


Abstract
Large expansions of the trinucleotide repeat GAA•TTC within the first intron of the X25 (frataxin) gene cause Friedreich’s ataxia, the most common inherited ataxia. Expansion leads to reduced levels of frataxin mRNA in affected individuals. Here we show that GAA•TTC tracts, in the absence of any other frataxin gene sequences, can reduce the amount of GAA-containing transcript produced in a defined in vitro transcription system. This effect is due to an impediment to elongation that forms in the GAA•TTC tract during transcription, a phenomenon that is exacerbated by both superhelical stress and increased tract length. On supercoiled templates the major truncations of the GAA-containing transcripts occur in the distal (3′) end of the GAA repeat. To account for these observations we present a model in which an RNA polymerase advancing within a long GAA•TTC tract initiates the transient formation of an R•R•Y intramolecular DNA triplex. The non-template (GAA) strand folds back creating a loop in the template strand, and the polymerase is paused at the distal triplex–duplex junction.
INTRODUCTION
Friedreich’s ataxia (FRDA), the most common inherited ataxia, is a progressive neurodegenerative disease caused by a large expansion of the trinucleotide repeat GAA•TTC within the first intron of the frataxin gene (1). Normal frataxin alleles have only a small number of GAA•TTC triplets (usually 7–21), while expanded alleles contain hundreds to thousands of these triplets (1,2). FRDA is recessive and a direct correlation between the length of expansion, particularly of the shorter allele, and disease severity has been described (3–5). The expanded repeat causes diminished expression of frataxin mRNA (6) and protein in patient tissues (7).
The GAA expansion in the frataxin primary transcript is not part of the spliced message, so diminished frataxin expression must result from an effect on transcription initiation, a block to transcription elongation, interference with proper splicing or some combination of these. Transcription inhibition by GAA•TTC tracts has been reproduced in transfected COS-7 cells (8) and in vitro with both HeLa nuclear extracts and T7 RNA polymerase (RNAP) (9). The results obtained with human RNA polymerase II (RNAP II) in extracts and T7 RNAP were qualitatively similar (9). A lack of evidence for improperly spliced products in RNA from FRDA patients (9) and the proper splicing of GAA•TTC bearing constructs transfected into COS-7 cells (8) all but rules out disrupted splicing as the major effect of expansion.
The GAA•TTC tract is a purine•pyrimidine (or R•Y) polymer and may adopt a number of unusual nucleic acid structures, including triple helices (10,11). Triplexes in general may take the form R•R•Y or Y•R•Y, depending on whether the third strand is purine-rich or pyrimidine-rich, and can be formed as intermolecular structures or as folded intramolecular structures (10–15). Models based on several different triplex variants have been suggested to explain the effects of GAA•TTC tract expansion as a possible block to transcription elongation in FRDA (8,9,16,17). However, the actual molecular mechanism by which the GAA•TTC repeat tract expansion reduces frataxin mRNA levels is still unknown.
We describe here our studies on the intrinsic biochemical properties of the GAA•TTC triplet repeat tract during transcription in the simple and well-defined in vitro transcription system of T7 RNAP. This polymerase is both highly processive in vitro and it requires no additional protein cofactors, simplifying the analysis of transcription biochemistry (18). We show that during transcription by T7 RNAP, an impediment to elongation is formed by the GAA•TTC template causing an extended pause in the distal end of the repeat. This is exacerbated by negative supercoiling but not by lower pH. Substantial pausing in the promoter proximal region of the GAA•TTC tract is not seen. These results are most consistent with a model in which an R•R•Y intramolecular triplex forms transiently during transcription of a long GAA•TTC tract, trapping the RNA polymerase.
MATERIALS AND METHODS
Plasmid construction
Templates containing uninterrupted (CAG•CTG)88 and (GAA•TTC)n repeats were made by a defined stepwise expansion of smaller units cloned into the pREX plasmid (a derivative of pSP72) using asymmetric type IIS restriction digests and ligation as previously described (19). Plasmids were prepared using a modified alkaline lysis procedure (20). Long GAA•TTC inserts are unstable in bacteria (19), so some templates were gel purified to enrich for those containing full-length inserts. To study transcription on supercoiled templates, an HpaI–XhoI fragment which included the T7 promoter and the repeats was excised from pREX derivatives and ligated into XbaI–XhoI digested pCMV-RiboCOP-250 (Sigma-Genosys, The Woodlands, TX). The RiboCOP plasmids contain a self-cleaving ribozyme sequence that cleaves the RNA transcript 226 bases 3′ to the XhoI recognition sequence.
In vitro transcription
RNA transcription from phage promoters was performed in T7 transcription buffer (50 mM HEPES pH 8.0, 100 mM NaCl, 20 mM MgCl2, 10 mM DTT and 0.5 mM each NTP) supplemented with 200 U/ml RNase Inhibitor (Ambion, Inc., Austin, TX) unless otherwise specified. T7 RNA polymerase (Amersham-Pharmacia Biotech, Piscataway, NJ) was used at a concentration of 500 U/ml, unless otherwise specified. Template concentrations were estimated by ethidium bromide fluorescence. Transcription was initiated by adding aliquots of a master mix of the above components to templates (usually ~20–200 ng/reaction), in a final volume of 20 µl, followed by incubation at 37°C for 20 min unless otherwise specified. The 5′ end was labeled by including [γ-32P]GTP (6000 Ci/mmol, NEN Life Science Products, Boston, MA) in the standard transcription reactions. RNA was purified where specified by isolation from a gel slice after electrophoresis, followed by phenol extraction and ethanol precipitation.
Gel electrophoresis
Agarose gel electrophoresis was performed in 1% gels in TAE buffer (40 mM Tris acetate, 2 mM EDTA at pH 8.0) and 1–2 µg/ml ethidium bromide (some gels were stained after electrophoresis). Radiolabeled reactions were stopped with 50 µl of stop buffer (96% deionized formamide, 10 mM EDTA and 10 µg/ml tRNA). The samples were precipitated with ethanol, resuspended in a denaturing gel loading buffer (96% formamide, 10 mM EDTA, and 0.05% bromophenol blue and xylene cyanol) heated to 90°C, and loaded on a prewarmed 6% polyacrylamide sequencing gel containing 8 M urea. An MspI digest of pBR322 (New England Biolabs, Beverly, MA) and a 10 bp ladder (Life Technologies, Rockville, MD) were used as size markers. Images of radioactive gels were obtained using Fujifilm type BAS III-S phosphorimaging screens and a Fujifilm BAS 1500 reader. Analysis and quantitation was performed with Fujifilm Image Gauge 3.0 (Mac) software.
Diethylpyrocarbonte (DEPC) analysis
An oligodeoxyribonucleotide with the sequence d[GTACGAATTCGAT(GAA)22(TTC)11GCATAGT] was synthesized using standard phosphoramidite chemistry and purified by polyacrylamide gel electrophoresis by IDT (Integrated DNA Technologies, Inc., Coralville, IA). The oligonucleotide (25 ng) was phosphorylated with [γ-32P]ATP (3000–6000 Ci/mmol, NEN Life Science Products) using T4 polynucleotide kinase (Epicentre Technologies, Inc., Madison, WI), in 50 mM Tris–HCl, pH 9.3, and 10 mM MgCl2. Samples (1 ng) of the labeled oligonucleotide were heated in TE (10 mM Tris–HCl pH 8.0, 1 mM EDTA) for 1 min at 90°C, then were adjusted to a final volume of 40 µl containing 50 mM HEPES pH 8.0, either with or without 6 mM MgCl2 as indicated, and incubated at 37°C for 5 min. DEPC (4 µl) was added to each tube and incubated for 20 min at 37°C. The DEPC reaction was terminated by precipitation with 1 ml butanol. Samples were resuspended in 1 M piperidine and cleavage at modified bases effected by incubation at 90°C for 30 min. Samples were precipitated twice with 1 ml butanol, dried under vacuum, dissolved in 20 µl of 42.5% (v/v) formamide, 5 mM EDTA (pH 9.5), 5 mM NaOH, 0.05% xylene cyanol, 0.05% bromophenol blue, denatured for 2 min at 90°C and the cleavage products resolved by electrophoresis on a 20% sequencing gel.
RESULTS
A long GAA•TTC repeat has an intrinsic ability to reduce transcription
The ethidium bromide stained gel in Figure 1 shows the linear templates containing the indicated number of GAA•TTC repeats and the products of their transcription by T7 RNA polymerase. The templates contained uninterrupted GAA•TTC tracts of defined length and specifically excluded any flanking frataxin sequences to ensure that any transcriptional effects were solely due to the repeat. A control template with no GAA•TTC insert (Fig. 1A, lane 1) and one with (GAA•TTC)11 (lane 2) produced similar levels of transcript, while templates containing 44 and 88 triplets showed progressive decreases in the amount of RNA produced (lanes 3 and 4). The heterogeneous appearance of the transcripts is due to secondary structure formation and is not observed if the RNA is denatured before electrophoresis (Fig. 2). The inverse relationship between the length of a transcribed GAA•TTC tract and the amount of GAA-containing RNA transcript produced recapitulates the effect of the GAA•TTC tract expansion seen in FRDA patients (6).
Figure 1.

GAA•TTC tracts inhibit transcript production, but not initiation. (A) Templates, digested with the restriction enzyme SspI which cuts 512 bp 3′ of the repeats, were transcribed as described in Materials and Methods. The site of T7 initiation is 55 bp 5′ of the repeat. After phenol extraction, precipitation with ethanol and resuspension in loading buffer the material was resolved on an agarose gel and then stained with ethidium bromide. The faint band above each template is an artifact of sample preparation. The number of GAA triplets in the transcripts is indicated above the lanes. Lane M is the 1 kb DNA ladder (Life Technologies). (B) Autoradiograph showing the products of abortive transcription reactions performed on the same templates used in (A). Only GTP and ATP were present (500 µM each) with [γ-32P]GTP (25 µCi per reaction) to label the 5′ end of the reaction products. Samples were taken after 2 and 20 min. The number of repeats in each template is indicated above the lane. The products were separated on a 23% denaturing polyacrylamide gel. The arrow indicates the position of the hexamer products. The GAA•TTC repeat tracts begin ∼50 bp beyond the termination of the abortive reactions and were not transcribed. The lane marked with a minus sign contains the product of a 20 min reaction with no template.
Figure 2.

Exogenous RNA has no effect on the transcription of (GAA•TTC)88 templates. Exogenous control (GAA)0 RNA was produced from a template digested with SspI (568 base transcript) and exogenous (GAA)88 RNA was prepared from an XhoI digested template (380 base transcript). The amounts added approximated the usual transcript yield obtained in this type of reaction for the (GAA•TTC)88 template (1×) and for the control (4×). Reactions were stopped with 2 vol formamide loading buffer, then heated to 65°C before loading to denature secondary structures. (A) Ethidium bromide stained agarose gel displays the transcripts (black arrow, 834 base transcript) produced from a (GAA•TTC)88 template linearized with SspI and transcribed in the presence of exogenous control RNA (lanes 2 and 3) and (GAA)88 RNA (lanes 4 and 5). (B) Ethidium bromide stained agarose gel displays the transcripts (black arrow, 773 base transcript) produced from a control template with no repeats linearized with XmnI and transcribed in the presence of exogenous control RNA (lanes 2 and 3) and (GAA)88 RNA (lanes 4 and 5).
To distinguish an effect on transcription initiation from one on elongation through the repeat tract, we performed transcription reactions in which GTP and ATP were the only ribose triphosphates present. The first eight bases of the T7 transcripts in these templates are GGGAGACC. In the presence of only GTP and ATP, a six base product is made, the accumulation of which has been shown to reflect relative promoter strength (21). The gamma phosphate is retained only by the initiating G, so inclusion of [γ-32P]GTP results in end-labeled RNA product. Promoter use as assessed by accumulation of labeled hexamer product (indicated with a black arrow in Fig. 1B) was the same on all templates at both early (lanes 1–4) and later (lanes 5–8) time points. These results suggest that transcription elongation is required for the inhibition.
(GAA)88 RNA does not inhibit transcription
One consequence of transcription elongation on GAA•TTC bearing templates is production of transcripts with (GAA)n tracts. The inverse relationship between GAA tract length and transcript yield seen in Figure 1 might reflect a concentration-dependent r(GAA) toxicity to T7 RNAP. It has also been suggested that transcription inhibition in FRDA may be mediated by the GAA RNA sequence binding to the GAA•TTC template as the third strand in an R•R•Y triplex (16). To test whether lower transcript yield resulted from either mechanism we carried out transcription reactions in the presence of exogenously synthesized RNA with a (GAA)88 tract. The exogenous RNAs were shorter than the transcripts produced in these experiments so that the two could be separated on a denaturing agarose gel.
A template containing 88 repeats produced about the same amount of product RNA whether or not purified RNA was present and without regard to GAA content of the added RNA (Fig. 2A, black arrow). The amount of RNA with a (GAA)88 tract (open arrow) added to the reaction shown in lane 4 was similar to the usual yield obtained in these experiments, and four times that amount was added to the reaction shown in lane 5. A similar lack of response to exogenous RNA was obtained with the control template (Fig. 2B). These results indicate that (GAA)88 RNA does not inhibit transcription by interacting with the (GAA•TTC)88 template or T7 RNA polymerase.
Transcription of the (GAA•TTC)88 template reduces transcription of a second template in trans
Several potential template-mediated mechanisms of inhibition can be evaluated by observing how a GAA•TTC bearing template and a control template compete for a limited pool of RNA polymerase. If transcription through a GAA•TTC tract causes a change in template conformation that alters the strength or availability of the promoter for subsequent rounds of initiation then the co-transcribed control template would become the preferred template for initiation. The disparity in yield between the two co-transcribed templates should at least equal the ratio obtained in separate reactions, and may exceed that at low concentrations of RNAP due to the competition. A second possibility, that promoter strength remains equal, but the GAA•TTC tract acts as a simple terminator, would result in the differential yield being maintained during co-transcription at all polymerase concentrations. Finally, if the primary defect in the GAA•TTC template is due to a reduction in the rate of transcription elongation, or to a long pause or arrest of the polymerase without termination and release, then the GAA•TTC template will act as a sink for RNAP, lowering the rate of RNAP recycling and hence the effective concentration of free polymerase. If promoter strength remains equal then the rate of RNAP reinitiation on either template will decline to the same degree during co-transcription with a limited pool of polymerase, reducing the yield of the control to approximately that of the repeat bearing template.
Figure 3 shows the results obtained when templates with 0 or 88 GAA•TTC triplets were transcribed separately (lanes 1–3 for control, 4–6 for 88 GAA•TTC triplets), or together (lanes 7–9) for 20 min. The reactions in each series of three lanes contained 5-fold dilutions of polymerase. When these templates are transcribed separately, the control template usually produces about four times as much transcript as the (GAA•TTC)88 template. However, when the templates are co-transcribed the amount of transcript produced from the control template is reduced to about that of the template with (GAA•TTC)88 (Fig. 3, compare lane 7 with lanes 4 and 1). Yields from either template during co-transcription (lane 7) more closely resemble that of the (GAA•TTC)88 template alone (lane 4) than of the control (lane 1). This is consistent with transcription through the repeat tract leading to a reduction in the amount of available polymerase, suggesting that RNAP is sequestered by the (GAA•TTC)88 template.
Figure 3.
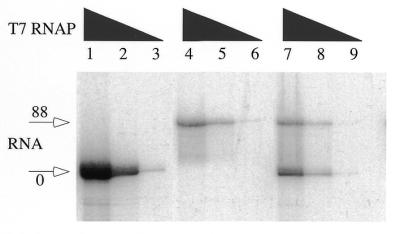
Transcription through a long GAA•TTC tract reduces transcription of a second template in trans. Transcription reactions were performed in the presence of [γ-32P]GTP (10 µCi per reaction) on aliquots of SspI linearized templates with no repeat (lanes 1–3), (GAA•TTC)88 (lanes 4–6) or both templates together (lanes 7–9). Arrows indicate the locations of the two transcripts in this denaturing gel. Serial 5-fold dilutions of T7 RNAP (10, 2 and 0.4 U per 20 µl reaction) were used for each set of three (indicated by a triangle). The concentration of each template was kept constant, in the mixed reactions the total template DNA concentration was therefore doubled. The smearing below the transcript with 88 repeats comes from deletions within the repeat in the template used for this experiment, and is not due to RNase activity.
Negative supercoils in (GAA•TTC)88 templates exacerbate transcription inhibition
If the decrease in RNAP turnover is due to a pause or slowing in the GAA•TTC tract, then transcription reactions that produce longer transcripts should diminish the apparent magnitude of the inhibition, since the increased time required to produce the longer transcripts decreases the relative contribution of a pause to the total RNAP turnover time. If the pause stems from structural properties of the template, then conditions that favor formation of the responsible structure should exacerbate the inhibition. Negative supercoiling can favor the formation of a number of non-B DNA structures that may interfere with transcription such as Z-DNA, cruciforms and intramolecular triplexes (10,22,23). To assess a role for structures such as these we tested the effects of negative supercoiling on transcription through a GAA•TTC repeat.
To facilitate direct comparison of transcription products from both long, linear and supercoiled templates, we made a series of constructs containing a self-cleaving ribozyme sequence 270 bp 3′ of the triplet repeat cloning site. Self-cleavage by the ribozyme yields a 5′ RNA product of 590 bases from templates containing 88 triplets regardless of the length of the primary transcript. This is the primary product labeled with [γ-32P]GTP. Ribozyme cleavage was usually >90%, and occurred equally efficiently on all templates. The control template contained 88 CTG•CAG triplets. RNA polymerase II has been shown to effectively transcribe all four CNG repeats (24). In other reactions we determined that T7 RNAP produced CUG-containing RNA from this template in yields similar to templates lacking repeats both in linear and supercoiled conformations (data not shown), indicating that the presence of a triplet repeat sequence per se does not account for impaired transcription.
To make linear templates for the experiment shown in Figure 4, the 3961 bp long supercoiled templates were linearized so that they would produce a run-off transcript of 2700 bases (after ribozyme cleavage this yields end-labeled 590 base and unlabeled 2110 base RNA products). About half as much (GAA)88 transcript was produced as the (CUG)88 control from these long linear transcription units during a 20 min reaction (Fig 4A, lanes 1 and 2). This difference is less than that obtained for the short linear templates used in the previous experiments, consistent with the idea of a pause on linear GAA•TTC templates. Supercoiled templates can produce even longer transcripts, thus one might expect that the difference in yield would be even less when transcription was performed on supercoiled GAA•TTC templates, all else being equal. However, far less full-length RNA (Fig. 4A. lane 4) was synthesized from a supercoiled (GAA•TTC)88 template than from either the linear form of the same template (lane 2) or the supercoiled control template (lane 3). In addition, prominent truncation products in the distal (3′) half of the (GAA)88 repeat tract were produced (Fig. 4A, lane 4). Most of the truncations are located in the second half of the repeat with the strongest peaks occurring at the very end of the repeat (Fig 4B). Longer exposures show a qualitatively similar distribution of truncations within the (GAA)88 tract on linear templates, indicating that negative supercoiling increases the frequency or efficiency of stopping.
Figure 4.

Negative supercoils exacerbate transcription inhibition by a (GAA•TTC)88 tract. (A) The templates used in these experiments contain the sequence for a self-cleaving ribozyme that cuts the transcript ∼270 bases 3′ to the end of the repeat tract, so the size of the full-length cleaved transcript (590 bases) is the same for both linear (L) and supercoiled (SC) templates. Linear templates were opened with restriction enzyme SspI and the primary transcript was 2698 bases. The templates produced RNA containing either (CUG)88 or (GAA)88 as indicated above the lanes. Templates were transcribed in the presence of [γ-32P]GTP (10 µCi per reaction). Bands immediately below the full-length transcripts extending to a length of ∼350 bases are due to deletions within the repeats in the templates. The numbers to the left indicate the size in bases of selected bands of the MspI digest of pBR322 used as a marker. (B) A scan of lane 4 aligned to the gel highlights the distribution of truncation products within the (GAA)88 tract. The bracket to the right of the scan labeled repeat tract indicates the location of the 88 triplets within the 5′ end-labeled transcripts in both the scan in (B) and the gel in (A).
The exacerbating effect of negative supercoiling suggests that a supercoil-enhanced structure in the template impeded elongation. Z-DNA causes supercoil-enhanced stops for Escherichia coli RNAP (23) but has been shown to be ineffective at stopping T7 RNAP (25) and, in any event, is unlikely to form with this sequence. Furthermore, the GAA•TTC tract is not G-rich enough to form a stable tetraplex, and does not form the hairpins needed for cruciform extrusion. However, R•Y sequences in general are known to form two major classes of intramolecular triplexes that may impede transcription. Examples of these are shown in Figure 5A. While formation of either type is enhanced by negative supercoiling, they do differ in other requirements. Formation of the R•R•Y form requires divalent cations but is relatively pH independent (11,13–15). The Y•R•Y triplex on the other hand, can form in the absence of divalent cations but is favored by low pH due to the need to protonate Cs in the third strand (10,11).
Figure 5.

A pH dependent inhibition of (UUC)n but not (GAA)n synthesis. (A) Line drawings of intramolecular R•R•Y and Y•R•Y triplexes. The purine (R) strand is black, the pyrimidine (Y) strand is gray. The single black dots indicate normal Watson–Crick base pairs and the smaller double dots indicate alternative hydrogen bonding interactions that are pH independent. Hoogsteen base pairs involving a protonated cytosine are indicated with a plus sign. (B) and (C) Ethidium bromide stained agarose gels display the products of transcription containing the indicated number of GAA or UUC triplets. Aliquots of the SspI linearized templates were transcribed in reactions buffered at pH 8.0 (left half of the gel) or pH 7.0 (right half). The reactions were stopped by adding an equal volume of loading buffer (25 mM EDTA, 200 mM Tris pH 8.0, 10% v/v glycerol) and loaded directly on a 1% agarose gel with TAE pH 8.0 as the running buffer. (B) GAA-containing transcript accumulation is similar at both pH 8.0 and pH 7.0. Transcribed templates directing GAA 44 and 88 transcription exhibit smearing at both pHs when loaded directly from the transcription reaction, but the same amounts were used in the reactions as the controls (compare to Fig.1). (C) UUC-containing transcript accumulation, from templates in which the orientation of the GAA•TTC repeat relative to the T7 promoter had been reversed, transcribed at pH 8.0 and pH 7.0. Templates producing 44 and 88 UUC triplets exhibit smearing after transcription at pH 7, and transcript production is reduced compared to transcription at pH 8. Lane M is the 1 kb DNA ladder
Inhibition of (UUC)n RNA synthesis is exacerbated at lower pH, but inhibition of (GAA)n RNA synthesis is not
To assess the role of the Y•R•Y triplex in transcription inhibition we compared the RNA yield of transcription reactions performed at pH 8.0 with those performed at pH 7.0. Production of GAA-containing transcripts from linear templates showed essentially the same length dependent inhibition at either pH 8.0 or pH 7.0 (Fig. 5B). In contrast, when the GAA•TTC tract was transcribed in the opposite direction to yield UUC-containing transcripts, the inhibition of transcription on linear templates containing 44 or 88 repeats was much greater at the lower pH (Fig. 5C). Production of RNA bearing 11 or 0 UUC triplets was not affected. That transcription at a lowered pH decreased accumulation of UUC-containing transcripts is consistent with inhibition by formation of a Y•R•Y triplex. The pH independence of the inhibition of GAA-containing transcripts is consistent with a role for the R•R•Y triplex but not for the Y•R•Y triplex.
GAA•TTC sequences can form a folded R•R•Y triplex
While the formation of an intramolecular Y•R•Y triplex by GAA•TTC tracts has been documented (16,26–28), the formation of the analogous folded R•R•Y triplex by GAA•TTC is controversial (17,27,28). The formation of an intramolecular R•R•Y triplex should leave part of the TTC-strand single-stranded. However, treatment of supercoiled molecules containing 44 or 88 repeats with the single strand specific nucleases S1 or P1 in the transcription buffer showed no specific cleavage, although cleavage in the GAA•TTC tract was readily detected at low pH (data not shown). This suggests that the R•R•Y structure does not form spontaneously in these plasmids at normal bacterial superhelical densities. However, such structures might be able to form when the template is unpaired during transcription.
We therefore tested the triplex forming potential of short stretches of unpaired GAA•TTC repeats in a simple model system. A single-stranded oligodeoxyribonucleotide containing 22 GAA repeats followed by 11 TTC repeats embedded in non-repetitive flanking sequence was treated with the chemical probe DEPC in the presence or absence of a divalent cation. DEPC carboxyethylates adenines, and to a lesser extent guanines (29). This modification occurs at adenine N6 and N7 and guanine N7 and is seen most readily when these bases are unpaired or in an unusual conformation. Subsequent treatment with piperidine cleaves at the modified bases.
In the absence of Mg2+, the 11 5′-most GAA repeats were DEPC reactive as were a small number of bases at the 3′ end of the GAA tract (Fig. 6A, gray arrow). This reactivity suggests that these bases are unpaired. The protection of the remaining GAA repeats is consistent with Watson–Crick hydrogen bonding to the TTC repeats with the reactive purines at the 3′ end of the tract forming part of the loop of the hairpin (Fig. 6B, arrow).
Figure 6.

DEPC reactivity of an oligonucleotide containing 5′-(GAA)22-(TTC)11-3′. (A) An autoradiograph of a denaturing 20% polyacrylamide gel showing the DEPC-specific piperidine cleavage of an end-labeled oligonucleotide containing 5′-(GAA)22-(TTC)11-3′ in the absence (lane 1) or presence (lane 2) of Mg2+ (6 mM). The numbers to the left of the gel indicate the GAA triplets from the 5′ end of the oligonucleotide. A line drawing to the right shows the general correspondence between bands on the gel and the linear oligonucleotide. The black portion indicates the (GAA)22 tract and the gray part corresponds to (TTC)11. Full-length, uncleaved material forms a dark band at the top of the gel in both lanes. The gray arrow indicates a region of Mg2+ independent DEPC hyper-reactivity; the black arrow indicates a region of Mg2+ reactivity. (B) and (C) Diagrammatic representation of the structure of the oligonucleotide in the absence (B) and presence (C) of Mg2+. The single black dots indicate Watson–Crick bonds and the smaller double dots indicate alternative hydrogen bonding interactions between different regions of the oligonucleotide. The gray arrow indicates the junction between the two tracts that is hyper-reactive with DEPC under these conditions. The black arrow in (C) indicates the DEPC reactive region seen in the middle of the GAA tract in the presence of Mg2+.
When Mg2+ is present the DEPC reactivity of the GAA tract becomes quite different. Most of the GAA triplets are now protected from DEPC, with reactive bases only in the middle of the GAA tract (Fig. 6A, black arrow) and at the 3′ end of the tract (gray arrow). These data are consistent with an R•R•Y triplex as shown in Figure 6C, in which the GAA repeats that were unpaired in the absence of Mg2+ have become associated with the Watson–Crick hairpin. This Mg2+ dependent transition from a hairpin to a triplex is consistent with a requirement for divalent cations in order for the R•R•Y triplex to form (13–15). Failure to observe the antiparallel R•R•Y structure formed by GAA•TTC in previous studies (27,28) may be due to the use of insufficiently long test sequences, the omission of divalent cations or the preferential formation of competing structures.
DISCUSSION
We have shown that GAA•TTC triplet repeats, in the absence of any other frataxin gene sequences, can reduce the yield of full-length transcripts made by T7 RNAP. The reduced yield was not the result of impaired promoter function, but due to impeded elongation. Since it required no proteins other than T7 RNA polymerase, or any of the sequences that normally flank the repeat in the frataxin gene, this elongation pause is an intrinsic biochemical property of long GAA•TTC tracts.
The properties of the pause site indicate that it is different from other intrinsic pausing and termination sites that have been described for T7 RNAP (30,31). Truncated transcripts accumulate throughout the distal half of the repeat tract with the most prominent truncations occurring near the junction of the repeat tract and the 3′ region flanking the repeat (Fig. 4). Pausing is exacerbated by GAA•TTC tract length and the presence of negative supercoiling in the template indicating a role for DNA structure.
The GAA•TTC tract, due to its asymmetrical strand distribution of purine and pyrimidine bases, has the potential to form triplex structures (10,11). This intrinsic potential has been invoked in models of GAA•TTC tract-mediated transcription impairment proposed for FRDA, the majority of which posit a structure, either a DNA triplex, combination RNA + DNA triplex, or a pair of DNA triplexes interacting between two templates, that stop RNA polymerase (8,9,16,17). These models do not explain our results with T7 RNAP.
In general, pre-existing triplex models do not fit our data, since these would result in truncations either promoter proximal to the tract, or within the promoter proximal half of the tract. Varying the template concentrations did not change the inhibition (data not shown), indicating that interactions between two templates are not likely to be involved. Because inhibition of the GAA-containing transcript was not exacerbated by lower pH we exclude the Y•R•Y triplex as the causal agent. An intermolecular R•R•Y triplex with the transcript as the third strand is not consistent with our data since formation of such a structure would not be enhanced by increased negative superhelicity, would predict promoter proximal rather than promoter distal stops and is incompatible with our observation that the addition of exogenous GAA-RNA did not affect transcription. While it is formally possible that RNA generated during transcription might behave differently, it has been shown that an RNA•DNA•DNA triplex of the R•R•Y type does not form even under conditions where the corresponding all DNA triplex is stable (32). Our data thus suggest a role for an intramolecular R•R•Y DNA triplex on an individual template.
We propose that a transient intramolecular R•R•Y triple helix is formed behind the polymerase during transcription, pausing the polymerase within the GAA•TTC tract as illustrated in Figure 7. The movement of RNAP along the template locally unpairs the DNA duplex and generates a wave of negative supercoiling in its wake (33,34) (Fig. 7A). This creates conditions favorable for triplex formation. At the transcription bubble the polymerase covers the Y (TTC) template strand but the single-stranded portion of the GAA non-template strand is available to initiate triplex formation, promoting formation of the R•R•Y structure (Fig. 7B). The initial folding may be analogous to the formation of the folded R•R•Y structure by an oligodeoxyribonucleotide (Fig. 5). The spread of triplex formation (Fig. 7C) is driven by the release of the standing wave of negative superhelical energy that had formed behind the polymerase (33,35,36). We suggest that the polymerase has trouble negotiating the junction between the triplex and the duplex in the distal end of the repeat tract (indicated by the black arrow in Fig. 7D). This can result in a transcript truncated at the 3′ end of the structure, an outcome not predicted by previously proposed models for GAA•TTC mediated transcription inhibition (8,9,16,17).
Figure 7.

An intramolecular R•R•Y triplex as a structural impediment to transcription through the GAA•TTC repeat tract. Ribbon diagram showing the model for transcription dependent triplex formation leading to a pause at the promoter distal end of the structure. The GAA (R) strand is shown as black, the TTC (Y) strand is shown as white, and the flanking DNA is gray. (A) A standing wave of negative supercoiling follows RNA polymerase as it enters the GAA•TTC repeat tract. Underwound DNA is shown as a widened helix, the direction of the rotation imparted by the motion of the polymerase is shown by the curved arrow. (B) The non-template (GAA) strand is available to fold back in an R•R•Y interaction; the template strand is covered by RNAP. (C) Relaxation of negative supercoils by rotation of the helix (curved arrow shows direction) as it winds in the third strand aids in the formation of the triplex. (D) RNA polymerase is paused at the triplex/duplex junction in the distal end of the GAA•TTC tract (black arrow).
Triplex formation as shown in Figure 7 results in an unpaired region on the template to which RNA can bind (Fig. 7D). RNA•DNA hybrid formation has been demonstrated for other R•Y sequences (35,36). A hybrid also forms in the GAA•TTC tract. However, transcription in the presence of RNase H1 suggests that hybrid formation is a consequence of a stalled T7 RNAP on an open template rather than a cause (E.Grabczyk and K.Usdin, unpublished data).
Although the model in Figure 7 shares features with previous models for transcription driven triplex formation (35,36), the effect on transcription differs substantially. For instance, the G•C-rich R•Y tract of the GAP-43 gene formed a strong block to T7 RNAP on linear templates with prominent truncations in the promoter proximal part of the tracts (36). In contrast, the frataxin GAA•TTC tract causes only a moderate decline in transcript levels when linear. This difference may be a function of the relative stabilities of the R•R•Y structure formed by the G-rich GAP-43 sequence and the structure that we propose here for the A-rich frataxin sequence. The GAP-43 sequence also traps what may be the structure initiating RNAP within the distal part of the R•Y tract (36), but forms a more stable triplex that stops ensuing polymerases and results in a second set of truncations in the promoter proximal regions of the repeat tract. The lack of prominent truncations on linear GAA•TTC-containing templates, and their absence from the promoter proximal portion of the repeat tract on supercoiled templates suggest a weaker or more transient structure for the frataxin sequence. Although we do not exclude the possibility that different mechanisms of transcription inhibition predominate on linear versus supercoiled templates, the simplest interpretation is that negative supercoiling in the GAA•TTC template exacerbates the inhibition because a relatively unstable intramolecular triplex can form more frequently, and have a longer half-life on a supercoiled template.
Lowering the pH of transcription from 8.0 to 7.0, which favors formation of the Y•R•Y triplex, specifically reduces accumulation of UUC-containing transcripts. This observation can also be accommodated in our model since an intramolecular Y•R•Y triplex could form in an analogous way from transcription in the opposite direction through the GAA•TTC tract. That the two forms of intramolecular triplex are apparently formed by, and affect, transcription in only one direction, is consistent with previous predictions (36).
T7 RNAP is a single subunit enzyme with counterparts in the organelles of many eucaryotes (37). Our findings may thus be directly applicable to transcription by these RNAPs as well. While the human RNAP II is a multi-subunit enzyme that requires a number of accessory proteins to accomplish essentially the same biosynthetic steps, it is possible that the biochemical properties of a long GAA•TTC tract during transcription remain fundamentally the same. That both human RNAP II in HeLa nuclear extracts and T7 RNAP show a tract length dependent decrease in RNA production without significant truncated products on linear GAA•TTC templates with (9) or without (data not shown) flanking frataxin sequences, supports this idea. Unfortunately, nicking/relaxing activities in eukaryotic nuclear extracts have precluded our analysis with RNAP II on supercoiled templates (data not shown).
The likelihood of triplex formation in vivo may be a complex function of the length of the triplex forming sequence, local superhelical density, transcriptional activity and the influence of DNA binding/modifying proteins. In FRDA patients the GAA•TTC tracts are often much longer than the ones we have used in this study, and may contain from hundreds to thousands of triplets (1,2). So the opportunities for triplex formation by these expanded GAA•TTC alleles in vivo might be considerably greater.
R•Y tracts are common in mammalian genomes comprising up to 1% of the total DNA (38–40). Therefore the potential for triplex mediated transcription inhibition is widespread, whether as part of the normal regulation of a gene, or as a pathological consequence of the insertion or expansion of an R•Y tract. Since this mechanism affects transcription elongation, triplex formation can reduce gene expression from anywhere within a transcription unit. The target site for such mutations is therefore immense, and it is possible that Friedreich’s ataxia is just the first of many disorders that may be caused by such a mechanism.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Drs Debbie Hinton, Anthony V. Furano and Nancy Nossal for their critical reading of the manuscript, helpful comments and support.
REFERENCES
- 1.Campuzano V., Montermini,L., Molto,M.D., Pianese,L., Cossee,M., Cavalcanti,F., Monros,E., Rodius,F., Duclos,F., Monticelli,A., et al. (1996) Science, 271, 1423–1427. [DOI] [PubMed] [Google Scholar]
- 2.Cossee M., Schmitt,M., Campuzano,V., Reutenauer,L., Moutou,C., Mandel,J.L. and Koenig,M. (1997) Proc. Natl Acad. Sci. USA, 94, 7452–7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Durr A., Cossee,M., Agid,Y., Campuzano,V., Mignard,C., Penet,C., Mandel,J.L., Brice,A. and Koenig,M. (1996) N. Engl. J. Med., 335, 1169–1175. [DOI] [PubMed] [Google Scholar]
- 4.Filla A., De Michele,G., Cavalcanti,F., Pianese,L., Monticelli,A., Campanella,G. and Cocozza,S. (1996) Am. J. Hum. Genet., 59, 554–560. [PMC free article] [PubMed] [Google Scholar]
- 5.Montermini L., Richter,A., Morgan,K., Justice,C.M., Julien,D., Castellotti,B., Mercier,J., Poirier,J., Capozzoli,F., Bouchard,J.P., Lemieux,B., Mathieu,J., Vanasse,M., Seni,M.H., Graham,G., Andermann,F., Andermann,E., Melancon,S.B., Keats,B.J., Di Donato,S. and Pandolfo,M. (1997) Ann. Neurol., 41, 675–682. [DOI] [PubMed] [Google Scholar]
- 6.Cossee M., Campuzano,V., Koutnikova,H., Fischbeck,K., Mandel,J.L., Koenig,M., Bidichandani,S.I., Patel,P.I., Molte,M.D., Canizares,J., De Frutos,R., Pianese,L., Cavalcanti,F., Monticelli,A., Cocozza,S., Montermini,L. and Pandolfo,M. (1997) Nature Genet., 15, 337–338. [DOI] [PubMed] [Google Scholar]
- 7.Campuzano V., Montermini,L., Lutz,Y., Cova,L., Hindelang,C., Jiralerspong,S., Trottier,Y., Kish,S.J., Faucheux,B., Trouillas,P., Authier,F.J., Durr,A., Mandel,J.L., Vescovi,A., Pandolfo,M. and Koenig,M. (1997) Hum. Mol. Genet., 6, 1771–1780. [DOI] [PubMed] [Google Scholar]
- 8.Ohshima K., Montermini,L., Wells,R.D. and Pandolfo,M. (1998) J. Biol. Chem., 273, 14588–14595. [DOI] [PubMed] [Google Scholar]
- 9.Bidichandani S.I., Ashizawa,T. and Patel,P.I. (1998) Am. J. Hum. Genet., 62, 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wells R.D., Collier,D.A., Hanvey,J.C., Shimizu,M. and Wohlrab,F. (1988) FASEB J., 2, 2939–2949. [PubMed] [Google Scholar]
- 11.Frank-Kamenetskii M.D. and Mirkin,S.M. (1995) Annu. Rev. Biochem., 64, 65–95. [DOI] [PubMed] [Google Scholar]
- 12.Htun H. and Dahlberg,J.E. (1988) Science, 241, 1791–1796. [DOI] [PubMed] [Google Scholar]
- 13.Kohwi Y. and Kohwi-Shigematsu,T. (1988) Proc. Natl Acad. Sci. USA, 85, 3781–3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Htun H. and Dahlberg,J.E. (1989) Science, 243, 1571–1576. [DOI] [PubMed] [Google Scholar]
- 15.Kohwi Y. and Kohwi-Shigematsu,T. (1993) J. Mol. Biol., 231, 1090–1101. [DOI] [PubMed] [Google Scholar]
- 16.Ohshima K., Kang,S., Larson,J.E. and Wells,R.D. (1996) J. Biol. Chem., 271, 16773–16783. [DOI] [PubMed] [Google Scholar]
- 17.Sakamoto N., Chastain,P.D., Parniewski,P., Ohshima,K., Pandolfo,M., Griffith,J.D. and Wells,R.D. (1999) Mol. Cell, 3, 465–475. [DOI] [PubMed] [Google Scholar]
- 18.McAllister W.T. (1993) Cell. Mol. Biol. Res., 39, 385–391. [PubMed] [Google Scholar]
- 19.Grabczyk E. and Usdin,K. (1999) Anal. Biochem., 267, 241–243. [DOI] [PubMed] [Google Scholar]
- 20.Baumann C.G. and Bloomfield,V.A. (1995) Biotechniques, 19, 884–890. [PubMed] [Google Scholar]
- 21.Jia Y. and Patel,S.S. (1997) Biochemistry, 36, 4223–4232. [DOI] [PubMed] [Google Scholar]
- 22.Lilley D.M. (1983) Cold Spring Harb. Symp. Quant. Biol., 47, 101–112. [DOI] [PubMed] [Google Scholar]
- 23.Peck L.J. and Wang,J.C. (1985) Cell, 40, 129–137. [DOI] [PubMed] [Google Scholar]
- 24.Parsons M.A., Sinden,R.R. and Izban,M.G. (1998) J. Biol. Chem., 273, 26998–27008. [DOI] [PubMed] [Google Scholar]
- 25.Droge P. and Pohl,F.M. (1991) Nucleic Acids Res., 19, 5301–5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanvey J.C., Shimizu,M. and Wells,R.D. (1988) Proc. Natl Acad. Sci. USA, 85, 6292–6296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gacy A.M., Goellner,G.M., Spiro,C., Chen,X., Gupta,G., Bradbury,E.M., Dyer,R.B., Mikesell,M.J., Yao,J.Z., Johnson,A.J., Richter,A., Melancon,S.B. and McMurray,C.T. (1998) Mol. Cell, 1, 583–593. [DOI] [PubMed] [Google Scholar]
- 28.Mariappan S.V., Catasti,P., Silks,L.A.,III, Bradbury,E.M. and Gupta,G. (1999) J. Mol. Biol., 285, 2035–2052. [DOI] [PubMed] [Google Scholar]
- 29.Leonard N.J., McDonald,J.J., Henderson,R.E. and Reichmann,M.E. (1971) Biochemistry, 10, 3335–3342. [DOI] [PubMed] [Google Scholar]
- 30.Hartvig L. and Christiansen,J. (1996) EMBO J., 15, 4767–4774. [PMC free article] [PubMed] [Google Scholar]
- 31.Lyakhov D.L., He,B., Zhang,X., Studier,F.W., Dunn,J.J. and McAllister,W.T. (1998) J. Mol. Biol., 280, 201–213. [DOI] [PubMed] [Google Scholar]
- 32.Escude C., Francois,J.C., Sun,J.S., Ott,G., Sprinzl,M., Garestier,T. and Helene,C. (1993) Nucleic Acids Res., 21, 5547–5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu L.F. and Wang,J.C. (1987) Proc. Natl Acad. Sci. USA, 84, 7024–7027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu H.Y., Shyy,S.H., Wang,J.C. and Liu,L.F. (1988) Cell, 53, 433–440. [DOI] [PubMed] [Google Scholar]
- 35.Reaban M.E. and Griffin,J.A. (1990) Nature, 348, 342–344. [DOI] [PubMed] [Google Scholar]
- 36.Grabczyk E. and Fishman,M.C. (1995) J. Biol. Chem., 270, 1791–1797. [DOI] [PubMed] [Google Scholar]
- 37.Cermakian N., Ikeda,T.M., Miramontes,P., Lang,B.F., Gray,M.W. and Cedergren,R. (1997) J. Mol. Evol., 45, 671–681. [DOI] [PubMed] [Google Scholar]
- 38.Birnboim H.C., Sederoff,R.R. and Paterson,M.C. (1979) Eur. J. Biochem., 98, 301–307. [DOI] [PubMed] [Google Scholar]
- 39.Manor H., Rao,B.S. and Martin,R.G. (1988) J. Mol. Evol., 27, 96–101. [DOI] [PubMed] [Google Scholar]
- 40.Tripathi J. and Brahmachari,S.K. (1991) J. Biomol. Struct. Dyn., 9, 387–397. [DOI] [PubMed] [Google Scholar]