

Significance
Cardiac muscle contraction is finely regulated by the concerted action of thin- and thick-filaments. Pathogenic variants in cardiac thin-filament (cTF) proteins associated with cardiomyopathies dysregulate physiological processes of cardiac muscle regulation. We provide structural and functional evidence that a pathogenic variant in cardiac troponin T loop-region of the N-terminal tail domain, which interacts with the actin backbone, destabilizes cTF relaxed state. This initial trigger culminates in multiple mechanical, sarcomere structural, and energetic alterations at the myofilament level. Integration of cTF structure coupled to muscle biophysics and biochemistry allows us to determine 1) the role of cardiac troponin T loop-region in muscle regulation; and 2) the intricate molecular mechanisms of a troponin T tail-domain variant in the development of cardiomyopathy.
Keywords: cardiac thin filament, cardiac troponin T tail domain, myosin SRX/DRX, TnT1 loop region, cardiomyopathy
Abstract
Missense variant Ile79Asn in human cardiac troponin T (cTnT-I79N) has been associated with hypertrophic cardiomyopathy and sudden cardiac arrest in juveniles. cTnT-I79N is located in the cTnT N-terminal (TnT1) loop region and is known for its pathological and prognostic relevance. A recent structural study revealed that I79 is part of a hydrophobic interface between the TnT1 loop and actin, which stabilizes the relaxed (OFF) state of the cardiac thin filament. Given the importance of understanding the role of TnT1 loop region in Ca2+ regulation of the cardiac thin filament along with the underlying mechanisms of cTnT-I79N-linked pathogenesis, we investigated the effects of cTnT-I79N on cardiac myofilament function. Transgenic I79N (Tg-I79N) muscle bundles displayed increased myofilament Ca2+ sensitivity, smaller myofilament lattice spacing, and slower crossbridge kinetics. These findings can be attributed to destabilization of the cardiac thin filament’s relaxed state resulting in an increased number of crossbridges during Ca2+ activation. Additionally, in the low Ca2+-relaxed state (pCa8), we showed that more myosin heads are in the disordered-relaxed state (DRX) that are more likely to interact with actin in cTnT-I79N muscle bundles. Dysregulation of the myosin super-relaxed state (SRX) and the SRX/DRX equilibrium in cTnT-I79N muscle bundles likely result in increased mobility of myosin heads at pCa8, enhanced actomyosin interactions as evidenced by increased active force at low Ca2+, and increased sinusoidal stiffness. These findings point to a mechanism whereby cTnT-I79N weakens the interaction of the TnT1 loop with the actin filament, which in turn destabilizes the relaxed state of the cardiac thin filament.
Contraction in striated muscle is a well-coordinated process governed by Ca2+/adenosine triphosphate (ATP)-dependent cyclic interactions between the two sarcomeric filamentous systems—thick and thin filaments (1). Actomyosin crossbridges generate force, causing thick and thin filaments to slide past each other and sarcomere shortening (1). The velocity of sarcomere shortening varies with load, where at zero load, the sarcomeres shorten at their maximal speed (2). Thick filaments are mainly comprised of myosin, myosin-binding protein C, and titin. Thin filaments are comprised of actin, tropomyosin, and the troponin complex. The troponin complex is formed by three distinct subunits: troponin C (TnC), the Ca2+-binding subunit; troponin I (TnI), the inhibitory subunit; and troponin T (TnT), the anchoring subunit. Recently, the three-dimensional organization of the cardiac thin filament has been resolved (Fig. 1A) (3, 4). Cardiac troponin T (cTnT) (Fig. 1A, cyan ribbons) is the largest troponin subunit and plays major structural roles: i) anchoring the troponin complex to the thin filament through interactions between its C-terminal region (TnT2) with TnC (Fig. 1A, black ribbons), TnI (Fig. 1A, blue ribbons), actin (Fig. 1A, tan ribbons), and Tm (Fig. 1A, green and yellow ribbons); ii) orienting the cTn core domain relative to the direction of the thin filament; and iii) stabilizing the tropomyosin head-to-tail region (Fig. 1A, black bracket) (3–10). Structural studies demonstrated that the loop region of TnT1 interacts with the actin backbone of the cardiac thin filament via ionic bonds (Fig. 1B) and hydrophobic interactions (Fig. 1C) that may stabilize the relaxed (OFF) state of the cardiac thin filament (10). Given the importance of understanding cTnT’s role in the regulatory properties of the cardiac thin filament, considerable effort has been directed toward investigating cTnT function, specifically the TnT2 region (residues 201 to 288) (11–14). However, less experimental scrutiny has been directed toward the TnT1 region (residues 1 to 168). In particular, the TnT1 loop region’s intimate contribution to cardiac thin-filament regulation remains poorly understood.
Fig. 1.
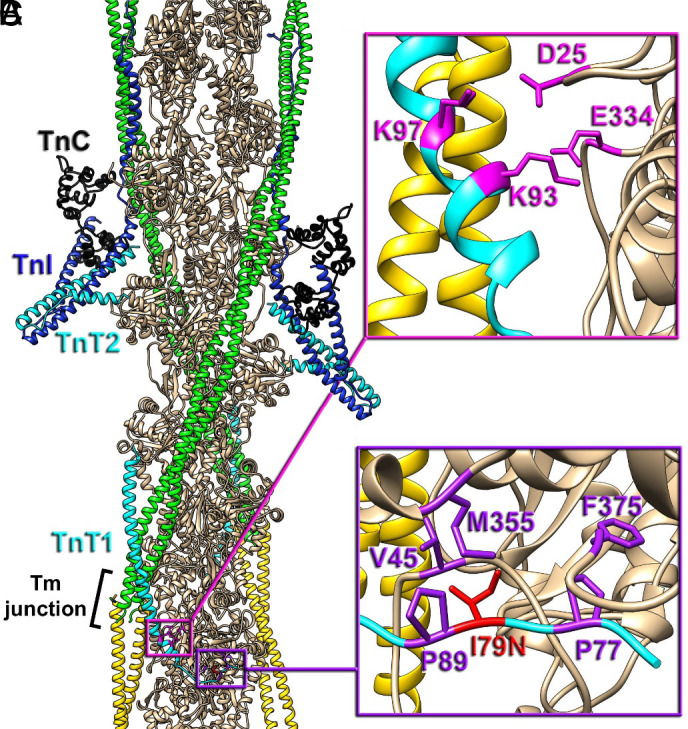
Positioning of cTnT-I79N within the cardiac TF. (A) The molecular organization of the cardiac TF in relaxed state (3, 10). Actin subunits (tan) are shown with a pair of Tm cables (green and yellow) and a pair of Tn complexes comprised of TnC N- and C-lobes (black), TnI (blue), and TnT (cyan) with its N-terminal region (TnT1) extended to the Tm junction region (black bracket). The TnT1 ionic (magenta box detailed in panel B) and hydrophobic interactions (purple box detailed in panel C with actin) anchor the Tm junction region to the actin filament to presumably stabilize the relaxed state of the thin filament (10). (B) Ionic interactions of TnT1 K93 and K97 with E334 and D25 of actin (magenta atoms) link TnT1 helix to the actin surface. (C) Positioning of TnT1 is further stabilized by the hydrophobic interactions between V45, M355, F375 actin residues and P77, I79, P89 TnT1 residues (purple atoms). cTnT-I79 (red atoms), when replaced to N79 presumably disrupts the hydrophobic cluster and weakens the interaction of TnT1 with actin.
Missense variants in sarcomeric protein genes are the main cause of familial hypertrophic cardiomyopathy (HCM), which is a Mendelian disorder characterized by structural and functional abnormalities in heart muscle (2, 15). Among the thin filament–related genes, variants in cardiac troponin T gene (TNNT2) are the main cause of familial HCM (16, 17). A recent meta-analysis concluded that TnT1 is an important region of pathologic and prognostic relevance (15). Patients bearing variants located in within the range of amino acids 73 to 80 showed significantly higher incidence of sudden cardiac death and/or ventricular fibrillation (15). The Ile79Asn (cTnT-I79N) (Fig. 1C, red atoms) is one of these devastating pathogenic variants (18, 19).
Several studies using reconstituted myofilament proteins, transgenic mouse models, and human-induced pluripotent stem cell–derived-cardiomyocytes (hiPSC-CMs) have demonstrated that cTnT-I79N leads to a broad spectrum of abnormalities (20–26). Permeabilized cardiac muscle preparations reconstituted with cTnT-I79N exhibited moderate increases in the Ca2+ sensitivity of force development (23). Knollmann et al. demonstrated that Tg-I79N hearts have significantly impaired diastolic function, increased diastolic chamber stiffness, and blunted response to isoproterenol-induced inotropic stimulation (21). In vitro motility assays showed that thin filaments containing cTnT-I79N moved faster than the control group at both low and saturating Ca2+ levels (24). Wang et al. investigated the effect of cTnT-I79N on cTnC’s affinity for Ca2+ in hiPSC-CMs and reported that cTnT-I79N exhibited an increase in myofilament Ca2+ buffering (25). Shafaattalab et al. later confirmed in human cardiac recombinant/reconstituted thin filaments that cTnT-I79N decreases the Ca2+ OFF-rate constant (kOFF) from thin filaments with a concomitant increase of Ca2+ binding affinity (26). Despite these seminal results, little is known about cTnT-I79N’s underlying mechanisms of pathogenesis. Here, we sought to investigate the mechanisms underlying abnormal contractility. Specifically, we provide detailed information concerning the primal biochemical and structural alterations at both thin- and thick-filament levels that together contribute to the elevated degree of contractile dysfunction and pathogenesis of HCM.
In a recent structural study, we showed that cTnT-I79 contributes to the hydrophobic interaction of the TnT1 loop with actin, stabilizing the relaxed state of the cardiac thin filament (10). To investigate the consequences of the structurally proposed cTnT-I79N-driven weakening of the I79 interaction with actin, we used permeabilized left-ventricular papillary muscle obtained from a cTnT-I79N-bearing transgenic mouse model. We interrogated the variant’s effects (and their respective mechanisms) on myofilament function, as a function of sarcomere length (SL), using mechanical, biochemical, and structural measurements. Our findings suggest that weakening of TnT1 loop region–actin interactions by the cTnT-I79N variant results in destabilization of the relaxed state of the cardiac thin filament, allowing more force-producing crossbridges to form at low Ca2+ levels. Comparing genotypes, the increase in crossbridge population detected by sinusoidal stiffness (SS) is associated with an increase in mobility of myosin heads as evidenced by the decrease of the intensity of the third-order myosin-based meridional reflection (IM3). This enhanced recruitment of myosin heads at low Ca2+ levels is associated with a shift in the crossbridge SRX↔DRX equilibrium toward greater numbers of disordered heads free to interact with actin. Enhanced crossbridge formation reduces the myofilament lattice spacing, slowing crossbridge kinetics. Based on our data, we propose that the cTnT-I79N’s primary underlying mechanism of myofilament dysfunction is the destabilization of the resting (OFF) state of the cardiac thin filament by a perturbation of the TnT1 loop region–actin interaction. This primary thin-filament dysregulation translates into a series of biochemical and structural alterations at the thick-filament level that contribute to the overall altered muscle function and pathogenesis of HCM.
Results
Tg-I79N Myocardium Displays Reduced Length-Dependent Activation and Increased Active Force at Low Ca2+.
It has been reported that Tg-I79N mice exhibit abnormal cardiac contractile and electrical properties, and no response to β-adrenergic stimulation (21, 22). To date, very little is known about the underlying mechanisms by which this pathogenic variant induces cardiac malfunction. Considering that altered length-dependent activation is a common feature in human hearts bearing HCM-related variants in myofilament proteins (27), we sought to investigate the effects of the I79N variant on permeabilized cardiac myofilament function with changes in SL.
It is expected that, under normal physiological conditions, both force production and myofilament Ca2+ sensitivity would increase upon stretch of the cardiac sarcomere [length-dependent activation (LDA)]. LDA is an intrinsic property of cardiomyocytes related to the Frank–Starling mechanism (28). We observed that Tg-I79N myocardium exhibited greater force generation at physiological Ca2+ levels (pCa 6.5 to 5.8) compared to NTg (Fig. 2 A and B). Although Tg-I79N showed a trend toward increased maximum absolute force, no statistically significant changes were observed between NTg and Tg-I79N at both SLs (Fig. 1C and SI Appendix, Table S1). In addition, we observed that both NTg and Tg-I79N exhibit significant increases in maximum steady-state isometric force production with increasing SL (from 2.1 µm to 2.3 µm) (Fig. 2C and SI Appendix, Table S1). Upon cardiac muscle stretch, both groups showed an increase in active force at low Ca2+ levels (pCa 8, resting condition). However, Tg-I79N displayed a significant increase in active force at low Ca2+ levels as compared to NTg at both SLs (Fig. 2 D and E and SI Appendix, Table S1).
Fig. 2.

Effects of the cTnT-I79N variant on length dependence of Ca2+ sensitivity. (A and B) pCa-force curves at different SL (2.1 µm and 2.3 µm). Force levels are normalized to the cross-sectional area of the cardiac muscle preparations. (C) Maximal steady-state isometric force values at different SLs. (D and E) Absolute active force levels at low Ca2+ levels (pCa 8). Absolute force values were normalized by the cross-sectional area of the cardiac muscle preparation. (F and G) pCa-relative force plots at different SLs: The force levels are relative to the maximum steady-state isometric force exhibited by each fiber. (H) Myofilament Ca2+ sensitivity response upon muscle stretch. Data are shown as average (AVG) ± standard error (SE) (NTg, n = 8; Tg-I79N, n = 8); statistical significance was determined using Student’s t test or one-way ANOVA with Tukey’s post-hoc analysis; *P < 0.05, **P < 0.01, ***P < 0.001 within the same genotype, and #P < 0.05, ##P < 0.01, ###P < 0.01 between genotypes.
Tg-I79N also displayed a significant increase in myofilament Ca2+ sensitivity of steady-state isometric force production as compared to NTg at both SLs (Fig. 2 F–H and SI Appendix, Table S1). Interestingly, Tg-I79N showed no significant increase in myofilament Ca2+ sensitivity upon stretch, i.e., SL 2.1 vs. 2.3 µm, while NTg showed a significant leftward shift in the pCa x force plot (Fig. 2 F–H and SI Appendix, Fig. S1 A and B and Table S1). There were no statistically significant differences in cooperativity of cardiac thin-filament activation (nHill) among groups (Fig. 2 F–H and SI Appendix, Table S1). These results suggest that the cTnT-I79N variant blunts LDA, disrupts the resting OFF state of the thin filament, and increases force production at low levels of Ca2+ activation.
Tg-I79N Displays Abnormal Cardiac Muscle Mechanics beyond Isometric Force.
Next, we used measurements of SS to determine whether the I79N variant could influence the overall recruitment of crossbridges. We observed that both groups, especially Tg-I79N, exhibited significant increase in SS upon SL variation (SI Appendix, Fig. S1 C and D). Compared to NTg, Tg-I79N showed a significant increase in overall number of crossbridges at all levels of Ca2+ activation (Fig. 3 A–D and SI Appendix, Figs. S1 C and D and S2 and Table S1). Then, we evaluated the effects of the cTnT-I79N variant on crossbridge cycling kinetics by measuring the kinetics of tension redevelopment (kTR). For each genotype, no changes in maximum kTR were observed upon cardiac muscle stretch. However, Tg-I79N exhibited significantly slower submaximum and maximum kTR when compared to NTg (Fig. 3 E–G and SI Appendix, Figs. S1 E and F and S3 and Table S1). In accordance with previous studies (29–33), we observed that NTg exhibits Ca2+ dependence of kTR at both SLs. Interestingly, Tg-I79N presented a flat Ca2+ dependence of kTR at both SLs (Fig. 3 E and F and SI Appendix, Figs. S1 E and F and S3).
Fig. 3.

Effects of the cTnT-I79N variant on the mechanics of cardiac muscle contraction. (A and B) Normalized steady-state isometric force vs. SS plots at different SLs. Bar plots of both (C) maximal and (D) low Ca2+ (pCa 8) stiffness values at different SLs. (E and F) Normalized steady-state isometric force vs. kinetics of tension redevelopment (kTR) plots at different SLs. (G) Bar plot of the maximum kTR values upon muscle stretch. All force levels are normalized to the maximum steady-state isometric force exhibited at SL 2.1 µm. Data are shown as AVG ± SE (NTg, n = 6 to 8; Tg-I79N, n = 7 to 8). Statistical significance was determined using one-way ANOVA with Tukey’s post-hoc analysis; *P < 0.05, **P < 0.01, ***P < 0.001 within the same genotype, and #P < 0.05, ##P < 0.01, ###P < 0.01 between genotypes.
Based on Brenner’s kinetic model (34) in which Ca2+ plays an important role on crossbridge kinetics by regulating fapp (transition from the nonforce-producing to the force-producing state) and with no effects on gapp (transition from the force-producing to the nonforce-producing state), combined with the observation that cTnT-I79N variant alters the ability of Ca2+ to modulate kTR, we hypothesized that cTnT-I79N could be influencing the crossbridge attachment rate (f). To test our hypothesis, we estimated the crossbridge attachment (f) and detachment rates (g), and thin-filament kOFF (Ca2+ OFF-rate constant) by using a three-state model of cardiac muscle contraction that models the relationship between steady-state isometric force and kTR (29, 35). This model is an adaptation of the four-state model that was originally developed for modeling force-kTR data obtained from skeletal muscle contraction (29, 36). Within each genotype, f increased as the SL increased. And as hypothesized, f was found to be altered (much lower) for Tg-I79N relative to NTg (Table 1). Within each genotype, g appeared to be essentially constant upon stretch with no major differences between groups (Table 1). Within each genotype, the increase of pCa50 upon stretch was associated with a decrease in kOFF, as expected. Comparing NTg vs. Tg-I79N, we observed that Tg-I79N displayed an approximately twofold reduction in kOFF. Interestingly, from SL 2.1 µm to 2.3 µm, NTg exhibited an ~18% decrease in kOFF, while Tg-I79N only decreased ~11%, which might explain Tg-I79N’s lack of myofilament Ca2+ sensitization (SI Appendix, Fig. S1 A and B and Table S1 and Table 1).
Table 1.
Optimized parameter predictions and estimates from the three-state model for force-kTR data depicted in Fig. 3
| Three-state model-fitted parameters | ||||||||
|---|---|---|---|---|---|---|---|---|
| Crossbridge | Regulatory unit | Three-state model predictions | ||||||
| SL (µm) | f (s−1) | g (s−1) | kON (M−1 s−1) (37) | kOFF (s−1) | pCa50 | Max force (norm) | Max kTR(s−1) | |
| NTg | 2.1 | 9.1 | 23.9 | 1.84 × 108 | 623.9 | 5.63 | 0.83 | 32.4 |
| 2.3 | 12.4 | 25.0 | 1.84 × 108 | 519.8 | 5.74 | 1.00 | 36.6 | |
| Tg-I79N | 2.1 | 1.1 | 24.2 | 1.84 × 108 | 266.9 | 5.87 | 0.90 | 24.7 |
| 2.3 | 1.4 | 26.8 | 1.84 × 108 | 238.6 | 5.92 | 1.00 | 26.8 | |
All best-fit parameter estimates for f (attachment rate), g (detachment rate), and kOFF (Ca2+ dissociation rate) were obtained in MatLab by using the simplex method for the force-kTR data shown in Fig. 3. The simplex method considers the values of myofilament Ca2+ sensitivity (pCa50) from steady-state isometric force measurements as reported in Fig. 2; values for kON were derived from measurements reported by Pinto et al. (37), and maximum steady-state isometric force at SL 2.3 µm was assumed to be 1.0 for each dataset. These parameter sets were used to fit and illustrate relations between kTR and steady-state isometric force in Fig. 3 (dashed lines in Fig. 3 E and F).
cTnT-I79N Variant Does Not Alter Cardiac Thin-Filament Length.
Considering that thin-filament length is key in maintaining the responsiveness of cardiac myofilaments to stretch, we hypothesized that the myofilament’s lack of responsiveness to stretch in Tg-I79N could be due to differences in thin-filament length between NTg and Tg-I79N. However, there were no statistical differences in thin-filament length between the groups (Fig. 4 A and B and SI Appendix, Fig. S2 A and B).
Fig. 4.

Lack of effect of the cTnT-I79N variant on the cardiac thin-filament length and sarcomere ultrastructure. (A) Representative image of the phalloidin staining. (B) Measurements of cardiac thin-filament length per myofibril. Data are shown as individual points; NTg, n = 4 animals; Tg-I79N, n = 4 animals. (C) Representative image of the sarcomere’s ultrastructure in longitudinal sections of left ventricular papillary muscle obtained from NTg and TG-I79N mice. SI Appendix, Table S2 contains the average of the width of the A-band, I-band, Z-line, and SL for each group. Sarcomere’s ultrastructure measurements: NTg, n = 150 to 193; Tg-I79N, n = 144 to 175.
Tg-I79N Exhibits Shortened Width of the A-Band, I-Band, and SL.
Ultrastructure analysis by electron microscopy revealed alterations in the Tg-I79N-containing sarcomeres compared to NTg (Fig. 4C and SI Appendix, Table S2). Analysis of the width of A-band and I-band from sarcomeres in longitudinal sections showed a statistically significant reduction in the width of both bands in Tg-I79N sarcomeres compared to NTg. Additionally, Tg-I79N-containing sarcomeres exhibited significantly shorter SL compared to NTg. No major changes were observed in the thickness of the Z-disk between the groups.
Tg-I79N Displays Reduced Interfilament Lattice Spacing and Alters the Relative Ordering of Myosin Heads.
Next, we sought to investigate whether Tg-I79N cardiac bundles exhibit altered spatial organization of the thick and thin filaments within the sarcomere using small-angle X-ray diffraction (Fig. 5A). Within each genotype, myofilament lattice spacing decreased upon stretch (Fig. 5B and SI Appendix, Table S3) as expected (38). In addition, Tg-I79N displayed a significantly smaller interfilament lattice spacing as compared to NTg at both SLs (Fig. 5B and SI Appendix, Table S3). Both NTg and Tg-I79N showed increases in the equatorial intensity ratio, I11/I10, with sarcomere stretch from 1.9 µm to 2.1 µm. However, no significant changes in I11/I10 were observed from SL 2.1 µm to 2.3 µm in either genotype (Fig. 5C and SI Appendix, Fig. S3 and Table S3). Furthermore, no changes in IM3 were seen upon sarcomere stretch. However, Tg-I79N exhibited a statistically significant reduction in IM3 as compared to NTg at both SLs (Fig. 5D and SI Appendix, Table S3), suggesting an increase in the axial angular dispersion of myosin heads (39).
Fig. 5.

Effects of the cTnT-I79N variant on myofilament lattice spacing and relative ordering of myosin heads. Measurements of myofilament lattice spacing in permeabilized cardiac muscle preparations at different SLs. (A) Small-angle X-ray diffraction patterns. (B) Myofilament lattice spacing measurements upon change in SL. (C) Intensity ratio of the equatorial reflections I1.1 and I1.0. (D) Intensity of the meridional M3 reflection. Data are shown as AVG ± SE; NTg, n = 19; Tg-I79N, n = 10; statistical significance was assessed by one-way ANOVA with Tukey’s post-hoc analysis; ***P < 0.001 within the same genotype, and #P < 0.05, ###P < 0.01 between genotypes.
Tg-I79N Exhibited No Major Alterations in Myofibril Orientation.
Recently, our group demonstrated that X-ray diffraction can be a powerful tool for directly assessing myofibril orientation in cardiac muscle under near-physiological conditions (40). A decrease in the angular divergence of the 1,0 equatorial reflection indicates a better alignment of the sarcomeres relative to the longitudinal axis of the cardiac muscle preparation. Here, we investigated the cardiac myofibril orientation upon stretch in permeabilized, relaxed, left ventricular papillary muscle from NTg and Tg-I79N by calculating the angular spread of the 1,0-equatorial reflection (angle σ). Within each genotype, σ decreased significantly upon sarcomere stretch (Fig. 6A and SI Appendix, Table S4). No significant differences were observed between the two groups at any SL. Next, we calculated the width of σ for the 1,0 reflection to assess the degree of heterogeneity in lattice spacing between myofibrils. Tg-I79N’s width of σ significantly increased when the sarcomeres were stretched from 2.1 µm to 2.3 µm (Fig. 6B and SI Appendix, Table S4). Altogether, these results indicate that both NTg and Tg-I79N exhibited greater myofibril alignment with the longitudinal axis upon muscle stretch. In addition, our data showed that the Tg-I79N myocardium does not suffer from increased myofibril disorientation as compared to NTg.
Fig. 6.

Effects of the cTnT-I79N variant on myofibril orientation. (A) The angular SD of 1,0 equatorial reflection as a function of SL. (B) The SD of the 1,0 equatorial reflections in radial direction as a function of SL. Data are shown as AVG ± SE; myofibril orientation: NTg, n = 23 to 25; Tg-I79N, n = 10. Statistical significance was determined using One-way ANOVA with Tukey’s post-hoc analysis; *P < 0.05, and *** P < 0.001 within the same genotype.
Tg-I79N Exhibits a Dysregulated SRX ↔ DRX Equilibrium.
We tested the impact of cTnT-I79N variant on the energetic states of myosin heads using previously described single-nucleotide turnover assays (41). Disordered-relaxed (DRX) and super-relaxed (SRX) states of myosin were determined by the pulse-chase turnover rate of fluorescent mant-ATP to nonfluorescent ATP in permeabilized left ventricular papillary muscle. Fig. 7A shows representative data illustrating displacement of mant-ATP by ATP, where the fast phase (P1) and slow phase (P2) of fluorescence decay represent mant-ATP displacement from myosin heads. Compared to NTg, the magnitude of P1 increased significantly in Tg-I79N (84% Tg-I79N vs. 71% NTg) (Fig. 7B and SI Appendix, Table S5). The magnitude of P2 was lower in Tg-I79N (12%) compared to that of NTg (23%) (Fig. 7C and SI Appendix, Table S5). The approximate percent of myosin heads occupying the DRX state was significantly higher in Tg-I79N (40%, Tg-I79N vs. 35%, NTg) (Fig. 7D and SI Appendix, Table S5), while SRX was significantly lower in Tg-I79N compared to NTg (60%, Tg-I79N vs. 65%, NTg) (Fig. 7E and SI Appendix, Table S5).
Fig. 7.

Effects of the cTnT-I79N variant on the fraction of DRX and SRX myosin populations. (A) Mant-ATP decay assay performed in permeabilized left ventricular papillary muscle of NTg and Tg-I79N mice at SL ~2.1 µm. Mant-ATP fluorescence decay in (B) fast phase (P1) and (C) slow phase (P2). Approximate percent of myosin heads in DRX (D) and SRX (E) was calculated using 0.48 as the correction factor to account for washout of nonmyosin-bound Mant-ATP (42): DRX (%) = P1 (%) * 0.48; SRX (%) = 100 – DRX (%). Data are shown as AVG ± SE (NTg, n = 7; Tg-I79N, n = 6 to 7). Statistical significance was assessed by one-way ANOVA with Tukey’s post-hoc analysis; ##P < 0.01 between genotypes.
Absence of Posttranslational Changes in Cardiac Microtubule Network, cTnI-Pser23/24, cMyBPC-Pser282, and RLC-P.
Contemporaneous studies have shown the importance of posttranslational modifications (PTMs) of the cardiac microtubule network—especially C-terminal detyrosination of α-tubulin—on cardiac muscle contractility and stiffness (43–45). Here, we tested whether the greater stiffness observed in the Tg-I79N myocardium could be due to alterations in PTMs in the microtubule network of the heart via western blots. Although we observed significant increase in both total α- and β-tubulin (when normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH)) in Tg-I79N myocardium, no changes in acetylated nor detyrosinated α-tubulin were observed compared to NTg (SI Appendix, Fig. S6). These findings support the proposed mechanism in which the greater stiffness seen in Tg-I79N at low Ca2+ is due to enhanced crossbridge population. Next, we sought to investigate whether cTnT-I79N variant affects the phosphorylation status of cTnI-Pser23/24, cMyBPC-Pser282, and regulatory light-chain (RLC, RLC-P). The total protein levels of both cTnI and cMyBP-C (when normalized to GAPDH) were elevated in Tg-I79N myocardium compared to NTg (SI Appendix, Fig. S7). However, no statistically significant differences were observed for cTnI-Pser23/24, and cMyBPC-Pser282 between genotypes (SI Appendix, Fig. S7). Phos-tag followed by immunoblotting analysis of RLC-P showed that the RLC phosphorylation ratio is unchanged between NTg and Tg-I79N (SI Appendix, Fig. S8).
Discussion
In this study, we used a transgenic mouse model of HCM (Tg-I79N) to determine the underlying mechanisms for how the cTnT-I79N variant leads to pathogenesis. In particular, we provide explicit evidence for structural, biochemical, and functional alterations caused by the cTnT-I79N variant that collectively culminate in myofilament dysfunction.
TnT1 and Its Pathologic Relevance.
Cardiac TnT is composed of the N-terminal domain (TnT1, residues 1 to 168), linker domain (residues 169 to 200), and C-terminal domain (TnT2, 201 to 288) (6). TnT is well known for its structural and modulatory roles as it binds to tropomyosin and actin, as well as tethering TnI and TnC to the thin filament (6, 46, 47). Previously, the TnT1 region has been shown to modulate thin-filament regulation as it inhibits the actin-tropomyosin-activated S1 ATPase by stabilizing the inhibited position of tropomyosin on F-actin (46). Consistently, TnT1 has been shown to be a fundamental component of the modulation of submaximal and maximal activation of cardiac myofilaments by its interaction with tropomyosin (48). Considering the evidence for a role of the TnT1 region in cardiac thin-filament regulation, it is not surprising that variants located in this region would disrupt this well-regulated system, and consequently lead to disease (49–51). Recently, a meta-analysis study showed that TnT1 is a region of pathologic and prognostic relevance. More specifically, patients bearing variants located in between the residues 73 and 80 showed significantly higher incidence of sudden cardiac death and ventricular fibrillation (15). Among this group of variants is cTnT-I79N, located specifically at the TnT1 loop region (Fig. 1 A–C, red atoms). A recent structural study directly showed that, in addition to TnT1 interactions with Tm, the TnT1 loop region makes hydrophobic interactions with actin, which presumably stabilize the intrinsic relaxed (OFF) state of the cardiac thin filaments. Here, we present a unified model for the pathogenesis of an HCM-associated TNNT2 variant (cTnT-I79N) located in the TnT1 loop region.
Primary Mechanism of Myofilament Dysfunction: cTnT-I79N Reduces Stabilization of the Relaxed (OFF) State of Cardiac Thin Filaments.
A recent structural study demonstrated that the cTnT-I79 is part of the hydrophobic interface between cTnT and actin, suggesting that the presence of the cTnT-I79N variant could disrupt the TnT1 loop–actin interaction (52). Consequently, it would lead to destabilization of the intrinsic relaxed (OFF) state of the cardiac thin filament. Structural data predict that cTnT-I79N-containing thin filaments should display higher sensitivity to Ca2+. Such effects have been previously observed in reconstituted myofilaments (22, 26), hiPSC-CMs (25), and a transgenic mouse model of cTnT-I79N (21). Here, we show that the cTnT-I79N variant increases the Ca2+ sensitivity of steady-state isometric force production regardless of the SL (Fig. 2). Our mathematical modeling predicts that Tg-I79N displays an approximately twofold reduction in the OFF rate of Ca2+ from cTnC compared to NTg (Table 1), which agrees with results reported in previous studies (22, 25, 26). The reduction in kOFF supports our findings that show an increase in myofilament Ca2+ sensitivity (29, 37). Previous in vitro experiments performed with reconstituted thin filaments also demonstrated an increase in myofilament Ca2+ sensitivity, and a slight reduction in kOFF in the presence of cycling myosin S1 using fluorescently labeled thin filament at 23 °C (53). TnT1’s regulatory role in cardiac thin filament inhibition has been previously demonstrated (46, 51), and reducing the stability of the relaxed (OFF) state of cardiac thin filaments leads to dysregulation of thin-filament activation (54, 55). Here, we show that Tg-I79N dysregulates the OFF state of the thin filament leading to a significant increased actomyosin interaction under low Ca2+ conditions, as evidenced by the observed increase in both active force and SS. Enhanced actomyosin interaction at low Ca2+ levels may be expected to lead to the observed increases in force under contracting conditions.
Previous studies showed that titin has phosphorylation sites that are substrates for protein kinase A (PKA) (56), G (PKG) (57), and D (PKD) (58), and that titin-mediated cardiac stiffness is intracellularly modulated by titin phosphorylation and oxidation, and/or titin isoform switching (58, 59). Moreover, there are two titin adult forms: N2B (shorter protein that results in the highest stiffness) and N2BA (longer protein that results in intermediate level of stiffness) (60). It has been demonstrated that the expression ratio between the isoforms N2B and N2BA determines the titin-mediated cardiac passive stiffness, with amplified N2B/N2BA ratios resulting in increased myocardium stiffness (61). Therefore, despite the evidence presented here that indicate an increase in force-generating crossbridge at diastolic Ca2+ levels, we do not exclude the possibility of alterations in titin’s phosphorylation status and/or isoform switch in Tg-I79N myocardium.
Destabilization of the Relaxed State of Cardiac Thin Filaments Is Associated with Increased Numbers of Disordered Myosin Heads Able to Bind to Actin.
Proper regulation of the intrinsic inhibitory properties of the cardiac thin filament is of monumental importance to prevent inappropriate formation of force-producing crossbridges. Consistent with the structural prediction that cTnT-I79N may destabilize the relaxed state of cardiac thin filaments, our data suggest that the reduced stability of the relaxed state allows more myosin heads to interact with actin by increasing the availability of activated regions of the thin filament. Our single-nucleotide turnover assays showed that myosin molecules in the cTnT-I79N variant–containing muscle bundles exhibit a shift in the SRX/DRX equilibrium toward increasing numbers of heads in the (biochemically defined) DRX state (Fig. 7). This result is consistent with our X-ray data that suggest that more myosin heads are leaving their helically ordered arrangement close to the thick-filament backbone, assumed to correspond to SRX heads, becoming disordered, and moving toward the vicinity of the thin filaments in the presence of the cTnT-I79N variant, consistent with increased numbers of DRX heads (Fig. 5 and SI Appendix, Fig. S3). Previous studies have predicted upon a structural model (62) and demonstrated the effects of thick-filament variants on the SRX↔DRX equilibrium (62–67). Here, we report using a mouse model, that variants in thin filament–associated proteins can result in altered energetic states of myosin heads.
It has been proposed that variants in other thin filament proteins (TnT and TnI) may affect the SRX/DRX equilibrium (68). Using human in-silico models of the variants TNNT2R92Q/+ and TNNI3R21C/+, Margara et al. (68) predicted that HCM-linked thin-filament variants lead to a decrease in DRX myosins in contrast to our experimental findings in murine myocardium–containing cTnT-I79N, which displays an increase in DRX myosins. A consistent finding from experimental X-ray studies of thick filament–based mutations (64, 69) associated with HCM, however, is an increase in DRX/ON state myosins under relaxed conditions, so the predictions of Margara et al. (68) are surprising. In silico predictions, however, at their present state of development, while very valuable as hypothesis-generating tools, do not take into consideration several aspects of the disease that can be captured in an animal model. One is tissue remodeling caused by cardiomyocyte death, fibrosis, fibrofatty infiltration, inflammatory response, and other events that altogether can result in altered muscle function. Another possible source of divergent results is the location of specific variants cTnT-R92Q, cTnI-R21C, and cTnT-I79N in different proteins or different residues of the same protein that effect SRX/DRX equilibrium in different ways. This hypothesis can be tested in future studies in SRX/DRX measurements using animal models of disease and human samples carrying thin-filament variants.
Proposed Mechanism for Increased DRX in HCM-Associated Thin-Filament Variants.
Myosin heads in the ON (DRX) state are constantly “checking” the thin filament in search of available binding sites on actin. Near the actin filament, DRX heads can be electrostatically attracted to and bind weakly to actin under relaxed conditions (70, 71). We show that disruption of an interaction between TnT and actin leads to diminished inhibition of the cardiac thin filament at low Ca2+. The latter allows DRX myosin heads in the vicinity of the thin filament to not only form weakly bound crossbridges, but to also progress to form force-generating crossbridges at low Ca2+. Enhanced strong crossbridge formation shrinks the myofilament lattice, which in turn, promotes weakly bound crossbridge formation that is driven by the electrostatic forces between actin and nearby myosin heads. As ON (DRX) state heads are being pulled out of the DRX head pool by binding to actin, this might be expected to shift the OFF (SRX)/ON (DRX) equilibrium toward the ON state, facilitating subsequent crossbridge formation and force generation. Fig. 8 summarizes this proposed mechanism.
Fig. 8.

Proposed mechanism on how cTnT-I79N variant increases DRX myosin population. The cTnT-I79N variant destabilizes the interaction between TnT1 loop and actin, leading to thin-filament disinhibition at low Ca2+ levels (Fig. 1). Under normal physiological circumstances, a few ON (DRX) heads are checking the thin filaments and will be close enough to be electrostatically attracted to actin-binding sites. As ON (DRX) state heads are being pulled out of the DRX head pool by binding to actin, this might be expected to shift the OFF (SRX)/ON (DRX) equilibrium toward the ON state, facilitating subsequent crossbridge formation and force generation, even at low Ca2+.
Smaller Myofilament Lattice Spacing Influences the Kinetics of Muscle Contraction.
As discussed above, cTnT-I79N muscle preparations exhibited slower crossbridge kinetics, and our mathematical modeling predicted a decrease in myosin-to-actin attachment rate (Fig. 3E and Table 1). These results can be explained by our X-ray data that showed a significantly reduced myofilament lattice spacing in cTnT-I79N myocardium (Fig. 5). Previously, we have demonstrated in a mouse model of HCM that crossbridge kinetics is directly modulated by the expansion of the myofilament lattice (the larger the distance between the myofilaments, the faster the crossbridges cycle) (29). Moreover, a study done in Drosophila flight muscle demonstrated that the crossbridge kinetics are slower when the myofilament lattice spacing is reduced (72). Therefore, destabilization of the relaxed state of cardiac thin filament could allow increased formation of crossbridges, which may bring the myofilaments closer to each other, and consequently slow the kinetics of crossbridge cycling. Our findings further support the idea that myofilament lattice spacing is a component of the regulation of the crossbridge kinetics.
Conclusion
We conclude that the primary mechanism by which the cTnT-I79N located in the TnT1 loop region leads to myofilament dysfunction is due to destabilization of the relaxed state of cardiac thin filaments (Fig. 8). Presumably, the latter arises from the destabilization of the interaction between TnT1 loop region with actin. This disruption of proper cardiac thin-filament regulation further results in a series of biochemical and structural modifications at the thick-filament level that contribute to altered muscle function, especially hypercontractility at physiologically relevant Ca2+ levels, and pathogenesis of HCM-linked cTnT-I79N (Fig. 8 and SI Appendix, Fig. S9).
Methods
Experimental Animals.
All protocols and experimental procedures followed both NIH and American Heart Association guidelines and were approved by Florida State University’s Animal Care and Use Committee. For this study, we used a previously established transgenic mouse model of HCM with overexpression of the mutated human cardiac TnT driven by the alpha-myosin heavy chain promoter (Tg-I79N, mouse line 8) and nontransgenic (NTg) littermates as its respective control (22). The age of the mice used from both the experimental and control groups ranged from 3 to 5 mo (males and females, all from the same genetic background).
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We would like to acknowledge Dr. Kenneth A. Taylor for the critical review of the manuscript. We would like to thank Elise Wight and Dr. Timothy Megraw for providing the α-tubulin detyrosinated antibody. This work was supported by the National Heart, Lung and Blood Institute HL128683 (to J.R.P.), HL160966 (to V.E. G., P.B.C., and J.R.P.), R35 HL144980 (to B.C.K.), and R01HL12150 (to C.A.C.O.) and the National Institute of Arthritis and Musculoskeletal and Skin Disease R21 AR077802 (to H.R. and J.R.P.). American Heart Association Fellowship training grants supported T.S. (19POST34380448) and M.L.-V. (2021AHAPRE216237). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. BioCAT was supported by grant P30 GM138395 (to T.C.I.) from the National Institute of General Medical Sciences of the NIH. S.S. has received support from NIH grants R01 AR079435, R01 AR079477, R01 AR078001, R01 HL130356, R01 HL105826, R38 HL155775, and R01 HL143490; the American Heart Association 2019 Institutional Undergraduate Student (19UFEL34380251) and Transformation (19TPA34830084 and 945748) awards; the Phospholamban (PLN) Foundation (PLN crazy idea); and the Leducq Foundation (Transatlantic Network 18CVD01).
Author contributions
M.L.-V., T.C.I., V.E.G., P.B.C., and J.R.P. designed research; M.L.-V., W.M., T.S., H.R., H.G., I.L.C., S.J.P.B., S.P.C., M.P., and V.E.G. performed research; C.A.C.O., H.S.H., B.C.K., S.S., T.C.I., P.B.C., and J.R.P. contributed new reagents/analytic tools; M.L.-V., W.M., T.S., H.R., H.G., I.L.C., C.A.C.O., T.C.I., P.B.C., and J.R.P. analyzed data; and M.L.-V., W.M., T.S., C.A.C.O., T.C.I., V.E.G., P.B.C., and J.R.P. wrote the paper.
Competing interests
J.R.P. provides consulting to Kate Therapeutics, but such work is unrelated to the content of this article. S.S. provides consulting and collaborative research studies to the Leducq Foundation (CURE-PLaN), Red Saree Inc., Greater Cincinnati Tamil Sangam, Novo Nordisk, Pfizer, AavantioBio, Affinia Therapeutics Inc., Cardiocare Genetics - Cosmogene Skincare Pvt. Ltd., AstraZeneca, MyoKardia, Merck and Amgen, but such work is unrelated to the content of this article. T.C.I. provides consulting and collaborative research studies to Edgewise Therapeutics, but such work is unrelated to the content of this article.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Gordon A. M., Homsher E., Regnier M., Regulation of contraction in striated muscle. Physiol. Rev. 80, 853–924 (2000). [DOI] [PubMed] [Google Scholar]
- 2.van der Velden J., Stienen G. J. M., Cardiac disorders and pathophysiology of sarcomeric proteins. Physiol. Rev. 99, 381–426 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Risi C. M., et al. , The structure of the native cardiac thin filament at systolic Ca(2+) levels. Proc. Natl. Acad. Sci. U.S.A. 118, e2024288118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yamada Y., Namba K., Fujii T., Cardiac muscle thin filament structures reveal calcium regulatory mechanism. Nat. Commun. 11, 153 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jackson P., Amphlett G. W., Perry S. V., The primary structure of troponin T and the interaction with tropomyosin. Biochem. J. 151, 85–97 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marston S., Zamora J. E., Troponin structure and function: A view of recent progress. J. Muscle Res. Cell Motil. 41, 71–89 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murakami K., et al. , Structural basis for tropomyosin overlap in thin (actin) filaments and the generation of a molecular swivel by troponin-T. Proc. Natl. Acad. Sci. U.S.A. 105, 7200–7205 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pearlstone J. R., Smillie L. B., The binding site of skeletal alpha-tropomyosin on troponin-T. Can J. Biochem. 55, 1032–1038 (1977). [DOI] [PubMed] [Google Scholar]
- 9.Perry S. V., Troponin T: Genetics, properties and function. J. Muscle Res. Cell Motil. 19, 575–602 (1998). [DOI] [PubMed] [Google Scholar]
- 10.Risi C. M., et al. , High-resolution cryo-EM structure of the junction region of the native cardiac thin filament in relaxed state. Proc. Natl. Acad. Sci. U.S.A. Nexus 2, pgac298 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Franklin A. J., Baxley T., Kobayashi T., Chalovich J. M., The C-terminus of troponin T is essential for maintaining the inactive state of regulated actin. Biophys. J. 102, 2536–2544 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson D., et al. , Eliminating the first inactive state and stabilizing the active state of the cardiac regulatory system alters behavior in solution and in ordered systems. Biochemistry 59, 3487–3497 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson D., Zhu L., Landim-Vieira M., Pinto J. R., Chalovich J. M., Basic residues within the cardiac troponin T C terminus are required for full inhibition of muscle contraction and limit activation by calcium. J. Biol. Chem. 294, 19535–19545 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnston J. R., et al. , The intrinsically disordered C terminus of troponin T binds to troponin C to modulate myocardial force generation. J. Biol. Chem. 294, 20054–20069 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tadros H. J., et al. , Meta-analysis of cardiomyopathy-associated variants in troponin genes identifies loci and intragenic hot spots that are associated with worse clinical outcomes. J. Mol. Cell Cardiol. 142, 118–125 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Messer A. E., et al. , Mutations in troponin T associated with hypertrophic cardiomyopathy increase Ca(2+)-sensitivity and suppress the modulation of Ca(2+)-sensitivity by troponin I phosphorylation. Arch. Biochem. Biophys. 601, 113–120 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pasquale F., et al. , Long-term outcomes in hypertrophic cardiomyopathy caused by mutations in the cardiac troponin T gene. Circ. Cardiovasc Genet. 5, 10–17 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Watkins H., et al. , Mutations in the genes for cardiac troponin T and alpha-tropomyosin in hypertrophic cardiomyopathy. N. Engl. J. Med. 332, 1058–1064 (1995). [DOI] [PubMed] [Google Scholar]
- 19.Thierfelder L., et al. , Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: A disease of the sarcomere. Cell 77, 701–712 (1994). [DOI] [PubMed] [Google Scholar]
- 20.Eschenhagen T., Mummery C., Knollmann B. C., Modelling sarcomeric cardiomyopathies in the dish: From human heart samples to iPSC cardiomyocytes. Cardiovasc Res. 105, 424–438 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knollmann B. C., et al. , Inotropic stimulation induces cardiac dysfunction in transgenic mice expressing a troponin T (I79N) mutation linked to familial hypertrophic cardiomyopathy. J. Biol. Chem. 276, 10039–10048 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Miller T., et al. , Abnormal contractile function in transgenic mice expressing a familial hypertrophic cardiomyopathy-linked troponin T (I79N) mutation. J. Biol. Chem. 276, 3743–3755 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Szczesna D., et al. , Altered regulation of cardiac muscle contraction by troponin T mutations that cause familial hypertrophic cardiomyopathy. J. Biol. Chem. 275, 624–630 (2000). [DOI] [PubMed] [Google Scholar]
- 24.Lin D., Bobkova A., Homsher E., Tobacman L. S., Altered cardiac troponin T in vitro function in the presence of a mutation implicated in familial hypertrophic cardiomyopathy. J. Clin. Invest. 97, 2842–2848 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang L., et al. , Hypertrophic cardiomyopathy-linked mutation in troponin T causes myofibrillar disarray and pro-arrhythmic action potential changes in human iPSC cardiomyocytes. J. Mol. Cell Cardiol. 114, 320–327 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shafaattalab S., et al. , Mechanisms of arrhythmogenicity of hypertrophic cardiomyopathy-associated Troponin T (TNNT2) variant I79N. Front. Cell Dev. Biol. 9, 787581 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sequeira V., et al. , Perturbed length-dependent activation in human hypertrophic cardiomyopathy with missense sarcomeric gene mutations. Circ. Res. 112, 1491–1505 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Tombe P. P., et al. , Myofilament length dependent activation. J. Mol. Cell Cardiol. 48, 851–858 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gonzalez-Martinez D., et al. , Structural and functional impact of troponin C-mediated Ca(2+) sensitization on myofilament lattice spacing and cross-bridge mechanics in mouse cardiac muscle. J. Mol. Cell Cardiol. 123, 26–37 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palmer S., Kentish J. C., Roles of Ca2+ and crossbridge kinetics in determining the maximum rates of Ca2+ activation and relaxation in rat and guinea pig skinned trabeculae. Circ. Res. 83, 179–186 (1998). [DOI] [PubMed] [Google Scholar]
- 31.Vannier C., Chevassus H., Vassort G., Ca-dependence of isometric force kinetics in single skinned ventricular cardiomyocytes from rats. Cardiovasc Res. 32, 580–586 (1996). [PubMed] [Google Scholar]
- 32.Wolff M. R., McDonald K. S., Moss R. L., Rate of tension development in cardiac muscle varies with level of activator calcium. Circ. Res. 76, 154–160 (1995). [DOI] [PubMed] [Google Scholar]
- 33.Marques M. A., et al. , Anomalous structural dynamics of minimally frustrated residues in cardiac troponin C triggers hypertrophic cardiomyopathy. Chem. Sci. 12, 7308–7323 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brenner B., Effect of Ca2+ on cross-bridge turnover kinetics in skinned single rabbit psoas fibers: Implications for regulation of muscle contraction. Proc. Natl. Acad. Sci. U.S.A. 85, 3265–3269 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loong C. K., Takeda A. K., Badr M. A., Rogers J. S., Chase P. B., Slowed dynamics of thin filament regulatory units reduces Ca(2+)-sensitivity of cardiac biomechanical function. Cell Mol. Bioeng. 6, 183–198 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Regnier M., Rivera A. J., Chase P. B., Smillie L. B., Sorenson M. M., Regulation of skeletal muscle tension redevelopment by troponin C constructs with different Ca2+ affinities. Biophys. J. 76, 2664–2672 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pinto J. R., et al. , Strong cross-bridges potentiate the Ca(2+) affinity changes produced by hypertrophic cardiomyopathy cardiac troponin C mutants in myofilaments: A fast kinetic approach. J. Biol. Chem. 286, 1005–1013 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Irving T. C., Konhilas J., Perry D., Fischetti R., de Tombe P. P., Myofilament lattice spacing as a function of sarcomere length in isolated rat myocardium. Am. J. Physiol. Heart Circ. Physiol. 279, H2568–2573 (2000). [DOI] [PubMed] [Google Scholar]
- 39.Huxley H., Reconditi M., Stewart A., Irving T., X-ray interference studies of crossbridge action in muscle contraction: Evidence from muscles during steady shortening. J. Mol. Biol. 363, 762–772 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Ma W., et al. , Myofibril orientation as a metric for characterizing heart disease. Biophys. J. 121, 565–574 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McNamara J. W., et al. , Ablation of cardiac myosin binding protein-C disrupts the super-relaxed state of myosin in murine cardiomyocytes. J. Mol. Cell Cardiol. 94, 65–71 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hooijman P., Stewart M. A., Cooke R., A new state of cardiac myosin with very slow ATP turnover: A potential cardioprotective mechanism in the heart. Biophys. J. 100, 1969–1976 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Caporizzo M. A., Chen C. Y., Bedi K., Margulies K. B., Prosser B. L., Microtubules increase diastolic stiffness in failing human cardiomyocytes and myocardium. Circulation 141, 902–915 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen C. Y., et al. , Suppression of detyrosinated microtubules improves cardiomyocyte function in human heart failure. Nat. Med. 24, 1225–1233 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robison P., et al. , Detyrosinated microtubules buckle and bear load in contracting cardiomyocytes. Science 352, aaf0659 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maytum R., Geeves M. A., Lehrer S. S., A modulatory role for the troponin T tail domain in thin filament regulation. J. Biol. Chem. 277, 29774–29780 (2002). [DOI] [PubMed] [Google Scholar]
- 47.Hinkle A., Goranson A., Butters C. A., Tobacman L. S., Roles for the troponin tail domain in thin filament assembly and regulation. A deletional study of cardiac troponin T. J. Biol. Chem. 274, 7157–7164 (1999). [DOI] [PubMed] [Google Scholar]
- 48.Chandra M., Montgomery D. E., Kim J. J., Solaro R. J., The N-terminal region of troponin T is essential for the maximal activation of rat cardiac myofilaments. J. Mol. Cell Cardiol. 31, 867–880 (1999). [DOI] [PubMed] [Google Scholar]
- 49.Tardiff J. C., Sarcomeric proteins and familial hypertrophic cardiomyopathy: Linking mutations in structural proteins to complex cardiovascular phenotypes. Heart Fail Rev. 10, 237–248 (2005). [DOI] [PubMed] [Google Scholar]
- 50.Morimoto S., Sarcomeric proteins and inherited cardiomyopathies. Cardiovasc. Res. 77, 659–666 (2008). [DOI] [PubMed] [Google Scholar]
- 51.Madan A., et al. , TNNT2 mutations in the tropomyosin binding region of TNT1 disrupt its role in contractile inhibition and stimulate cardiac dysfunction. Proc. Natl. Acad. Sci. U.S.A. 117, 18822–18831 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Risi C. M., et al. , High-resolution cryo-EM structure of the junction region of the native cardiac thin filament in relaxed state. Proc. Natl. Acad. Sci. U.S.A. 2, pgac298 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sommese R. F., et al. , Effects of troponin T cardiomyopathy mutations on the calcium sensitivity of the regulated thin filament and the actomyosin cross-bridge kinetics of human beta-cardiac myosin. PLoS One 8, e83403 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Davis J., Wen H., Edwards T., Metzger J. M., Thin filament disinhibition by restrictive cardiomyopathy mutant R193H troponin I induces Ca2+-independent mechanical tone and acute myocyte remodeling Circ. Res. 100, 1494–1502 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Wen Y., et al. , Functional consequences of the human cardiac troponin I hypertrophic cardiomyopathy mutation R145G in transgenic mice. J. Biol. Chem. 283, 20484–20494 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamasaki R., et al. , Protein kinase A phosphorylates titin’s cardiac-specific N2B domain and reduces passive tension in rat cardiac myocytes. Circ. Res. 90, 1181–1188 (2002). [DOI] [PubMed] [Google Scholar]
- 57.Kruger M., et al. , Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ. Res. 104, 87–94 (2009). [DOI] [PubMed] [Google Scholar]
- 58.Herwig M., et al. , Modulation of titin-based stiffness in hypertrophic cardiomyopathy via protein kinase D. Front. Physiol. 11, 240 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roe A. T., et al. , Increased passive stiffness promotes diastolic dysfunction despite improved Ca2+ handling during left ventricular concentric hypertrophy. Cardiovasc Res. 113, 1161–1172 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Linke W. A., Hamdani N., Gigantic business: Titin properties and function through thick and thin. Circ. Res. 114, 1052–1068 (2014). [DOI] [PubMed] [Google Scholar]
- 61.Lahmers S., Wu Y., Call D. R., Labeit S., Granzier H., Developmental control of titin isoform expression and passive stiffness in fetal and neonatal myocardium. Circ. Res. 94, 505–513 (2004). [DOI] [PubMed] [Google Scholar]
- 62.Alamo L., et al. , Effects of myosin variants on interacting-heads motif explain distinct hypertrophic and dilated cardiomyopathy phenotypes. Elife 6, e24634 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sitbon Y. H., et al. , Cardiomyopathic mutations in essential light chain reveal mechanisms regulating the super relaxed state of myosin. J. Gen. Physiol. 153, e202012801 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Anderson R. L., et al. , Deciphering the super relaxed state of human beta-cardiac myosin and the mode of action of mavacamten from myosin molecules to muscle fibers. Proc. Natl. Acad. Sci. U.S.A. 115, E8143–E8152 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Toepfer C. N., et al. , Myosin sequestration regulates sarcomere function, cardiomyocyte energetics, and metabolism, informing the pathogenesis of hypertrophic cardiomyopathy. Circulation 141, 828–842 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nag S., Trivedi D. V., To lie or not to lie: Super-relaxing with myosins. Elife 10, e63703 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schmid M., Toepfer C. N., Cardiac myosin super relaxation (SRX): A perspective on fundamental biology, human disease and therapeutics. Biol. Open 10, bio057646 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Margara F., et al. , Mechanism based therapies enable personalised treatment of hypertrophic cardiomyopathy. Sci. Rep. 12, 22501 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yuan C. C., et al. , Molecular basis of force-pCa relation in MYL2 cardiomyopathy mice: Role of the super-relaxed state of myosin. Proc. Natl. Acad. Sci. U.S.A. 119, e2110328119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ma W., et al. , Structural OFF/ON transitions of myosin in relaxed porcine myocardium predict calcium-activated force. Proc. Natl. Acad. Sci. U.S.A. 120, e2207615120 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brenner B., Schoenberg M., Chalovich J. M., Greene L. E., Eisenberg E., Evidence for cross-bridge attachment in relaxed muscle at low ionic strength. Proc. Natl. Acad. Sci. U.S.A. 79, 7288–7291 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tanner B. C., et al. , Thick-to-thin filament surface distance modulates cross-bridge kinetics in Drosophila flight muscle. Biophys J. 103, 1275–1284 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix.