

Significance
Tuft cells detect parasitic worms in the intestine and orchestrate type 2 immunity. The metabolite succinate engages the receptor SUCNR1 and activates tuft cells. However, SUCNR1 is dispensable for antiparasitic immunity, making the purpose of succinate sensing by tuft cells unclear. Here, we showed that tuft cell stimulation by microbial-derived succinate and the resulting type 2 immune response leads to increased Paneth cell numbers and profound shifts in small intestinal antimicrobial peptide expression. These antimicrobial changes, in turn, alter the bacterial microbiota composition. Our findings challenge the conventional thinking that tuft cells and type 2 immunity solely play antiparasitic roles. Instead, they suggest that tuft cells may utilize SUCNR1 and type 2 immunity to modulate intestinal bacterial homeostasis.
Keywords: tuft cell, Paneth cell, antimicrobial peptides, succinate, type 2 immunity
Abstract
Succinate produced by the commensal protist Tritrichomonas musculis (T. mu) stimulates chemosensory tuft cells, resulting in intestinal type 2 immunity. Tuft cells express the succinate receptor SUCNR1, yet this receptor does not mediate antihelminth immunity nor alter protist colonization. Here, we report that microbial-derived succinate increases Paneth cell numbers and profoundly alters the antimicrobial peptide (AMP) landscape in the small intestine. Succinate was sufficient to drive this epithelial remodeling, but not in mice lacking tuft cell chemosensory components required to detect this metabolite. Tuft cells respond to succinate by stimulating type 2 immunity, leading to interleukin-13-mediated epithelial and AMP expression changes. Moreover, type 2 immunity decreases the total number of mucosa-associated bacteria and alters the small intestinal microbiota composition. Finally, tuft cells can detect short-term bacterial dysbiosis that leads to a spike in luminal succinate levels and modulate AMP production in response. These findings demonstrate that a single metabolite produced by commensals can markedly shift the intestinal AMP profile and suggest that tuft cells utilize SUCNR1 and succinate sensing to modulate bacterial homeostasis.
The small intestine (SI) provides a massive surface area for the digestion and absorption of ingested nutrients. At the same time, it contends with continuous interactions with numerous pathogenic and commensal microbes, where indigenous bacterial densities can reach as high as 107 to 108 cells per gram of contents in the distal SI or ileum (1, 2). A mucus layer permeated with antimicrobial peptides (AMPs) forms a chemical barrier that prevents these microbes from overgrowing, contacting the epithelium, and/or translocating across mucosal tissues (3–5). Loss of this barrier results in excessive inflammatory responses to the microbiota and is a pathophysiological feature of many gastrointestinal diseases (6).
While mucus and AMPs influence gut microbiota composition and localization (5, 7–9), microbes reciprocally play a critical role in forming and maintaining proper barrier function within the intestinal tract. Germfree (GF) animals have fewer mucus-producing goblet cells, a thinner mucus layer, and reduced production of specific AMPs (10, 11). Strikingly, colonization of GF mice with a conventional (CV) microbiome is sufficient to restore normal mucus properties and Paneth cell expression of specific AMPs within these animals (10, 12–14). This induction of specific AMP production by the microbiota occurs via pattern-recognition receptor signaling (15) or through sensing microbial-derived metabolites, such as short-chain fatty acids (16).
Much of our knowledge about how the gut microbiota modulates barrier function and intestinal physiology derives from studies on commensal bacteria. However, this microbial ecosystem also includes archaea, fungi, viruses, and protists (17, 18). Recently, commensal protists from the genus Tritrichomonas have emerged as important members of the gut microbiota of wild and laboratory rodents (19). One of these protist species, Tritrichomonas musculis (T. mu), induces profound changes to the murine gut epithelium and immune system. Type 2 immunity, the hallmark immune response mounted against parasitic worm infections, is activated in the distal SI in response to T. mu, concomitant with a notable expansion of specialized epithelial cells, including chemosensory tuft cells and mucus-producing goblet cells (20). Succinate, a metabolic by-product of commensal tritrichomonads, activates tuft cell taste–chemosensory signal transduction by engaging the G-protein-coupled receptor SUCNR1 (21–23). In response to succinate, tuft cells release the cytokine interleukin-25 (IL-25), which activates group 2 innate lymphoid cells (ILC2s) in the lamina propria (LP). Subsequently, ILC2s produce the type 2 cytokine interleukin-13 (IL-13), which initiates type 2 immune responses within the gut. In addition, IL-13 biases intestinal stem cells to differentiate into tuft and goblet cells, thereby amplifying this tuft cell–ILC2 circuit (20, 24, 25).
Succinate is an intriguing tuft cell–activating ligand, as commensal bacteria, tritrichomonads, parasitic worms, and mammalian host cells produce this metabolite (22, 23, 26–28). Thus, luminal accumulation of succinate does not distinguish between the host or microbe, nor commensal or pathogen. In fact, intestinal type 2 immunity triggered by T. mu–produced succinate does not reduce or enhance protist colonization, and the succinate receptor SUCNR1 is dispensable for antiparasitic immunity (20–22). These observations suggest that tuft cell detection of succinate did not evolve for antiparasitic responses. Interestingly, SI tuft cells express higher levels of Sucnr1 than colonic tuft cells (22, 29), and succinate induces greater tuft cell hyperplasia in the distal SI (ileum) compared to the proximal SI (22), suggesting that ileal tuft cells are particularly sensitive to this metabolite. However, why ileal tuft cells orchestrate type 2 immunity in response to succinate is still not well understood.
To address this question, we profiled the global effects of microbial-derived succinate on ileal physiology. We used spatial transcriptomics (ST) as an unbiased approach to characterize the impact of T. mu, a major succinate producer, on distal SI gene expression in situ. T. mu induced substantial transcriptional changes in the SI epithelium and LP, including increased goblet and Paneth cell numbers and profound alterations to the antimicrobial expression profile. Furthermore, we demonstrated that these shifts in AMP production depend on tuft cell detection of T. mu–produced succinate and subsequent activation of type 2 immunity. Induction of type 2 immunity significantly decreased the total number of mucosa-associated bacteria in the distal SI, consistent with increased microbial killing and restructuring of the gut community by up-regulated AMPs. Finally, we found that short-term bacterial dysbiosis induced by polyethylene glycol (PEG), which leads to a spike in luminal succinate levels in the gut, activates SI tuft cells and shifts AMP production in a similar manner to T. mu colonization or succinate feeding. Collectively, our work highlights the significant impact succinate exerts on the cellular composition of the gut epithelium and host–microbiota interactions through activating tuft cells and suggests that tuft cells utilize succinate sensing as a metric for bacterial homeostasis in the distal SI.
Results
The Commensal Protist T. mu Induces Significant Remodeling of the Intestinal Epithelium.
To map the impact of microbial-derived succinate on the SI landscape, we conducted spatial transcriptomics (ST) on uncolonized and T. mu-colonized mice. T. mu colonization induced the most pronounced tuft cell hyperplasia in the distal SI (ileum) (SI Appendix, Fig. S1A); therefore, we focused on the ileum for our ST analysis. We harvested ileal tissue from one uncolonized mouse and one T. mu-colonized mouse (3 wk) and affixed 10-µm sections to Visium slides (10× Genomics). These slides have thousands of spots containing mRNA-binding probes that capture the gene expression profiles of ~5 to 15 cells within the tissue section (Fig. 1A). This approach produced complete transcriptomes of the cell populations within each ST spot and allowed us to map these gene expression signatures in situ (SI Appendix, Fig. S1B).
Fig. 1.
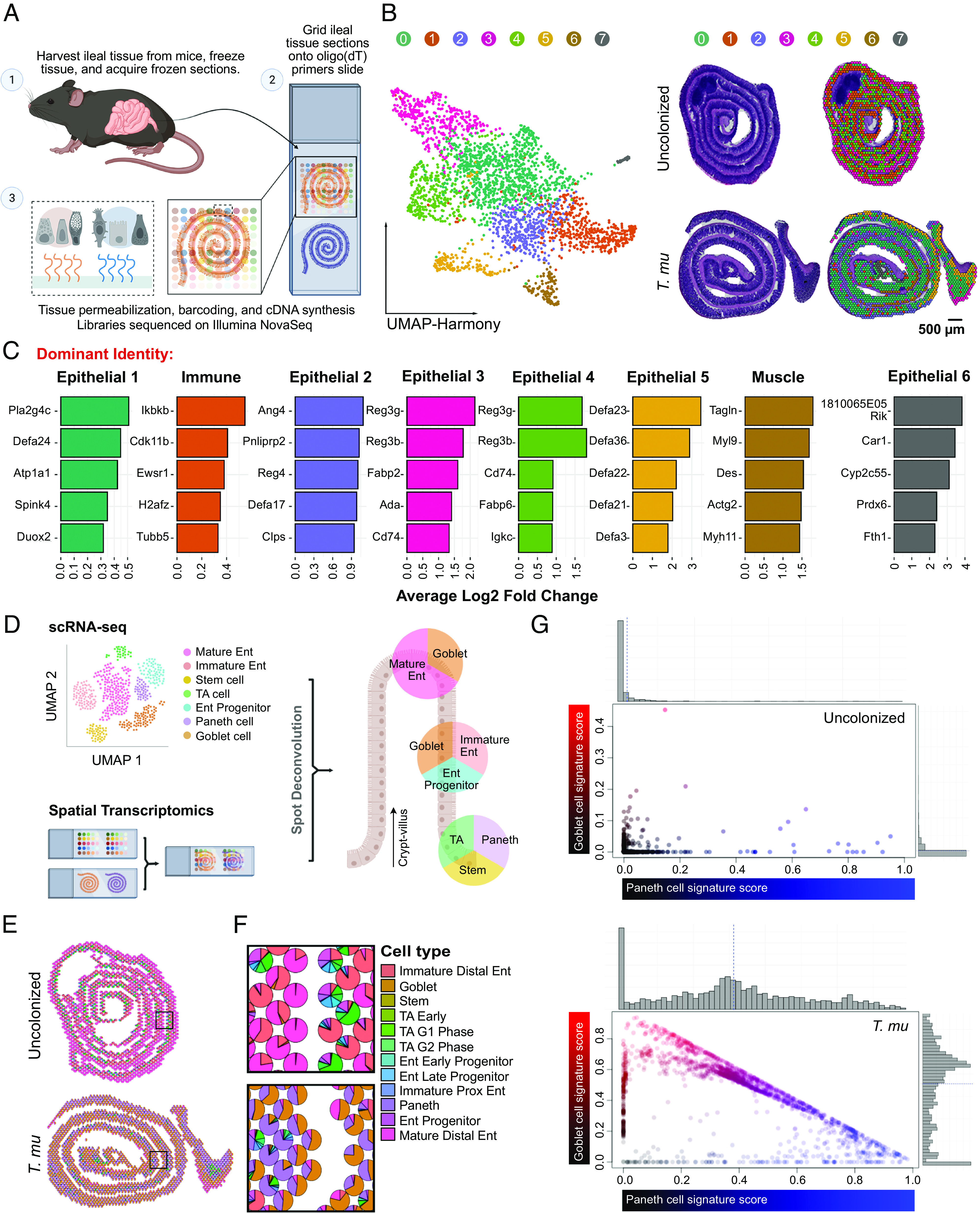
ST analysis of the T. mu–colonized SI reveals goblet and Paneth cell expansion. (A) Schematic of the ST pipeline. (B) Harmonized UMAP plots of ST spots organized by cluster identity (Left); corresponding hematoxylin and eosin (H&E)-stained tissue scans with overlaid Seurat clustering (Right). (C) Top 5 highly expressed genes for each cluster shown by log 2-fold change compared to other clusters. Clusters annotated based on dominance of gene expression associated with epithelial, immune, or muscle cells. (D) Schematic of the Seurat anchoring method to integrate a single-cell RNA sequencing (scRNA-seq) small intestinal epithelial dataset (30) with the ST dataset. (E and F) Scatter pie plots of the ileum depicting predicted epithelial cellular identities (E), with the corresponding zoomed regions of interest (F). Ent, enterocyte; TA, transit-amplifying; G1, G1/S cell-cycle phase; G2, G2/M cell-cycle phase; Prox, proximal. (G) Visualization of goblet (red) and Paneth cell (blue) signature scores of ST spots colored by identity. Gray histograms on top and right show the distribution of spots along each axis.
Using the ST analysis tool, STUtility (31), we manually annotated the tissue-associated spots with one of three identities: 1) Muscularis (Muscle), 2) Peyer’s Patch (PP), or 3) Epithelium/Lamina Propria (Epi/LP) (SI Appendix, Fig. S1C). We excluded the muscularis and PP compartments for downstream analysis to focus on epithelial and immune changes induced by T. mu. Next, we performed nonnegative matrix factorization to create gene and cell topic modules that described the two samples (SI Appendix, Fig. S1D). The sequencing data from the two samples were integrated using Harmony (32) and partitioned into eight distinct clusters, as visualized by Uniform Manifold Approximation and Projection (UMAP); we also mapped these clusters back onto the tissue array to visualize their spatial location (Fig. 1B). Since each dot on the UMAP projection represents an ST spot comprising 5 to 15 cells, we could not assign a specific cellular identity to each cluster. Therefore, we annotated the clusters based on their dominant expression of genes associated with epithelial, immune, or muscle function (Fig. 1C). T. mu colonization highly altered the abundance and composition of all clusters, except for Cluster 6, which is enriched for muscle and stromal genes (Fig. 1 B and C and SI Appendix, Fig. S1E). In addition, we found an increased number of ST spots containing tuft cell–specific genes Trpm5 and Dclk1 and an increased number of DCLK1-positive cells in the T. mu–colonized tissue relative to the uncolonized sample (SI Appendix, Fig. S1 F and G), confirming that tuft cell hyperplasia occurred in response to the protist.
Integration of Single-Cell RNA Sequencing (scRNA-seq) with ST Identifies Hybrid Goblet–Paneth Cell Signatures during T. mu Colonization.
Six of the seven clusters altered by T. mu were epithelial-dominant (Fig. 1C). To identify the epithelial composition of each ST spot within our samples, we anchored a previously published reference scRNA-seq dataset of the SI epithelium onto our spatial analysis (30). This allowed us to deconvolute the transcriptomes of the ST spots with the 15 annotated epithelial cell types present in the reference dataset (Fig. 1D). We then calculated the probability of a specific epithelial cell identity within each spot; non-epithelial cell identities were excluded from this analysis. The deconvoluted spots were visualized on the tissue array as pie charts to represent the proportion of epithelial cell types per spot (Fig. 1 E and F). Immature and mature distal enterocyte identities predominated the spots in the uncolonized sample, whereas goblet and Paneth cell identities were the most prevalent in the T. mu–colonized sample (SI Appendix, Fig. S2 A–C). The increased goblet cell expression signatures were expected, as tritrichomonad colonization induces goblet cell hyperplasia (20, 22, 23). However, an increase in Paneth cell expression signatures in response to T. mu has not been described.
Paneth cells are typically restricted to the intestinal crypts, while goblet cells are distributed throughout the crypt–villus axis (33, 34). Nonetheless, because of the highly enriched Paneth cell and goblet cell expression signatures in the T. mu–colonized sample (Fig. 1F and SI Appendix, Fig. S2 A–C), we investigated whether the two cell types share the same cellular neighborhoods. We plotted each ST spot in a two-dimensional space, where the axes represent the percentage of Paneth or goblet cell signature per spot. Most spots from the uncolonized ileum aligned along either the X or Y axis, indicating that these spots contained either pure goblet cells or Paneth cells colocalizing with other epithelial cell types, but rarely with each other. In contrast, spots from the T. mu–colonized ileum were distributed across the spectrum of shared identities between goblet and Paneth cells, meaning that goblet and Paneth cell signatures were more frequently found together in the same ST spot (Fig. 1G). In addition, these hybrid goblet–Paneth cell spots localized throughout the crypt–villus axes (SI Appendix, Fig. S2A). This indicates that T. mu colonization may 1) increase the number of Paneth cells residing outside of the crypts among villus goblet cells or 2) increase the abundance of a cellular subtype expressing both Paneth and goblet cell features. While our ST data cannot distinguish between either possibility, we observed cells above the crypts in T. mu–colonized tissue that are reminiscent of “intermediate cells” (SI Appendix, Fig. S3 A and B). These cells are characterized by the presence of granules within mucus globules, morphological features that are both Paneth and goblet cell–like (SI Appendix, Fig. S3 A and B) (35). While rare at homeostasis, intermediate cells expand in the SI during helminth infections (36, 37). Whether these intermediate cells represent a transitionary state between Paneth and goblet cells or whether they form a stable epithelial subset with specialized functions during type 2 immunity remains unclear. Altogether, these results demonstrate that T. mu colonization favors a secretory epithelial cell program, including a significant increase in goblet and Paneth cell gene expression signatures.
T. mu Colonization Broadly Alters the Antimicrobial Landscape in the SI.
Comparing gene expression between the Epi/LP-associated clusters revealed that AMPs are some of the most differentially regulated genes between the uncolonized and T. mu–colonized ilea. Despite increased goblet and Paneth cell signatures, not all AMPs were globally up-regulated in the T. mu–colonized sample. A select group of AMP-encoding genes was down-regulated by T. mu colonization (Fig. 2 A–C), with the most dramatic effects on Reg3b and Reg3g. These two AMPs belong to the Regenerating islet-derived (Reg) family of proteins (38) and play important roles in host–microbe interactions (5, 39–41).
Fig. 2.

T. mu colonization alters the small intestinal AMP repertoire. (A) Heatmap of AMP genes clustered by sample condition and ranked with the corresponding Z-score. Each column represents a cluster from the uncolonized (pink) or T. mu–colonized mouse (aqua). (B and C) Gene features overlay plots on ST spots throughout ileal tissue of the uncolonized or T. mu–colonized mouse (B), with corresponding violin plots depicting global gene expression (C).
Numerous AMP-encoding genes were also highly up-regulated in response to T. mu colonization, including various α-defensin-encoding genes, Ang4, Sprr2a3, and Retnlb (Fig. 2 A–C). The α-defensin family of AMPs and ANG4 are specifically produced by Paneth cells in the SI epithelium and have broad-spectrum antimicrobial activity (14, 42). Sprr2a3 is produced by both Paneth and goblet cells, kills gram-positive bacteria, and is deployed during helminth infection (43). Alternatively, Retnlb is expressed only by goblet cells (44, 45) and encodes the protein Resistin-like molecule β (RELMβ), which has both antibacterial and antihelminthic properties (8, 44, 46). Taken together, these data illustrate that T. mu profoundly alters the epithelial and immune compartments of the distal SI, with broad shifts in transcriptional AMP programs.
T. mu Alters Antimicrobial Production in the SI by Activating Tuft Cell Taste–Chemosensory Pathways.
To investigate whether T. mu alters the abundance and morphology of secretory epithelial cells, we colonized wild-type (WT) mice with the protist for 3 wk (SI Appendix, Fig. S4A) and examined the ileum by microscopy. In line with previous observations (20, 22, 23), T. mu colonization resulted in tuft and goblet cell hyperplasia (SI Appendix, Fig. S4 B and C), but we also observed a significant increase in Paneth cell numbers in protist-colonized samples (Fig. 3 A and B). Furthermore, Paneth cells in T. mu–colonized mice were on average much larger (Fig. 3C) and possessed ultrastructural changes. Normal Paneth cells, as seen in mice lacking T. mu, contained large, electron-dense secretory granules concentrated at the apical end of the cell, with each granule surrounded by a thin, electron-lucent halo (Fig. 3D and SI Appendix, Fig. S4D). These halos were previously shown to contain MUC2 mucins (47). Paneth cells in T. mu–colonized mice, however, possessed features that are intermediate between Paneth and goblet cells—smaller, electron-dense granules surrounded by larger, electron-lucent mucin globules (Fig. 3D and SI Appendix, Fig. S4D). We also visualized lysozyme (LYZ), an AMP specifically produced by Paneth cells, in the ilea by microscopy (33). In uncolonized mice, LYZ localized within distinct secretory granules at the apical ends of Paneth cells. However, the LYZ signal within the Paneth cells of T. mu–colonized mice was dimmer and dispersed throughout the entirety of the cell (Fig. 3E and SI Appendix, Fig. S4E). Collectively, these results show that T. mu induces Paneth cell hyperplasia, morphological changes to Paneth cell granules, and abnormal AMP localization.
Fig. 3.
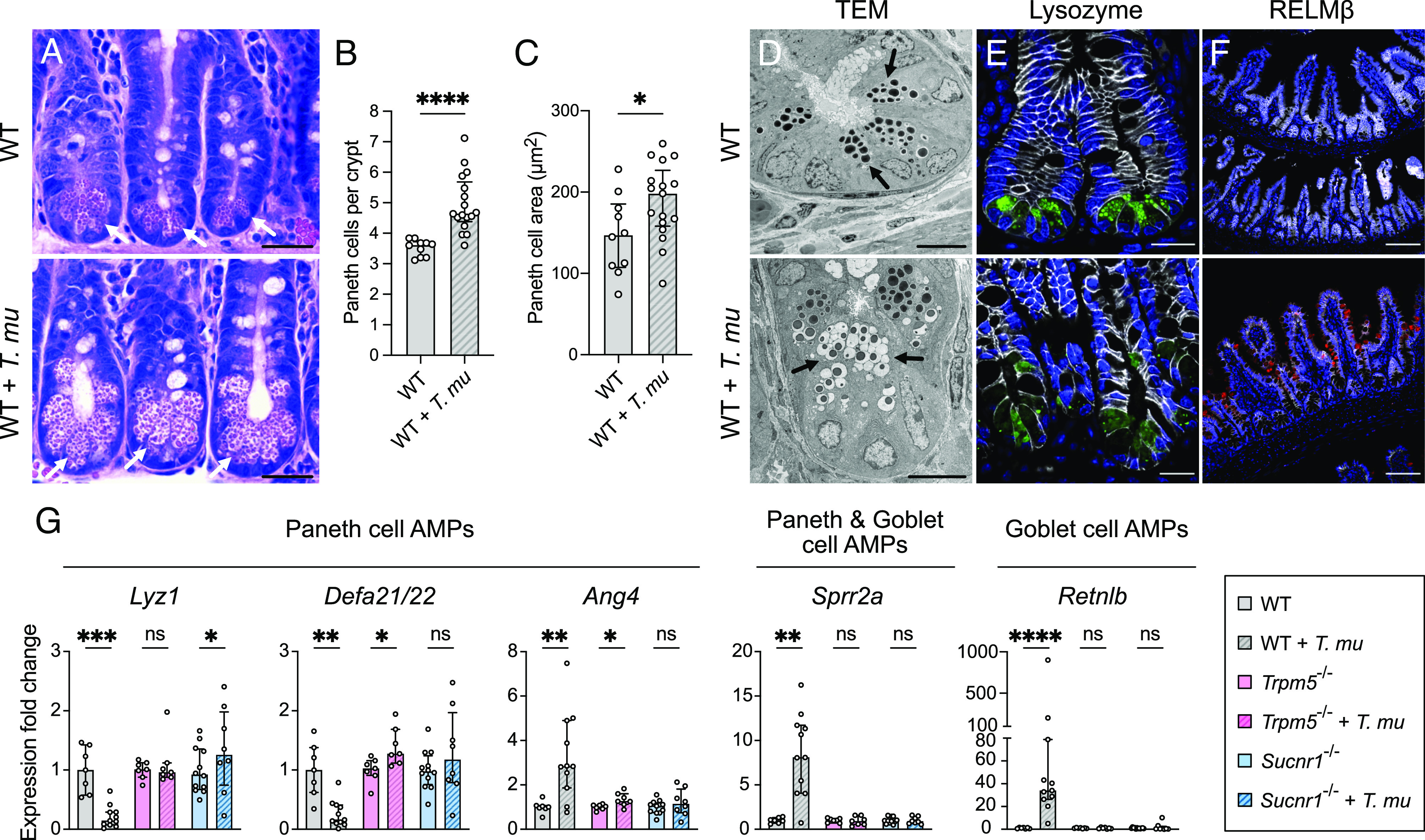
T. mu induces goblet and Paneth cell hyperplasia and alters AMP production in the distal SI via tuft cell stimulation. (A) Representative images of H&E-stained sections of the ileal crypts from uncolonized or T. mu–colonized WT mice. White arrows indicate representative Paneth cells. (Scale bar: 25 µm.) (B) Average number of Paneth cells per crypt in the ilea of uncolonized and T. mu–colonized WT mice (n = 10 to 17 mice per group). (C) Average size of individual Paneth cells (µm2) in the ilea of uncolonized and T. mu–colonized WT mice (n = 10 to 17 mice per group). (D) Representative transmission electron microscopy images of the ileal crypts from uncolonized or T. mu–colonized WT mice. Black arrows indicate representative Paneth cell secretory granules. (Scale bar: 10 µm.) (E and F) Representative fluorescence microscopy images of the ilea from uncolonized or T. mu–colonized WT mice. Nuclei (blue), E-cadherin (white), LYZ (Paneth cell AMP) (green), RELMβ (goblet cell AMP) (red). (E) Scale bar: 20 µm. (F) Scale bar: 100 µm. (G) Expression of representative AMP genes determined by qRT-PCR in the ileal epithelial fraction from uncolonized or T. mu–colonized WT, Trpm5−/−, and Sucnr1−/− mice (n = 7 to 12 mice per group). Relative expression normalized to Gapdh. In panels B, C, and G, center values = median; error bars = interquartile range (IQR). Significance was determined using the Mann–Whitney U test. ns = no significance, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To determine whether tuft cell sensing of the protist metabolite succinate was required to alter AMPs in Paneth and goblet cells, we colonized WT mice and mice either lacking TRPM5, a calcium-gated ion channel that is critical for the taste pathway, or SUCNR1, a G-protein-coupled receptor that detects succinate (22, 23, 48, 49)(SI Appendix, Fig. S4 A and B). We measured the relative expression of a panel of representative AMP genes produced by Paneth and/or goblet cells via qRT-PCR of the ileal epithelium. T. mu colonization altered AMP gene expression in the ileal epithelium, with decreased expression of Lyz1 and Defa21/22 and increased expression of Ang4, Sprr2a, and Retnlb (Fig. 3G). These transcriptional changes are consistent with ileal LYZ and RELMβ protein levels (encoded by Retnlb) in T. mu–colonized mice (Fig. 3F and SI Appendix, Fig. S4 F and G). In contrast to WT mice, these AMP gene expression changes were abrogated in T. mu–colonized Trpm5−/− and Sucnr1−/− mice compared to uncolonized controls (Fig. 3G and SI Appendix, Fig. S4F). Altogether, these results suggest that T. mu alters AMP production in the distal SI by stimulating tuft cell taste–chemosensory pathways via the metabolite succinate. These Paneth cell ultrastructural and AMP expression changes were still present in WT mice after 8 wk of T. mu colonization (SI Appendix, Fig. S4 H and I), demonstrating that these Paneth cell phenotypes are maintained with long-term tuft cell activation. Given that Paneth cells turn over approximately every 57 d (50), this also suggests that newly differentiated Paneth cells may adopt this altered phenotype as long as the tuft cell stimulus remains present.
Succinate Alters Intestinal AMP Production by Stimulating Tuft Cells and Activating Type 2 Immunity.
Succinate is sufficient to induce tuft and goblet cell hyperplasia in the distal SI (22, 23); however, little is known about its effect on Paneth cells. To determine whether succinate is sufficient to drive changes in Paneth cell numbers, morphology, and AMP production in the ileum, we treated GF mice and CV mice with 100 mM succinate in their drinking water for 1 wk, after which tissues were collected for microscopy, qRT-PCR, and western blot analysis. Succinate increased both tuft and goblet cell numbers and led to a striking increase in MUC2 throughout the crypt–villus axes of GF and CV mice (SI Appendix, Fig. S5 A–D). In addition, Paneth cell numbers were significantly increased in both GF and CV mice fed succinate compared to respective controls (Fig. 4 A and B), indicating that this metabolite is sufficient to drive Paneth cell hyperplasia in the distal SI.
Fig. 4.
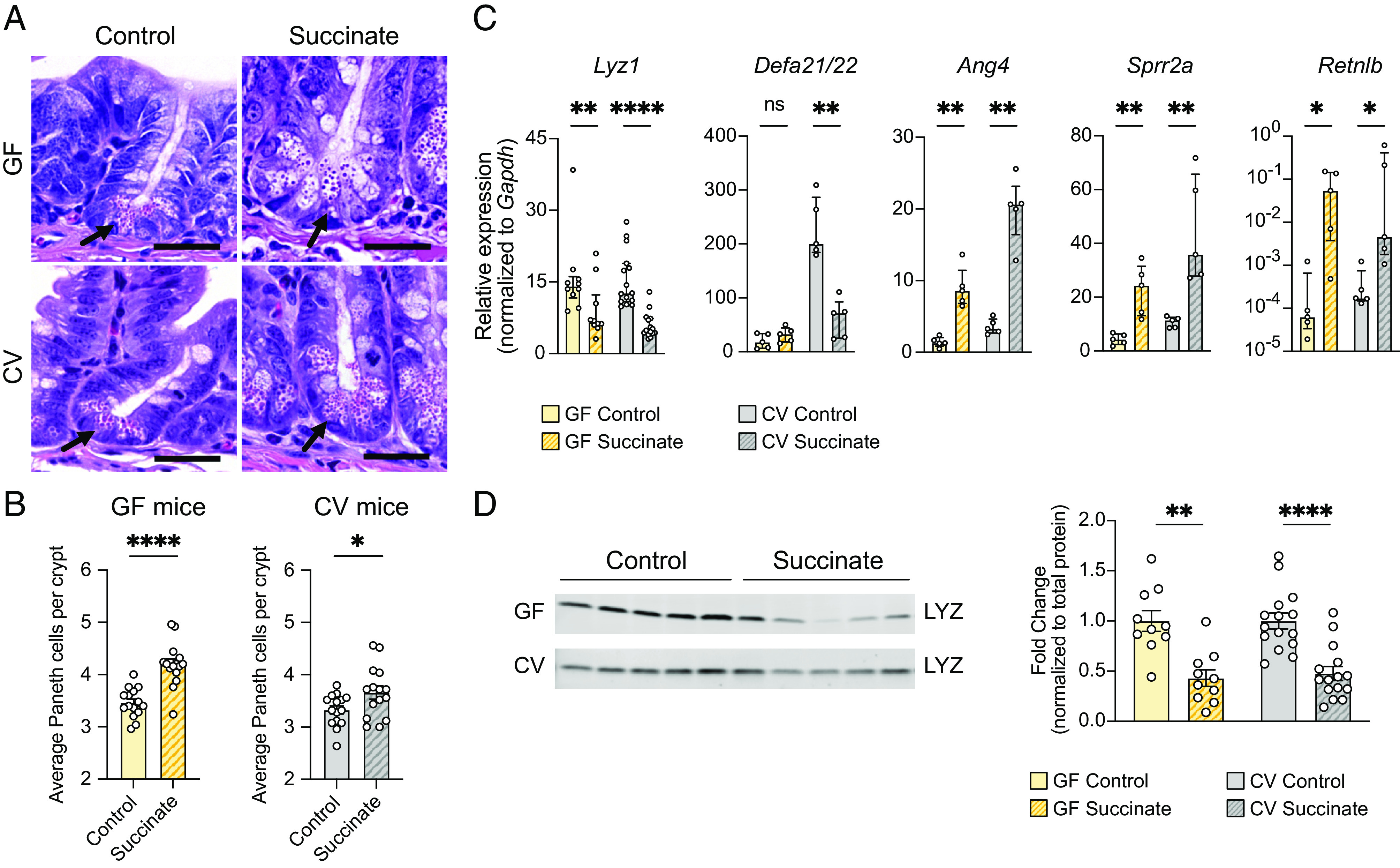
Oral administration of succinate results in similar changes to small intestinal AMP production as T. mu colonization. (A) Representative images of H&E-stained sections of the ileal crypts from control or succinate-treated GF (top row) or CV mice (bottom row). (Scale bar: 25 µm.) (B) Average number of Paneth cells per crypt in the ilea of control or succinate-treated GF or CV WT mice (n = 15 mice per group). Center values = arithmetic mean; error bars = SEM. Significance was determined using Student’s t test. (C) Expression of representative AMP genes determined by qRT-PCR in the ilea of control or succinate-treated GF and CV mice (n = 5 to 15 mice per group). Relative expression normalized to Gapdh. Center values = median; error bars = IQR. Significance was determined using the Mann–Whitney U test. (D) Representative western blot images and quantitative analysis of intracellular LYZ levels in the ilea of control or succinate-treated GF and CV mice. Each band or symbol represents an individual mouse. LYZ levels normalized to REVERT total protein stain (SI Appendix, Fig. S5F) (n = 10 to 15 mice per group). Center values = arithmetic mean; error bars = SEM. A linear mixed model was used to determine significance. ns = no significance, *P < 0.05, **P < 0.01, ****P < 0.0001.
Succinate treatment also altered expression of representative Paneth and goblet cell AMP genes in both GF and CV mice in a manner similar to T. mu colonization, increasing Ang4, Sprr2a, and Retnlb expression while decreasing Lyz1 expression (Fig. 4C and SI Appendix, Fig. S5E). These transcriptional changes are consistent with decreased LYZ protein levels (Fig. 4D and SI Appendix, Fig. S5 F and G) and increased RELMβ protein levels in response to succinate (SI Appendix, Fig. S5G). While Defa21/22 expression was decreased in succinate-treated CV mice, it was not significantly altered in succinate-treated GF mice in comparison to GF controls (Fig. 4C). However, Defa21/22 baseline expression in GF mice is much lower than in CV mice (Fig. 4C), likely due to a lack of induction by the microbiota, which could explain why Defa21/22 expression is not decreased further in succinate-treated GF mice. Overall, these results show that succinate is sufficient to drive the same changes to AMP production observed in T. mu–colonized mice, independent of the resident microbiota.
To confirm that succinate sensing through the taste–chemosensory signal transduction pathway in tuft cells is critical for changes in AMP production, we treated Trpm5−/− and Sucnr1−/− mice with 100 mM succinate for 1 wk in their drinking water. We found that LYZ protein levels decreased in succinate-treated WT mice, but not in succinate-treated Trpm5−/− and Sucnr1−/− mice compared to their respective controls (SI Appendix, Fig. S5H). This suggests that succinate is sufficient to remodel ileal AMP production by stimulating tuft cell taste–chemosensory pathways.
Upon activation of taste–chemosensory signal transduction, SI tuft cells release IL-25, which induces ILC2s to produce type 2 cytokines such as IL-13 (20, 22–25). This cytokine biases the differentiation of SI stem cells toward tuft and goblet cells. Additionally, helminth-induced type 2 immunity has been shown to alter expression of AMP-encoding genes in an IL-13-dependent manner (43, 44, 51). To determine whether succinate-induced type 2 immunity and IL-13 are responsible for modulating ileal AMP production, we treated WT mice or mice lacking Il13 (Il13−/−) with succinate for 1 wk. Succinate induced tuft cell hyperplasia in WT mice but not in Il13−/− mice (SI Appendix, Fig. S6 A and B). Moreover, decreased Lyz1 and Defa21/22 expression and increased Ang4, Sprr2a, and Retnlb expression found in succinate-treated WT mice were abrogated in succinate-treated Il13−/− mice (Fig. 5A). Consistent with these results, we found no difference in intracellular LYZ protein levels between control and succinate-treated Il13−/− mice (SI Appendix, Fig. S6C), indicating that changes to AMP production in response to succinate are dependent on the cytokine IL-13.
Fig. 5.
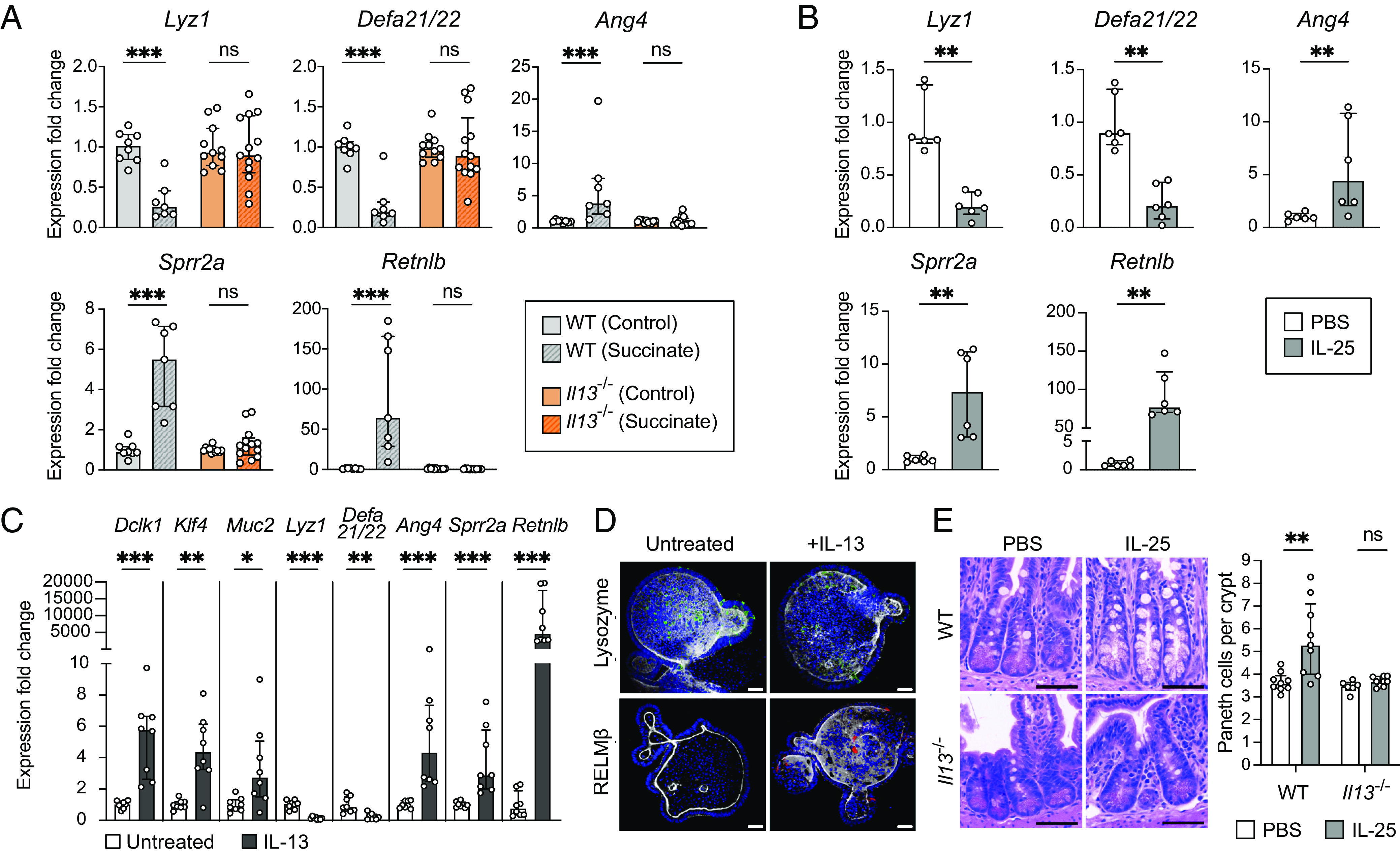
The type 2 cytokine IL-13 is critical for small intestinal Paneth cell hyperplasia and changes to AMP expression downstream of tuft cell stimulation. (A) Expression of representative AMP genes determined by qRT-PCR in the ileal epithelial fraction of control or succinate-treated WT and Il13−/− mice (n = 7 to 13 mice per group). (B) Expression of representative AMP genes determined by qRT-PCR in the ileal epithelial fraction of WT mice IP-injected with phosphate-buffered saline (PBS) or IL-25 (n = 6 mice per group). (C) Expression of Dclk1 (tuft cell marker), Klf4 and Muc2 (goblet cell markers), and representative AMP genes determined by qRT-PCR in untreated or IL-13-treated ileal organoids (n = 8 samples per group). (D) Representative fluorescence microscopy images of untreated or IL-13-treated ileal organoids. Nuclei (blue), F-actin (white), LYZ (green), RELMβ (red). (Scale bar: 30 µm.) (E) Left: Representative images of H&E-stained sections of the ileal crypts from PBS- or IL-25-injected WT or Il13−/− mice. (Scale bar: 50 µm.) Right: Average number of Paneth cells per crypt in the ilea of PBS- or IL-25-injected WT or Il13−/− mice (n = 6 to 9 mice per group). For qRT-PCR data, relative expression normalized to Gapdh. For all panels, center values = median; error bars = IQR. Significance was determined using the Mann–Whitney U test. ns = no significance, *P < 0.05, **P < 0.01, ***P < 0.001.
To determine whether type 2 immune induction in the ileum is sufficient to alter host AMP production, we bypassed tuft cell stimulation and injected recombinant IL-25 intraperitoneally (IP) into WT mice (SI Appendix, Fig. S6D). IL-25 injections induced tuft cell hyperplasia (SI Appendix, Fig. S6 E and F) and altered the expression of our representative panel of AMP genes similarly to T. mu colonization or succinate administration (Fig. 5B). These AMP expression changes correlate with alterations in LYZ organization within Paneth cells and increased RELMβ protein levels in IL-25-injected mice (SI Appendix, Fig. S6 G and H). This demonstrates that type 2 immune induction without initial tuft cell stimulation by succinate is sufficient to drive these changes to the SI epithelium.
To test whether IL-13 directly remodels epithelial AMP expression, we used an in vitro primary intestinal organoid system (52, 53). We confirmed that IL-13 treatment of ileal organoids promoted tuft and goblet cell differentiation (20, 54) by measuring the expression of tuft and goblet cell markers (Fig. 5C). We also found that IL-13 treatment alone decreased epithelial expression of Lyz1 and Defa21/22, while increasing expression of Ang4, Sprr2a, and Retnlb (Fig. 5 C and D). These effects are specific to IL-13 as IL-5 and IL-9, two other cytokines produced by ILC2s (55), do not alter AMP expression in ileal organoids (SI Appendix, Fig. S6I). Altogether, these results suggest that IL-13 signals directly to the epithelium to alter host AMP production.
Because IL-13 directly influences Paneth cell AMP expression, we reasoned that IL-13 may also induce Paneth cell hyperplasia downstream of tuft cell stimulation. To test this hypothesis, we IP-injected recombinant IL-25 into WT or Il13−/− mice and quantified the number of Paneth cells per crypt. While IL-25 injections resulted in Paneth cell hyperplasia in WT mice, there were no significant differences in Paneth cell numbers between IL-25 and mock-injected Il13−/− mice (Fig. 5E ). These results demonstrate that IL-13 is crucial in inducing Paneth cell hyperplasia downstream of tuft cell activation and suggest that this cytokine may also bias SI stem cells to differentiate into Paneth cells.
Type 2 Immune Induction Alters the Microbiome Composition and Depletes Mucosa-Associated Microbes in the Ileum.
AMPs play crucial roles in regulating the composition of the intestinal microbiota, controlling the extent of contact between microbes and the epithelium, and protecting the host from pathogen colonization (7, 33). Because succinate alters the expression of a wide range of AMP-encoding genes by stimulating type 2 immunity, we hypothesized that activation of type 2 immune responses would alter the composition of the ileal microbiota.
To induce type 2 immunity without confounding variables such as succinate feeding or T. mu colonization, which could independently alter the microbiota, we IP-injected IL-25 into mice every other day for 1 wk (SI Appendix, Figs. S6D and S7A), harvested the ileal luminal and mucosal microbial populations, and quantified the total number of bacterial 16S rRNA gene copies as a proxy for abundance. We saw no differences in 16S rRNA gene copy number between the luminal populations of IL-25-injected mice and mock-injected controls, indicating that luminal bacterial numbers were not altered by type 2 immunity (Fig. 6A). However, we observed significantly fewer 16S rRNA gene copies in the mucosal populations from IL-25-injected mice compared to controls (Fig. 6A), indicating that type 2 immune induction reduces the load of mucosa-associated bacteria.
Fig. 6.
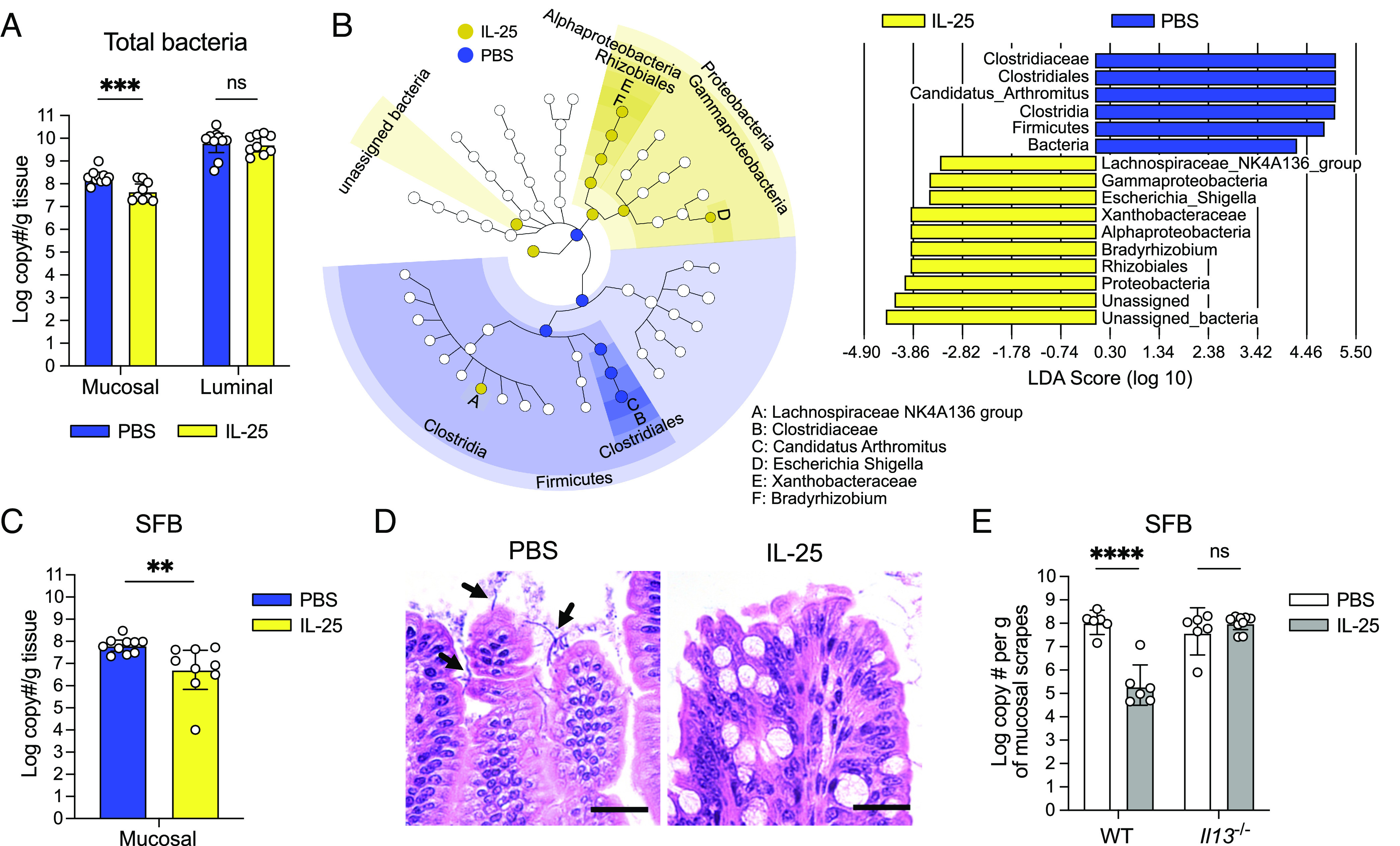
Type 2 immune induction depletes mucosa-associated bacteria in the SI. (A) Absolute quantification of total bacterial 16S rRNA gene copies in the ileal mucosal and luminal fractions of PBS- or IL-25-injected mice (n = 9 to 10 mice per group). (B) Linear discriminant analysis (LDA) Effect Size (LEfSe) analysis using a LDA threshold score of 2 to identify ileal mucosa-associated bacterial taxa in PBS- or IL-25-injected mice. The cladogram (Left) highlights taxonomic relatedness of bacteria, while the LDA plot (Right) is an ordered list of enriched bacteria. (C) Absolute quantification of SFB 16S rRNA gene copies in the ileal mucosal fractions of PBS- or IL-25-injected mice (n = 9 to 10 mice per group). (D) Representative images of Carnoy’s-fixed, H&E-stained sections of ileal villi from PBS- or IL-25-injected mice showing SFB (black arrows). (Scale bar: 25 µm.) (E) Absolute quantification of SFB 16S rRNA gene copies in the ileal mucosal fractions of PBS- or IL-25-injected WT and Il13−/− mice (n = 6 to 9 mice per group). In Panels A, C, and E, center values = geometric mean; error bars = 95% CI. Significance was determined using a generalized linear model. ns = no significance, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To obtain a broader view of the changes in the ileal microbiota composition in the face of these AMP alterations, we conducted 16S rRNA gene sequencing analysis on these mucosal and luminal population samples. We performed β-diversity analysis to assess whether treatment conditions were associated with distinct communities. Weighted UniFrac analysis, which accounts for both feature abundance and phylogenetic relatedness, showed that the mucosal populations of IL-25-injected mice are distinct from those of mock-injected mice (SI Appendix, Fig. S7B). Next, we used LDA effect size (LEfSe) analysis to determine which bacterial taxa are most likely to explain differences between the mock- and IL-25-injected mucosal populations. Candidatus arthromitus, commonly referred to as segmented filamentous bacteria (SFB), were enriched in the mucosa of control mice relative to those injected with IL-25 (Fig. 6B). SFB adhere to the ileal epithelium (56, 57) and are the dominant members of mucosal communities in mice that harbor them (43). Conversely, taxa that were enriched in the IL-25-injected mucosal populations include Lachnospiraceae and Proteobacteria (Fig. 6B). However, upon further examination, these Proteobacteria (i.e., Bradyrhizobium, Escherichia) are often found as contaminants in low biomass microbiota samples (58). Given that the total bacterial 16S rRNA gene copy number was reduced about 10-fold with IL-25 injections (Fig. 6A), these mucosal samples may have been susceptible to detecting these contaminants. LEfSe analysis of the luminal populations revealed many unique taxa enriched in the mock-injected group. Intriguingly, all these enriched taxa are gram-positive bacteria, including SFB (SI Appendix, Fig. S7 C and D). This suggests that despite no difference in the overall luminal bacterial numbers between both experimental groups (Fig. 6A), type 2 immunity can alter the luminal microbiota composition. Altogether, these data show that SFB are differentially abundant between mock- and IL-25-injected mice and that type 2 immune induction may disproportionally impact the abundance of specific gram-positive commensals in the ileal lumen.
To confirm SFB depletion in the ileal mucosa, we used qPCR to measure the number of SFB 16S rRNA gene copies in the mucosal samples as a proxy for SFB abundance. There were significantly fewer SFB 16S rRNA gene copies in IL-25-injected samples in comparison to their respective controls (Fig. 6C); we further confirmed this by microscopy (Fig. 6D). Given the importance of IL-13 in altering AMP expression during type 2 immunity, we also wanted to determine whether this cytokine is required to decrease SFB abundance with IL-25 stimulation. We IP-injected IL-25 into Il13−/− mice (SI Appendix, Fig. S7E) and quantified SFB 16S rRNA gene copy number in the mucosal fraction. Unlike in WT mice, SFB 16S rRNA gene copies did not decrease in Il13−/− mice with IL-25 injections (Fig. 6E), suggesting that IL-13 is important for altering SFB abundance downstream of tuft cell stimulation. We further showed that a tuft cell stimulus such as succinate also decreases SFB numbers (SI Appendix, Fig. S7F). Finally, we investigated whether type 2 immune-induced AMP alterations would decrease the abundance of other sensitive microbes, such as the gram-positive human gut commensal Enterococcus faecalis (Ef). Ef is killed in vitro by recombinant ANG4 or SPRR2A (14, 43), AMPs that are up-regulated with type 2 immunity. We found that IL-25 injections reduced Ef abundance specifically in the SI (where Paneth cells are found) but not other gastrointestinal sites (SI Appendix, Fig. S8), which could reflect an inhospitable AMP environment for this bacterium. Altogether, these results indicate that tuft cell activation by sensing microbial metabolites has the capacity to alter intestinal immunity and the composition of the bacterial microbiota.
Tuft Cells Sense Bacterial Dysbiosis in the Ileum and Alter AMP Production in Response.
Given that type 2 immune induction alters AMP production and the composition of the ileal bacterial microbiota, we wondered whether tuft cells use succinate levels to monitor bacterial homeostasis in the distal SI. Succinate is a common metabolic by-product produced by commensal bacteria during carbohydrate fermentation. However, it is present at relatively low concentrations in the gut lumen due to rapid consumption by other microbes (26, 27). Thus, succinate accumulation in the gut lumen is generally considered abnormal and is associated with dysbiotic events such as antibiotic treatment and gut motility disturbances (21, 59).
To test whether bacterial dysbiosis alters AMP production through stimulating tuft cells, we treated mice with PEG, which has previously been shown to cause mild watery diarrhea, increase luminal succinate levels due to disturbance of the bacterial microbiota, and induce tuft cell hyperplasia in a SUCNR1-dependent manner (21, 59). Indeed, we confirmed previous observations that PEG treatment does increase tuft cell numbers (Fig. 7A and SI Appendix, Fig. S9). Intriguingly, we found that PEG treatment also alters AMP expression in the same pattern as T. mu colonization or succinate, with the exception of Retnlb (Fig. 7B). Although Retnlb expression is increased with PEG treatment relative to untreated controls, this difference was not statistically significant, although it trended toward significance (P = 0.095). Furthermore, we show that these AMP changes are dependent on tuft cells, as mutant mice that completely lack tuft cells (Pou2f3−/−) do not show the same AMP production changes with PEG treatment (Fig. 7B). These results suggest that tuft cells can sense a succinate spike in the gut lumen during bacterial dysbiosis and alter the AMP repertoire in response, perhaps in an effort to restore bacterial homeostasis.
Fig. 7.
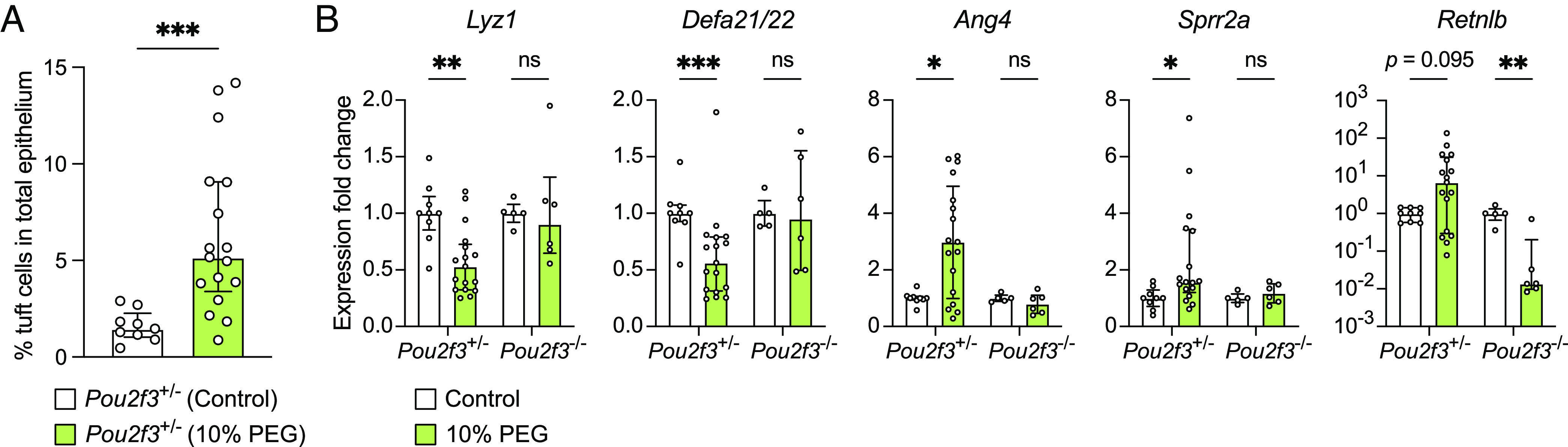
Tuft cells are required to induce AMP expression changes in response to bacterial dysbiosis in the distal SI. (A) Ileal tuft cell frequency in control or PEG-treated (12 d) Pou2f3+/− mice as determined by flow cytometry (n = 9 to 17 mice per group). (B) Expression of representative AMP genes determined by qRT-PCR in the ileal epithelial fraction of control or PEG-treated Pou2f3+/− and Pou2f3−/− mice (n = 5 to 17 mice per group). Relative expression normalized to Gapdh. Center values = median; error bars = IQR. Significance was determined using the Mann–Whitney U test. ns = no significance, *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
Our results demonstrate that succinate produced by the commensal protist T. mu biases epithelial differentiation toward the secretory lineage and alters AMP expression in the distal SI. Succinate drives these changes by activating tuft cells, resulting in a type 2 immune response in the distal SI. Changes in AMP production in response to type 2 immunity alter the SI microbiota composition, with a specific reduction in the abundance of mucosa-associated bacteria such as SFB (SI Appendix, Fig. S10) but not the abundance of protists (SI Appendix, Fig. S4A). Finally, we found that oral treatment with PEG, which can increase luminal succinate levels in the gut due to the resulting bacterial dysbiosis (21, 59), leads to the same AMP expression changes that we observed with T. mu colonization or succinate. Moreover, tuft cells are required to initiate these shifts in AMP production in response to PEG. Altogether, our study suggests that tuft cell SUCNR1 may be attuned to sense bacterial-derived succinate rather than having evolved to detect protists in the distal SI.
Previous studies have shown that commensal tritrichomonad colonization leads to tuft and goblet cell hyperplasia (20, 22, 23). However, through ST, we also identified an increase in Paneth cell gene expression signatures in the T. mu–colonized ileum, which reflects a previously unknown response of this epithelial cell to T. mu. We confirmed that Paneth cell hyperplasia in the distal SI was induced by T. mu colonization and, specifically, by its metabolic by-product succinate. In addition, T. mu colonization also altered Paneth cell secretory granule morphology and AMP localization. In comparison to Paneth cells in uncolonized mice, Paneth cells in T. mu–colonized mice contain smaller, electron-dense granules surrounded by larger, electron-lucent mucin globules. Furthermore, LYZ is dispersed throughout the Paneth cell in T. mu–colonized mice as opposed to being concentrated within secretory granules. This altered granule morphology and LYZ distribution is reminiscent of Paneth cells observed in mice with mutations in susceptibility genes linked to Crohn’s disease in humans—in particular, Atg16L1, which encodes an autophagy protein (60–62). Loss of Atg16L1 in mice led to abnormal LYZ distribution, indicating defects in packaging AMPs into secretory granules (60). Furthermore, mutations in Atg16L1 disrupt the release of AMPs through the secretory autophagy pathway during bacterial infection (63). The morphological similarities between Paneth cells in Atg16L1 mutant mice and T. mu–colonized mice raised the possibility that this protist alters Paneth cell autophagy and can potentially affect their secretory function. However, type 2 immune induction significantly reduced mucosa-associated bacteria, suggesting that these changes to Paneth cell granule morphology and AMP localization do not interfere with overall AMP secretion or microbial killing. Determining how succinate and type 2 immunity may impact Paneth cell functions and AMP secretion will be important for future investigation.
We also observed that T. mu profoundly shifted ileal AMP expression and production in the distal SI. Intriguingly, these transcriptional changes did not solely upregulate all AMP-encoding genes, as one might expect with Paneth cell hyperplasia. AMPs such as Reg3b, Reg3g, Lyz1, and Defa21/22 dramatically decreased, indicating that the host selectively remodeled the antimicrobial landscape rather than uniformly increasing AMP production. Succinate and the type 2 cytokine IL-13 were sufficient to elicit these same changes in AMP expression.
Given that AMPs are critical mediators of host–bacterial interactions, we investigated how type 2 immunity impacted the overall numbers and composition of the luminal and mucosal microbiota in the ileum. Type 2 immune induction significantly decreased the total number of mucosa-associated bacteria, while luminal bacterial numbers largely remained unaffected. This was surprising because T. mu colonization down-regulates Reg3g, an AMP that is known to be important for segregating commensals from the gut epithelium (5). Therefore, other AMPs up-regulated by type 2 immunity likely compensate for reduced Reg3g and enhance the killing of mucosal microbes. Indeed, compared to controls, the absolute abundance of SFB is reduced in the mucosal populations of IL-25-injected mice, and this requires IL-13. SFB are often the dominant taxa at the ileal mucosa (43), but previous studies showed that SFB abundance can be reduced by mucosal immunity and AMPs. For instance, SFB numbers decrease during helminth infection (51) or in transgenic mice expressing a human Paneth cell defensin (9), while SFB numbers increase in Sprr2a-deficient mice (43). These observations are consistent with our results, as IL-25 injections up-regulate the AMP Sprr2a and reduce SFB abundance concurrently. In addition, type 2 immune induction appeared to disproportionately impact the abundance of specific gram-positive bacteria in the luminal microbiota. This is intriguing given that the global AMP expression changes with T. mu colonization did not follow a specific pattern of bacterial susceptibility. While decreases in Lyz1 and Reg3g expression may suggest less killing of gram-positive bacteria (7), we also saw increases in Sprr2a expression, which kills gram-positive bacteria (43), and Retnlb expression, which preferentially targets gram-negative bacteria (8). Collectively, these data suggest that type 2 immunity can restructure the bacterial microbiota composition. AMPs are likely involved in these population changes, as these peptides are embedded within the mucus layer close to the mucosal surface (4). The AMP repertoire resulting from type 2 immune induction may also selectively target specific gram-positive bacteria in the lumen, which warrants future investigation.
Succinate is produced as a metabolic by-product of redox homeostasis in protists belonging to the class Tritrichomonadea (64), but colonization abundance by tritrichomonads like T. mu is not affected by succinate-driven type 2 immunity (20, 22). Furthermore, T. mu does not damage tissue during chronic colonization like parasitic helminths, implying that fortification of the intestinal barrier by increasing mucus production and altering AMP expression is for a different purpose. Instead, our findings with PEG-induced dysbiosis suggest that tuft cell sensing of succinate and the subsequent AMP expression changes may have evolved to respond to bacterial disturbances. Succinate is produced by numerous commensal bacteria during carbohydrate fermentation as a metabolic by-product. However, succinate is not found at high concentrations in the gut lumen because it is rapidly consumed by other microbes (26, 27). Thus, succinate accumulation in the intestinal lumen is generally associated with gut microbiota disturbances (i.e., antibiotic treatment, watery diarrhea, or inflammatory bowel disease) (21, 26, 59). In addition, the loss of goblet and Paneth cells leads to a bloom in succinate-producing bacteria, further reinforcing the connection between secretory epithelial cells and this metabolite (29). Thus, it is possible that distal SI tuft cells have evolved to respond to increased luminal succinate secondary to bacterial dysbiosis, triggering type 2 immunity and altering AMP production to restructure the microbiota and restore homeostasis.
Materials and Methods
Mouse Strains.
All mouse experiments were performed with a mixture of male and female mice (5 to 8 wk old) on the C57BL/6 background. Detailed information on mouse strains used is provided in SI Appendix, Materials and Methods.
Isolation of T. mu for Mouse Colonization.
A colony of WT C57BL/6J mice colonized with T. mu is maintained at Stanford University as a protist source. To obtain protists for colonization experiments, T. mu was isolated from source animals and cultured overnight at 37 °C in an anaerobic cabinet (Coy Lab Products), as previously described (20, 65). The following day, 1 × 105 T. mu were orally administered to each mouse. T. mu–colonized mice were killed at specified time points post-colonization.
10× Visium ST.
The distal 5 cm of the SI from uncolonized and T. mu–colonized mice were harvested. The luminal contents were flushed out with ice-cold sterile PBS using a 19-gauge feeding needle, and the tissue was opened longitudinally and swiss-rolled. The tissue was then covered in optimal cutting temperature (OCT) compound (VWR) and immediately frozen in an isopentane and liquid nitrogen bath. The frozen tissues were stored at −80 °C until further processing.
To perform gene expression analysis, 10-µm-thick sections were cut and placed onto the Visium Gateway slide (10× Genomics, Pleasanton, CA). The manufacturer’s protocol was followed without any significant alterations. Briefly, sections were fixed with methanol for 30 min at −20 °C, followed by hematoxylin and eosin (H&E) staining and imaging using a BZ X-800 microscope (Keyence). Sections were subjected to tissue permeabilization for 10 min—permeabilization time was established using a Tissue Optimization slide (10× Genomics) and the manufacturer’s protocol. This was followed by reverse transcription for cDNA synthesis, second strand synthesis, cDNA amplification (18 cycles), and next-generation sequencing library preparation. Libraries were quantified using Bioanalyzer (Agilent) and qPCR analysis (Bio-Rad). Libraries were pooled and sequenced on the NovaSeq 6000 instrument (Illumina Inc.), and data were processed through the Space Ranger pipeline (10× Genomics). Detailed information on ST data analysis is provided in SI Appendix, Materials and Methods.
Succinate Treatment.
Mice were provided with 100 mM succinic acid disodium salt (Sigma-Aldrich; Cat# 224731-500G) in their drinking water for 1 wk. To match sodium molarity, 200 mM sodium chloride (Sigma-Aldrich; Cat# S9888-500G) was used as a control, except for experiments with GF and CV mice in Fig. 4 and SI Appendix, Fig. S5 A–E, where water was used as a control.
PEG Treatment.
PEG 3350 (Miralax) was administered to mice at a 10% concentration in the drinking water for 12 d. The distal 3 cm of the SI was removed and processed for flow cytometry and qRT-PCR as described in SI Appendix, Materials and Methods.
Other Procedures.
Details about ST data analysis, transmission electron microscopy, T. mu enumeration via qPCR, epithelial cell isolation and flow cytometry, histology and fluorescence microscopy, Paneth and tuft cell enumeration in intestinal tissues, RNA extractions and qRT-PCR, immunoblotting, small intestinal organoid culture and imaging, in vivo IL-25 injections, 16S rRNA gene sequencing sample collection and data analysis, total bacterial or SFB 16S rRNA gene copy enumeration, E. faecalis culture, and in vivo bacterial abundance assays are provided in SI Appendix, Materials and Methods. All primer sequences are listed in SI Appendix, Table S1. All antibodies used are listed in SI Appendix, Table S2.
Statistical Analysis.
Details about statistical analyses on experimental data are provided in SI Appendix, Materials and Methods.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank the Howitt Lab, Salzman Lab, and Dr. K. Ng for comments on the manuscript and helpful discussions. We thank Dr. R. Margolskee, Amgen, Dr. C.-H. Lee, and Dr. C. Wilen for generously sharing the Trpm5−/− mice, Sucnr1−/− mice, Il13−/− mice, and Pou2f3−/− mice, respectively. We thank C. Mechler for her help with mouse colony maintenance and genotyping. We thank A. Torres and H. Tang for their help with cryosectioning tissue. We thank P. Chu at Stanford’s Pathology Department Histology Service Center and the Core Histology Facility at the Children’s Research Institute (CRI), Children’s Wisconsin for processing tissue samples, and Dr. S. Kumar at the CRI Core Imaging Facility for scanning microscopy slides. We thank J. Perrino at the Stanford Cell Sciences Imaging Facility (CSIF) for transmission electron microscopy (TEM). We thank Drs. D. Wagh and J. Coller from the Stanford Functional Genomics Facility (SFGF) for their help with spatial transcriptomics (ST). We thank the UW-Madison Biotechnology Center for 16S rRNA gene sequencing. Cartoon schematics were created with BioRender.com, except for SI Appendix, Fig. S10. This research was supported by the Stanford School of Medicine Dean’s Postdoctoral Fellowship, Stanford Maternal & Child Health Research Institute (MCHRI) Postdoctoral Fellowship, and A.P. Giannini Foundation Postdoctoral Fellowship (to C.F.); NIH R01DK128292 and R21AI171222 (to M.R.H.); the Medical College of Wisconsin’s Digestive Disease Center, the Advancing a Healthier Wisconsin Endowment, and NIH R35GM122503 (to N.H.S.); NIH T32 AI00729037 (to G.M.B. and K.F.N.); the Stanford MCHRI Pediatric Inflammatory Bowel Disease and Celiac Disease Research Postdoctoral Fellowship (to E.R.G.); the NSF Graduate Research Fellowship and Stanford Graduate Fellowship (to M.D.W.T.); and the Stanford Bio-X Undergraduate Summer Research Program Award (to L.B.D.). ST sequencing data were generated with instrumentation at the SFGF, which were purchased with NIH awards S10OD025212 and 1S10OD021763. TEM done by Stanford’s CSIF was supported, in part, by NIH S10 Award Number 1S10OD028536-01 from the Office of Research Infrastructure Programs. The data/contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Center for Research Resources or the NIH.
Author contributions
C.F., L.M.F., N.H.S., and M.R.H. designed research; C.F., L.M.F., G.M.B., M.B.G., E.R.G., M.H., J.Z., J.A.L., K.F.N., M.D.W.T., and L.B.D. performed research; C.F., L.M.F., G.M.B., M.B.G., and S.N.A. analyzed data; A.P. conducted statistical analyses of all experimental data; and C.F., L.M.F., G.M.B., N.H.S., and M.R.H. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
Preprint Servers: This manuscript was deposited into bioRxiv, and the licensing information states that the copyright holder for this preprint is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse is allowed without permission.
This article is a PNAS Direct Submission.
Contributor Information
Nita H. Salzman, Email: nsalzman@mcw.edu.
Michael R. Howitt, Email: mhowitt@stanford.edu.
Data, Materials, and Software Availability
The following data and code are publicly available as of the date of publication. ST data are deposited at Stanford Digital Repository (https://purl.stanford.edu/pr220nz2789) (66). Original code for ST analysis is deposited at https://github.com/Howittlab/tmu-smallintestine-visium (67). The 16S rRNA gene sequencing data are deposited at the National Center for Biotechnology Information (NCBI) Sequence Read Archive (BioProject ID PRJNA873680) (68). Original code for 16S rRNA gene sequencing analysis is deposited at https://github.com/MCW-CMR/Salzman-Howitt_collaboration (69). All other data are included in the manuscript and/or SI Appendix.
Supporting Information
References
- 1.Sekirov I., Russell S. L., Antunes L. C. M., Finlay B. B., Gut microbiota in health and disease. Physiol. Rev. 90, 859–904 (2010). [DOI] [PubMed] [Google Scholar]
- 2.de Vos W. M., Tilg H., Van Hul M., Cani P. D., Gut microbiome and health: Mechanistic insights. Gut 71, 1020–1032 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johansson M. E. V., Hansson G. C., Immunological aspects of intestinal mucus and mucins. Nat. Rev. Immunol. 16, 639–649 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meyer-Hoffert U., et al. , Secreted enteric antimicrobial activity localises to the mucus surface layer. Gut 57, 764–771 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Vaishnava S., et al. , The antibacterial lectin RegIIIγ promotes the spatial segregation of microbiota and host in the intestine. Science 334, 255–258 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johansson M. E. V., Sjövall H., Hansson G. C., The gastrointestinal mucus system in health and disease. Nat. Rev. Gastroenterol. Hepatol. 10, 352–361 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gallo R. L., Hooper L. V., Epithelial antimicrobial defence of the skin and intestine. Nat. Rev. Immunol. 12, 503–516 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Propheter D. C., Chara A. L., Harris T. A., Ruhn K. A., Hooper L. V., Resistin-like molecule β is a bactericidal protein that promotes spatial segregation of the microbiota and the colonic epithelium. Proc. Natl. Acad. Sci. U.S.A. 114, 11027–11033 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salzman N. H., et al. , Enteric defensins are essential regulators of intestinal microbial ecology. Nat. Immunol. 11, 76–82 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johansson M. E. V., et al. , Normalization of host intestinal mucus layers requires long-term microbial colonization. Cell Host Microbe 18, 582–592 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sommer F., Bäckhed F., The gut microbiota — masters of host development and physiology. Nat. Rev. Microbiol. 11, 227–238 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Cash H. L., Whitham C. V., Behrendt C. L., Hooper L. V., Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science 313, 1126–1130 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hooper L. V., et al. , Molecular analysis of commensal host-microbial relationships in the intestine. Science 291, 881–884 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Hooper L. V., Stappenbeck T. S., Hong C. V., Gordon J. I., Angiogenins: A new class of microbicidal proteins involved in innate immunity. Nat. Immunol. 4, 269–273 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Vaishnava S., Behrendt C. L., Ismail A. S., Eckmann L., Hooper L. V., Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc. Natl. Acad. Sci. U.S.A. 105, 20858–20863 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao Y., et al. , GPR43 mediates microbiota metabolite SCFA regulation of antimicrobial peptide expression in intestinal epithelial cells via activation of mTOR and STAT3. Mucosal. Immunol. 11, 752–762 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berg G., et al. , Microbiome definition re-visited: Old concepts and new challenges. Microbiome 8, 103 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matijašić M., et al. , Gut microbiota beyond bacteria—mycobiome, virome, archaeome, and eukaryotic parasites in IBD. Int. J. Mol. Sci. 21, 2668 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baker D. G., Flynn’s Parasites of Laboratory Animals (John Wiley & Sons, 2008). [Google Scholar]
- 20.Howitt M. R., et al. , Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science 351, 1329–1333 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lei W., et al. , Activation of intestinal tuft cell-expressed Sucnr1 triggers type 2 immunity in the mouse small intestine. Proc. Natl. Acad. Sci. U.S.A. 115, 5552–5557 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nadjsombati M. S., et al. , Detection of succinate by intestinal tuft cells triggers a Type 2 innate immune circuit. Immunity 49, 33–41.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schneider C., et al. , A metabolite-triggered tuft cell-ILC2 circuit drives small intestinal remodeling. Cell 174, 271–284.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gerbe F., et al. , Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature 529, 226–230 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.von Moltke J., Ji M., Liang H.-E., Locksley R. M., Tuft-cell-derived IL-25 regulates an intestinal ILC2–epithelial response circuit. Nature 529, 221–225 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fernández-Veledo S., Vendrell J., Gut microbiota-derived succinate: Friend or foe in human metabolic diseases? Rev. Endocr. Metab. Disord. 20, 439–447 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koh A., De Vadder F., Kovatcheva-Datchary P., Bäckhed F., From dietary fiber to host physiology: Short-chain fatty acids as key bacterial metabolites. Cell 165, 1332–1345 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Müller M., et al. , Biochemistry and evolution of anaerobic energy metabolism in eukaryotes. Microbiol. Mol. Biol. Rev. 76, 444–495 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Banerjee A., et al. , Succinate produced by intestinal microbes promotes specification of tuft cells to suppress ileal inflammation. Gastroenterology 159, 2101–2115.e5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haber A. L., et al. , A single-cell survey of the small intestinal epithelium. Nature 551, 333–339 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bergenstråhle J., Larsson L., Lundeberg J., Seamless integration of image and molecular analysis for spatial transcriptomics workflows. BMC Genomics 21, 1–7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Korsunsky I., et al. , Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bevins C. L., Salzman N. H., Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat. Rev. Microbiol. 9, 356–368 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Peterson L. W., Artis D., Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 14, 141–153 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Merzel J., Leblond C. P., Origin and renewal of goblet cells in the epithelium of the mouse small intestine. Am. J. Anatomy 124, 281–305 (1969). [DOI] [PubMed] [Google Scholar]
- 36.Kamal M., et al. , Mucosal T cells regulate Paneth and intermediate cell numbers in the small intestine of T. spiralis-infected mice. Clin. Exp. Immunol. 126, 117–125 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kamal M., Dehlawi M. S., Brunet L. R., Wakelin D., Paneth and intermediate cell hyperplasia induced in mice by helminth infections. Parasitology 125, 275–281 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Chen Z., Downing S., Tzanakakis E. S., Four decades after the discovery of regenerating islet-derived (Reg) proteins: Current understanding and challenges. Front. Cell Dev. Biol. 7, 235 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Ampting M. T. J., et al. , Intestinally secreted c-type lectin Reg3b attenuates salmonellosis but not listeriosis in mice. Infect. Immun. 80, 1115–1120 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dessein R., et al. , Toll-like receptor 2 is critical for induction of Reg3β expression and intestinal clearance of Yersinia pseudotuberculosis. Gut 58, 771–776 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Keilbaugh S. A., et al. , Activation of RegIIIβ/γ and interferon γ expression in the intestinal tract of SCID mice: An innate response to bacterial colonisation of the gut. Gut 54, 623–629 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ouellette A. J., Paneth cell α-defensins in enteric innate immunity. Cell. Mol. Life Sci. 68, 2215–2229 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu Z., et al. , Small proline-rich protein 2A is a gut bactericidal protein deployed during helminth infection. Science 374, eabe6723 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Artis D., et al. , RELMβ/FIZZ2 is a goblet cell-specific immune-effector molecule in the gastrointestinal tract. Proc. Natl. Acad. Sci. U.S.A. 101, 13596–13600 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.He W., et al. , Bacterial colonization leads to the colonic secretion of RELMβ/FIZZ2, a novel goblet cell-specific protein. Gastroenterology 125, 1388–1397 (2003). [DOI] [PubMed] [Google Scholar]
- 46.Herbert D. R., et al. , Intestinal epithelial cell secretion of RELM-β protects against gastrointestinal worm infection. J. Exp. Med. 206, 2947–2957 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stahl M., et al. , The Muc2 mucin coats murine Paneth cell granules and facilitates their content release and dispersion. Am. J. Physiol. Gastrointest. Liver Physiol. 315, G195–G205 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Damak S., et al. , Trpm5 Null mice respond to bitter, sweet, and umami compounds. Chem. Senses 31, 253–264 (2006). [DOI] [PubMed] [Google Scholar]
- 49.Toma I., et al. , Succinate receptor GPR91 provides a direct link between high glucose levels and renin release in murine and rabbit kidney. J. Clin. Invest. 118, 2526–2534 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ireland H., Houghton C., Howard L., Winton D. J., Cellular inheritance of a Cre-activated reporter gene to determine paneth cell longevity in the murine small intestine. Dev. Dyn. 233, 1332–1336 (2005). [DOI] [PubMed] [Google Scholar]
- 51.Fricke W. F., et al. , Type 2 immunity-dependent reduction of segmented filamentous bacteria in mice infected with the helminthic parasite Nippostrongylus brasiliensis. Microbiome 3, 40 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miyoshi H., Stappenbeck T. S., In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat. Protoc. 8, 2471–2482 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sato T., et al. , Single Lgr5 stem cells build crypt–villus structures in vitro without a mesenchymal niche. Nature 459, 262–265 (2009). [DOI] [PubMed] [Google Scholar]
- 54.Waddell A., Vallance J. E., Hummel A., Alenghat T., Rosen M. J., IL-33 induces murine intestinal goblet cell differentiation indirectly via innate lymphoid cell IL-13 secretion. J. Immunol. 202, 598–607 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fallon P. G., et al. , Identification of an interleukin (IL)-25–dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J. Exp. Med. 203, 1105–1116 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Davis C. P., Savage D. C., Habitat, succession, attachment, and morphology of segmented, filamentous microbes indigenous to the murine gastrointestinal tract. Infect. Immun. 10, 948–956 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ivanov I. I., et al. , Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Salter S. J., et al. , Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12, 87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ferreyra J. A., et al. , Gut microbiota-produced succinate promotes C. difficile infection after antibiotic treatment or motility disturbance. Cell Host Microbe 16, 770–777 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cadwell K., et al. , A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456, 259–263 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lassen K. G., et al. , Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc. Natl. Acad. Sci. U.S.A. 111, 7741–7746 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu B., et al. , Irgm1-deficient mice exhibit Paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. Am. J. Physiol.-Gastrointest. Liver Physiol. 305, G573–G584 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bel S., et al. , Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science 357, 1047–1052 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gerrick E. R., et al. , Metabolic diversity in commensal protists regulates intestinal immunity and trans-kingdom competition. bioRxiv [Preprint]. 10.1101/2022.08.26.505490 (Accessed 27 August 2022). [DOI] [PMC free article] [PubMed]
- 65.Howitt M. R., et al. , The taste receptor TAS1R3 regulates small intestinal tuft cell homeostasis. ImmunoHorizons 4, 23–32 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fung C., Fraser L. M., Fung_Fraser_Tuft_Paneth. Stanford Digital Repository. https://purl.stanford.edu/pr220nz2789. Deposited 2 August 2022.
- 67.Fung C., Fraser L. M., tmu-smallintestine-visium. Howitt Lab Github. https://github.com/Howittlab/tmu-smallintestine-visium. Deposited 15 May 2023.
- 68.Fung C., Fraser L. M., SubmissionID: SUB11971783 BioProject ID: PRJNA873680. NCBI sra. https://www.ncbi.nlm.nih.gov/bioproject/PRJNA873680. Deposited 22 August 2022.
- 69.Fung C., Fraser L. M., Salzman-Howitt collaboration. MCW-CMR Github. https://github.com/MCW-CMR/Salzman-Howitt_collaboration. Deposited 22 August 2022.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
The following data and code are publicly available as of the date of publication. ST data are deposited at Stanford Digital Repository (https://purl.stanford.edu/pr220nz2789) (66). Original code for ST analysis is deposited at https://github.com/Howittlab/tmu-smallintestine-visium (67). The 16S rRNA gene sequencing data are deposited at the National Center for Biotechnology Information (NCBI) Sequence Read Archive (BioProject ID PRJNA873680) (68). Original code for 16S rRNA gene sequencing analysis is deposited at https://github.com/MCW-CMR/Salzman-Howitt_collaboration (69). All other data are included in the manuscript and/or SI Appendix.