

Significance
HIV-1, a human pandemic pathogen, relies on host machinery to express its RNA. HIV-1 unspliced RNA plays two critical roles in generating infectious particles: It is translated to produce viral proteins and it is packaged into nascent particles as genomic RNA to transfer genetic information. HIV-1 uses a host enzyme to start transcription at neighboring sites to generate two major RNA species that are 99.9% identical in sequence but differ functionally. Here, we show that when one of the major RNA species is depleted, HIV-1 can still replicate but exhibits fitness defects. These findings reveal the intricate regulation of HIV-1 replication and how this pathogen usurps host machinery to maximize replication fitness.
Keywords: HIV-1, RNA, transcription start site, genome packaging, replication fitness
Abstract
HIV-1 relies on host RNA polymeraseII (Pol II) to transcribe its genome and uses multiple transcription start sites (TSS), including three consecutive guanosines located near the U3-R junction, to generate transcripts containing three, two, and one guanosine at the 5′ end, referred to as 3G, 2G, and 1G RNA, respectively. The 1G RNA is preferentially selected for packaging, indicating that these 99.9% identical RNAs exhibit functional differences and highlighting the importance of TSS selection. Here, we demonstrate that TSS selection is regulated by sequences between the CATA/TATA box and the beginning of R. Furthermore, we have generated two HIV-1 mutants with distinct 2-nucleotide modifications that predominantly express 3G RNA or 1G RNA. Both mutants can generate infectious viruses and undergo multiple rounds of replication in T cells. However, both mutants exhibit replication defects compared to the wild-type virus. The 3G-RNA-expressing mutant displays an RNA genome–packaging defect and delayed replication kinetics, whereas the 1G-RNA-expressing mutant exhibits reduced Gag expression and a replication fitness defect. Additionally, reversion of the latter mutant is frequently observed, consistent with sequence correction by plus-strand DNA transfer during reverse transcription. These findings demonstrate that HIV-1 maximizes its replication fitness by usurping the TSS heterogeneity of host RNA Pol II to generate unspliced RNAs with different specialized roles in viral replication. The three consecutive guanosines at the junction of U3 and R may also maintain HIV-1 genome integrity during reverse transcription. These studies reveal the intricate regulation of HIV-1 RNA and complex replication strategy.
The pandemic pathogen HIV-1 hijacks host machinery to express its genome. After integration, the HIV-1 provirus becomes part of the host cell genome and host RNA polymerase II (Pol II) transcribes the provirus to generate viral RNA (1). Some viral transcripts undergo alternative splicing to produce multiple RNA forms to express discrete viral proteins, whereas others remain unspliced (2, 3). HIV-1 unspliced RNA (referred to as HIV-1 RNA hereafter) plays critical roles during viral replication: it is packaged as an RNA dimer into nascent virions to serve as the viral genome, and it is translated to produce Gag/Gag-Pol polyproteins required for virus assembly and replication (4, 5). How HIV-1 RNA balances these two essential functions has been a long-standing question in the field.
Recent studies showed that HIV-1 uses several neighboring sequences as transcription start sites (TSS), generating multiple transcripts that only differ by a few nucleotides (nt) (6–9). There are three consecutive guanosines at the U3–R junction in the HIV-1 long terminal repeat (LTR) and transcription can start at any of the three positions, generating RNAs that contain three, two, or one guanosines at the 5′ end, referred to 3G, 2G, and 1G RNAs, respectively. Critically, the TSS usage affects RNA function(s). For example, even though 3G RNA is often more abundant in cells, 1G RNA is selected over 3G RNA for packaging into virions (6–9). Therefore, HIV-1 can distinguish a 2-nt difference between two 9-kb RNAs and selectively package 1G RNA as the viral genome. We and others showed that the 5′ untranslated regions (UTRs) of 3G and 1G RNAs fold into distinct structures (7, 9–11). Our studies demonstrated that 1G RNA tends to form structures that expose the RNA dimerization signal and the Gag-binding sites, thereby facilitating RNA dimerization, Gag:RNA interactions, and packaging into the virion. In contrast, 3G RNA tends to form structures that sequester the dimerization signal and Gag-binding sites; consequently, this RNA is not favored for packaging (9).
Multiple TSS usage and selective 1G RNA packaging are conserved features in all HIV-1 variants that we have examined (12). These striking features raised multiple questions including how the TSS usage is regulated and whether there is an advantage for HIV-1 to use multiple TSS but only package a selected RNA species. Host RNA Pol II can often initiate transcription at more than one site in host genes (13). It is possible that the multiple TSS usage in HIV-1 simply reflects the nature of the host RNA Pol II and does not confer a replication advantage. Alternatively, HIV-1 may utilize the imprecise initiation of RNA Pol II to generate distinct RNA species and gain an advantage in viral replication. Indeed, it was suggested that the 5′-5′-triphosphate-linked 7-methylguanosine 5′ cap is sequestered in the 1G RNA and therefore not accessible to the translation machinery, whereas the 5′ cap is exposed in the 3G RNA, which is used as the translation template (10, 11).
In this report, we examined whether HIV-1 gains a replication advantage by using different TSS to generate distinct RNA species such as 3G and 1G RNA. We first determined elements that regulate HIV-1 TSS usage, and then constructed two HIV-1 mutants each containing a 2-nt modification that altered the TSS usage. One mutant primarily generates 1G RNA and does not transcribe 3G RNA. Another mutant predominantly generates 3G RNA and 1G RNA is severely depleted. We found that both mutants can produce infectious viruses and undergo multiple rounds of replication in T cells, indicating that coexpression of 1G and 3G RNA is not required for HIV-1 replication. However, compared to the unmodified wild-type (WT) virus, both mutants exhibited replication fitness defects, indicating that utilizing multiple TSSs to express both 1G and 3G RNA increases viral replication fitness. Furthermore, the rapid reversion of a mutated base in one mutant revealed the unexpected role of the three guanosines at the U3–R junction in maintaining the integrity of the viral genome.
Results
Next-Generation Sequencing (NGS)–Based 5′ Rapid Amplification of cDNA Ends (5′ RACE).
We have previously used 5′ RACE to examine the TSS of HIV-1 RNA (9). Briefly, a gag-specific primer was used to prime cDNA synthesis to ensure that only unspliced HIV-1 RNAs were studied. During cDNA synthesis, an oligonucleotide was added to the 3′ end of minus-strand DNA, the resulting product was amplified by PCR, cloned into a plasmid, and analyzed by sequencing multiple plasmid clones. Here, we adapted the 5′ RACE method so that the readout is based on NGS and not sequencing multiple plasmid clones. We compared the two methods using the same RNA samples isolated from HIV-1-infected cells and HIV-1 particles and found that the NGS-based 5′ RACE generated similar results on HIV-1 RNA ends compared with the standard 5′ RACE (SI Appendix, Fig. S1A). Consistent with previous studies, both methods showed that 3G RNA is more abundant than 1G RNA in the cytoplasm, whereas 1G RNA is preferentially packaged into particles (6, 7, 9). These studies also showed that the NGS-based 5′ RACE is highly reproducible; three independent replicates using RNA samples isolated from the same cell lines and virions produced from these cell lines generated similar results (SI Appendix, Fig. S1B). Additionally, we have used in vitro transcribed RNAs harboring HIV-1 sequences and encoding hammerhead ribozymes (HHRs) to ensure defined homogeneous 5′ ends as templates and showed that the NGS-based 5′ RACE is highly accurate (SI Appendix, Fig. S1C).
Deletion of Guanosines Shifts the TSS Usage to Downstream Sequences.
To explore the mechanisms that regulate HIV-1 TSS usage, we first deleted one or two guanosines at the U3–R junction and examined transcripts generated from these mutant proviruses. Traditionally, the R region is defined by the repeated sequences at the two ends of the retroviral RNA (1, 14). As HIV-1 uses multiple TSSs, various RNAs have different amounts of redundant sequences. For simplicity, in this report, the 5′ most base of the three consecutive guanosines at the U3–R junction is denoted as the start of the R region (Fig. 1A, position 1). Using a previously described single-cycle HIV-1 construct, BH0 (15), we generated mutants BH0-delG and BH0-delGG that contain deletions of one or two guanosines at the U3–R junction in both LTRs, respectively. BH0 is an NL4-3-based construct that contains two intact LTRs, 5′ UTR, and all cis-acting elements required for replication. BH0 also expresses Gag/Gag-Pol, Tat, Rev, and a heat stable antigen (HSA) gene in nef (Fig. 1B) but contains inactivating deletions in env, vif, vpr, and vpu. To analyze the TSS usage, we generated cell pools containing BH0, BH0-delG, or BH0-delGG proviruses. Briefly, BH0, BH0-delG, or BH0-delGG plasmids were cotransfected into 293T cells with pCMV-VSVG, a plasmid expressing the G protein of vesicular stomatitis virus (VSV-G) (16), and the resulting viruses were used to infect fresh 293T cells. Cells were infected at a multiplicity of infection (MOI) between 0.14 and 1.05, as determined by the expression of HSA marker genes. Each infection was pooled to generate a cell pool containing more than 129,000 independently infected cells. We isolated RNA from cell pool lysates and virions in the supernatant and performed NGS-based 5′ RACE to examine the TSSs of the HIV-1 RNAs.
Fig. 1.

System used to study HIV-1 TSS usage and the effects of guanosine deletions. (A) Sequence alignment of the region between CATA/TATA box and the start of R of NL4-3 (BH0) and delG and delGG mutants. CATA/TATA box is denoted by underline; conserved guanosines at the start of R are shown in bold with underline and labeled TSS; position from the start of R is indicated by the numbering on top of the sequence; red dashes denote deletions introduced to mutants. (B) General structure of NL4-3-based construct BH0 and experimental workflow. (C) Proportions of HIV-1 RNA species in infected cells and in virions. WT, delG, delGG refer to BH0, BH0-delG, and BH0-delGG, respectively. Data from three independent experiments were combined and contain at least 269,000 (cell) or 194,000 (virion) total reads. Labeling for CATA/TATA box, conserved guanosines, and color-coding of RNA species are the same throughout all figures in this report.
As shown in Fig. 1C, we found that instead of starting from the guanosine(s) at the U3–R junction, most transcripts from BH0-delG and BH0-delGG mutants were initiated from a downstream cytosine (denoted as C5 in Fig. 1A). The C5 transcript abundance is also reflected in the virion RNAs from BH0-delG and BH0-delGG. These results showed that deleting the guanosines caused the transcription initiation to shift downstream of the original TSS. Therefore, HIV-1 cannot be forced to produce mostly 1G RNA by simply removing guanosines at the U3–R junction; rather, a complex regulation is likely at play to select HIV-1 TSS.
Distance between CATA/TATA Box and R Affects Transcription Start Site Selection.
The transcripts of BH0-delG and BH0-delGG mutants predominantly initiate at the C5 position. This phenotype is reminiscent of that of two primary subtype B isolates, CH058 and CH106, in which a large proportion of unspliced RNA initiated from the C5 position (12). Compared to NL4-3, these two isolates have a 1-nt deletion between CATA/TATA box and R (Fig. 2A). These findings led us to propose that the distance between CATA/TATA box and R affects TSS selection. To test this hypothesis, we generated a BH0-based construct, BH0-dT(-10), that mimics the 1-nt deletion of CH058 and CH106 in both LTRs (Fig. 2A). We generated a pool of 293T cells infected with BH0-dT(-10) and examined RNA expressed from these proviruses and the virion RNA in the supernatants (Fig. 2B). Our results showed that indeed, most of the unspliced transcripts were initiated from C5 and the 1-nt deletion altered TSS selection.
Fig. 2.

The impact of the distance between CATA/TATA box and R on TSS usage. (A) Sequence alignments of the HIV-1, SIV, and NL4-3-based mutants with shorter (Upper) and longer (Lower) distances between CATA/TATA box and R. Compared to NL4-3 (BH0), naturally occurring polymorphisms are shown in blue, whereas mutations introduced into NL4-3 are shown in red. (B) Proportions of HIV-1 RNA species in infected cells and in virions. WT BH0 results from Fig. 1C are shown here for comparison. dT(-10), rcmCATA, and plusAC refer to BH0-dT(-10), BH0-rcmCATA, and BH0-plusAC, respectively. For each mutant, data from four independent experiments were combined and represent at least 107,000 (cell) or 76,000 (virion) reads.
We have previously observed that the simian immunodeficiency virus isolated from red-capped mangabey (SIVrcmGAB1) mostly generates 3G RNA (12). SIVrcmGAB1 differs from NL4-3 in the length and the sequence of the region between CATA/TATA box and R. Thus, we generated an NL4-3-based construct, BH0-rcmCATA, in which the sequences between CATA/TATA box and R were replaced with that of SIVrcmGAB1 in both LTRs (Fig. 2A). We performed 5′ RACE on cellular RNA and virion RNA isolated from BH0-rcmCATA-infected cells and found that most of the HIV-1 transcripts were 3G RNA with severe depletion of 1G RNA. The latter constituted <1.2% of all detected RNAs, indicating that the substituted SIVrcmGAB1 sequence indeed altered the TSS usage to mimic that of SIVrcm. There were seven differences between NL4-3 and SIVrcmGAB1 in the substituted region: 5 substitutions and a 2-nt insertion (AC) in the SIVrcm sequence. To test whether the 2-nt insertion affects TSS usage, we generated BH0-plusAC in which the AC dinucleotide sequence was added immediately upstream of R in both LTRs of BH0 (Fig. 2A). We found that the TSS usage of BH0-plusAC is similar to that of the BH0-rcmCATA mutant: most of the unspliced transcripts are 3G RNA with a minimal level of 1G RNA (Fig. 2B). Importantly, 1G RNA is also depleted in virions generated from cells infected with BH0-rcmCATA and BH0-plusAC. Thus, the 2-nt insertion upstream of R is sufficient to alter the TSS usage in HIV-1. Together, the results from BH0-dT(-10) and BH0-plusAC mutants demonstrate that the TSS usage is affected by the distance between the CATA/TATA box and R.
The Sequence Context of the HIV-1 TSS Affects Its Usage.
Host cell RNA Pol II has preferred initiation sequences (13). We reasoned that altering the guanosines at the beginning of R can affect the frequency of transcription initiation. To test this hypothesis, we generated two mutants, BH0-TGG and BH0-TTG, in which the three guanosines are changed to TGG or TTG, respectively (Fig. 3A), and examined their TSS usage as described in Fig. 1B. Our results indicated that in the TGG mutant, most of the transcription initiated from the second, third, and fifth positions of the R region, generating 2G, 1G, and C5 RNAs (Fig. 3B), respectively. Of these, 1G RNA is preferentially packaged. In the BH0-TTG mutant, transcription mostly started from the third position, making 1G RNA the dominating species in the cell and in the virion. These results demonstrated that the sequence context of the TSS affects its usage. Furthermore, the TTG mutant produces mostly 1G RNA without any 3G RNA detected, making it an excellent candidate for independently examining the role of 1G and 3G RNA in HIV-1 replication.
Fig. 3.

The effects of nucleotide sequence on HIV-1 TSS usage. (A) Sequence alignment of the region between CATA/TATA box and the start of R for NL4-3 (BH0), BH0-TGG, and BH0-TTG. Mutations introduced into NL4-3-based BH0 are shown in red. (B) Proportions of HIV-1 RNA species in infected cells and in virions. WT BH0 results from Fig. 1C are shown here for comparison. TGG and TTG refer to BH0-TGG and BH0-TTG, respectively. For TGG and TTG, 3G and 2G refer to transcripts starting from position 1 and 2, respectively. Results from four independent experiments were combined and represent at least 120,000 (cell) or 72,000 (virion) reads.
Characterization of Mutants That Transcribe Predominantly 1G or 3G RNA.
To better understand the impact of altering TSS on HIV-1 replication, we characterized the phenotypes of the BH0-TGG and BH0-plusAC mutants including Gag expression, virus production, RNA genome packaging, and viral infectivity. The cell pools we used to analyze TSS usage of BH0 and the two mutants contained uninfected cells, which do not interfere with the TSS analyses but complicate Gag expression quantification that uses a host gene product as an internal control. To establish a direct comparison and better define the mutant phenotypes, we generated cell lines infected with each mutant at a low MOI (MOI < 0.1) to ensure that most infected cells contained only one provirus. These cells then underwent several rounds of cell sorting until >90% of the cell population expressed the HSA marker encoded within each BH0 construct to minimize the proportion of uninfected cells. We then analyzed Gag expression and virus production from these cell lines using western blotting (SI Appendix, Fig. S2). Compared to the BH0-WT, BH0-TTG, which predominantly transcribes 1G RNA, expressed Gag at a slightly lower level (Fig. 4A), whereas BH0-plusAC, which predominantly transcribes 3G RNA, expressed Gag at a comparable level. Additionally, we measured the efficiency of virus release by comparing the amounts of Gag products (p55 plus p24) in the supernatant with total (cellular and supernatant) Gag products and found that virus release occurred at similar efficiencies for all the three constructs (Fig. 4B). Additionally, BH0-plusAC exhibited a genome-packaging defect (Fig. 4C), whereas BH0-TTG and WT BH0 displayed similar level of RNA packaging. It was previously shown that when both RNAs are present, 1G RNA is selected for packaging over 3G RNA (6, 7, 9). Our current results show that even without the competing 1G RNA, 3G RNA is not packaged efficiently into viral particles. We have also introduced VSV-G into these provirus-containing cell lines and measured the infectivity of the resulting viral particles using TZM-Bl indicator cells (Fig. 4D). Our results showed that particles produced by BH0-plusAC exhibited an infectivity defect, consistent with the observation that this mutant has a genome-packaging defect. In contrast, particles produced by the BH0-TTG construct have infectivity comparable to that of WT BH0. Thus, our studies show that HIV-1 mutants that predominantly express 3G RNA or 1G RNA can express Gag and produce infectious viruses. However, HIV-1 that mostly expresses 3G RNA exhibits a defect in genome packaging and infectivity, whereas HIV-1 that mostly expresses 1G RNA has a slight defect in Gag expression.
Fig. 4.

Characterization of BH0-based HIV-1 constructs that produce predominantly 1G (BH0-TTG) or predominantly 3G RNAs (BH0-plusAC) compared to BH0 (WT). Gag expression levels (A), virion release efficiency (B), HIV-1 RNA packaging efficiency (C), and infectivity (D) of virions produced from cell lines containing BH0, BH0-TTG, or BH0-plusAC proviruses. Data from at least three independent experiments are shown with the SD indicated. In all assays, the values for WT BH0 were set at 100%. *P < 0.05, **P < 0.005, ***P < 0.001, ns, not significant.
Replication Kinetics of Mutants That Transcribe Predominantly 3G or 1G RNA in T Cells.
To determine whether HIV-1 with altered TSS can undergo spreading infections in T cells, we modified NL4-3 to contain either the TTG or the plusAC mutation in both LTRs and examined their replication in CEM-174 T cells. We have previously shown that HIV-1 has the same TSS usage in T cells and 293T cells (12). Human 293T cells were transfected with plasmid encoding NL4-3 or its mutants, and viruses were harvested, quantified, and equal p24 amounts of viruses were used to infect CEM-174 cells. Virus replication was monitored by p24 amounts in the supernatant, and the replication kinetics are shown in Fig. 5. Levels of p24 detected in the supernatant peaked at 7 to 9 d postinoculation for WT NL4-3 and NL4-3-TTG, indicating that the TTG mutation did not cause detectable delays of viral replication in CEM-174 cells (Fig. 5 A and B). In contrast, the p24 production for NL4-3-plusAC peaked later, at 9 to 11 d (Fig. 5C). Thus, HIV-1 mutants with altered TSSs can replicate in T cells, albeit the mutant expressing mostly 3G RNA exhibits delayed replication kinetics.
Fig. 5.

Replication kinetics of NL4-3- and NL4-3-based mutants in T cells. Replication kinetics of wild-type NL4-3 (A), NL4-3-TTG that predominantly produces 1G RNA (B), and NL4-3-plusAC that predominantly produces 3G RNA (C) in CEM-174 T cells. Each line represents an independent experiment. The amounts of viruses released into the supernatant were monitored by p24 ELISA assay.
Replication Fitness of the TTG Mutant That Predominantly Transcribes 1G RNA.
NL4-3-TTG displayed replication kinetics similar to that of WT virus. To better understand the effect of the TTG mutation on viral replication, we performed a viral fitness assay that allowed the TTG mutant to compete against WT NL4-3 in the same culture (17). Briefly, WT NL4-3 and mutant NL4-3-TTG viruses were mixed at either a 1:1 or 1:9 ratio and the virus mixtures were used to infect CEM-174 cells. We then harvested the supernatant from the culture at various time points and analyzed the proportion of these two viruses by converting HIV-1 RNA to cDNA and sequencing the polymorphic sites. The proportion of the WT and the mutant viruses was determined by peak intensities of the polymorphic first base of the R region in sequencing chromatograms.
We have performed 12 competition assays: in 6 experiments, cells were infected with WT and TTG NL4-3 mixed at ratios close to 1:1, and in the other 6 experiments, cells were infected with viruses mixed at a 1:9 ratio. In all except one experiment, WT NL4-3 overtook the culture; representative experiments are shown in Fig. 6 A and B. In the one exception, cells were infected with a 1:1 mixture of WT:mutant viruses, and NL4-3-TTG overtook the cell culture (Fig. 6C). We calculated the relative fitness of the TTG mutant using results from the 12 experiments and found that the TTG mutant has a relative fitness of 0.91 ± 0.04 (mean ± SE) compared to the fitness of the WT NL4-3 (P = 0.0185; unpaired t test). Taken together, these results showed that compared to NL4-3, NL4-3-TTG has a replication fitness defect.
Fig. 6.
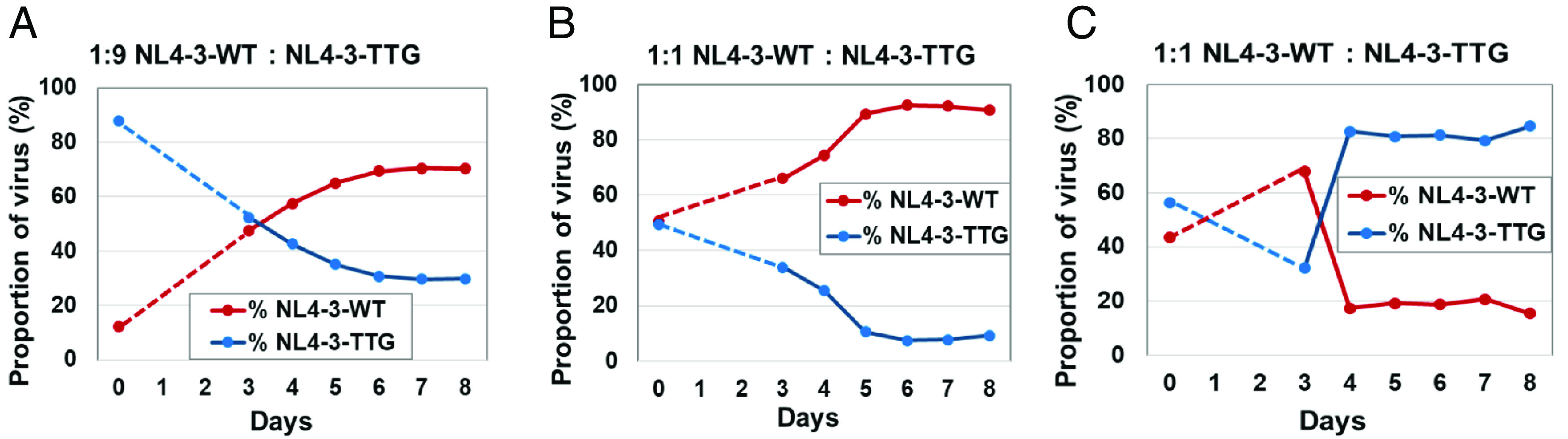
Determining the replication fitness of mutant NL4-3-TTG by competing against WT NL4-3. Representative experiments in which cell cultures were inoculated with virus mixtures containing NL4-3 and NL4-3-TTG at 1:9 (A) or 1:1 (B) ratios. In one of the 12 experiments, the mutant overtook the WT (C). Dashed line connects the data for input virus on day of infection (day 0) to the data on day 3 when sample collection began.
Reversion of the TTG Mutation during HIV-1 Replication.
The fitness defects of NL4-3-TTG led us to examine whether reversion can occur during its replication. For this purpose, we transfected the NL4-3-TTG plasmid into 293T cells to generate viruses (referred to as passage 0 or P0 virus), infected CEM-174 cells, monitored virus replication, and harvested supernatant at the peak of virus production (referred to as P1 virus). P1 viruses were used to infect fresh T cells to allow another round of replication and the supernatant was harvested at the peak of virus production, referred to as P2 virus. Similarly, P2 virus was used to infect fresh T cells to generate P3 virus. We then isolated RNA from these viruses and examined sequences near the U3–R junction to determine whether reversion had occurred by RT-PCR and sequencing of the PCR products (Fig. 7A). For clarity, the 5′ and the 3′ thymidines of the TTG are referred to as T1 and T2, respectively (Fig. 7A). We performed 5 independent passage experiments and observed T-to-G reversion in the T2 position in 4 of 5 cultures of the P1 viruses and all of the cultures in P2 viruses. Furthermore, the proportion of the revertant increased with passaging (Fig. 7B). In contrast, we did not observe detectable reversion of the T1 position in P1, P2, and P3 viruses (Fig. 7B). These results indicate that T1 and T2 mutations are maintained at very different rates: T2 is rapidly reverted to guanosine, whereas T1 is maintained during short passages of HIV-1. Furthermore, we only observed T2 reversion to guanosine and not to other bases even though T-to-G transversion mutations occur less frequently during reverse transcription compared to transition mutations (18, 19). We hypothesize that the instability of the T2 substitution is caused by stand-transfer events rather than mis-incorporation of nt during reverse transcription.
Fig. 7.

Detection of NL4-3-TTG revertant after HIV-1 replication. (A) A representative sequencing chromatogram illustrating mixed T and G signals that were detected at the T2 position. (B) The detection of reversion at the T2 but not T1 position after serial passaging of NL4-3-TTG mutant in T cells. Gray and black circles denote T1 reversions, whereas red, orange, and pink triangles depict T2 reversions.
Because 1G RNA is preferentially packaged, we show the strand-transfer process using 1G RNA to illustrate our hypothesis (Fig. 8). HIV-1 reverse transcriptase (RT) uses human tRNALys3, which is complementary to the primer-binding site, as a primer to initiate minus-strand DNA synthesis; in this step, RT copies U5 and R sequences. At the end of R, if RT copies the guanosine cap, an additional, nonencoded cytosine would be added to the minus-strand strong-stop DNA (indicated by yellow highlight in Fig. 8). After RNase H degrades the RNA template, minus-strand strong-stop DNA anneals to the 3′ R region of the viral RNA and RT continues DNA synthesis. For the WT NL4-3, the additional cytosine can anneal to the guanosine in the 3′ R and DNA synthesis continues. For the TTG mutant, the additional cytosine cannot base pair with the uridine, generating a mismatch at the end of the RNA:DNA hybrid such that mismatch extension must occur to continue DNA synthesis. During plus-strand DNA synthesis, RT uses the minus-strand DNA as a template and incorporates a guanosine opposite to the cytosine base, thereby generating the T-to-G substitution. Because 1G RNA is preferentially packaged, the T2 position is rapidly reverted. This hypothesis provides an explanation for the instability of the T2 position and the specific T-to-G conversion at this position.
Fig. 8.

The proposed mechanism for T-to-G reversion at the T2 position during NL4-3-TTG reverse transcription. RNA sequences are shown in black, with the polymorphic sites between WT and mutant shown in red; guanosine cap and its derived sequences are shown in yellow highlight. Mismatched base pairs are indicated by boxes. Minus- and plus-strand DNA sequences are in blue and gray, respectively.
Discussion
HIV-1 LTRs contain multiple cis-acting elements that regulate the transfer of genetic information encoded in the viral genome. In this study, we have shown that a very short stretch of sequence (~32 nt) in the LTR, from the CATA/TATA box to the beginning of the R region, regulates TSS selection. Mutational analyses reveal that both the sequence context of the start sites and the distance between the CATA/TATA box and the start sites are important elements that affect TSS selection. Importantly, minor modifications of this region can drastically change TSS usage. By introducing two different 2-nt mutations, we generated HIV-1 mutants that predominantly express 1G RNA without any 3G RNA, or predominantly express 3G RNA with depleted 1G RNA. We found that both mutants can undergo multiple rounds of replication in T cells, indicating that HIV-1 has the flexibility to replicate without 3G RNA, which is the major transcript in WT NL4-3–infected cells, or without 1G RNA, the preferred packaging substrate. However, both mutants have replication defects compared to WT virus. The mutant without 3G RNA has reduced Gag expression, whereas the mutant with very little 1G RNA exhibits a genome-packaging defect. These findings demonstrate that HIV-1 uses TSS heterogeneity to generate RNAs with almost identical sequences that have specialized functions. Furthermore, neither mutant can replicate as well as the WT virus, indicating that the expression of both 1G and 3G RNAs is essential for optimal HIV-1 replication fitness. Thus, HIV-1 utilizes a feature of the host enzyme RNA Pol II, TSS heterogeneity, to maximize replication fitness.
Although many primate lentiviruses use multiple TSS and select a specific unspliced RNA species for packaging, there are exceptions. SIVrcmGAB1, the inspiration of the plusAC mutant, mainly generates 3G RNA, and the unspliced viral RNA content in the virions mirrors that of the cells (12). HIV-1 plusAC mutant exhibits a packaging defect because the 3G RNA is not an ideal substrate for genome packaging. However, SIVrcmGAB1 3G RNA may not have a similar packaging defect. The 5′ UTRs of SIVrcmGAB1 and NL4-3 only share 42% sequence identity, and it is possible that the 3G RNA of SIVrcmGAB1 can be packaged efficiently by the homologous Gag. Regardless, SIVrcmGAB1 does not appear to generate two RNA species with distinct functions, which is different than HIV-1. Additional studies are needed to understand how SIVrcmGAB1, and other retroviruses that generate one major transcript, regulates the unspliced RNA functions and optimizes replication fitness.
Most eukaryotic mRNAs contain a modified guanosine cap that is added during transcription (20). Because host RNA Pol II transcribes proviruses, most retroviral RNAs also contain a guanosine cap, although the modification of the cap may differ (21, 22). Thus, having an added guanosine cap is a complication for most if not all retroviruses during reverse transcription of their RNA genomes. Most HIV-1 variants have three guanosines at the U3–R junction and all HIV-1 variants examined to date preferentially package 1G RNA (6, 7, 10, 12, 23). These features allow HIV-1 to handle the addition of a nonencoded guanosine from the cap as described in Fig. 8. However, this is not the case in many other retroviruses. Two HIV-2 molecular clones, ST and ROD, use multiple TSSs and the −1 nt immediately upstream of the TSS is a cytosine for the preferentially packaged RNA in each case (12). Thus, if the guanosine cap is copied in these viruses, it would generate a mismatched base pair during minus-strand DNA transfer much like the TTG mutant in Fig. 8. The boundary of the R region in many viruses has been defined by characterizing the minus-strand strong-stop DNA generated by the virions and using primer extension to identify the viral sequences upstream of the polyA tail (24–29). Intriguingly, the −1 nt immediately upstream of the defined R region in Rous Sarcoma virus and in Friend murine leukemia virus is also a cytosine (25–27). Thus, consecutive guanosines near the TSS in HIV-1 may help maintain its genome integrity; however, this does not appear to be a conserved feature among retroviruses.
The mechanisms by which non-HIV-1 retroviruses handle the added cap during reverse transcription is unknown. One possibility is that the guanosine cap is not copied efficiently during reverse transcription in these viruses because of intrinsic properties of their RTs or modifications to the cap. Alternatively, other mechanisms may be in play to maintain genome integrity. Further studies will be needed to address this question.
Materials and Methods
Molecular Cloning of HIV-1 Constructs.
NL4-3 (30), a generous gift from Malcom Martin (NIAID), was obtained through NIH HIV Reagent Program. Previously described pON-H0 is referred to as BH0 for simplicity (15). BH0 contains an hsa gene, internal ribosomal entry site (IRES), and a nonfunctional green fluorescent protein (gfp) gene in the nef gene (ref); for simplicity, IRES-gfp is not described in text or shown in figures. All LTR modifications in BH0 were introduced to both the 5′ and the 3′ LTRs by replacing the AatI-to-SpeI or XhoI-to-NgoMIV fragments, respectively, with synthesized DNA fragments containing mutations (IDT). To generate NL4-3 with modified LTRs, the AgeI-to-XhoI fragments of the BH0 mutants were replaced with that from NL4-3. Cloning was performed using NEBuilder Gibson Assembly kit (NEB) or by standard molecular cloning techniques. All modified regions were verified by Sanger sequencing to confirm modifications and avoid inadvertent mutations.
Cell Culture, Virus Infections, and Flow Cytometry.
Human 293T cells and CEM-174 were obtained from American Type Culture Collection (catalog nos. CRL-3216 and CRL-1992, respectively). TZM-bl cells are HeLa cell derivatives that stably express CD4, CXCR4, and CCR5, and express beta-galactosidase and firefly luciferase upon HIV-1 infection (31, 32). TZM-bl, 293T, and CEM-174 cells were maintained as previously described (15, 33).
Transfection of 293T cells was carried out using TransIT-LT1 reagent (Mirus Bio). To generate pools of infected cells, VSV-G-pseudotyped viruses were used to infect 293T cells and HSA expression was determined 3 d postinfection by flow cytometry using phycoerythrin-conjugated anti-HSA antibodies (BioLegend) as previously described (15). Cell pools were infected with BH0 or BH0-derivatives at MOI 0.14–1.25 and contained >129,000 independent infection events. To generate cell lines in which most cells contain one provirus, VSV-G-pseudotyped viruses were used to infect 293T cells at MOI < 0.1 and each cell line contained >52,000 independent infection events. Infected cells were enriched by multiple rounds of cell sorting until >90% of cells expressed the HSA marker. Flow cytometry was performed using an LSR II system (BD Biosciences), cell sorting was performed on BD FACSAria™ II Cell Sorter (Becton Dickinson), and data analyses were performed using FlowJo software (TreeStar, LLC).
Virus supernatants were collected, clarified through 0.45-µm-pore-size filters, and quantified by p24 ELISA (XpressBio). To examine replication kinetics, 100 pg of virus was used to infect 1 million CEM-174 cells, and input virus was removed 6 h later and replaced with fresh media. Culture fluid was removed at various time points and replenished with fresh media. Removed fluid was centrifuged and supernatant was collected; virus replication was monitored by p24 ELISA.
RNA Isolation and Quantification.
Supernatant from infected cells was collected, clarified through a 0.45-µm-pore-size filter (Millex), when necessary, concentrated by centrifugation, and used for RNA isolation with Mini Viral RNA kit (Qiagen). Total cellular RNA from infected cells was isolated using RNeasy Plus mini kit (Qiagen). A previously described RT-PCR sequencing method was used to determine the sequences of replicating viruses (34). RNA samples were treated with TURBO DNA-free™ Kit (Invitrogen) and subjected to RT-PCR using SuperScript™ II One-Step RT-PCR System with Platinum™ Taq (ThermoFisher) with primers NL9301S and NL9591R (SI Appendix, Table S1). PCR products were gel-purified and sequenced with primer NL9345S (SI Appendix, Table S1). The relative proportion of the mutant sequences was calculated by the height of mutant peak in the chromatogram divided by the sum of the heights of the mutant and the WT peaks. A standard curve was generated by mixing the WT and mutant plasmids at various ratios, sequencing the DNA mixtures, and measuring the chromatogram peaks of polymorphic sequences. Measurements within the linear dynamic range of the assay were used.
To quantify HIV-1 RNA in viral particles, RNA samples were serially diluted, and gag copy numbers were quantified using gag-specific primers Spe-WT1495F and Spe1564R (SI Appendix, Table S1) and iTaq Universal SYBR® Green One-Step kit (Bio-Rad). Potential DNA contamination was examined in all samples by performing the amplification without RT and found to be negligible.
In Vitro RNA Transcription.
Previously described plasmids p2GHH400 and p4GHH402 (9) were modified by replacing the BssHII-to-PciI fragment with the BssHII-to-SpeI fragment from BH0. The resulting plasmids, p2GHH1054 and p4GHH1056, contain a T7 promoter followed by a 5′ hammerhead ribozymes (HHR) (35) and encode 1,054- and 1,056-nt NL4-3 sequence, respectively, including the 5′ UTR and part of the gag gene. p2GHH1054 and p4GHH1056 were linearized with AccI restriction enzyme, purified by ethanol precipitation, and used for in vitro transcription with MEGAscript™ T7 Transcription Kit (Invitrogen) followed by RNA purification with MEGAclear™ Transcription Clean-Up Kit (Invitrogen). HHRs were designed to cotranscriptionally cleave the transcript, leaving either two 5′ guanosines to mimic the cap-1G RNA, or four 5′ guanosines to mimic the cap-3G RNA.
NGS-Based 5′ RACE.
To determine the 5′ end of HIV-1 unspliced transcript, RNA was converted into cDNA using a gag-specific primer BH01310to1329R (SI Appendix, Table S1) and the SMARTer RACE 5′/3′ kit (Takara). Next, a mixture of three different forward primers (RNGS-F1, RNGS-F4, RNGS-F6) and a mixture of three different reverse primers (RNGS-911-R5, RNGS-911-R6, RNGS-911-R7) were used to PCR amplify the 5′ RACE product and add NGS adaptors. These custom NGS PCR primers contained sections that anneal to SMART oligo (forward primers) or gag (reverse primers) regions of cDNA separated by variable-length spacers from the adaptor sequences for Illumina Indexes (SI Appendix, Table S1). The NGS adaptor PCR was performed using Q5® High-Fidelity DNA Polymerase (NEB); the PCR products were gel-purified using NucleoSpin Gel and PCR Clean-up Mini kit (Macherey-Nagel) and quantified using Qubit® dsDNA HS (High Sensitivity) Assay Kit (ThermoFisher). At least 50 ng of purified PCR product was used for library preparation with Q5® High-Fidelity DNA Polymerase (NEB) and Nextera® XT Index kit v2 Set B (Illumina). The indexed NGS libraries were gel-purified, quantified as above, and pooled for sequencing using MiSeq® Micro kit v2 (Illumina).
Western Immunoblotting.
HIV-1-infected cells were washed and resuspended in CellLytic M lysis buffer (Sigma) containing cOmplete ULTRA Proteinase inhibitor cocktail (Roche). Supernatants were clarified, pelleted through 20% sucrose by centrifugation, and resuspended in CellLytic lysis buffer. Loading buffer (4xNuPAGE) was added to lysates and heated at 99 °C for 5 min prior to loading. Immunoblots were probed for HIV-1 Gag with a mouse anti-p24 CA antibody (36) (a kind gift from Michael H. Malim through NIH AIDS Reagent Program, NIAID, NIH), followed by a secondary goat anti-mouse antibody (IRDye®-680RD, LI-COR). The host protein glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was probed using rabbit anti-GAPDH antibodies (ab128915, Abcam) followed by goat anti-rabbit antibody (IRDye®-800CW, LI-COR). Western blots were imaged and quantitated using the Odyssey infrared imaging system (LI-COR).
Single-Cycle Infectivity Assay.
Cell lines in which most cells contain one provirus were transfected with HIV-1 envelope–expressing plasmid pNL(AD8env)III (37). Viruses were harvested 24 h later, clarified, quantified using p24 ELISA assay, and used to infect TZM-bl cells. Luciferase activity was measured 48 h postinfection using Britelite plus Ultra-High Sensitivity Luminescence Reporter kit (PerkinElmer) in a 96-well luminometer (LUMIstar Galaxy; BMG LABTECH, Cary, NC, USA). Average luminescence from uninfected cells was subtracted from luminescence from infected cells followed by normalization to p24 virus input; WT values were set to 100%.
Competition Fitness Assays.
WT and mutant viruses were mixed at different ratios, and the relative proportion of the two viruses in the mixture was determined by the RT-PCR sequencing method described above. CEM-174 cells were infected with 100 pg of virus mixtures, and culture fluid was collected at various time points. Viral RNA from cleared supernatants was isolated, treated with DNAse, and subjected to RT-PCR sequencing.
Relative fitness of mutants (s) was calculated as follows (38):
where
H(T): The ratio of M(T)/W(T) on day T of measurement.
M(T): The quantity of mutant virus on day T was calculated as % of mutant in the mix × total viral load measured by p24 on day T
W(T): The quantity of WT virus on day T was calculated as % of WT in the mix × total viral load by p24 on day T
Tf: The final day of measurements
Ti: The initial day of measurements
δ: The death rate at 0.5 according to previous estimates for HIV-1 (>50% of CD4+ T cells are depleted in 2 d) (39).
A custom Python script was used to calculate the relative fitness s for each pair-wise time interval (e.g., day 3 vs. day 4; day 3 vs. day 5), and all pair-wise values were used to calculate s mean for the experiment.
Bioinformatics and Statistical Analysis.
MiSeq NGS reads were analyzed using a custom bioinformatics pipeline. Briefly, after trimming adaptors, NGS reads were filtered for quality (q20p90) and converted into fasta files; the number of reads corresponding to different HIV-1 RNA species was counted by a custom script.
One-way ANOVA with Dunnett’s posttest for multiple comparisons and unpaired t tests were performed in GraphPad Prism v9.2.0 (GraphPad Software, LLC).
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank E. Freed, S. Hughes, and J. Coffin for helpful discussions. This work is supported in part by Intramural Research, National Cancer Institute, NIH; Innovation funds from Office of AIDS Research, NIH, to W.-S.H. and to V.K.P.; supplemental funds from Center for Cancer Research to W.-S.H. and to V.K.P.; Intramural AIDS Research Fellowship to J.P.K.; NIH grants R01AI153216 and U54 AI170855 to K.M.-F.
Author contributions
O.A.N., J.P.K., A.D., V.K.P., and W.-S.H. designed research; O.A.N., S.I., J.P.K., A.D., Z.C., Y.L., and J.M.O.R. performed research; O.A.N., S.I., J.P.K., Z.C., Y.L., W.S., M.N., M.F.K., F.M., and K.M.-F. contributed new reagents/analytic tools; O.A.N., S.I., J.P.K., A.D., Z.C., Y.L., J.M.O.R., V.K.P., and W.-S.H. analyzed data; and O.A.N. and W.-S.H. wrote the paper; all contributing authors revised the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
Olga A. Nikolaitchik, Email: nikolaio@mail.nih.gov.
Wei-Shau Hu, Email: Wei-Shau.Hu@nih.gov.
Data, Materials, and Software Availability
Supporting Information
References
- 1.Ott M., Freed E. O., “Human immunodeficiency viruses: Replication” in Fields Virology , P. M. Howley, Knipe D. M., Eds. (Wolters Kluwer, Philadelphia, PA, USA, 2023), vol. 3, chap. 17, pp. 558–617. [Google Scholar]
- 2.Stoltzfus C. M., Regulation of HIV-1 alternative RNA splicing and its role in virus replication. Adv. Virus Res. 74, 1–40 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Emery A., Swanstrom R., HIV-1: To splice or not to splice, that is the question. Viruses 13, 181 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Butsch M., Boris-Lawrie K., Destiny of unspliced retroviral RNA: Ribosome and/or virion? J. Virol. 76, 3089–3094 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuzembayeva M., Dilley K., Sardo L., Hu W. S., Life of psi: How full-length HIV-1 RNAs become packaged genomes in the viral particles. Virology 454–455, 362–370 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Masuda T., et al. , Fate of HIV-1 cDNA intermediates during reverse transcription is dictated by transcription initiation site of virus genomic RNA. Sci. Rep. 5, 17680 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kharytonchyk S., et al. , Transcriptional start site heterogeneity modulates the structure and function of the HIV-1 genome. Proc. Natl. Acad. Sci. U.S.A. 113, 13378–13383 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pollpeter D., et al. , Deep sequencing of HIV-1 reverse transcripts reveals the multifaceted antiviral functions of APOBEC3G. Nat. Microbiol. 3, 220–233 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nikolaitchik O. A., et al. , Selective packaging of HIV-1 RNA genome is guided by the stability of 5’ untranslated region polyA stem. Proc. Natl. Acad. Sci. U.S.A. 118, e2114494118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown J. D., et al. , Structural basis for transcriptional start site control of HIV-1 RNA fate. Science. 368, 413–417 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ding P., et al. , 5’-Cap sequestration is an essential determinant of HIV-1 genome packaging. Proc. Natl. Acad. Sci. U.S.A. 118, e2112475118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rawson J. M. O., et al. , Transcription start site heterogeneity and preferential packaging of specific full-length RNA species are conserved features of primate lentiviruses. Microbiol. Spectr 10, e0105322 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haberle V., Stark A., Eukaryotic core promoters and the functional basis of transcription initiation. Nat. Rev. Mol. Cell Biol. 19, 621–637 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goff S. P., Roth M. J., “Retroviridae” in Fields Virology, Howley P. M., Knipe D. M., Eds. (Wolters Kluwer, Philadelphia, PA, USA, 2023), vol. 3, chap. 15, pp. 465–526. [Google Scholar]
- 15.Rhodes T. D., Nikolaitchik O., Chen J., Powell D., Hu W. S., Genetic recombination of human immunodeficiency virus type 1 in one round of viral replication: Effects of genetic distance, target cells, accessory genes, and lack of high negative interference in crossover events. J. Virol. 79, 1666–1677 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yee J. K., et al. , A general method for the generation of high-titer, pantropic retroviral vectors: Highly efficient infection of primary hepatocytes. Proc. Natl. Acad. Sci. U.S.A. 91, 9564–9568 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y., et al. , The roles of five conserved lentiviral RNA structures in HIV-1 replication. Virology 514, 1–8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pathak V. K., Temin H. M., Broad spectrum of in vivo forward mutations, hypermutations, and mutational hotspots in a retroviral shuttle vector after a single replication cycle: Substitutions, frameshifts, and hypermutations. Proc. Natl. Acad. Sci. U.S.A. 87, 6019–6023 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mansky L. M., Forward mutation rate of human immunodeficiency virus type 1 in a T lymphoid cell line. AIDS Res. Hum. Retroviruses 12, 307–314 (1996). [DOI] [PubMed] [Google Scholar]
- 20.Galloway A., Cowling V. H., mRNA cap regulation in mammalian cell function and fate. Biochim. Biophys. Acta Gene Regul. Mech. 1862, 270–279 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yedavalli V. S., Jeang K. T., Trimethylguanosine capping selectively promotes expression of Rev-dependent HIV-1 RNAs. Proc. Natl. Acad. Sci. U.S.A. 107, 14787–14792 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh G., et al. , HIV-1 hypermethylated guanosine cap licenses specialized translation unaffected by mTOR. Proc. Natl. Acad. Sci. U.S.A. 119, e2105153118 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang Y. L., Kawai G., Hasegawa A., Kannagi M., Masuda T., Impact of 5’-end nucleotide modifications of HIV-1 genomic RNA on reverse transcription. Biochem. Biophys. Res. Commun. 516, 1145–1151 (2019), 10.1016/j.bbrc.2019.06.152. [DOI] [PubMed] [Google Scholar]
- 24.Gilboa E., Mitra S. W., Goff S., Baltimore D., A detailed model of reverse transcription and tests of crucial aspects. Cell 18, 93–100 (1979). [DOI] [PubMed] [Google Scholar]
- 25.Coffin J. M., Haseltine W. A., Terminal redundancy and the origin of replication of Rous sarcoma virus RNA. Proc. Natl. Acad. Sci. U.S.A. 74, 1908–1912 (1977). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haseltine W. A., Maxam A. M., Gilbert W., Rous sarcoma virus genome is terminally redundant: The 5’ sequence. Proc. Natl. Acad. Sci. U.S.A. 74, 989–993 (1977). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coffin J. M., Hageman T. C., Maxam A. M., Haseltine W. A., Structure of the genome of Moloney murine leukemia virus: A terminally redundant sequence. Cell 13, 761–773 (1978). [DOI] [PubMed] [Google Scholar]
- 28.Mitra S. W., Goff S., Gilboa E., Baltimore D., Synthesis of a 600-nucleotide-long plus-strand DNA by virions of Moloney murine leukemia virus. Proc. Natl. Acad. Sci. U.S.A. 76, 4355–4359 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shank P. R., et al. , Mapping unintegrated avian sarcoma virus DNA: Termini of linear DNA bear 300 nucleotides present once or twice in two species of circular DNA. Cell 15, 1383–1395 (1978). [DOI] [PubMed] [Google Scholar]
- 30.Adachi A., et al. , Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59, 284–291 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Derdeyn C. A., et al. , Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J. Virol. 74, 8358–8367 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wei X., et al. , Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 46, 1896–1905 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delviks-Frankenberry K. A., et al. , Development of lentiviral vectors for HIV-1 gene therapy with vif-resistant APOBEC3G. Mol. Ther. Nucleic Acids 18, 1023–1038 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nikolaitchik O. A., Hu W. S., Deciphering the role of the Gag-Pol ribosomal frameshift signal in HIV-1 RNA genome packaging. J. Virol. 88, 4040–4046 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Birikh K. R., Heaton P. A., Eckstein F., The structure, function and application of the hammerhead ribozyme. Eur. J. Biochem. 245, 1–16 (1997). [DOI] [PubMed] [Google Scholar]
- 36.Simon J. H., et al. , The Vif and Gag proteins of human immunodeficiency virus type 1 colocalize in infected human T cells. J. Virol. 71, 5259–5267 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang M., Orenstein J. M., Martin M. A., Freed E. O., p6Gag is required for particle production from full-length human immunodeficiency virus type 1 molecular clones expressing protease. J. Virol. 69, 6810–6818 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maree A. F., Keulen W., Boucher C. A., De Boer R. J., Estimating relative fitness in viral competition experiments. J. Virol. 74, 11067–11072 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gandhi R. T., et al. , HIV-1 directly kills CD4+ T cells by a Fas-independent mechanism. J. Exp. Med. 187, 1113–1122 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)