

Summary
Transforming growth factor-β-activated kinase 1 (TAK1) is a central regulator of innate immunity, cell death, inflammation, and cellular homeostasis. Therefore, many pathogens carry TAK1 inhibitors (TAK1i). As a host strategy to counteract this, inhibition or deletion of TAK1 induces spontaneous inflammatory cell death, PANoptosis, through the RIPK1-PANoptosome complex, containing the NLRP3 inflammasome and caspase-8/FADD/RIPK3 as integral components; however, PANoptosis also promotes pathological inflammation. Therefore, understanding molecular mechanisms that regulate TAK1i-induced cell death is essential. Here, we report a genome-wide CRISPR screen in macrophages that identified TAK1i-induced cell death regulators, including polypyrimidine tract-binding (PTB) protein 1 (PTBP1), a known regulator of RIPK1, and a previously unknown regulator RAVER1. RAVER1 blocked alternative splicing of Ripk1, and its genetic depletion inhibited TAK1i-induced, RIPK1-mediated inflammasome activation and PANoptosis. Overall, our CRISPR screen identified several positive regulators of PANoptosis. Moreover, our study highlights the utility of genome-wide CRISPR-Cas9 screens in myeloid cells for comprehensive characterization of complex cell death pathways to discover therapeutic targets.
Subject areas: Biological sciences, Molecular biology, Immunology, Cell biology
Graphical abstract
Highlights
-
•
Whole-genome CRISPR screen identifies TAK1 inhibition-induced PANoptosis regulators
-
•
CRISPR screen validated by identifying known regulators TNFR1, RIPK1, and caspase-8
-
•
RAVER1, PTBP1 also identified in CRISPR screen to activate TAK1i-induced PANoptosis
-
•
RAVER1 and PTBP1 regulate RIPK1 pre-mRNA splicing and canonical RIPK1 expression
Biological sciences; Molecular biology; Immunology; Cell biology
Introduction
The host innate immune system forms the first line of defense against invading infectious and sterile agents and maintains homeostatic equilibrium. To carry out these functions, pattern recognition receptors sense perturbations and initiate inflammatory responses, including mitogen-activated protein kinase (MAPK) signaling and cell death pathways. The MAPK kinase kinase 7 (MAP3K7), also known as transforming growth factor-β (TGF-β)-activated kinase 1 (TAK1), is an essential kinase that acts as a central node downstream of a wide range of innate immune, cytokine, and cell death receptors to drive rapid and robust immune activation for host defense.1,2,3,4,5,6,7,8,9,10
Due to its role in integrating immune signaling cascades, TAK1 is a target for pathogen-mediated immune evasion strategies.11 For instance, Yersinia has evolved to use its type-III secretion system to release the highly virulent Yersinia outer protein J (YopJ) into the host macrophage to inhibit TAK1 and facilitate systemic spread.12,13,14 This strategy for TAK1 inhibition likely contributes to Yersinia’s high mortality rates and ability to cause pandemics.15,16,17,18,19 In response to YopJ, or pharmacological inhibition of TAK1 through 5z-7-oxozeaenol (5z7), TAK1 inhibition induces a RIPK1-dependent alternate cell death pathway, both in mouse and human macrophages.20,21,22,23 This cell death can occur in the absence21 or presence of innate immune priming.20,22,23 Mechanistically, in the absence of external innate immune priming, TAK1 inhibition induces RIPK1-dependent PANoptosis,20,21 a unique innate immune inflammatory, lytic cell death pathway regulated by PANoptosomes, which integrate components from other cell death pathways. TAK1 inhibition-mediated PANoptosis is characterized by the activation of the NLRP3 inflammasome as well as caspase-1, caspase-8, caspase-3, caspase-7, and MLKL,20,21 and the RIPK1-PANoptosome contains the NLRP3 inflammasome along with caspase-8, FADD, and RIPK3 as integral components.24 This RIPK1-dependent cell death phenocopies the homeostatic aspects of the TNFR1-RIPK1 signaling axis-driven inflammatory mechanism and disease.25,26,27,28,29,30,31,32 In contrast, in the presence of LPS-mediated innate immune priming, TAK1 inhibition drives responses that are regulated by the Rag-Regulator complex at the cross-roads of cellular energy metabolism and activation of RIPK1-dependent cell death.33 However, our understanding of the regulation of TAK1 inhibition-mediated cell death and inflammation remains incomplete, and there is an urgent and unmet need to define the regulators of this pathway to advance the identification of strategies for therapeutic targeting of infectious and inflammatory diseases.
Therefore, in this study, we conducted a whole-genome CRISPR-based knockout screen to identity key regulators of TAK1 inhibition-mediated cell death using immortalized bone marrow-derived macrophages (iBMDMs) expressing Cas9. Our screen identified well-known activators (positive regulators) of the RIPK1-dependent cell death pathway, including TNFR1, RIPK1, and caspase-8, providing an intrinsic validation of the screen results. Beyond these activators, the screen also identified polypyrimidine tract-binding (PTB) protein 1 (PTBP1) and RAVER1, two components of mRNA splicing; these molecules were required for the formation of the functional, full-length Ripk1 mRNA by suppressing a non-canonical alternative splicing variant. Overall, our screen identified a comprehensive list of both known and previously unknown regulators of the TNFR1-RIPK1-caspase-8 pathway of inflammatory signaling and the RIPK1-PANoptosome and PANoptosis in macrophages, suggesting unique therapeutic targets and illustrating the utility of CRISPR screens for mechanistic discovery.
Results
Whole-genome CRISPR screen identifies essential regulators of TAK1 inhibition-mediated cell death
Given the critical role of TAK1 at the intersection of multiple innate immune sensing and cytokine signaling pathways, we sought to discover and characterize key regulators of TAK1 functions. To do this, we first developed a genome-wide CRISPR-based genetic screen to identify the genes that regulate TAK1 inhibition-mediated cell death in iBMDMs.34,35 We selected iBMDMs since they are a well-established and routinely used cell type for studying innate immune mechanisms of cell death and inflammation, and they provide a virtually unlimited supply of cells for large scale studies. We infected Cas9-expressing iBMDMs with validated lentiviral particles prepared using a whole-genome CRISPR-Brie library (#73633, Addgene) (Figure 1A). The Brie library carries four gRNAs for each gene, covering the entire genome.36 The pooled Cas9-iBMDMs stably expressing the library of gene-specific gRNAs were then subjected to TAK1 inhibitor (TAK1i) treatment for 24 h to induce the RIPK1-dependent cell death. We collected the surviving cells for genomic DNA isolation and further analyses (Figure 1A).
Figure 1.

Whole-genome CRISPR screen identifies essential regulators of TAK1 inhibition-mediated cell death
(A) Schematic representation of the genome-wide CRISPR screen workflow, showing the immortalized bone marrow-derived macrophage (iBMDM) pool, lentiviral transduction to induce gRNA expression for gene deletions, and TAK1 inhibitor (TAK1i; 5z-7-oxozeaenol) treatment for 24 h before collection for next-generation sequencing (NGS) and enrichment analyses.
(B) Visual representation of the CRISPR data using a Volcano plot. The selected list of top-ranked gRNA-targeted genes that represent known and previously unknown regulators of TAK1 inhibition-mediated cell death are indicated.
(C) A scatterplot depicting the distribution of the normalized gRNA count, presented in log scale, for each of the individual gRNAs targeting the enriched genes highlighted in the Volcano plot presented in panel B.
(D) Reactome pathway analysis of the top 25 genes enriched from the TAK1i-induced cell death screen presented in panel B. Data are shown from two independent replicates, and the log10(p value) and mean gRNA counts were calculated by the MAGeCK algorithm (B–D). See also Figure S1.
Next-generation sequencing (NGS) of the PCR-amplified, barcoded gRNAs allowed us to quantitatively identify gRNAs that were enriched in the surviving pool of cells (BioProject: PRJNA973658). Because the presence of a gRNA should delete the corresponding gene in that cell, enriched gRNAs are expected to represent genes that, when deleted, rescue the cells from TAK1i-mediated cell death, i.e., genes that are positive regulators of the cell death. Performing CRISPR MAGeCK analysis followed by plotting the log-fold change against the p value identified gRNAs targeting TNFR1, RIPK1, and caspase-8 among the most highly enriched gRNAs from the screen, with a positive enrichment ranking of 1, 2, and 5, respectively (Figures 1B and 1C). TNFR1, RIPK1, and caspase-8 are known to be key regulators of TAK1 inhibition-mediated cell death, affirming the quality and integrity of the screen. Moreover, TNF signaling and TNFR1-induced signaling were found to be the most significantly enriched pathways in the screen based on the Reactome pathway analysis of the top 25 genes from the screen (Figure 1D) and the GO term analysis of the full set of enriched genes (Figure S1), providing further validation of the screen results.
In addition to TNFR1, RIPK1, and caspase-8, the screen also identified the spliceosome components PTBP1 and RAVER1 as significantly enriched molecules (Figures 1B and 1C), suggesting that they may also play a role in the regulation of TAK1 inhibition-mediated cell death. PTBP1 was previously implicated in RIPK1 splicing,37 and a role for RAVER1 in the TAK1i-mediated cell death pathway has not been found before. Together these results show that CRISPR-based genetic screening in iBMDMs can identify both known and previously uncharacterized regulators of TAK1i-induced cell death in the absence of innate immune priming.
PTB splicing factors are required for TAK1 inhibition-mediated cell death
To further validate the cell death function of select genes corresponding to the significantly enriched gRNAs from our CRISPR screen, we used siRNA to knockdown the expression of Tnfr1, Ripk1, Ptbp1, and Raver1 in BMDMs, as these primary cells are considered optimal representatives of the myeloid cell lineage that are suitable for studying innate immune cell death mechanisms.38 The siRNA-transfected BMDMs were treated with TAK1i in the absence of any external priming. Consistent with the whole-genome screen findings from iBMDMs (Figure 1), we observed that TAK1i treatment induced robust cell death in BMDMs (Figure 2A). Compared to BMDMs transfected with the NT (non-targeting) siRNA, we found that BMDMs transfected with siRNA targeting Tnfr1, Ripk1, Ptbp1, or Raver1 had significantly reduced cell death (Figures 2A and 2B). We also confirmed the efficiency of the siRNA-based knockdown using qPCR analysis with two individual pairs of PCR primers for each gene and found consistent and significant knockdown efficiencies (Figures S2A–S2D). Together, these data provide validation for our whole-genome CRISPR screen and suggest that TNFR1, RIPK1, PTBP1, and RAVER1 are key regulators of TAK1 inhibition-mediated cell death.
Figure 2.

PTB splicing factors are required for TAK1 inhibition-mediated cell death
(A and B) Bone marrow-derived macrophages (BMDMs) treated with vehicle control or transfected with NT (non-targeting) siRNA or the indicated gene-specific siRNAs were stimulated with the pharmacological inhibitor of TAK1 kinase activity (5z-7-oxozeaenol, TAK1i). (A) Cell death images at 12 h post-treatment with TAK1i are shown (scale bar, 50 μm). Cells positive for PI (propidium iodide) uptake were pseudo-colored in red. (B) Quantification of PI-positive dead cells at 12 h post-treatment with TAK1i. All data are presented as mean ± SEM (B). p < 0.05 is considered statistically significant. ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001 (two-tailed t test [B]). Data are representative of three independent experiments with n = 2–3 in each repeat (A–B). See also Figure S2.
PTB splicing factors promote TAK1 inhibition-mediated PANoptosis
To further understand the functional requirement of the PTB proteins in TAK1 inhibition-mediated cell death, we next examined the biochemical markers of PANoptosis in siRNA-transfected BMDMs. Consistent with our previous findings,20,21 TAK1i treatment induced activation of the inflammasome and pyroptotic molecules as demonstrated by cleavage of caspase-1, gasdermin D (GSDMD), and GSDME in NT siRNA-transfected cells (Figure 3A). Additionally, we observed cleavage of the apoptotic molecules caspase-8, caspase-3, and caspase-7 (Figure 3B), as well as the phosphorylation of the necroptotic molecule MLKL (Figure 3C) and RIPK1 (Figure 3D), in response to TAK1i treatment. These results are consistent with the known activation of PANoptosis in response to TAK1i treatment.20,21 In line with the observed protection from cell death (Figure 2), we found that genetic knockdown of the PTB splicing components PTBP1 and RAVER1 resulted in reduced activation of the inflammasome and pyroptotic, apoptotic and necroptotic components of PANoptosis as measured by immunoblotting (Figures 3A–3C) and further confirmed by quantification (Figures S3A–S3G). Additionally, siRNA-based knockdown of Ptbp1 or Raver1 expression also abrogated the S166 phosphorylation of RIPK1 (Figure 3D). The reduction in cell death, as well as reduction in biochemical activation of PANoptosis markers, observed in the BMDMs transfected with siRNA targeting Tnfr1 or Ripk1 (Figures 2 and 3A–3D), provided further validation of our results and established siRNA-mediated knockdown as an efficient strategy for the validation of whole-genome screen results in BMDMs. Together, these findings suggest a requirement for the regulators identified in our screen, PTBP1 and RAVER1, in TAK1 inhibition-mediated, RIPK1-dependent PANoptosis (Figure 3E).
Figure 3.

PTB splicing factors promote TAK1 inhibition-mediated PANoptosis
(A–D) Bone marrow-derived macrophages (BMDMs) transfected with NT (non-targeting) siRNA or the indicated gene-specific siRNAs were stimulated with the pharmacological inhibitor of TAK1 kinase activity (5z-7-oxozeaenol, TAK1i), and the samples were collected for immunoblot analyses at 12 h post-treatment. (A) Immunoblot analysis of pro- (P45) and activated (P20) caspase-1 (CASP1), pro- (P53) and activated (P30) gasdermin D (GSDMD), pro- (P53) and activated (P34) gasdermin E (GSDME), and β-Actin (P42) as the internal control. (B) Immunoblot analysis of activated caspase-8 (CASP8, P43 and P18), pro- (P35) and activated (P19/17) caspase-3 (CASP3), pro- (P35) and activated (P20) caspase-7 (CASP7), and β-Actin (P42) as the internal control. (C) Immunoblot analysis of the total (tMLKL) and phosphorylated (pMLKL) MLKL, and β-Actin (P42) as the internal control. (D) Immunoblot analysis of phosphorylated RIPK1 (pRIPK1), total RIPK1 (tRIPK1), and β-Actin (P42) as the internal control.
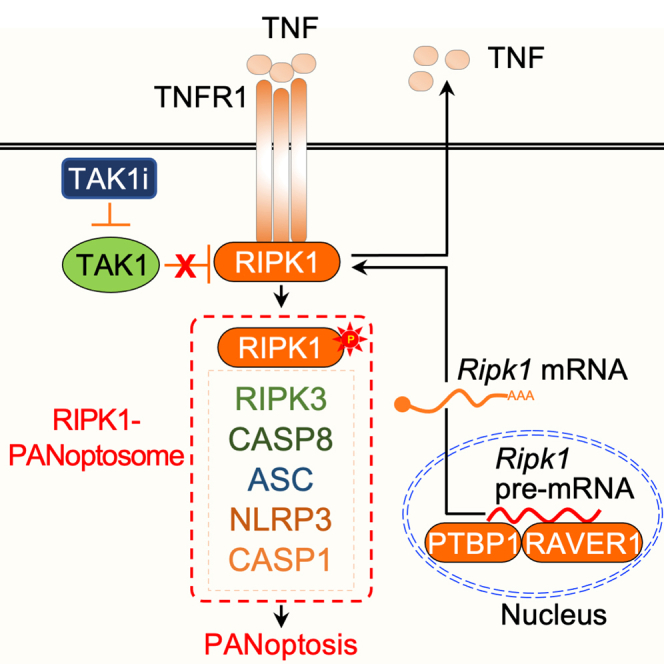
(E) Graphical representation of the key findings of the study, showing TNFR1-RIPK1 induced cell death. The uppercase “P” in immunoblots indicates protein molecular weight and the lower case “p” indicates phosphorylated form of the proteins. Data are representative of three independent experiments with n = 2–3 in each repeat (A–D). See also Figure S3.
PTBP1 and RAVER1 suppress the alternative splicing of Ripk1 to promote expression of the canonical Ripk1 transcript
We next sought to understand how the splicing factors PTBP1 and RAVER1 could regulate TAK1 inhibition-mediated PANoptosis. RIPK1 is known to be regulated at the level of splicing37; production of the canonical Ripk1 mRNA allows the induction of RIPK1-dependent cell death pathways, while alternative Ripk1 splicing results in a frameshift that introduces a stop codon and produces a non-functional version of the protein (Figures 4A and S4). Structural and sequence alignment of the full-length human RIPK1 protein sequence and the alternatively spliced murine homolog confirmed that the alternative splicing leads to a truncation of the C-terminal region and suggested there may be a change in the active site residues (Figure S5); these differences may result in structural rearrangements around the active site that could affect substrate binding specificities. A previous CRISPR screen focused on necroptosis regulators identified PTBP1 as a key splicing factor regulating the expression of the canonical RIPK1 transcript and protein.37 Since PTBP1 can interact with RAVER1 (another PTB domain-containing protein)39,40,41 and both are required for pre-mRNA splicing and alternative splicing, we hypothesized that PTBP1 and RAVER1 may act together in the Ripk1 mRNA splicing process to regulate the production of the canonical Ripk1 mRNA to induce cell death. To test this, we designed a pair of qPCR primers to amplify the alternate exon (alt-exon)-containing mRNA transcript (alt-transcript) of Ripk1 (Figures 4A and 4B), which is enriched in the absence of PTBP1.37 We found that there was a basal level of expression of the non-functional Ripk1 alt-transcript in control cells, and this alt-transcript expression was significantly increased in BMDMs treated with siRNA targeting Ptbp1, as expected (Figure 4B). Similarly, we found that siRNA-mediated knockdown of Raver1 also resulted in increased expression of the non-functional Ripk1 transcript, similar to that observed in PTBP1-deficient cells (Figures 4B and 4C). Consistently, siRNA-based knockdown of Ptbp1 or Raver1 also resulted in abrogation of expression and activation of RIPK1 (Figure 3D and Figure S3H). Together, these findings suggest that PTBP1 and RAVER1 regulate TAK1 inhibition-mediated PANoptosis by suppressing the alternative splicing of the Ripk1 pre-mRNA, thereby allowing the expression of the functional RIPK1 protein.
Figure 4.

PTBP1 and RAVER1 suppress the alternative splicing of Ripk1 to promote expression of the canonical Ripk1 transcript
(A) Diagrammatic representation of the mouse Ripk1 pre-mRNA exons and splicing variants, resulting in the production of the canonical and alternative mRNAs for RIPK1 translation. Primer binding sites for amplification of the alternative transcript are shown.
(B) qPCR-based quantification of the expression of the alternative Ripk1 transcript using two independent pairs of qPCR primers that are depicted as Forward primer, Reverse primer 1, and Reverse primer 2 in panel A. The primer pairs used in the qPCR reactions were: Primer pair 1, Forward primer + Reverse primer 1; and Primer pair 2, Forward primer + Reverse primer 2. The data are presented as relative expression normalized to the internal control of Actb mRNA levels.
(C) The Ripk1 canonical mRNA (spanning exons 4 and 5) and alternative mRNA (spanning exon 4 and the Alt-Exon) were PCR-amplified, and the PCR products were subjected to Sanger sequencing analyses. The resulting chromatograms are presented side-by-side for comparison. All data are presented as mean ± SEM, from three replicate samples (n = 3) (B). p < 0.05 is considered statistically significant. ∗∗p < 0.01; ∗∗∗p < 0.001 (two-tailed t test [B]). Data are representative of three independent experiments (B–C). See also Figures S4 and S5.
Discussion
The regulation of cell death pathways is critical to prevent infection, inflammatory syndromes, and cancers. TAK1 is a central hub in this regulation and acts upstream of RIPK1-dependent PANoptosis,20,21 which is of particular importance to fight against the highly pathogenic Gram-negative bacteria Yersinia. Understanding the molecular mechanisms of Yersinia-induced pathogenesis is crucial, as this pathogen has caused repeated pandemics over the millennia, resulting in the largest loss of human life to a pathogen in written history, and it remains a major threat with the emergence of antibiotic resistance.15,16,17,18,19 Yersinia carries virulence factors such as YopJ, which can inactivate TAK1 through deubiquitinating42 and/or acetyltransferase activities.13,43 However, host cells have evolved to sense this evasion mechanism and activate an alternate protective response; TAK1 inhibition unleashes RIPK1’s cell death function to drive inflammatory cell death, PANoptosis.20,21,22,23,24,44,45
RIPK1 itself is also a key regulator of innate immune processes, including TNFR1 signaling,46,47,48,49,50 TLR responses,51,52,53 inflammation, cellular homeostasis, and organismal development.47,54,55,56 As such, RIPK1 dysregulation often drives a spectrum of inflammatory diseases associated with sterile and infectious triggers.25,26,27,28,29,30,31,32,57,58,59,60,61 These disease connections make RIPK1 an attractive target for developing therapeutics. However, these efforts are complicated by RIPK1’s vital role in maintaining cell survival and organismal development, suggesting other regulators in the TAK1 pathway may serve as better targets. Our findings of critical roles for PTBP1 and RAVER1 in regulating TAK1 inhibition-mediated PANoptosis suggest these molecules could be viable alternatives for therapeutic targeting. Because PTBP1 and RAVER1 regulate the recently identified alt-splicing of Ripk1, targeting these molecules may reduce the total amount of canonical RIPK1 present to prevent inflammatory signaling and cell death without completely blocking the basal expression of RIPK1, which is essential for cellular and organismal homeostasis. A recent study reported that PTBP1 is associated with atherogenic inflammation and the plaque burden in patients; furthermore, PTBP1-dependent RIPK1 splicing contributes to enhanced TNF signaling and endothelial priming to promote vascular inflammation,62 further highlighting its potential as a therapeutic target. Moreover, our results also indicate that PTBP1 and RAVER1 may be recruited together to form a single spliceosome complex or function in the same pathway at different levels to regulate the alternative splicing of Ripk1. These results, together with the established roles of RIPK1 in a range of inflammatory diseases, make PTBP1 and RAVER1 highly promising alternative targets both in basic and clinical studies. However, targeting splicing factors may also affect the expression of other genes, and a cautious approach is warranted.
Our findings also demonstrated the utility of iBMDMs derived from Cas9-transgenic mice as a reliable and powerful macrophage model system for large-scale CRISPR-based whole-genome screens to identify regulators of innate immune signaling pathways and cell death. The iBMDM system, combined with other previous CRISPR screens focused on myeloid or macrophage cell types,33,63,64,65 provides an important avenue for continued investigations into innate immune pathways. Given the central roles of TAK1, RIPK1, and other regulators at the intersection of diverse innate immune functions and disease processes, it is critical to improve our mechanistic understanding of these pathways and identify specific strategies for modulation to achieve therapeutic benefits.
Limitations of the study
Our study identified several molecules required for positive regulation of TNFR1-RIPK1-dependent cell death. RIPK1 is known to critically regulate inflammation and cell death in diverse cell types, including both immune and non-immune cells, and in response to many sterile and infectious triggers. The focus of the current study was on macrophages, to establish the utility and validity of whole-genome CRISPR screening to identify regulators in both primary and immortalized macrophages, but it is likely that the splicing-dependent regulatory role of RAVER1 on full-length RIPK1 expression to control PANoptosis may also occur in other cell types and in response to diverse stimuli. Additionally, the expression patterns and functional implications of the alt-spliced Ripk1 mRNA product that results from loss of PTBP1 or RAVER1 should be further evaluated. Alt-splicing of Ripk1 results in the insertion of a stop codon that can produce a RIPK1 protein containing a non-functional truncated kinase domain. Our structural model indicated that this truncated kinase domain may form a complex with full-length RIPK1 to regulate its functions, and additional studies would be needed to conclusively validate this possibility. Moreover, the function of RAVER1 in RIPK1-dependent cell death and inflammation in vivo remains unknown and requires the generation of RAVER1-deficent mice, either conditionally or at the whole-genome level, to understand RAVER1’s specific functions in different tissues and cell types during homeostasis and disease.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-caspase-3 | Cell Signaling Technology | Cat# 9662; RRID:AB_331439 |
| anti-cleaved caspase-3 | Cell Signaling Technology | Cat# 9661; RRID:AB_2341188 |
| anti-caspase-7 | Cell Signaling Technology | Cat# 9492; RRID:AB_2228313 |
| anti-cleaved caspase-7 | Cell Signaling Technology | Cat# 9491; RRID:AB_2068144 |
| anti-caspase-8 | Adipogen | Cat# AG-20T-0138-C100; RRID:AB_2490519 |
| anti-cleaved caspase-8 | Cell Signaling Technology | Cat# 8592; RRID:AB_10891784 |
| anti-pMLKL | Cell Signaling Technology | Cat# 37333; RRID:AB_2799112 |
| anti-MLKL | Abgent | Cat# AP14272b; RRID:AB_11134649 |
| anti-pRIPK1 | Cell Signaling Technology | Cat# 31122; RRID:AB_2799000 |
| anti-RIPK1 | BD Transduction Laboratories | Cat# 610458; RRID:AB_397832 |
| anti-GSDMD | Abcam | Cat# ab209845; RRID:AB_2783550 |
| anti-GSDME | Abcam | Cat# ab215191; RRID:AB_2737000 |
| anti-caspase-1 | AdipoGen | Cat# AG-20B-0044; RRID: AB_2490253 |
| anti-β-actin | Santa Cruz | Cat# sc-47778 HRP; RRID:AB_2714189 |
| HRP-conjugated secondary anti-rabbit | Jackson ImmunoResearch Laboratories | Cat# 111-035-047; RRID:AB_2337940 |
| HRP-conjugated secondary anti-mouse | Jackson ImmunoResearch Laboratories | Cat# 315-035-047; RRID:AB_2340068 |
| Chemicals, peptides, and recombinant proteins | ||
| IMDM | Thermo Fisher Scientific | Cat# 12440053 |
| BMDM media | This paper | N/A |
| Fetal bovine serum | Biowest | Cat# S1620 |
| Non-essential amino acids | Thermo Fisher Scientific | Cat# 11140-050 |
| Penicillin and streptomycin | Thermo Fisher Scientific | Cat# 15070-063 |
| 5 mM penicillin and streptomycin | Gibco | Cat# 30-001-Cl |
| DPBS | Thermo Fisher Scientific | Cat# 14190-250 |
| Protease inhibitor | Roche | Cat# 11697498001 |
| Phosphatase inhibitor | Roche | Cat# 04906837001 |
| Forte western HRP substrate | Millipore | Cat# WBLUF055 |
| TAK1 inhibitor ((5Z)-7-Oxozeaenol) | Caymanchem | Cat# 17459 |
| Critical commercial assays | ||
| NucleoSpin Blood L | Macherey-Nagel | Cat# 740954.2 |
| Experimental models: Cell lines | ||
| Cas9-expressing iBMDMs | Current study | N/A |
| Experimental models: Organisms/strains | ||
| CRISPR-Cas9 Knockin mice | Platt et al., 2014 | JAX stock #024858 |
| Deposited data | ||
| NGS results from CRISPR screen | This study | BioProject; accession number PRJNA973658 |
| Software and algorithms | ||
| GraphPad Prism 8.0 and 9.0 | GraphPad Software, Inc. | https://www.graphpad.com/ |
| Seurat R package v3.1.4 | Satija et al., 2015 | N/A |
Resource availability
Lead contact
Further information and requests for reagents may be directed to, and will be fulfilled upon reasonable request by, the lead contact Thirumala-Devi Kanneganti (thirumala-devi.kanneganti@stjude.org).
Materials availability
All unique reagents generated in this study are available upon reasonable request from the lead contact.
Experimental model and study participant details
Mice
Male and female CRISPR-Cas9 Knockin mice (Rosa26-Cas9 knock in mouse, JAX stock #024858)66 and C57/BL6J control mice (WT) used in the current study at 6–10 weeks of age were bred at the Animal Resources Center at St. Jude Children’s Research Hospital and maintained under specific pathogen-free conditions. Mice were maintained with a 12 h light/dark cycle and were fed standard chow. Animal studies were conducted under protocols approved by the St. Jude Children’s Research Hospital Committee on the Use and Care of Animals (protocol 482).
Method details
Generation of whole-genome iBMDM-Brie cellular library
The management of the lentiviral Brie library and generation of the Brie-iBMDM libraries were discussed previously.35 In brief, the Mouse Brie CRISPR-KO library was a gift from David Root and John Doench (#73632 and #73633, Addgene36). The plasmid library was amplified and validated in the Center for Advanced Genome Engineering at St. Jude as described in the Broad GPP protocol, the only exception being the use of Endura DUOs electrocompetent E. coli cells for high efficiency transformation and reducing the gRNA dropouts. The St. Jude Hartwell Center Genome Sequencing Facility provided all NGS sequencing. Single end 100 cycle sequencing was performed on a NovaSeq 6000 (Illumina). Validation to check gRNA presence and representation was performed using calc_auc_v1.1.py (https://github.com/mhegde/) and count_spacers.py.67 Viral particles were produced by the St. Jude Vector Development and Production laboratory using the standard transfection methods involving the co-transfection of the lentiviral packaging plasmids along with the Brie library in the 293T cells. CRISPR KO screens were analyzed using MAGeCK-VISPR (version 0.5.7).68
Genome-wide CRISPR-Cas9 screen
Cas9-expressing iBMDMs were generated from CRISPR-Cas9 Knockin mice.66 A total of 300 × 106 Cas9-iBMDMs were distributed across 12 of the 15 cm2 tissue culture dishes (25 × 106 cells per dish) and infected with the Brie library of lentiviral particles at an MOI of 0.3 in 25 ml of complete DMEM (DMEM supplemented with 10% heat-inactivated fetal bovine serum (HI-FBS; S1620, Biowest)) supplemented with 100 μl of LentiBOOST transduction reagent (SB-P-LV-101-02, Lentivirus Transduction Enhancer Solution, Siron Biotech) and incubated for 24 h for efficient transduction. These transduced cells were expanded with intermittent passaging to avoid overcrowding of the cells and to generate a sufficient number of cells for the downstream whole-genome CRISPR-KO screens. Two replicates of an adequate number of cells were used as a control to obtain a representation (screen depth) of > 500 cells for each sgRNA of the library, and a similar number of iBMDMs from the same preparation of cells were treated with vehicle control (DMSO) or the TAK1 inhibitor 5z-7-oxozeaenol (5z7) (#17459, Cayman Chemical) at 100 nM final concentration for 24 h. The vehicle-treated control cell population and the surviving cells from the TAK1i-treated samples were subjected to CRISPR screen enrichment analysis. The total genomic DNA (gDNA) was isolated using NucleoSpin® Blood kits (740954 and 740950, Takara Bio Inc., USA) and the concentrations of gDNA were measured using NanoDrop (Thermo Fisher Scientific, USA). The MAGeCK pipeline69 was used to robustly estimate the log2fold change (FC) and significance of the enriched gRNA/genes from the cell death-based CRISPR screens. The ‘fgsea’ package in R was used to analyze the pathways enrichments. The pathways as gene sets were obtained from multiple databases including Biocarta, Reactome, Kegg and Wikipathway. The positive fold change of the gRNAs indicates the requirement of the corresponding genes in theTAK1i-induced cell death pathway.
Primary macrophage differentiation and stimulation
Freshly isolated murine bone marrow cells were plated into three 15 cm2 tissue culture plates, each containing a total of 20 ml of macrophage differentiation medium (BMDM medium). BMDM medium was prepared by supplementing IMDM (12440053, Thermo Fisher Scientific) with 30% L929 cell-conditioned medium, 10% HI-FBS, 1% nonessential amino acids (11140-050, Thermo Fisher Scientific) and 1% penicillin-streptomycin (15070-063, Thermo Fisher Scientific). An additional 5 ml of fresh BMDM medium was added to each plate on Day 3 and Day 5 to provide better growth and differentiation conditions. The fully matured macrophages on Day 6 were used for siRNA knockdown experiments and/or counted and seeded at 106cells per well in 12-well culture plates in DMEM containing 10% HI-FBS, 1% nonessential amino acids and 1% penicillin-streptomycin for the experimental stimulations on the following day. The Cas9-iBMDMs were maintained in complete DMEM as described above. Stimulations were performed with TAK1i (100 nM final concentration) for the indicated times as detailed in the figure legends.
Transfection
The nucleofector machine was used to transfect the fully differentiated BMDMs from WT mice (Neon™ Transfection System, 100 μL / reaction Kit, MPK10025, Thermo Scientific). For each of the knockdown reactions, a total of 1 × 106 BMDMs were transfected with 50 μM of siGENOME SMARTpool siRNA using the nucleofector settings of 1500 V and a single pulse at 20 mS to deliver the siRNA to the BMDMs. The cells were immediately transferred to 1 ml of warm complete DMEM and seeded in a well of the 12-well tissue culture plate to conduct the experimental stimulations. At 2 h post-transfection, each well was supplemented with an equal volume of warm BMDM medium to promote the recovery and growth. The BMDM medium was replaced with fresh medium at 24 h post-transfection, and the cells were rested for another 24 h, for a total of 48 h of siRNA transfection time, before subjecting them to experimental stimulations. The siGENOME SMARTpool siRNA specific for mouse Tnfrsf1a (M-060201-01-0005), Ripk1 (M-040150-00-0005), Ptbp1 (M-042865-01-0005) and Raver1 (M-064602-01-0005) were used in this study along with a non-targeting (NT) control siGENOME SMARTpool siRNA (D-001206-14-20).
Western blotting
The samples for Western blot analyses were prepared as previously described70: for caspase-1 immunoblotting, the samples were prepared by combining the cell lysates with culture supernatants (lysis buffer: 5% NP-40 solution in water supplemented with 10 mM DTT and protease inhibitor solution at 1× final concentration). The lysate samples for immunoblot analysis of all other proteins were prepared by lysing the cells in RIPA buffer, without combining with the supernatants. All these samples were mixed and denatured in loading buffer containing SDS and 100 mM DTT and boiled for 12 min. SDS-PAGE–separated proteins were transferred to PVDF membranes (IPVH00010, Millipore) using the Trans-Blot® Turbo™ system. Immunoblotting was performed with primary antibodies against caspase-1 (AG-20B-0042; Adipogen, 1:1000), caspase-3 (#9662, Cell Signaling Technology [CST], 1:1000), cleaved caspase-3 (#9661, CST, 1:1000), caspase-7 (#9492, CST, 1:1000), cleaved caspase-7 (#9491, CST, 1:1000), caspase-8 (#4927, CST, 1:1000), cleaved caspase-8 (#8592, CST, 1:1000), GSDMD (ab209845, Abcam, 1:1000), GSDME (ab19859, Abcam, 1:1000), pMLKL (#37333, CST, 1:1000), tMLKL (AP14242B, Abgent, 1:1000), tRIPK1 (#610458, BD Transduction Laboratories, 1: 1000), pRIPK1 (#31122, CST, 1: 1000), and β-Actin (sc-47778 HRP, Santa Cruz, 1:5000). Appropriate horseradish peroxidase (HRP)–conjugated secondary antibodies (anti-Armenian hamster [127-035-099], anti-mouse [315-035-047], and anti-rabbit [111-035-047], Jackson ImmunoResearch Laboratories) were used as described previously.71 Immunoblot images were acquired on an Amersham Imager using Immobilon® Forte Western HRP Substrate (WBLUF0500, Millipore).
Cell death analysis
Time-course analyses of cell death were performed using a two-color IncuCyte S3/5 incubator imaging system.71 Fully differentiated BMDMs either un-transfected or transfected with NT or gene-specific siRNAs were seeded in 12-well (1.0 × 106 cells/well) or 24-well (0.5 × 106 cells/well) tissue culture plates in the presence of propidium iodide (PI) (P3566, Life Technologies), which leaks into the dying cells and marks them as positive for lytic cell death. The cells were treated with vehicle control or TAK1 inhibitor as indicated in the figure legends. Time course image acquisition was carried out using the 20× objective, and the dead cells positive for PI-uptake were marked by the mask-application provided in the IncuCyte software. A minimum of four images per well for each condition and time point were acquired for the quantitative studies. The dead cells positive for PI-uptake were pseudo colored in red for the presentation of original imaging data in the figures. The percent dead cell counts for each of conditions were plotted using GraphPad Prism version 9.0 software.
RNA isolation and qPCR analyses
Total RNA was isolated using TRIzol® reagent (#15596018, Ambion). The concentration of the total RNA from each sample was measured using the NanoDrop (Thermo Fisher Scientific, USA), and 500 ng of total RNA from each sample was reverse transcribed to cDNA using High-capacity cDNA synthesis kit (#4368813, Applied Biosystems), as per the manufacturer’s instructions. The resulting cDNA was diluted in nuclease-free water to a final volume of 200 μl, and the cDNA was used at 2 or 4 μl/reaction in 10 or 20 μl qPCR reaction volumes, respectively. The cDNAs corresponding to the genes of interest were individually quantified using qPCR analyses based on the Sybr® Green chemistry (#4367659, Applied Biosystems) and the Quant Studio™ 7 Flex Real-Time PCR machine (Applied Biosystems). Primer sequences used are detailed in Table S1. The gene-specific expression levels were normalized to β-actin and presented as fold change in arbitrary units.
Structural modeling of RIPK1 isoforms
The murine mmRIPK1-X3 protein model was generated on the Robetta online portal (https://robetta.bakerlab.org) and superimposed with the human RIPK1-KD structure (PDB ID-7FD0) using Chimera. The sequence similarity representation was generated using Chimera.
Quantification and statistical analysis
GraphPad Prism version 8.0 and 9.0 software packages were used for data analyses. ImageJ, a Java-based image processing program from the National Institutes of Health, was used for quantification of the Western blot data. Data are presented as mean ± SEM. Statistical significance was determined by t tests (two-tailed) for two groups.
Acknowledgments
We thank all the members of the Kanneganti laboratory for their comments and suggestions during the development of this manuscript. We thank R. Tweedell, PhD, for scientific editing and writing support, and K. Combs and L. Kneeland for mouse colony support. Work from our laboratory is supported by the US National Institutes of Health (AI101935, AI124346, AI160179, AR056296, and CA253095 to T.-D.K.) and the American Lebanese Syrian Associated Charities (to T.-D.K.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author contributions
R.K.S.M. and T.-D.K. designed the study. R.K.S.M., R.B.R., R.M., and J.P.C. performed experiments. R.K.S.M., R.B.R., R.M., J.P.C., S.M.P.-M., and T.-D.K. analyzed the data. E.K.N. performed the structural and sequence alignment. R.K.S.M. and R.B.R. wrote the manuscript with input from all authors. T.-D.K. oversaw the project.
Declaration of interests
T.-D.K. was a consultant for Pfizer.
Published: May 22, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106938.
Supplemental information
Data and code availability
-
•
Next generation sequencing results from the CRISPR screen are deposited in BioProject: PRJNA973658. All other datasets generated or analyzed during this study are included in the published article and will be shared by the lead contact upon reasonable request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.
References
- 1.Aashaq S., Batool A., Andrabi K.I. TAK1 mediates convergence of cellular signals for death and survival. Apoptosis. 2019;24:3–20. doi: 10.1007/s10495-018-1490-7. [DOI] [PubMed] [Google Scholar]
- 2.Adhikari A., Xu M., Chen Z.J. Ubiquitin-mediated activation of TAK1 and IKK. Oncogene. 2007;26:3214–3226. doi: 10.1038/sj.onc.1210413. [DOI] [PubMed] [Google Scholar]
- 3.Ajibade A.A., Wang H.Y., Wang R.F. Cell type-specific function of TAK1 in innate immune signaling. Trends Immunol. 2013;34:307–316. doi: 10.1016/j.it.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 4.Ninomiya-Tsuji J., Kishimoto K., Hiyama A., Inoue J., Cao Z., Matsumoto K. The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature. 1999;398:252–256. doi: 10.1038/18465. [DOI] [PubMed] [Google Scholar]
- 5.Sato S., Sanjo H., Takeda K., Ninomiya-Tsuji J., Yamamoto M., Kawai T., Matsumoto K., Takeuchi O., Akira S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 2005;6:1087–1095. doi: 10.1038/ni1255. [DOI] [PubMed] [Google Scholar]
- 6.Schuman J., Chen Y., Podd A., Yu M., Liu H.H., Wen R., Chen Z.J., Wang D. A critical role of TAK1 in B-cell receptor-mediated nuclear factor kappaB activation. Blood. 2009;113:4566–4574. doi: 10.1182/blood-2008-08-176057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shinohara H., Kurosaki T. Comprehending the complex connection between PKCbeta, TAK1, and IKK in BCR signaling. Immunol. Rev. 2009;232:300–318. doi: 10.1111/j.1600-065X.2009.00836.x. [DOI] [PubMed] [Google Scholar]
- 8.Tang M., Wei X., Guo Y., Breslin P., Zhang S., Zhang S., Wei W., Xia Z., Diaz M., Akira S., Zhang J. TAK1 is required for the survival of hematopoietic cells and hepatocytes in mice. J. Exp. Med. 2008;205:1611–1619. doi: 10.1084/jem.20080297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wan Y.Y., Chi H., Xie M., Schneider M.D., Flavell R.A. The kinase TAK1 integrates antigen and cytokine receptor signaling for T cell development, survival and function. Nat. Immunol. 2006;7:851–858. doi: 10.1038/ni1355. [DOI] [PubMed] [Google Scholar]
- 10.Wang C., Deng L., Hong M., Akkaraju G.R., Inoue J., Chen Z.J. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 11.Arthur J.S.C., Ley S.C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013;13:679–692. doi: 10.1038/nri3495. [DOI] [PubMed] [Google Scholar]
- 12.Mukherjee S., Keitany G., Li Y., Wang Y., Ball H.L., Goldsmith E.J., Orth K. Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science. 2006;312:1211–1214. doi: 10.1126/science.1126867. [DOI] [PubMed] [Google Scholar]
- 13.Paquette N., Conlon J., Sweet C., Rus F., Wilson L., Pereira A., Rosadini C.V., Goutagny N., Weber A.N.R., Lane W.S., et al. Serine/threonine acetylation of TGFbeta-activated kinase (TAK1) by Yersinia pestis YopJ inhibits innate immune signaling. Proc. Natl. Acad. Sci. USA. 2012;109:12710–12715. doi: 10.1073/pnas.1008203109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chung L.K., Bliska J.B. Yersinia versus host immunity: how a pathogen evades or triggers a protective response. Curr. Opin. Microbiol. 2016;29:56–62. doi: 10.1016/j.mib.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bos K.I., Schuenemann V.J., Golding G.B., Burbano H.A., Waglechner N., Coombes B.K., McPhee J.B., DeWitte S.N., Meyer M., Schmedes S., et al. A draft genome of Yersinia pestis from victims of the Black Death. Nature. 2011;478:506–510. doi: 10.1038/nature10549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drancourt M., Raoult D. Molecular history of plague. Clin. Microbiol. Infect. 2016;22:911–915. doi: 10.1016/j.cmi.2016.08.031. [DOI] [PubMed] [Google Scholar]
- 17.Gilbert M.T.P. Yersinia pestis: one pandemic, two pandemics, three pandemics, more? Lancet Infect. Dis. 2014;14:264–265. doi: 10.1016/S1473-3099(14)70002-7. [DOI] [PubMed] [Google Scholar]
- 18.Klunk J., Vilgalys T.P., Demeure C.E., Cheng X., Shiratori M., Madej J., Beau R., Elli D., Patino M.I., Redfern R., et al. Evolution of immune genes is associated with the Black Death. Nature. 2022;611:312–319. doi: 10.1038/s41586-022-05349-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wagner D.M., Klunk J., Harbeck M., Devault A., Waglechner N., Sahl J.W., Enk J., Birdsell D.N., Kuch M., Lumibao C., et al. Yersinia pestis and the plague of Justinian 541-543 AD: a genomic analysis. Lancet Infect. Dis. 2014;14:319–326. doi: 10.1016/S1473-3099(13)70323-2. [DOI] [PubMed] [Google Scholar]
- 20.Malireddi R.K.S., Gurung P., Kesavardhana S., Samir P., Burton A., Mummareddy H., Vogel P., Pelletier S., Burgula S., Kanneganti T.D. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity-independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J. Exp. Med. 2020;217 doi: 10.1084/jem.20191644. jem.20191644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malireddi R.K.S., Gurung P., Mavuluri J., Dasari T.K., Klco J.M., Chi H., Kanneganti T.D. TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J. Exp. Med. 2018;215:1023–1034. doi: 10.1084/jem.20171922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Orning P., Weng D., Starheim K., Ratner D., Best Z., Lee B., Brooks A., Xia S., Wu H., Kelliher M.A., et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science. 2018;362:1064–1069. doi: 10.1126/science.aau2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sarhan J., Liu B.C., Muendlein H.I., Li P., Nilson R., Tang A.Y., Rongvaux A., Bunnell S.C., Shao F., Green D.R., Poltorak A. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl. Acad. Sci. USA. 2018;115:E10888–E10897. doi: 10.1073/pnas.1809548115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malireddi R.K.S., Kesavardhana S., Karki R., Kancharana B., Burton A.R., Kanneganti T.D. RIPK1 distinctly regulates yersinia-induced inflammatory cell death. Immunohorizons. 2020;4:789–796. doi: 10.4049/immunohorizons.2000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fritsch M., Günther S.D., Schwarzer R., Albert M.C., Schorn F., Werthenbach J.P., Schiffmann L.M., Stair N., Stocks H., Seeger J.M., et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature. 2019;575:683–687. doi: 10.1038/s41586-019-1770-6. [DOI] [PubMed] [Google Scholar]
- 26.Lalaoui N., Boyden S.E., Oda H., Wood G.M., Stone D.L., Chau D., Liu L., Stoffels M., Kratina T., Lawlor K.E., et al. Mutations that prevent caspase cleavage of RIPK1 cause autoinflammatory disease. Nature. 2020;577:103–108. doi: 10.1038/s41586-019-1828-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lukens J.R., Vogel P., Johnson G.R., Kelliher M.A., Iwakura Y., Lamkanfi M., Kanneganti T.D. RIP1-driven autoinflammation targets IL-1alpha independently of inflammasomes and RIP3. Nature. 2013;498:224–227. doi: 10.1038/nature12174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Newton K., Wickliffe K.E., Dugger D.L., Maltzman A., Roose-Girma M., Dohse M., Kőműves L., Webster J.D., Dixit V.M. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature. 2019;574:428–431. doi: 10.1038/s41586-019-1548-x. [DOI] [PubMed] [Google Scholar]
- 29.Newton K., Wickliffe K.E., Maltzman A., Dugger D.L., Reja R., Zhang Y., Roose-Girma M., Modrusan Z., Sagolla M.S., Webster J.D., Dixit V.M. Activity of caspase-8 determines plasticity between cell death pathways. Nature. 2019;575:679–682. doi: 10.1038/s41586-019-1752-8. [DOI] [PubMed] [Google Scholar]
- 30.Speir M., Nowell C.J., Chen A.A., O'Donnell J.A., Shamie I.S., Lakin P.R., D'Cruz A.A., Braun R.O., Babon J.J., Lewis R.S., et al. Ptpn6 inhibits caspase-8- and Ripk3/Mlkl-dependent inflammation. Nat. Immunol. 2020;21:54–64. doi: 10.1038/s41590-019-0550-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tao P., Sun J., Wu Z., Wang S., Wang J., Li W., Pan H., Bai R., Zhang J., Wang Y., et al. A dominant autoinflammatory disease caused by non-cleavable variants of RIPK1. Nature. 2020;577:109–114. doi: 10.1038/s41586-019-1830-y. [DOI] [PubMed] [Google Scholar]
- 32.Varfolomeev E.E., Schuchmann M., Luria V., Chiannilkulchai N., Beckmann J.S., Mett I.L., Rebrikov D., Brodianski V.M., Kemper O.C., Kollet O., et al. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 1998;9:267–276. doi: 10.1016/s1074-7613(00)80609-3. [DOI] [PubMed] [Google Scholar]
- 33.Zheng Z., Deng W., Bai Y., Miao R., Mei S., Zhang Z., Pan Y., Wang Y., Min R., Deng F., et al. The lysosomal rag-ragulator complex licenses RIPK1 and caspase-8-mediated pyroptosis by Yersinia. Science. 2021;372:eabg0269. doi: 10.1126/science.abg0269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spera I., Sánchez-Rodríguez R., Favia M., Menga A., Venegas F.C., Angioni R., Munari F., Lanza M., Campanella A., Pierri C.L., et al. The J2-immortalized murine macrophage cell line displays phenotypical and metabolic features of primary BMDMs in their M1 and M2 polarization state. Cancers. 2021;13:5478. doi: 10.3390/cancers13215478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karki R., Lee S., Mall R., Pandian N., Wang Y., Sharma B.R., Malireddi R.S., Yang D., Trifkovic S., Steele J.A., et al. ZBP1-dependent inflammatory cell death, PANoptosis, and cytokine storm disrupt IFN therapeutic efficacy during coronavirus infection. Sci. Immunol. 2022;7:eabo6294. doi: 10.1126/sciimmunol.abo6294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Callow M.G., Watanabe C., Wickliffe K.E., Bainer R., Kummerfield S., Weng J., Cuellar T., Janakiraman V., Chen H., Chih B., et al. CRISPR whole-genome screening identifies new necroptosis regulators and RIPK1 alternative splicing. Cell Death Dis. 2018;9:261. doi: 10.1038/s41419-018-0301-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor P.R., Martinez-Pomares L., Stacey M., Lin H.H., Brown G.D., Gordon S. Macrophage receptors and immune recognition. Annu. Rev. Immunol. 2005;23:901–944. doi: 10.1146/annurev.immunol.23.021704.115816. [DOI] [PubMed] [Google Scholar]
- 39.Keppetipola N.M., Yeom K.H., Hernandez A.L., Bui T., Sharma S., Black D.L. Multiple determinants of splicing repression activity in the polypyrimidine tract binding proteins, PTBP1 and PTBP2. RNA. 2016;22:1172–1180. doi: 10.1261/rna.057505.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rideau A.P., Gooding C., Simpson P.J., Monie T.P., Lorenz M., Hüttelmaier S., Singer R.H., Matthews S., Curry S., Smith C.W.J. A peptide motif in Raver1 mediates splicing repression by interaction with the PTB RRM2 domain. Nat. Struct. Mol. Biol. 2006;13:839–848. doi: 10.1038/nsmb1137. [DOI] [PubMed] [Google Scholar]
- 41.Wagner E.J., Garcia-Blanco M.A. Polypyrimidine tract binding protein antagonizes exon definition. Mol. Cell Biol. 2001;21:3281–3288. doi: 10.1128/MCB.21.10.3281-3288.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou H., Monack D.M., Kayagaki N., Wertz I., Yin J., Wolf B., Dixit V.M. Yersinia virulence factor YopJ acts as a deubiquitinase to inhibit NF-kappa B activation. J. Exp. Med. 2005;202:1327–1332. doi: 10.1084/jem.20051194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma K.W., Ma W. YopJ family effectors promote bacterial infection through a unique acetyltransferase activity. Microbiol. Mol. Biol. Rev. 2016;80:1011–1027. doi: 10.1128/MMBR.00032-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peterson L.W., Philip N.H., DeLaney A., Wynosky-Dolfi M.A., Asklof K., Gray F., Choa R., Bjanes E., Buza E.L., Hu B., et al. RIPK1-dependent apoptosis bypasses pathogen blockade of innate signaling to promote immune defense. J. Exp. Med. 2017;214:3171–3182. doi: 10.1084/jem.20170347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Philip N.H., Dillon C.P., Snyder A.G., Fitzgerald P., Wynosky-Dolfi M.A., Zwack E.E., Hu B., Fitzgerald L., Mauldin E.A., Copenhaver A.M., et al. Caspase-8 mediates caspase-1 processing and innate immune defense in response to bacterial blockade of NF-kappaB and MAPK signaling. Proc. Natl. Acad. Sci. USA. 2014;111:7385–7390. doi: 10.1073/pnas.1403252111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hsu H., Huang J., Shu H.B., Baichwal V., Goeddel D.V. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity. 1996;4:387–396. doi: 10.1016/s1074-7613(00)80252-6. [DOI] [PubMed] [Google Scholar]
- 47.Micheau O., Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 48.Shu H.B., Takeuchi M., Goeddel D.V. The tumor necrosis factor receptor 2 signal transducers TRAF2 and c-IAP1 are components of the tumor necrosis factor receptor 1 signaling complex. Proc. Natl. Acad. Sci. USA. 1996;93:13973–13978. doi: 10.1073/pnas.93.24.13973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stanger B.Z., Leder P., Lee T.H., Kim E., Seed B. RIP: a novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell. 1995;81:513–523. doi: 10.1016/0092-8674(95)90072-1. [DOI] [PubMed] [Google Scholar]
- 50.Vince J.E., Pantaki D., Feltham R., Mace P.D., Cordier S.M., Schmukle A.C., Davidson A.J., Callus B.A., Wong W.W.L., Gentle I.E., et al. TRAF2 must bind to cellular inhibitors of apoptosis for tumor necrosis factor (tnf) to efficiently activate nf-kappab and to prevent tnf-induced apoptosis. J. Biol. Chem. 2009;284:35906–35915. doi: 10.1074/jbc.M109.072256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Balachandran S., Thomas E., Barber G.N. A FADD-dependent innate immune mechanism in mammalian cells. Nature. 2004;432:401–405. doi: 10.1038/nature03124. [DOI] [PubMed] [Google Scholar]
- 52.Cusson-Hermance N., Khurana S., Lee T.H., Fitzgerald K.A., Kelliher M.A. Rip1 mediates the Trif-dependent toll-like receptor 3- and 4-induced NF-kappaB activation but does not contribute to interferon regulatory factor 3 activation. J. Biol. Chem. 2005;280:36560–36566. doi: 10.1074/jbc.M506831200. [DOI] [PubMed] [Google Scholar]
- 53.Meylan E., Burns K., Hofmann K., Blancheteau V., Martinon F., Kelliher M., Tschopp J. RIP1 is an essential mediator of Toll-like receptor 3-induced NF-kappa B activation. Nat. Immunol. 2004;5:503–507. doi: 10.1038/ni1061. [DOI] [PubMed] [Google Scholar]
- 54.Delanghe T., Dondelinger Y., Bertrand M.J.M. RIPK1 kinase-dependent death: a symphony of phosphorylation events. Trends Cell Biol. 2020;30:189–200. doi: 10.1016/j.tcb.2019.12.009. [DOI] [PubMed] [Google Scholar]
- 55.van Loo G., Bertrand M.J.M. Death by TNF: a road to inflammation. Nat. Rev. Immunol. 2023;23:289–303. doi: 10.1038/s41577-022-00792-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vandenabeele P., Declercq W., Van Herreweghe F., Vanden Berghe T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci. Signal. 2010;3:re4. doi: 10.1126/scisignal.3115re4. [DOI] [PubMed] [Google Scholar]
- 57.Place D.E., Lee S., Kanneganti T.D. PANoptosis in microbial infection. Curr. Opin. Microbiol. 2021;59:42–49. doi: 10.1016/j.mib.2020.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu D., Jin T., Zhu H., Chen H., Ofengeim D., Zou C., Mifflin L., Pan L., Amin P., Li W., et al. TBK1 suppresses RIPK1-driven apoptosis and inflammation during development and in aging. Cell. 2018;174:1477–1491. doi: 10.1016/j.cell.2018.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang J., Jin T., Aksentijevich I., Zhou Q. RIPK1-Associated inborn errors of innate immunity. Front. Immunol. 2021;12:676946. doi: 10.3389/fimmu.2021.676946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Karki R., Kanneganti T.D. The 'cytokine storm': molecular mechanisms and therapeutic prospects. Trends Immunol. 2021;42:681–705. doi: 10.1016/j.it.2021.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tummers B., Green D.R. The evolution of regulated cell death pathways in animals and their evasion by pathogens. Physiol. Rev. 2022;102:411–454. doi: 10.1152/physrev.00002.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hensel J.A., Nicholas S.A.E., Kimble A.L., Nagpal A.S., Omar O.M.F., Tyburski J.D., Jellison E.R., Ménoret A., Ozawa M., Rodriguez-Oquendo A., et al. Splice factor polypyrimidine tract-binding protein 1 (Ptbp1) primes endothelial inflammation in atherogenic disturbed flow conditions. Proc. Natl. Acad. Sci. USA. 2022;119 doi: 10.1073/pnas.2122227119. e2122227119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Evavold C.L., Hafner-Bratkovič I., Devant P., D'Andrea J.M., Ngwa E.M., Boršić E., Doench J.G., LaFleur M.W., Sharpe A.H., Thiagarajah J.R., Kagan J.C. Control of gasdermin D oligomerization and pyroptosis by the Ragulator-Rag-mTORC1 pathway. Cell. 2021;184:4495–4511. doi: 10.1016/j.cell.2021.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Parnas O., Jovanovic M., Eisenhaure T.M., Herbst R.H., Dixit A., Ye C.J., Przybylski D., Platt R.J., Tirosh I., Sanjana N.E., et al. A genome-wide CRISPR screen in primary immune cells to dissect regulatory networks. Cell. 2015;162:675–686. doi: 10.1016/j.cell.2015.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schmid-Burgk J.L., Chauhan D., Schmidt T., Ebert T.S., Reinhardt J., Endl E., Hornung V. A genome-wide CRISPR (clustered regularly interspaced short palindromic repeats) screen identifies NEK7 as an essential component of NLRP3 inflammasome activation. J. Biol. Chem. 2016;291:103–109. doi: 10.1074/jbc.C115.700492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Platt R.J., Chen S., Zhou Y., Yim M.J., Swiech L., Kempton H.R., Dahlman J.E., Parnas O., Eisenhaure T.M., Jovanovic M., et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159:440–455. doi: 10.1016/j.cell.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Joung J., Konermann S., Gootenberg J.S., Abudayyeh O.O., Platt R.J., Brigham M.D., Sanjana N.E., Zhang F. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 2017;12:828–863. doi: 10.1038/nprot.2017.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li W., Köster J., Xu H., Chen C.H., Xiao T., Liu J.S., Brown M., Liu X.S. Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol. 2015;16:281. doi: 10.1186/s13059-015-0843-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li W., Xu H., Xiao T., Cong L., Love M.I., Zhang F., Irizarry R.A., Liu J.S., Brown M., Liu X.S. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014;15:554. doi: 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Malireddi R.K.S., Bynigeri R.R., Kancharana B., Sharma B.R., Burton A.R., Pelletier S., Kanneganti T.D. Determining distinct roles of IL-1alpha through generation of an IL-1alpha knockout mouse with no defect in IL-1beta expression. Front. Immunol. 2022;13:1068230. doi: 10.3389/fimmu.2022.1068230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tweedell R.E., Malireddi R.K.S., Kanneganti T.D. A comprehensive guide to studying inflammasome activation and cell death. Nat. Protoc. 2020;15:3284–3333. doi: 10.1038/s41596-020-0374-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Next generation sequencing results from the CRISPR screen are deposited in BioProject: PRJNA973658. All other datasets generated or analyzed during this study are included in the published article and will be shared by the lead contact upon reasonable request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.