation assay. As shown in Figure 3, 8401 RNAP formed one obviously observable complex with labeled DNA at the temperatures tested (16, 28 and 37°C, lanes 3–5) except 0°C (lane 2); the amounts of this RNAP–DNA complex increased with rising incubation temperature (lanes 2–5), suggesting that formation of the complex is temperature dependent. Furthermore, this complex was stable with high concentrations of heparin (300 µg/ml) present in the reaction mixtures, indicating that the complex is also resistant to heparin. The temperature dependence and heparin resistance of the RNAP–DNA complex imply that it might be an open complex (43). Transcription can be initiated by adding rNTPs to an RNAP–DNA open complex, therefore the result that addition of rNTPs to the binding reaction led to smearing of the retarded bands further supports the hypothesis that this complex is an open complex (Fig. 3, lane 6). An additional discrete heparin-resistant complex migrating a bit more slowly than the major one could also be observed at 37°C (Fig. 3, lane 5). This minor heparin-resistant complex might be a complex of RNAP bound to another promoter or an isomer of RNAP bound to the same promoter (44).
Figure 3.
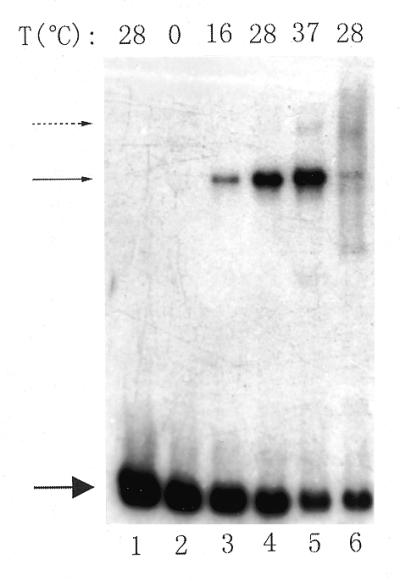
Gel retardation to show that 8401 RNAP forms an open complex with the ES fragment. RNAP (1.0 µg) and labeled DNA (∼4 ng) were incubated at different temperatures, then further incubated with heparin (3.0 µg) (lanes 2–5). For lane 6, 1.0 µl of 5 mM rNTPs was added to the reaction mixture after formation of the binary complex and then was competed with heparin. Lane 1 contains the free radiolabeled DNA. The incubation temperatures are shown above each lane. The upper solid arrow indicates the RNAP–DNA open complex, the lower solid arrow indicates the free DNA and the dotted arrow denotes another heparin-resistant complex.
The ES fragment contains two overlapping promoters, nodA and nodD, which are transcribed in opposite orientations (Fig. 1A). In order to show that the open complex was formed by RNAP binding at the nodD promoter rather than at the nodA promoter, an in vitro transcription assay was performed. Two different DNA fragments were used as templates for transcription: one (EB) was released from pUC18AD with EcoRI and BamHI digestion, the other (ES) with EcoRI and SalI digestion (Fig. 4B). The two fragments were basically identical: they shared the same nodA end, but ES was 6 nt longer than EB at the nodD end. Therefore, nodA-oriented transcripts would be of the same length using either ES or EB as template, whereas nodD-oriented transcripts produced from the ES fragment would be 6 nt longer than those from the EB fragment. As shown in Figure 4A, in vitro transcription on the two types of template produced three major transcripts (lanes 1 and 2). All three transcripts produced from the ES fragment (lane 2) were longer than that from the EB fragment (lane 1). Based on their lengths, the two longest transcripts were end-to-end read-throughs (rt) and the other two pairs of transcripts (nodDp1 and nodDp2) were products from the nodD promoter. In this in vitro transcription system no nodA-oriented transcript was detected.
Figure 4.

Run-off in vitro transcription. (A) The transcripts synthesized by different RNAPs with different fragments as template. Lane 1, 8401 RNAP with EB fragment; lane 2, 8401 RNAP with ES fragment; lane 3, 8401 RNAP preincubated with anti-E.coli σ70 antibody and with ES fragment; lane 4, R.leguminosarum 248 RNAP with ES fragment; lane 5, E.coli RNAP with ES fragment; lane 6, 8401 RNAP preincubated with anti-NodD antibody and with EB fragment; lane 7, 8401 RNAP with PE fragment; lane 8, 8401 RNAP with EB fragment; lane 9, E.coli RNAP with PE fragment. The size markers shown on the right are RNA transcripts produced by E.coli RNAP with the 180 bp PE fragment as template. In the absence of CRP–cAMP, E.coli RNAP initiated transcription from the lacZp2 start site, giving rise to an 80 nt transcript and a 180 nt read-through. (B) Restriction fragments as template. The numbers on the right indicate the size of each fragment.
nodD transcription requires the 91 kDa σ factor
The 91 kDa subunit of R.leguminosarum RNAP, a homolog of E.coli σ70, is absolutely required for in vitro nodD transcription. This was shown by the fact that there were no detectable specific transcripts in the in vitro transcription system when 8401 RNAP was preincubated with a specific anti-σ70 antibody (Fig. 4A, lane 3). As a control, the 8401 RNAP was preincubated with another antibody, anti-NodD. The specific transcripts did not disappear (Fig. 4A, lane 6). The RNAP–DNA open complex was excised from the polyacrylamide gel and then loaded onto SDS–PAGE, analysis of the protein components of the open complex revealed that it contained all the RNAP components, including the 91 kDa subunit (data not shown).
It is known that σ70 of E.coli RNAP recognizes promoters that contain –35/–10 consensus sequences. To investigate whether such consensus sequences within the nodD promoter exist, primer extension was used to determine the transcription start site of nodD both in vitro and in vivo. A primer complementary to the region of 21–45 nt downstream of the nodD translational start codon was used for both reverse transcription assay and dideoxy sequencing. Figure 5 shows that there is an in vitro nodD transcription start site located 60 bp upstream of the nodD translation start codon. Moreover, this in vitro start site is at the same position as the in vivo nodD transcription start site (Fig. 5, lanes vt and vi), implying that in R.leguminosarum the nodD gene is transcribed by an RNAP containing a σ70-like subunit. In the run-off transcription system, two specific transcripts oriented toward nodD (nodDp1 and nodDp2) were found, but under our experimental conditions, the in vivo transcription start site of nodDp2 was not detected. Therefore, the transcription start site of nodDp1 is considered as the start site of the nodD gene, whereas the nature of the in vitro nodDp2 promoter and the reason why the p2 transcript was produced in vitro need further investigation.
Figure 5.
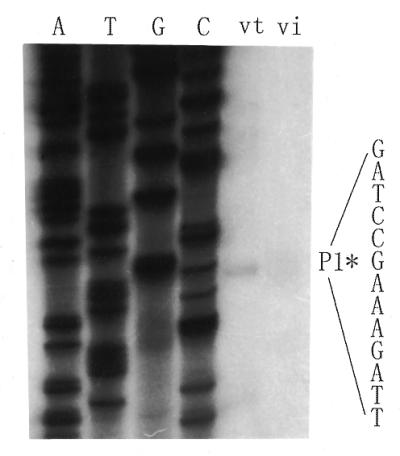
Primer extension to determine the in vitro and in vivo transcriptional start sites of the nodD gene. RNA synthesized in vitro (lane vt) or extracted from bacteria (lane vi) was used as template for reverse transcription. The asterisk denotes the position of the transcription start site.
Analysis of sequences revealed that two short sequences (TATAAG at –10 and TATTGA at –35) existed upstream of the nodDp1 promoter (Fig. 1B). These two sequences are similar to the –10/–35 consensus sequences (TATAAT and TTGACA) found in promoters recognized by E.coli σ70. Therefore, we were interested in whether the nodDp1 promoter could be recognized and transcribed by E.coli RNAP in vitro. As shown in Figure 4A (lane 5), transcription with the ES fragment as template could be initiated by E.coli RNAP, producing similar transcription patterns as that by R.leguminosarum RNAP. Those transcripts longer than read-through may have resulted from turn-back transcription by E.coli RNAP (45). Reciprocally, a well-characterized 180 bp PE fragment (Fig. 4B) containing the lacZ promoter was also recognized by R.leguminosarum RNAP, giving rise to an 80 bp transcript from the start site of lacZp2 (Fig. 4A, lane 7). These results were different from those obtained in R.meliloti (26). There it was reported that the R.meliloti RNAP transcribed the E.coli trpPOL promoter, but the E.coli RNAP did not transcribe the R.meliloti nodD1 promoter. This difference may be caused by less conservation in the –35/–10 region of the R.meliloti nodD1 promoter (5′-TCTAATN17TGATTC). It was revealed that in B.japomnicum, the TTG motif in the –35 region and the TAT sequence in the –10 region are both crucial for housekeeping genes to be transcribed by RNAP containing the σ70 homologous sigma factor (23). These two DNA sequence motifs also exist in the nodDp1 promoter of R.leguminosarum, which is very likely the reason why the nodD gene is constitutively expressed, no matter whether rhizobia grow in the presence or absence of flavonoids.
NodD binding is correlated with nodD repression
To see if NodD binding to DNA in vitro correlates with its repressive function in vivo, we carried out site-specific mutagenesis of nodD–nodA intergenic DNA. The nod box within the nodA–nodD interval is considered to contain the binding sites for NodD protein and it carries a T-N11-A motif which is involved in specific binding to LysR-type proteins (16). We converted the invariant nucleotide A of the T-N11-A motif to a C by site-directed mutagenesis. Another nucleotide A within the T-N11-A motif was also replaced by the same approach (Fig. 1B). The two mutant fragments (m1 and m2) and the wild-type nodD–nodA intergenic DNA fragment (ES) were recloned into reporter plasmid pMP220 with the nodD promoter preceding the lacZ gene (Materials and Methods). The constructs (pMP220ADm1, pMP220ADm2 and pMP220ADw) were introduced in R.leguminosarum 8401(nodD–) and R.leguminosarum 8401 (pIJ1518 containing nodD) by triparental conjugation and β-galactosidase levels were measured in the absence of inducer. As shown in Figure 6A, all the constructs in R.leguminosarum 8401 showed high levels of β-galactosidase, indicating that the two mutant nodD promoters remained active in transcription in the absence of NodD protein. However, in the presence of NodD protein [assaying in R.leguminosarum 8401 (pIJ1518)], the β-galactosidase levels of the wild-type and m2 constructs were reduced to ∼25% of that obtained in the absence of NodD protein. In contrast, the level of the m1 construct was reduced only slightly, to 80% of that of the wild type. Gel retardation was then performed to test the ability of the two mutant fragments to bind Form II NodD protein. As shown in Figure 6B, the binding of NodD protein to m1 was almost abolished compared with the complex formed between NodD and the wild-type fragment, whereas NodD binding to m2 was not destroyed; indeed, binding was enhanced compared to binding of NodD to the wild-type fragment (Fig. 6B). Therefore, in R.leguminosarum the NodD repression function depends on its binding to DNA.
Figure 6.

NodD repression correlates with NodD binding. (A) β-Galactosidase activities for the nodD promoter. In each pair of columns, the left column represents β-galactosidase activity in the absence of the nodD gene (assayed in R.leguminosarum 8401), whereas the right stands for β-galactosidase activity in the presence of the nodD gene [assayed in R.leguminosarum 8401 (pIJ1518)]. (B) NodD binding to wild-type and mutant nodD promoters. Lanes 1, 3 and 5, migration of labeled wild-type and two mutant DNA fragments in the absence of NodD protein; lanes 2, 4 and 6, as lanes 1, 3 and 5 but in the presence of 0.3 µg Form II NodD protein and 100 ng ctDNA. The intensities of free and shifted DNA bands were scanned on a Shimadzu CS-930 TLC scanner and were analyzed for the percent of shifted DNA against total free DNA. Wild-type, 40%; m1, 6%; m2, 65%.
nodD autoregulation is realized through competition for binding sites
In R.leguminosarum bv. viciae, NodD protein is bifunctional. It activates transcription of other nod genes in the presence of flavonoids and represses its own transcription independently of flavonoids (20). Here we provide evidence that this autoregulation is via NodD competing with RNAP for binding sites within the nodD promoter.
The DNA sequences protected by RNAP and NodD within the nodD–nodA intergenic region were determined using the DNase I footprinting technique. As shown in Figure 7A, RNAP covered the –31 to about the +43 region relative to the nodDp1 transcription start site on the nodD transcriptional strand (nodD strand). A similar length was protected on the complementary strand (nodA strand). Small gaps and several hypersensitive sites were also observed within the footprint (summarized in Fig. 1B). NodD protein protected a segment of ∼56 bp on both strands with a cluster of hypersensitive sites in the middle (Fig. 7B, and summarized in Fig. 1B). Similarly to the protected region with the NodD1 and NodD3 proteins of R.meliloti on the nod promoters (46), the region protected by R.leguminosarum NodD essentially covered the nod box, with a gap in the middle.
Figure 7.

DNase I footprinting to determine the DNA sequences within the nodA–nodD intergenic region protected by RNAP or NodD. (A and B) Autoradiographs to show the pattern of protection from DNase I digestion by RNAP (3.0 µg) (A) and by NodD (3.0 µg of Form I) (B), respectively, on the ES fragment. The sequence is numbered with respect to the nodDp1 transcription start site. – and +, absence and presence of proteins, respectively. The regions protected by RNAP and NodD are also summarized in Figure 1B.
The sequences protected by RNAP and by NodD largely overlap each other. It is thus reasonable to assume that NodD blocks its own transcription by its binding to DNA and that autoregulation is via its competition with RNAP for binding sites in the promoter. We tested this idea by gel retardation and run-off transcription assays. Figure 8A (lanes 1–10) shows competition between RNAP and NodD for binding to nodA–nodD intergenic DNA. With a constant concentration of RNAP in the binding reaction, the intensities of the band representing the RNAP–DNA open complex decreased with increasing NodD concentration (lanes 2–6). Thus NodD protein appears to prevent RNAP binding to its cognate sites. Reciprocally, RNAP prevented NodD protein binding to the nodA–nodD intergenic DNA (lanes 7–10). Correspondingly, in the run-off transcription assays (Fig. 8B) it was found that the intensity of the nodDp1 transcript decreased with increasing NodD concentration in the reaction. Almost complete inhibition of the p1 transcript was observed when NodD protein was added at 1.2 µg.
Figure 8.


(A) Competition between RNAP and NodD binding to the ES fragment. In each lane from 2 to 6, equal amounts of RNAP and different amounts of NodD protein were present in the reaction. From lane 7 to 10, the amount of NodD protein was kept constant and the amount of RNAP was different. Lanes 12–14, RNAP formed more complexes in the absence of heparin. Lane 15, NodD and RNAP were simultaneously added to the reaction mixture in the presence of heparin. Lanes 1 and 11 contain the free radiolabeled DNA. The amounts of RNAP and NodD are given above each lane. (B) The repression effect of NodD on transcription of nodD. The amounts of RNAP and NodD are the same as in lanes 4–6 of the competition assay and the ES fragment was used in this assay at ∼10 ng.
In the competition assay the reaction mixtures were not treated with heparin, therefore there were other complexes in addition to the RNAP–DNA open complex and NodD–DNA complex. These complexes were caused by RNAP alone being bound to DNA, as shown in Figure 8A, lanes 11–14. With heparin added to the reaction mixtures, no third complex band appeared in addition to the NodD–DNA and RNAP–DNA open complex bands (Fig. 8A, lane 15), suggesting that NodD and RNAP compete with each other for binding sites and do not bind cooperatively to the nodD promoter under such conditions.
High concentrations of naringenin have effects on NodD footprints
In R.leguminosarum bv. viciae, NodD in the presence of an inducer such as naringenin activates the expression of other inducible nod genes. However, the molecular basis for the effect of the inducer on this activation is still unknown. Interestingly, we found that in R.leguminosarum, the inducer naringenin affected the NodD–DNA complex at high concentrations. Figure 9 shows the effects of naringenin on the NodD footprints. As the final concentration of naringenin was increased from 0.01 to 1 mM, the protection by NodD on DNA gradually disappeared (i.e. the amount of NodD protein bound to the nod box was gradually reduced). With naringenin at 1 mM, the NodD protein no longer bound to DNA. This result is in line with our earlier observation that naringenin specifically dissociated the NodD–DNA complex at 1 mM (47). The hypersensitive cleavage site (–48, at the center of the nod box) also disappeared with increasing concentrations of naringenin.
Figure 9.
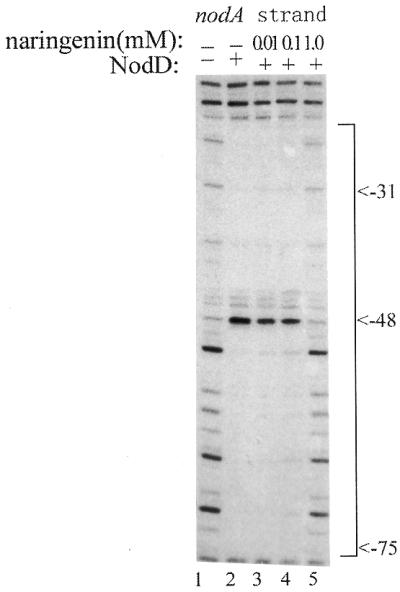
The relieving effect of naringenin on NodD footprints. Lane 1, free DNA cleavage by DNase I; lane 2, the region protected by NodD on the nodA transcriptional strand in the absence of naringenin. From lane 3 to 5, increasing amounts of naringenin (final concentrations 0.01, 0.1 and 1 mM) were added to the NodD–DNA complex before DNase I digestion. The sequence is numbered with respective to the transcription start site of nodA.
DISCUSSION
In R.leguminosarum bv. viciae, the regulatory gene nodD is subject to negative regulation by its own product independent of inducers (20). The mutant strains defective in this autoregulation nodulate their hosts with low efficiency (12), suggesting that repression of nodD transcription by NodD is an important step in the establishment of normal nodules.
In this report, a molecular mechanism has been elucidated whereby the autoregulation of nodD expression occurs. We first determined the transcription start site of the nodD gene using purified RNAP from R.leguminosarum and revealed that a –35/–10 consensus within the nodD promoter could be recognized by a σ70-like subunit in the RNAP. The formation of stable open complexes between RNAP and the nodD promoter implied that RNAP alone was responsible for nodD transcription, which is also in agreement with the fact that nodD is expressed constitutively. The specific binding of NodD to its promoter blocked RNAP access to the nodD promoter, leading to the repression of nodD expression. This mechanism of autoregulation is shared with other LysR family members, such as IlvY, TrpI and OccR (8,48,49), and efficiently maintains the proper cellular levels of NodD protein to control expression of other nod genes. We earlier put forward a DNA loop model for explaining nodD autoregulation (50), but we did not detect the suggested binding of NodD to the inverted repeat sequence A3 (Fig. 1A) in later experiments. Therefore, the role of A3 in nodD repression remains unknown and needs further investigation.
Burn et al. (12) have described four classes of mutation in the nodD gene which were specifically affected in autoregulation or in flavonoid-dependent activation. Extracts from Rhizobium strains containing these NodD mutants formed either no protein–DNA complex or formed a complex with altered mobility compared to that obtained with extracts from wild-type strains (9). Therefore, the precise relationship between NodD binding and its activation and/or repression functions need to be determined. Here, we have used purified wild-type NodD protein and a mutant nod box to prove that the binding of NodD to DNA is necessarily required for its repressive function (Fig. 6A and B).
In R.leguminosarum bv. viciae, NodD is bifunctional. It constitutively represses its own transcription and in the presence of inducer flavonoids activates the expression of inducible nod genes. The conversion of NodD protein from a repressor to an activator needs the presence of inducer flavonoid molecules. Recently Rhee et al. (48) reported that activation and repression by IlvY protein (a LysR-type protein) were mediated by the same regulatory locus. Binding of inducer to the IlvY protein–DNA complex evoked conformational changes in the DNA which are functionally correlated with transcriptional activation. The ES and EB fragments used in this study both contain two divergently overlapping promoters, the nodD promoter and the inducible nodA promoter, so we also examined protection by NodD on the nodA promoter in the absence of the inducer naringenin. NodD protein protected a 56 bp segment, located from –20 to –75 relative to the transcription start site of nodA with a gap in the middle (Fig. 7B). Fisher and Long (46) have identified the region protected by R.meliloti NodD1 and NodD3 on the nodA promoter using DNase I cleavage. Compared to their results, the protection pattern with R.leguminosarum NodD on the nodA promoter was more similar to the protection by NodD1 than that by NodD3. In R.meliloti, the two regulatory proteins NodD1 and NodD3 differ in their activating behavior: NodD1 requires the presence of an inducer to cause nod gene induction in vivo; NodD3, on the other hand, activates nod gene expression in the absence of inducer. The identical protection patterns of R.leguminosarum NodD and R.meliloti NodD1 on the nodA promoter imply that they might share the same activating mechanism.
However, in R.meliloti, addition of the inducer luteolin to a NodD1–DNA reaction mixture had no effect on the NodD1 footprint (46). In this study, we have shown that the inducer naringenin affects the R.leguminosarum NodD footprints at high concentrations (Fig. 9). It should be noted that this effect occurred only when the inducer naringenin was at a high concentration (0.01–1 mM; the concentration of naringenin usually used in the medium is ∼1 µM). However, this may be significant for the following reasons. First, naringenin could accumulate locally in the cytoplasmic membrane of R.leguminosarum cells to a level ∼80-fold higher than in the medium (51). Thus, the in vitro result that binding of NodD to DNA was relieved by naringenin at high concentrations, for example at 0.1 mM, may in fact mimic the conditions in vivo. Second, NodD protein can induce a bend in nod promoters (15). The appearance of a DNase I hypersensitive site (–48) in the protected region of NodD footprints (Fig. 9) is also indicative of NodD-induced DNA bends. Therefore, loss of hypersensitive site –48 implies a decrease in DNA bending due to a decrease in NodD binding. The change in bend may lead to DNA taking on an activation-competent state capable of directing RNAP to initiate transcription of the nodA promoter. However, we need to establish the correlation between the naringenin-induced change in the NodD–DNA complex and transcriptional activation of the nodA gene. At this stage we are still unable to rule out the possibility that some other factors are involved in the activation process. We previously reported that an HU-like protein of R.leguminosarum, Px, binds to specific sites within and induces a bend in the nod promoters (29). The great conformational changes in the DNA resulting from such a bend may facilitate the binding of RNAP and/or NodD to the nod promoters or RNAP–NodD interaction.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr A. Ishihama for providing us with antibodies to E.coli σ70 and Dr H. R. M Schlaman for sending us R.leguminosarum bv. viciae 248 and the anti-NodD antibody. We are also indebted to our laboratory members, especially Fudi Ni, Huafeng Lu, Jie Feng and Youyi Lu, for useful discussions. This work was supported by Pan-Deng Plan of China to G.H.
REFERENCES
- 1.Pueppke S.G. (1996) Crit. Rev. Biotechnol., 16, 1–51. [DOI] [PubMed] [Google Scholar]
- 2.van Rhijn P. and Vanderleyden,J. (1995) Microbiol. Rev., 59, 124–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Denarie J., Debelle,F. and Prome,J.-C. (1996) Annu. Rev. Biochem., 65, 503–535. [DOI] [PubMed] [Google Scholar]
- 4.Schlaman H.R.M., Okker,R.J.H. and Lugtenberg,B.J.J. (1992) J. Bacteriol., 174, 5177–5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Rhijn P., Feys,B., Verreth,C. and Vanderleyden,J. (1993) J. Bacteriol., 175, 438–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spaink H.P., Wijffelman,C.A., Pees,E., Okker,R.J.H. and Lugtenberg,B.J.J. (1987) Nature, 328, 337–340. [DOI] [PubMed] [Google Scholar]
- 7.Henikoff S., Haughn,G.W., Calvo,J.M. and Wallace,J.C. (1988) Proc. Natl Acad. Sci. USA, 85, 6602–6606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schell M.A. (1993) Annu. Rev. Microbiol., 47, 597–626. [DOI] [PubMed] [Google Scholar]
- 9.Hong G.F., Burn,J.E. and Johnston,A.W.B. (1987) Nucleic Acids Res., 15, 9677–9690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fisher R.F., Egelhoff,T.T., Mulligan,J.T. and Long,S.R. (1988) Genes Dev., 2, 282–293. [DOI] [PubMed] [Google Scholar]
- 11.Kondorosi E., Gyuris,J., Schmidt,J., John,E., Hoffmannm,D.B., Schell,J. and Kondorosi,A. (1989) EMBO J., 8, 1331–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burn J., Rossen,L. and Johnston,A.W.B. (1987) Genes Dev., 1, 456–464. [Google Scholar]
- 13.McIver J., Djordjevic,M.A., Weinman,J.J., Bender,G.L. and Rolfe,B.G. (1989) Mol. Plant Microbe Interact., 2, 97–106. [DOI] [PubMed] [Google Scholar]
- 14.Spaink H.P., Okker,R.J.H., Wijffelman,C.A., Tak,T., Goosen-deRoo,L., Dees,E., vanBrussel,A.A.N. and Lugtenberg,B.J.J. (1989) J. Bacteriol., 171, 4045–4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fisher R.F. and Long,S.R. (1993) J. Mol. Biol., 233, 336–348. [DOI] [PubMed] [Google Scholar]
- 16.Goethals K., VanMontagu,M. and Holsters,M. (1992) Proc. Natl Acad. Sci. USA, 89,1646–1650. [Google Scholar]
- 17.Cren M., Kondorosi,A. and Kondorosi,E. (1995) Mol. Microbiol., 15, 733–747. [DOI] [PubMed] [Google Scholar]
- 18.Swanson J.A., Mulligan,J.T. and Long,S.R. (1993) Genetics, 134, 435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Banfalvi Z., Nieuwkoop,A., Schell,M., Besl,L. and Stacey,G. (1988) Mol. Gen. Genet., 214, 420–424. [DOI] [PubMed] [Google Scholar]
- 20.Rossen L., Shearman,C.A., Johnston,A.W.B. and Downie,J.A. (1985) EMBO J., 4, 3369–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morett E. and Buck,M. (1989) J. Mol. Biol., 210, 65–77. [DOI] [PubMed] [Google Scholar]
- 22.Merrick M.J. (1993) Mol. Microbiol., 10, 903–909. [DOI] [PubMed] [Google Scholar]
- 23.Beck C., Marty,R., Klausli,S., Hennecke,H. and Gottfert,M. (1997) J. Bacteriol., 179, 364–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luka S., Patharca,E.J., Riccio,A., Iaccarino,M. and Defez,R. (1996) J. Bacteriol., 178, 7138–7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rushing B.G. and Long,S.R. (1995) J. Bacteriol., 177, 6952–6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fisher R.F., Brierley,H.L., Mulligan,J.T. and Long,S.R. (1987) J. Biol. Chem., 262, 6849–6855. [PubMed] [Google Scholar]
- 27.Spaink H.P., Okker,R.J.H., Wijffelman,C.A., Pees,E. and Lugtenberg,B.J.J. (1987) Plant Mol. Biol., 9, 27–39. [DOI] [PubMed] [Google Scholar]
- 28.Lotz W., Fees,H., Wohlleben,W. and Burkardt,H.J. (1981) J. Gen. Microbiol., 125, 301–309. [Google Scholar]
- 29.Liu S.T., Chang,W.Z., Cao,H.M., Hu,H.L., Chen,Z.H., Ni,F.D., Lu,H.F. and Hong,G.F. (1998) J. Biol. Chem., 273, 20568–20574. [DOI] [PubMed] [Google Scholar]
- 30.Lamb J.W., Hombrecher,G. and Johnston,A.W.B. (1982) Mol. Gen. Genet., 186, 449–452. [Google Scholar]
- 31.Josey D.P., Beynon,J.L., Johnston,A.W.B. and Beringer,J.E. (1979) J. Appl. Microbiol., 46, 343–350. [Google Scholar]
- 32.Shearman C.A., Rossen,L., Johnston,A.W.B. and Downie,J.A. (1986) EMBO J., 5, 647–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beringer J.E. (1974) J. Gen. Microbiol., 84, 188–198. [DOI] [PubMed] [Google Scholar]
- 34.Jishage M. and Ishihama,A. (1995) J. Bacteriol., 177, 6832–6835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu S.T. and Hong,G.F. (1998) Anal. Biochem., 255, 158–159. [DOI] [PubMed] [Google Scholar]
- 36.Schlaman H.R.M., Spaink,H.P., Okker,R.J.H. and Lugtengerg,B.J.J. (1989) J. Bacteriol., 171, 4686–4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 38.Babst M., Henncke,H. and Fischer,H.M. (1996) Mol. Microbiol., 19, 827–839. [DOI] [PubMed] [Google Scholar]
- 39.Kunkel T.A., Roberts,J.D. and Zakour,R.A. (1987) Methods Enzymol., 154, 367–382. [DOI] [PubMed] [Google Scholar]
- 40.Figurski D.H. and Helinski,D.R. (1979) Proc. Natl Acad. Sci. USA, 76, 1648–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Regensburger B. and Hennecke,H. (1983) Arch. Microbiol., 135, 103–109. [DOI] [PubMed] [Google Scholar]
- 42.Nielsen B.L. and Brown,L.R. (1985) J. Bacteriol., 162, 645–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zinkel S.S. and Crothers,D.M. (1991) J. Mol. Biol., 219, 201–215. [DOI] [PubMed] [Google Scholar]
- 44.Straney D.C. and Crothers,D.M. (1985) Cell, 43, 449–459. [DOI] [PubMed] [Google Scholar]
- 45.Kumar A., Malloch,R.A., Fujita,N., Smillie,D., Ishihama,A. and Hayward,R. (1993) J. Mol. Biol., 232, 406–418. [DOI] [PubMed] [Google Scholar]
- 46.Fisher R.F and Long,S.R. (1989) J. Bacteriol., 171, 5492–5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hong G.F. and Cao,H.M. (1993) Chin. J. Biotechnol., 9, 85–88. [PubMed] [Google Scholar]
- 48.Rhee K.Y., Senear,D.F. and Hatfield,G.W. (1998) J. Biol. Chem., 273, 11257–11266. [DOI] [PubMed] [Google Scholar]
- 49.Olekhnovich I. and Gussin,G.N. (1998) Gene, 223, 247–255. [DOI] [PubMed] [Google Scholar]
- 50.Mao C., Downie,J.A. and Hong,G. (1994) Gene, 144, 87–89. [DOI] [PubMed] [Google Scholar]
- 51.Recourt K., vanBrussel,A.A.A., Driessen,A.J.M. and Lugtenberg,B.J.J. (1989) J. Bacteriol., 171, 4370–4377. [DOI] [PMC free article] [PubMed] [Google Scholar]