

SUMMARY
G protein-coupled receptors (GPCRs) represent a ubiquitous membrane protein family and are important drug targets. Their diverse signaling pathways are driven by complex pharmacology arising from a conformational ensemble rarely captured by structural methods. Here, fluorine nuclear magnetic resonance spectroscopy (19F NMR) is used to delineate key functional states of the adenosine A2A receptor (A2AR) complexed with heterotrimeric G protein (Gαsβ1γ2) in a phospholipid membrane milieu. Analysis of A2AR spectra as a function of ligand, G protein, and nucleotide identifies an ensemble represented by inactive states, a G protein-bound activation intermediate, and distinct nucleotide-free states associated with either partialor full-agonist-driven activation. The Gβγ subunit is found to be critical in facilitating ligand-dependent allosteric transmission, as shown by 19F NMR, biochemical, and computational studies. The results provide a mechanistic basis for understanding basal signaling, efficacy, precoupling, and allostery in GPCRs.
INTRODUCTION
One-third of current pharmaceuticals target G protein-coupled receptors (GPCRs) (Hauser et al., 2017), the largest family of membrane proteins in the human genome and mediators of diverse biological processes through signal transduction across the cell membrane. The adenosine A2A receptor (A2AR) is a prototypical class A GPCR and a target for the treatment of respiratory and cardiovascular diseases (Guerrero, 2018), inflammation and cancer (Effendi et al., 2020; de Lera Ruiz et al., 2014; Yu et al., 2020), and diseases of the central nervous system (Zheng et al., 2019). Upon activation, A2AR engages the heterotrimeric stimulatory G protein Gsαβγ, resulting in nucleotide exchange, dissociation of the α and βγ subunits, and downstream activation of effector proteins. This theme is repeated in sensory signaling (vision, taste, smell, pain), neurotransmission, cardiovascular function, and immune response in over 800 other GPCRs (Fredriksson et al., 2003). The recent renaissance in X-ray crystallography and cryoelectron microscopy (cryo-EM) has generated high-resolution structures of many GPCRs, including A2AR, in both their inactive states and G protein-complexed active states (Carpenter et al., 2016;Garcıa-Nafrıa et al., 2018; Liu et al., 2012; Weis and Kobilka, 2018). This has spurred structure-based drug design (Congreve et al., 2020) and provided a greater understanding of the mechanism of activation.
Solution-state nuclear magnetic resonance (NMR) spectroscopy builds upon static crystallography and cryo-EM by capturing the entire conformational ensemble, using native constructs under physiological conditions. If the representative states are sufficiently resolved, relaxation experiments can provide additional insights into dynamics and lifetimes of states spanning nanoseconds to seconds. Many groups have taken the approach of isotopic labeling for one- or two-dimensional NMR of GPCRs in detergent micelles (Clark et al., 2017; Eddy et al., 2018; Frei et al., 2020; Isogai et al., 2016; Manglik et al., 2015; Nygaard et al., 2013; Solt et al., 2017; Sounier et al., 2015; Wu et al., 2020; Ye et al., 2016) and model membrane systems (Casiraghi et al., 2016; Kofuku et al., 2014). These studies, along with single-molecule fluorescence spectroscopy (Bockenhauer et al., 2011; Gregorio et al., 2017), electron paramagnetic resonance spectroscopy (Van Eps et al., 2017), and molecular dynamics (MD) simulations (Dror et al., 2011; Provasi et al., 2011), suggest that activation proceeds through complex allosteric pathways and multiple intermediate states. However, it is difficult to interpret the observed “active” states unless they can be measured in the presence of the heterotrimeric G protein.
The series of steps during activation (i.e., G protein binding, nucleotide exchange, and dissociation of the subunits) may all be a consequence of specific states within the active ensemble, which can be further complicated by the effects of the orthosteric ligand. Resolving the active ensemble, therefore, is key to understanding the mechanism of activation in both the receptor and the G protein.
Here, we use NMR to resolve the conformational ensemble of A2AR, free of any stabilizing mutations, reconstituted in lipid bilayers and complexed to the full-length heterotrimeric G protein. Fluorine (19F) NMR is a particularly sensitive technique to electrostatic and van der Waals environments and exhibits a large chemical shift dispersion, enabling improved resolution of states (Ye et al., 2015). Critically, as a one-dimensional method, potentially dynamic states that manifest as broad lines are also resolvable and quantifiable, whereas their observation might be obscured by multidimensional NMR methods that are susceptible to relaxation during insensitive nuclei enhancement by polarization transfer (INEPT) times (Cavanagh et al., 2007). Previous 19F NMR studies of A2AR in detergent micelles (Ye et al., 2016, 2018) identified two inactive-state conformers in fast exchange and two active-like conformers stabilized by partial or full agonists, respectively. Using the same construct (A2AR (2–317) with a single cysteine mutation, V229C, for 19F -labeling on transmembrane helix 6 [TM6]), the current work examines receptor states associated with G protein coupling and nucleotide exchange in discoidal phospholipid-containing reconstituted high-density-lipoprotein particles (rHDLs, commonly known as nanodiscs) and in the presence of the stimulatory heterotrimeric G protein (composed of human Gsα-short, β1, and γ2, henceforth referred to as Gsαβγ or Gαβγ) (Figures S1 and S2). By recording A2AR spectra as a function of orthosteric ligand, G protein, and nucleotide, the representative states in the ensemble can be monitored along the entire activation pathway—capturing signatures of both pre-coupling and nucleotide release. We discovered that the Gβγ subunit not only anchors Gα to the membrane but also is critical in transducing ligand efficacy. Through biochemical, biophysical, and computational methods, we seek to connect the observed conformational states and allosteric pathways of a receptor to the downstream pharmacological effects of basal signaling and partial agonism. Building upon previously published work on dynamical aspects of GPCRs, this study enables a more complete characterization of the signaling process through heterotrimeric G proteins and provides a basis for understanding receptor pharmacology from an ensemble perspective.
RESULTS AND DISCUSSION
A2AR adopts a precoupled activation intermediate and two distinct active states that mediate nucleotide exchange
The classical view of GPCR activation involves an equilibrium between an inactive (R) and an active (R*) pose (Park et al., 2008). The agonist induces an allosteric response, shifting the receptor population to the R* state to enable binding and coupling to the G protein. This idea is recapitulated in the cubic ternary complex model (Weiss et al., 1996) that encompasses precoupling, a phenomenon wherein the receptor-G protein complex is assembled prior to activation by ligands (Neubig, 1994; Rebois and Hebert, 2003).
19F NMR provides a more detailed perspective of the R-R* transition, wherein inactive and active signatures of A2AR can be resolved as a function of ligands and Gα (Figure 1). The spectra not only reveal a clear pattern of activation featuring a population shift to the active ensemble but also reveal subtleties regarding the conformational ensemble that imply a more complex response. For example, the resonance associated with the inactive pose not only decreases in population but also shifts downfield in response to activating conditions. This is likely a consequence of conformational exchange between two inactive conformers in which a salt bridge (also known as ionic lock) between R1023.50 of the highly conserved E(D)RY motif on TM3 and E2286.30on TM6 is either intact or broken. Here, superscripts denote the Ballesteros-Weinstein numbering for GPCRs (Ballesteros and Weinstein, 1995). This ionic lock motif stabilizes the inactive state of many class A receptors (Ballesteros et al., 2001; Vogel et al., 2008). In A2AR, both the ionic lock “on” and “off” conformers are part of the inactive ensemble, and higher efficacy ligands shift this equilibrium toward the ionic lock “off” state (Dore et al., 2011). Interestingly, the inactive state chemical shift of the inverse agonist saturated receptor is nearly coincident with those of apo (ligand-free)-A2AR, implying an unchanged ionic lock equilibrium between the two conditions. In the case of the partial agonist, there is a subtle inflection point in the inactive state resonance that aligns with the ionic lock “off” state. This suggests that the two conformers are in relatively slow exchange (ke≤~400 s−1) in lipid bilayers. By contrast, prior studies of β2AR and A2AR in detergent micelles suggested that ionic lock fluctuations occurred on a relatively fast NMR timescale (Manglik et al., 2015; Ye et al., 2016).
Figure 1. A2AR adopts an ensemble of conformational states and an activation mechanism consistent with conformational selection.
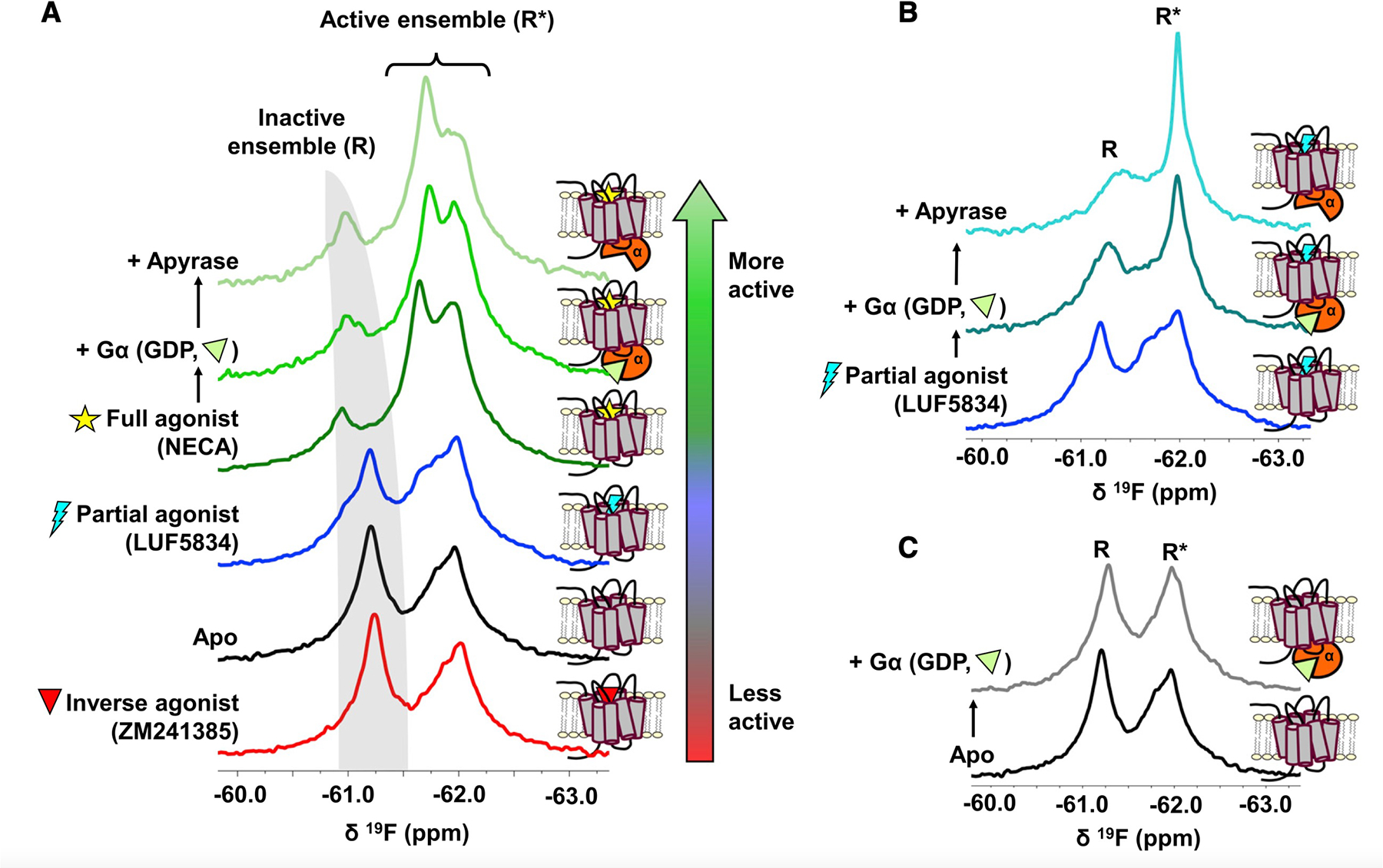
(A) 19F NMR spectra of nanodisc-reconstituted A2AR-V229C as a function of ligand, Gα, and nucleotide. Addition of apyrase removes nucleotide (GDP) from G proteins. The receptor was placed under increasingly activating conditions, as indicated by the color gradient bar. The apo receptor (black trace) samples both inactive (R, gray band) and active (R*) states, whose populations are modulated through the binding of ligands (antagonist, partial agonist, and full agonist), Gα, and GDP, in a lipid environment. (B and C) Spectra of partial agonist-bound (B) or apo (C) receptor with and without Gα.
Closer inspection of the spectral series in Figure 1 reveals that active pose is also represented by multiple states. Here, two distinct resonances, at 61.7 and 61.9 ppm, were stabilized by the addition of either agonist (Figure 1A) or partial agonist in the presence of Gα (Figure 1B). These effects are more pronounced upon removal of guanosine diphosphate (GDP) by apyrase. In the absence of an orthosteric ligand (Figure 1C), addition of Gα resulted in modest change. The results show that even under basal conditions, the receptor samples multiple active-like states including those that facilitate GDP release. The propensity for establishing a particular conformer is influenced by ligands and Gα while the same basis set of states are observed across a range of conditions, suggesting that conformational selection plays a significant role in receptor activation.
The overlap among resonances in the active ensemble poses challenges to robust spectral deconvolutions since nothing a priori is known of the respective line widths. An attempt to deconvolve the 19F NMR spectrum in the presence of the full agonist 5’-N-ethylcarboxamidoadenosine (NECA) is shown in Figure S3. In the absence of transverse relaxation time (T2) measurements, it is sufficient to obtain a fitted spectrum closely matching the experimental result by considering one inactive state (peak 1) and two active-like states (peaks 2 and 3). However, T2 relaxation measurements predict significantly narrower line widths for peaks 1 and 3. The observed broadening of peak 1 is likely a consequence of slow (millisecond) exchange between two inactive-state signatures, discussed above. Similarly, inhomogeneous broadening in the vicinity of peak 3 must arise from one or more additional nearby resonances, indicating that the active ensemble as a whole consists of at least three distinct and interconverting conformations. This is in contrast to 19F NMR spectra of A2AR reconstituted in detergent micelles, which gives rise to two active-state conformers (referred to as S3 and S3’ in our previous work) and an inactive ensemble with faster exchange dynamics (Ye et al., 2016). The lipid-stabilized receptors display slower exchange dynamics, a higher fraction of inactive states, and an overall improved spectral resolution (Figure 2).
Figure 2. A2AR exhibits a different distribution of states and slower exchange dynamics in a lipid bilayer environment than in detergent micelles.
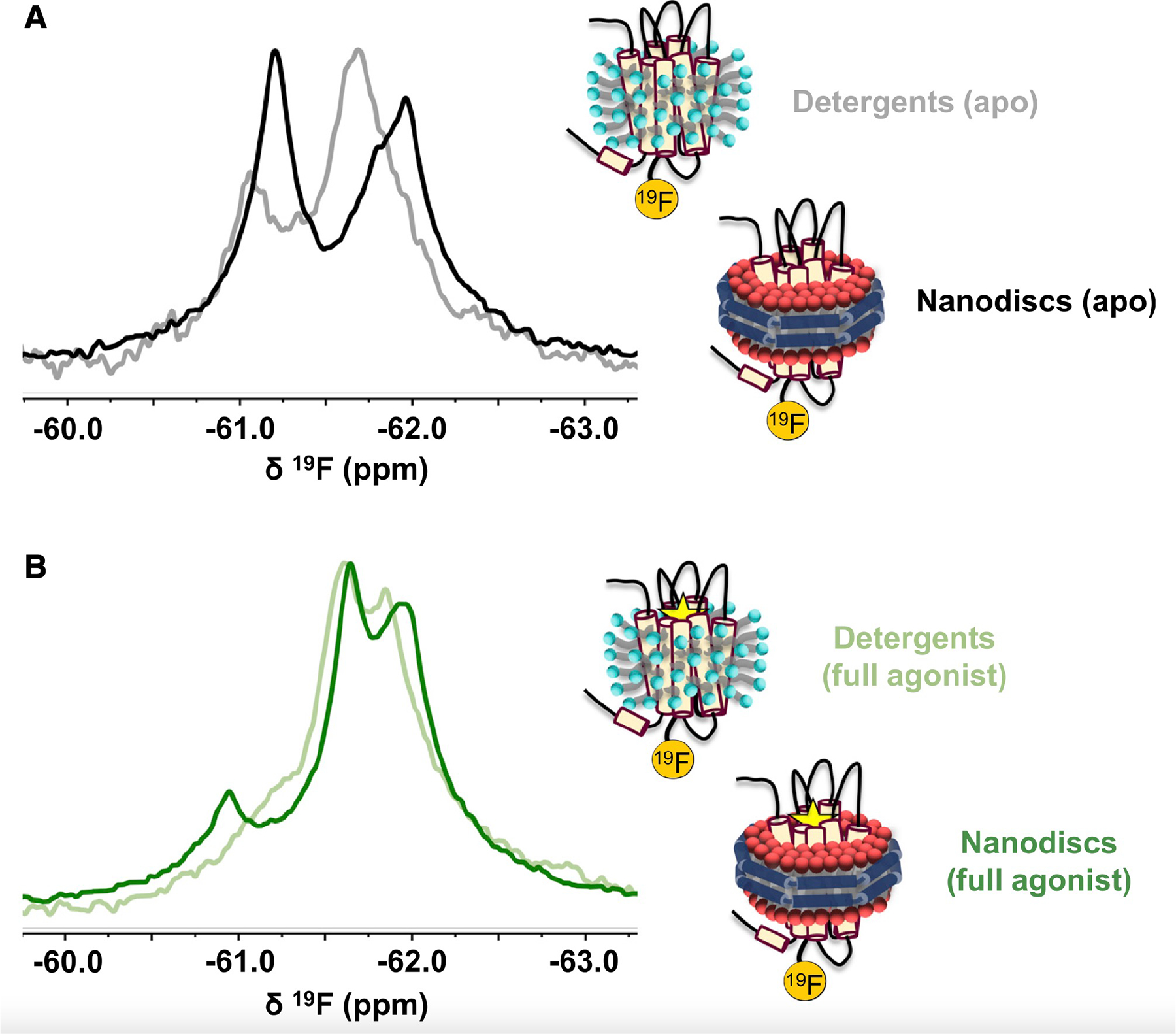
(A and B) Comparison of the 19F NMR spectra of apo (A) or agonist-bound (B) A2AR reconstituted in either lauryl maltose-neopentyl glycol (LMNG) micelles or phospholipid nanodiscs. Data for the detergent spectra were obtained from Ye et al. (2016) with permission from the authors.
The presence of the G protein heterotrimer is critical for both the assignment of functional states and understanding their role in facilitating signaling, as shown in Figure 3. Generally, the addition of Gsαβγ-GDP shifts the receptor equilibrium to a predominantly active ensemble. However, the inactive fraction persists in the presence of the inverse agonist ZM241385, whose chemical shift is upfield (ionic lock “on”) from that observed for the full-agonist-bound receptor (Figure 3A). This is consistent with the inverse agonist stabilizing the ionic lock “on” equilibrium, resulting in a reduced affinity to G protein and transitions to the active ensemble. Interestingly, the addition of Gsαβγ to the inverse- and partial-agonist-bound receptor results in an upfield shift of the inactive state. This is likely a result of millisecond timescale averaging between inactive and active conformers, which would contribute to line broadening and coalescence of the respective signatures. Furthermore, the addition of GDP-saturated Gsαβγ is generally accompanied by line broadening of the active states, particularly in the case of partial-and full-agonist-stabilized receptors. We interpret this as evidence of intermediate timescale exchange between conformers representing the active ensemble.
Figure 3. The precoupled state and nucleotide-free states are key facets of activation.
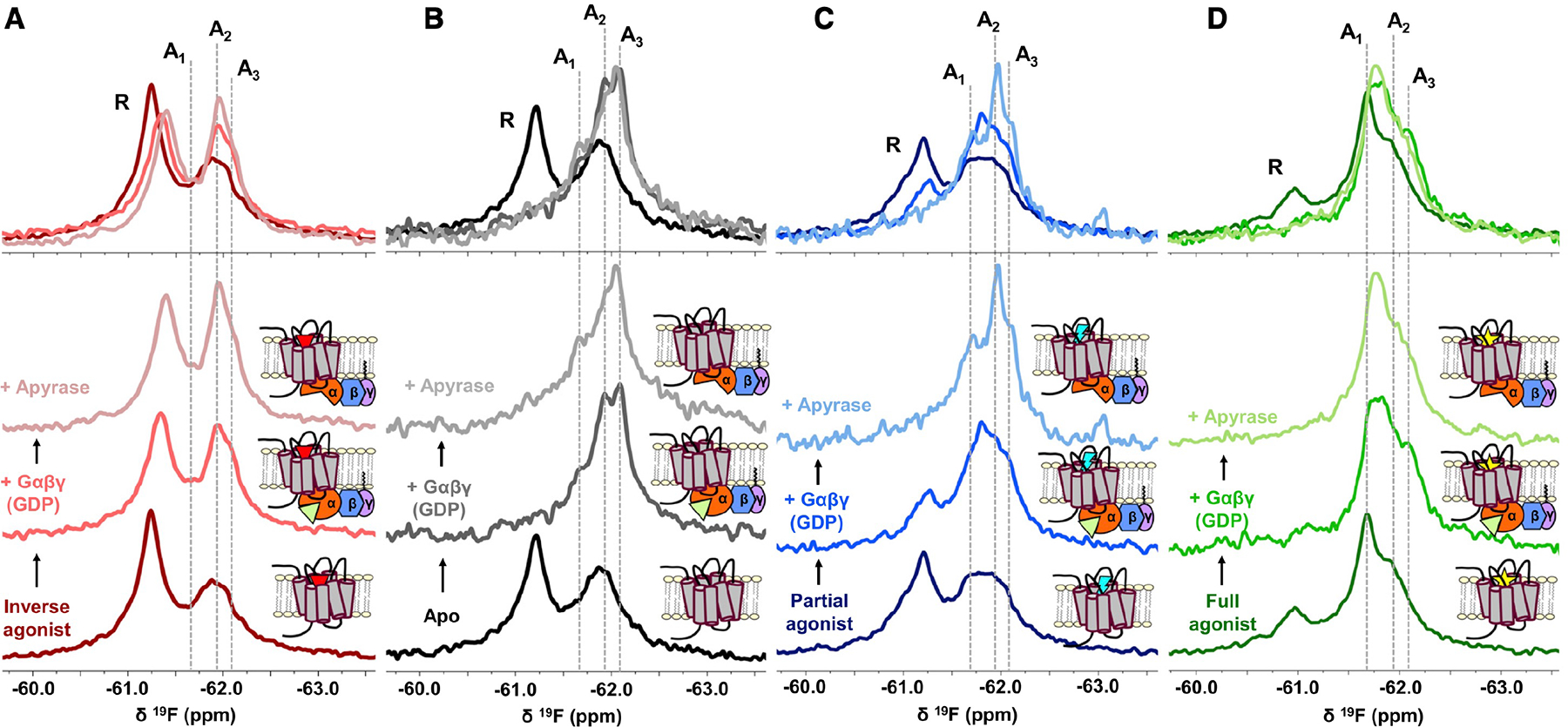
(A–D) 19F NMR spectra of A2AR-V229C as a function of ligands, Gαβγ, and GDP. The addition of Gs heterotrimer (Gαβγ) and subsequently apyrase to inverse agonist-bound (A), apo (B), partial-agonist-bound (C), and full-agonist-bound (D) A2AR enabled the assignment of at least three unique active state conformers as indicated by the gray dashed lines at 61.70 ppm (A1), 61.95 ppm (A2), and 62.10 ppm (A3). Stabilization of representative states by the GDP-bound Gαβγ and nucleotide-free Gαβγ can be directly visualized in the overlaid spectra.
A global analysis of the NMR spectra suggests that the active ensemble encompasses at least three distinct states, designated A1, A2, and A3 (spectral assignment details provided in STAR Methods). For the apo receptor (Figure 3B), addition of GDP-bound Gsαβγ dramatically shifts the equilibrium toward A3, a change that is recapitulated in the inverse-agonist-bound (Figure 3A), partial-agonist-bound (Figure 3C), and full-agonist-bound (Figure 3D) spectra. We attribute A3 to a precoupled conformation, which is likely responsible for the initial recognition between receptor and G protein. Precoupling has previously been shown for A2AR and other GPCRs (Braun and Levitzki, 1979; Galès et al., 2006; Nanoff and Stiles, 1993; Nobles et al., 2005) in addition to work supporting a G protein-bound intermediate prior to formation of the high-affinity complex associated with GDP release (Dror et al., 2011; Du et al., 2019; Lee et al., 2019; Liu et al., 2019; Sounier et al., 2015). The current NMR spectra corroborate these findings while at the same time revealing the response of the entire conformational ensemble to both ligand and G protein. As discussed below, spectral deconvolutions suggest the precoupled state is dynamic in the absence of G protein, which may be part of a mechanism for G protein recognition.
The redistribution of states upon formation of the complex with G protein is accompanied by changes in local dynamics. Spectral deconvolutions (Figures S4 and S5) reveal that A3 is represented by a broad resonance in the absence of G protein, consistent with intermediate timescale sampling of a broad range of conformational sub-states (within the A3 state). Addition of the G protein effectively limits exchange dynamics, resulting in a sharper, well-defined A3 resonance. The narrower line width indicates restricted sampling of sub-states and an overall convergence into more stable configurations. This precoupled population likely does not contribute significantly to nucleotide exchange given its higher occurrence in the apo spectra. It is conceivable, however, that complexes adopting stable intermediate states are more allosterically enabled to transition into active conformers capable of GDP release leading to either basal or ligand-induced G protein activation.
A comparison of the activation series for partial- and full-agonist-bound A2AR suggests that A1 and A2 are two unique active states associated with G protein activation. While full agonist preferentially stabilizes A1 (Figure 3D), the addition of either GDP-bound Gαβγ or partial agonist enhances the A2 fraction (Figures 3A–3D). Importantly, removal of GDP by apyrase further enhances the respective populations associated with A1 and A2, suggesting that these two states specifically facilitate nucleotide exchange and are favored by either a full agonist (A1) or a partial agonist (A2). The addition of an engineered mini-G protein (mini-Gs), previously designed to bind and stabilize the active state in the presence of agonist and without the use of the full heterotrimer (Carpenter and Tate, 2016), also stabilized A1, albeit with little dependence on nucleotide (Figure S6). Together, the results suggest that A2AR can adopt a unique precoupled/intermediate state (A3) that enables binding to the GDP-bound heterotrimer, in addition to distinct active-state conformers (A1 and A2) that facilitate GDP release through stabilization of a nucleotide-free G protein.
The above-mentioned interpretation provides a basis for understanding the ceiling effect in partial agonism. In the presence of heterotrimer, the precoupled fraction is in fact larger for the apo receptor than that observed with either partial or full agonist. However, the full-agonist-bound spectrum is distinguished by the A1 state, while the partial-agonist-bound spectrum is distinguished by the A2 state with an apparent A1 fraction that is comparable to that of the apo receptor. The results imply that both A1 and A2 are signaling competent, while the A1 conformer more efficiently enables GDP release and activation. Thus, a partial agonist will fail to promote maximal biological response at saturating concentrations of ligand, as the nucleotide exchange step is primarily driven by a less efficacious receptor state (A2). The observation addresses a long-standing phenomenon in pharmacology, namely partial agonism, from a molecular ensemble perspective. Prior NMR studies in detergent micelles have identified that partial and full agonists establish unique activation intermediates using G protein mimetics (Frei et al., 2020; Solt et al., 2017; Ye et al., 2016). Using full heterotrimeric G proteins, we are now able to understand efficacy from the perspective of the effect of the ligand on the precoupling and nucleotide exchange steps. Note that upon removal of nucleotide through apyrase, the apo (+ G protein) spectrum also reveals a small increase in the A1 fraction, which alongside A2 would presumably be responsible for basal signaling (Figure 3A).
While A1 and A2 designate two different conformations that are distinguished by chemical shifts, the mechanism for their varied efficacy is likely complex and kinetically driven. A recent single-molecule fluorescence study of the β2-adrenergic receptor showed that partial- and full-agonist-bound receptors facilitate nucleotide exchange through different intermediates that affect rates of G protein binding and GDP release (Gregorio et al., 2017). The current NMR data demonstrate that the entire activation process is defined by a basis set of activation states (A1–A3) wherein equilibria and exchange kinetics are influenced by ligand, heterotrimer, and nucleotide. This builds upon recent advances in receptor NMR that used conformation-selective nanobodies (Frei et al., 2020; Solt et al., 2017; Staus et al., 2016), a valuable tool for stabilizing specific receptor states, but that does not capture the critical steps that define a GPCR’s ability to facilitate nucleotide exchange. We note that while the current work suggests that the process is driven by conformational selection through three principle activation states, the true conformational landscape may be more complex and induced fit likely plays an important role especially at binding interfaces (Susac et al., 2018).
Gβγ plays a key role in reinforcing allosteric pathways and signal transmission
The spectra in Figure 3 point to the importance of Gβγ as an allosteric chaperone in signal transduction. Compared with spectra in the presence of Gα alone (Figure 1), the intact heterotrimer significantly enhances the fraction of active conformers while allowing better delineation of the individual active states. To assess the relationship between states observed by NMR and the receptor’s biological output, we conducted GTP hydrolysis assays using either Gsα or Gsαβγ in the presence of A2AR bound to various ligands (Figure 4). As a guanine nucleotide exchange factor (GEF), A2AR mediates nucleotide exchange and thereby accelerates GTP turnover. As expected, the presence of equimolar A2AR resulted in an increase in cumulative GTP turnover by both Gsα and Gsαβγ over a 90-min period, in comparison with that of with G protein alone. Additionally, the effect of this enhancement is substantially higher for Gsαβγ than Gsα (Figure 4A), implying a greater level of receptor-mediated nucleotide exchange in the presence of Gβγ.
Figure 4. Gβγ enhances receptor-mediated nucleotide exchange and ligand dependence of GEF action.
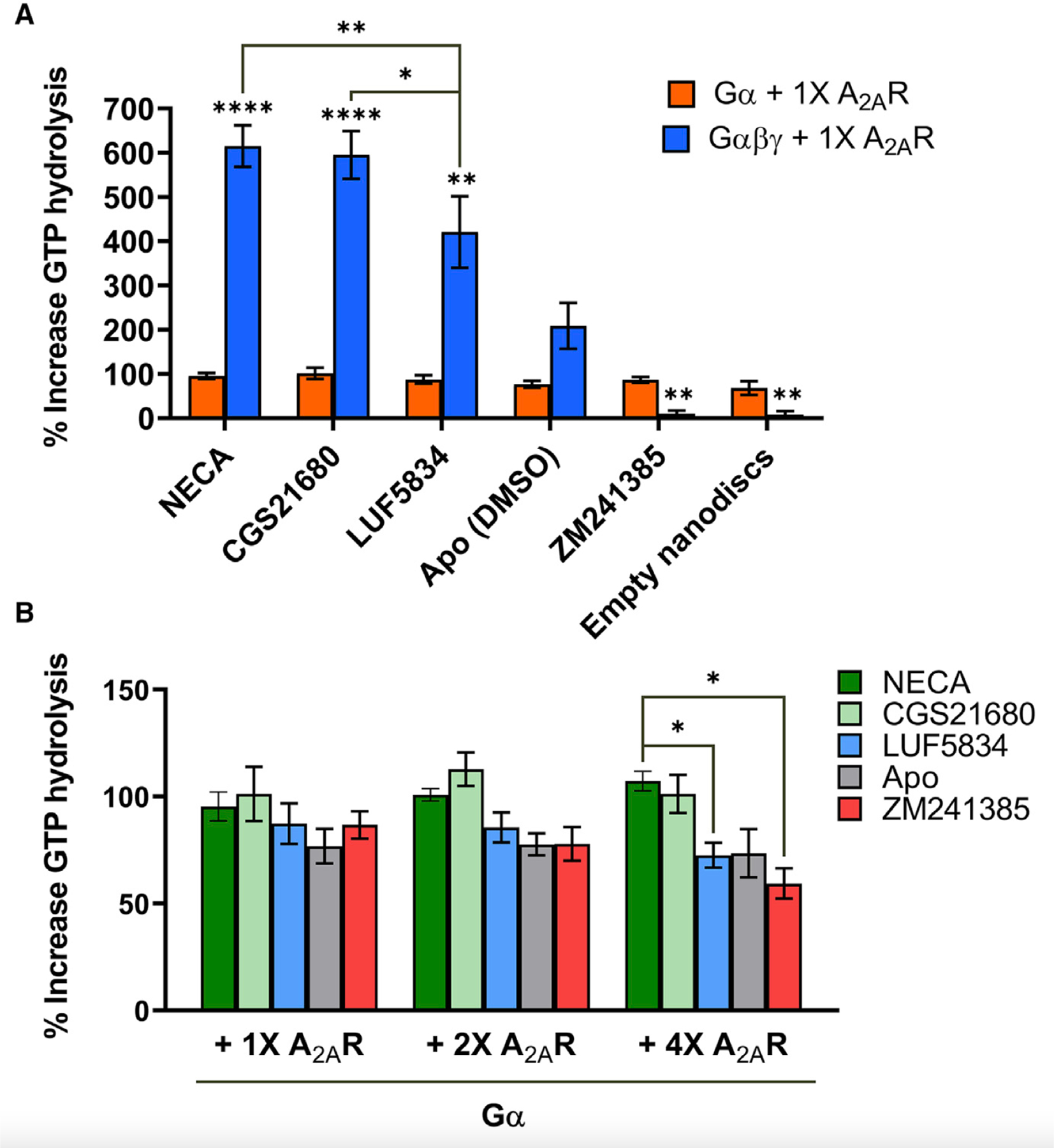
(A) Percent increase in GTP hydrolysis by either Gα or Gαβγ in the presence of one stoichiometric equivalence of A2AR bound to full agonists (NECA and CGS21680), partial agonist (LUF5834), inverse agonist (ZM241385), or no ligand, relative to the amount of GTP hydrolyzed by Gα or Gαβγ alone in the absence of A2AR over a 90-min period. Data represent mean ± SEM (n=3). Asterisks directly above the bars represent statistical significance relative to the apo condition. Statistical significance was determined by two-way ANOVA followed by the Bonferroni (comparison of Gα and Gαβγ for each ligand) or the Tukey test (comparison of each ligand condition to each other). In the case of Gα, there is no significant difference be-tween each ligand. (B) Percent increase in GTP hydrolysis by Gα in the presence of one, two, or four stoichiometric equivalence of A2AR bound to full agonists (NECA and CGS21680), partial agonist (LUF5834), inverse agonist (ZM241385), or no ligand, relative to the amount of GTP hydrolyzed in the absence of A2AR over a 90-min period. Data represent mean ± SEM (n=3). Statistical significance was determined by multiple t test using the Holm-Sidak method. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001.
The magnitude of GEF activity, shown in Figure 4A, correlates expectedly with ligand efficacy (full agonist > partial agonist > apo > inverse agonist). Comparing these with respective 19F NMR spectra challenges the classic two-state activation model (Figures S4). In general, the proportion of R* states (summation of the entire active ensemble) is not a predictor of efficacy. For example, while 92% of the apo-A2AR-Gαβγ (nucleotide-free) spectrum is represented by R*, only 84% of the spectrum is represented by R* in the presence of the partial agonist. The discrepancy speaks to the weakness of a two-state (or three-state) model and underscores the importance of resolving the ensemble. Direct comparison of the 19F NMR spectra and associated deconvolutions (Figures 3 and S7) show that efficacy in A2AR is determined by the propensity of the receptor to populate states that are competent for nucleotide exchange (A1 being more important than A2). This mechanism will allow ligands with different molecular signatures and binding modes to reshape the receptor equilibrium and the distribution of A1/A2, resulting in a wide range of possible efficacy values.
Ligand dependence of GEF activity is only observed with the intact heterotrimer, as shown in Figure 4A. While the increase in GTP turnover correlates with ligand efficacy for Gsαβγ, the trend is absent for Gsα alone regardless of receptor concentration (Figure 4B). Hence, Gβγ appears to be important in translating ligand efficacy from the receptor orthosteric pocket to the action of nucleotide exchange at Gα. While structures of GPCR-G protein complexes reveal weak (Garcıa-Nafrıa et al., 2018) to no direct contact between receptor and Gβγ, indirect interactions can be established via the Gα N-terminal helix that engages both ICL2 of the receptor and Gβ. A recent D2 dopamine receptor-Gi structure in lipid bilayer revealed significant electrostatic interactions of Gβ with the charged phospholipid head group moieties, in addition to contacts with acyl moieties on Gγ (Yin et al., 2020). Gβγ can therefore be considered a conformational scaffold important for stabilizing the receptor-Gα interface, particularly in the context of the phospholipid bilayer (Oldham and Hamm, 2008; Smrcka and Fisher, 2019).
We evaluated the effects of Gβγ on ternary complex stability through native-polyacrylamide gel electrophoresis (native-PAGE). As expected, affinity between A2AR and G protein was strengthened in the presence of Gβγ (Figure 5A). GDP-bound Gsαβγ migrated as a single band with partial- and full-agonist-bound A2AR, indicating the formation of tight complex in both cases. Note that due to negatively charged phospholipids, the bands for A2AR-Gsαβγ complexes migrated further down the gel than Gsαβγ alone. The similar migration pattern observed between the full- and partial-agonist-bound ternary complex is consistent with surface plasmon resonance (SPR) data, which showed binding kinetics associated with receptor-Gsαβγ interactions to be similar between the two ligands (Figures 5B–5D). On the other hand, the migration patterns of Gsαβγ with apo receptor, as well as that of Gsα alone with receptor bound to either partial or full agonist, appear to be smeared, indicating weaker receptor-G protein interactions. Taken together, the data suggest that the difference in functional output provided by a partial-agonist-bound and a full-agonist-bound receptor is not due to differences in receptor affinity to Gsαβγ. Rather, efficacy is a consequence of the allosteric interplay between ligand, receptor, and the intact heterotrimer.
Figure 5. The A2AR-Gsαβγ interaction is characterized by similar affinity and binding kinetics when bound to a full agonist or a partial agonist.
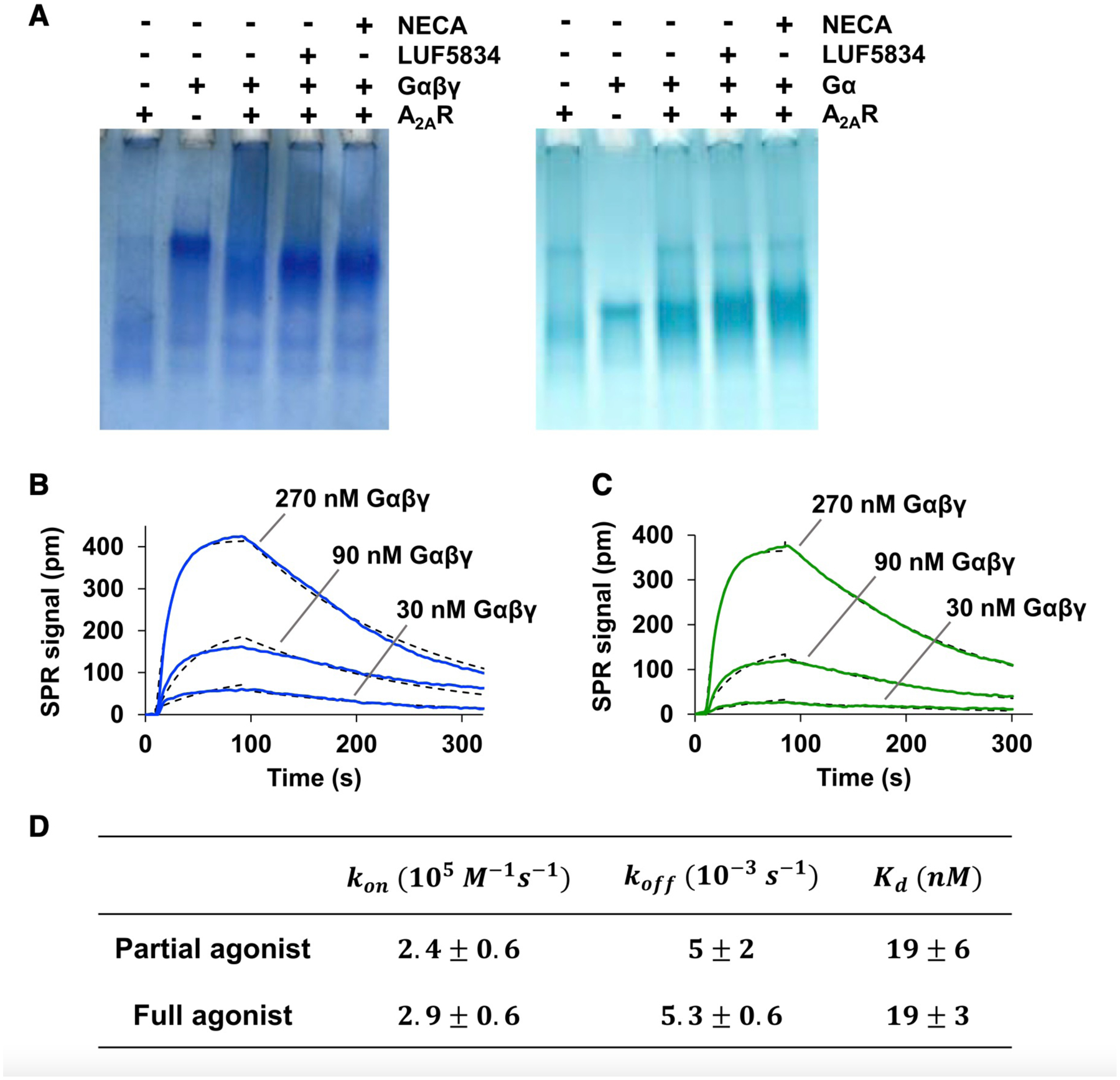
(A) A2AR-G protein interactions assessed by native-PAGE. Note that due to negatively charged lipids and the natural size distribution of nanodiscs, A2AR migrated further down the gel and the corresponding band appears smeared relative to that in the presence of Gα and Gαβγ. Consequently, complexation with receptor resulted in a band for the A2AR-Gαβγ complex that appeared lower on the gel than Gαβγ alone. This effect is absent in the case of Gα. (B and C) Representative SPR binding curves (solid lines) for the interaction of Gαβγ with immobilized A2AR saturated with either partial agonist (B) or full agonist (C). Curves obtained at the three indicated concentrations were simultaneously fitted to a one-to-one binding model (dotted lines). (D) SPR-derived Kd values and on/off rates for the interaction between A2AR and Gαβγ in the presence of indicated ligands. Data represent mean ± SD (n = 3).
Building on prior computational studies that have identified allosteric pathways between the receptor and G protein (Fanelli et al., 2016; Lee et al., 2019), we focused specifically on how Gβγ facilitates signal transmission, using rigidity-transmission allostery (RTA) algorithms (Jacobs et al., 2001; Sljoka, 2021). This graph-theory-based analysis explores potential allosteric networks within the system. In this case, the system consists of a receptor-heterotrimer complex in the presence of agonist and GDP. Upon rigidification of the agonist NECA, a well-defined pathway emerges across the ternary complex (Figure 6A). Atoms within these regions are characterized by significant changes in their degrees of freedom, a sign of allosteric communication to the orthosteric binding site. This pathway passes through the Gα binding interface encompassing ICL2 of the receptor and the N- and C-terminal helices of Gα, via an allosteric network propagating across extracellular loops 1 and 2, TM2, TM3, TM7, regions of the Gα Ras domain, and regions of Gβ.
Figure 6. Gβγ plays a key role in reinforcing allosteric pathways and signal transmission.
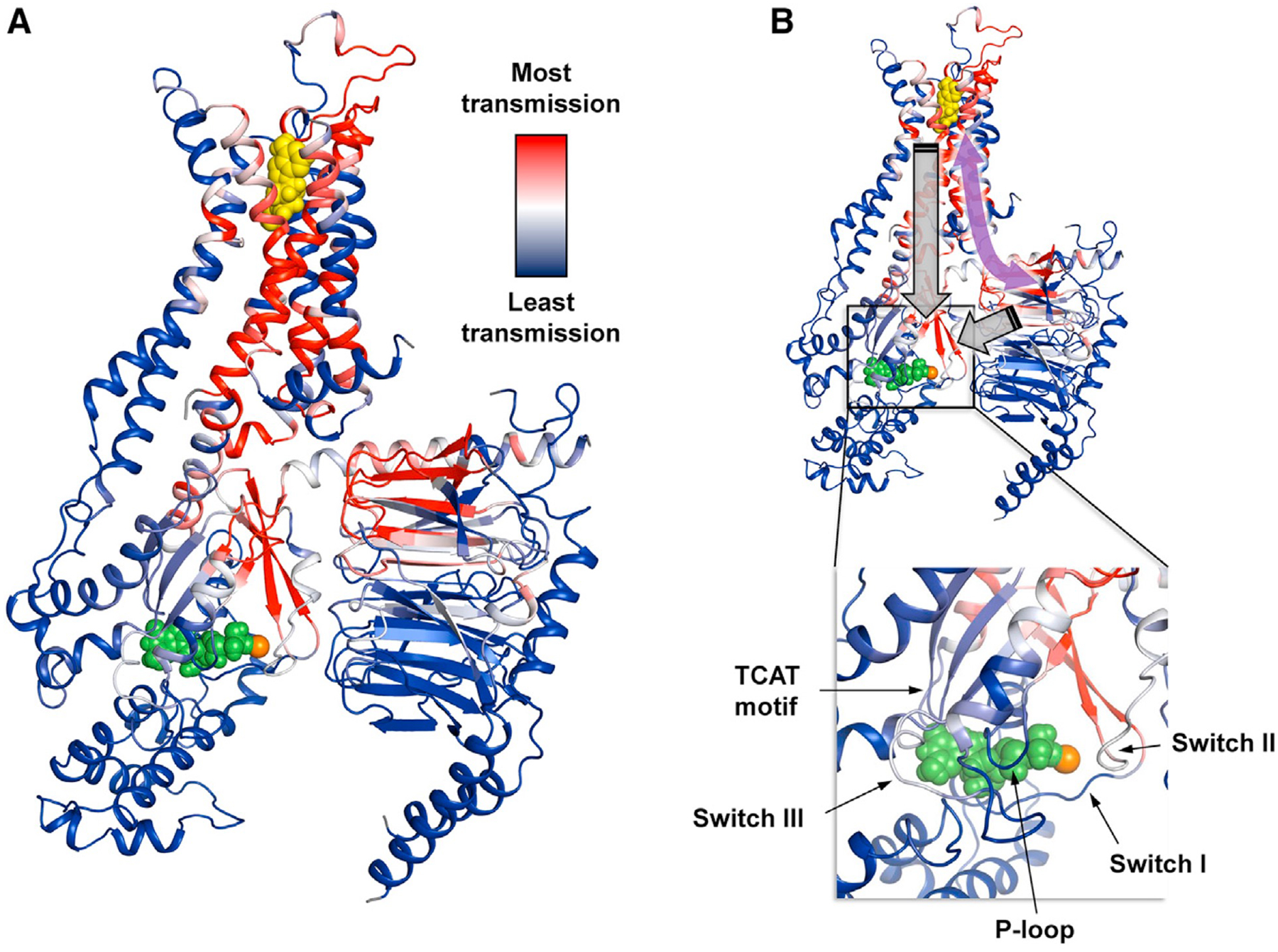
(A) The allosteric network within the ternary complex is revealed through rigidity theory analysis. Here, allosteric transmission is measured by regiospecific changes in degrees of freedom (red/blue color gradient bar) experienced upon rigidification of the agonist NECA (yellow spheres). An allosteric pathway can be defined between the orthosteric pocket and Gsαβγ that, in turn, connects with the nucleotide-binding region. Green spheres designate GDP, and the orange sphere represents Mg2+. (B) The symmetric property of allosteric transmission means that Gβγ, despite not being in direct contact with the receptor, may impart allosteric effects on remote regions in the pathway such as the orthosteric binding site (curved purple arrow). Nucleotide exchange involves structural rearrangement of Gα facilitated by movements of conserved motifs (annotated inset). This likely requires a concerted interplay between receptor and both the Gα and Gβγ subunits acting on the nucleotide-binding pocket (gray block arrows).
Remarkably, strong allosteric transmission is observed in three of the seven beta propellers and the helical region of Gβ that forms a coiled-coil interaction with the g subunit (Figure 6A). These allosteric hotspots form multiple contacts with Gα, suggesting a means for Gβγ to contribute to signal transmission and serve as an allosteric chaperone. Given that ligand dependence of nucleotide exchange requires an intact heterotrimer, regulation of this process likely involves a concerted effort from both the receptor and the Gβγ subunit over the nucleotide-binding site. This would involve allosterically engaging conserved motifs and switch regions, many of which are part of this allosteric pathway (Figure 6B). Interestingly, phylogenetic analysis showed that Gα and Gβγ initially co-evolved as a signaling module apart from the GPCRs (Bradford et al., 2013). Therefore, it is likely that the receptor-specific regulatory role of Gβγ observed in our current data emerged later in organisms that adopted GPCR-dependent G protein signaling.
As the largest family of membrane proteins, GPCRs are the gateways to diverse cellular processes and are among the most important drug targets. While the accumulation of structures over the past two decades enabled efficient design of lead compounds, success of these drug discovery efforts has been limited. Subtype selectivity, efficacy, and signaling bias are major factors to be considered in designing drugs with low side effects, and it is increasingly recognized that understanding the dynamical aspects of receptor signaling is key to grasping these processes. While it is tempting to string together structural snapshots from crystallography and cryo-EM in formulating a mechanistic model for G protein coupling,
NMR teaches us that activation is not a linear sequence of events. Rather, the functional states that we imagine to be necessary to equip the receptor for G protein coupling and subsequent activation are in fact sampled by the apo receptor. The equilibrium distribution of functional states, provided by 19F NMR spectra in the presence of G protein with and without nucleotide, reflects the receptor’s energy landscape at different steps along its activation pathway. Ligands and nucleotide not only remodel the landscape but also allosterically imbue the capacity for efficient exchange between functional states. A collection of such energy landscapes is illustrated in Figure 7 for A2AR, where the receptor is envisaged to reversibly sample states along “reaction coordinates.” Here, 19F NMR reveals a conformational ensemble comprised of at least five key functional states—two inactive states (S1 and S2) differentiated by the switching of a conserved ionic lock and three active states associated with recognition and precoupling (A3) and nucleotide exchange (A1 and A2). Ligands modulate both energies and lifetimes of these states, and multiple allosteric pathways are undoubtedly a common feature of GPCRs. In this study of A2AR, A1 is observed to be more efficacious and is preferentially stabilized by full agonist, while A2 is preferentially stabilized by partial agonist. Although nucleotide exchange is achieved in the Gsα subunit, the entire heterotrimer plays a role in signal transmission where distinct allosteric pathways are suggested to traverse the nucleotide binding site via the Gsα-A2AR and the Gsα-Gβ interfaces.
Figure 7. A2AR populates a dynamic energy landscape encompassing key functional states associated with activation, G protein coupling, and nucleotide exchange.
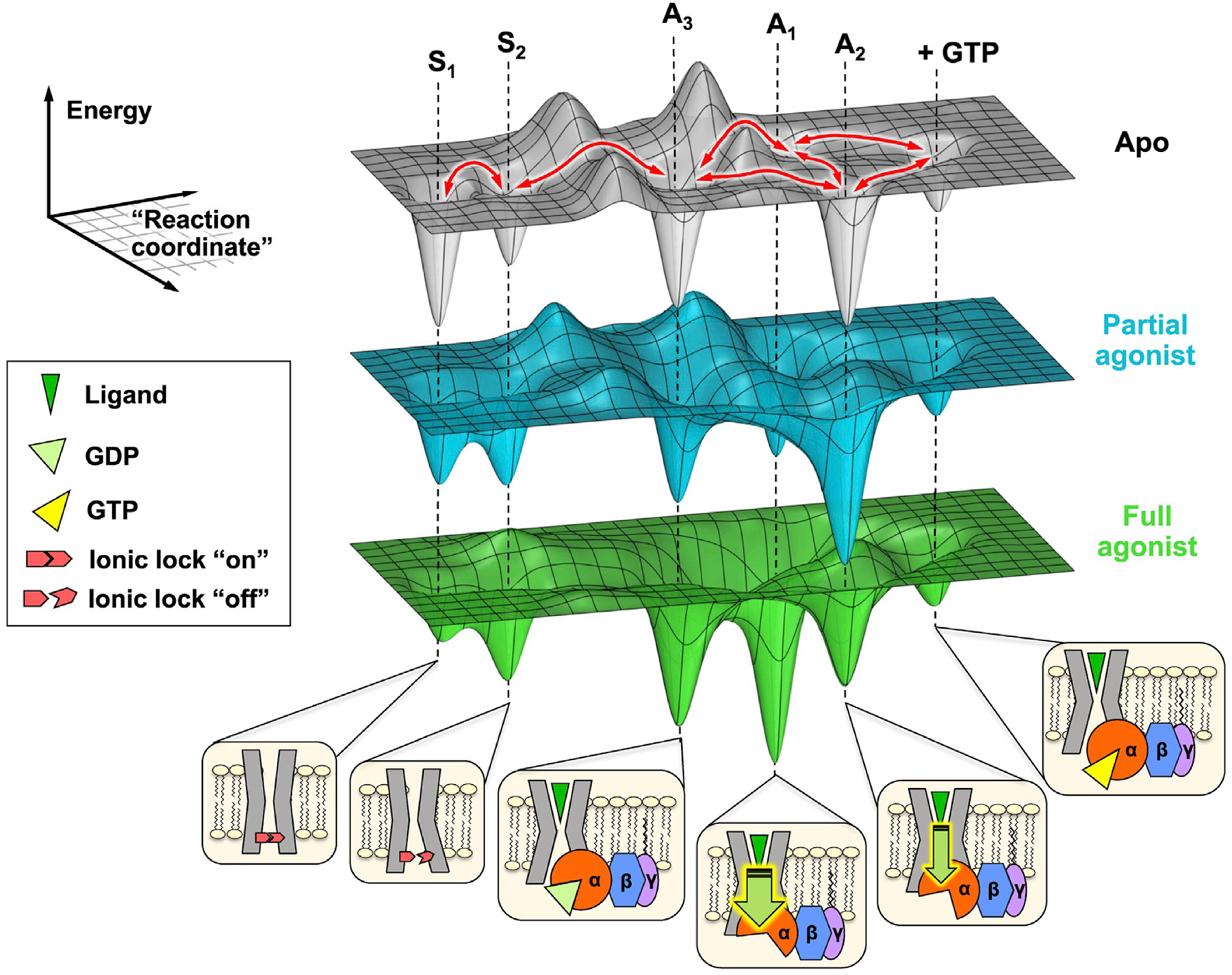
The conformational ensemble of A2AR is represented by five key functional states—two inactive states (S1 and S2) differentiated by the switching of a conserved ionic lock and three active states (A1, A2, and A3) associated with G protein coupling. A3, an intermediate or precoupled state, plays a role in the recognition and binding of the G protein. A1 and A2, on the other hand, are responsible for GDP release and stabilization of the nucleotide-free complex. While A1 is more efficacious (thicker downward arrow) and stabilized to a larger extent by the full agonist, A2 is less efficacious (thinner downward arrow) and is preferentially stabilized by a partial agonist. Although not included in this work, we also envision a state where the receptor forms a transiently stable complex with a GTP-bound G protein. The activation pathway can be considered as a series of reversible transformations between states (red arrows), whose populations and lifetimes are modulated through the presence of ligands, G protein, nucleotides, and other allosteric factors.
The energy landscape perspective provides the opportunity to advance our understanding of the mechanism of action for both orthosteric and allosteric drugs in A2AR and possibly other GPCRs. For example, the action of an inverse agonist can be precisely studied in terms of its capacity to alter the strength of the ionic lock and the inactive/active equilibrium. Similarly, the efficacy of both partial and full agonists in addition to allosteric modulators can be understood in terms of their capacity to stabilize individual activation intermediates. Future NMR relaxation studies may also add kinetic details and thus energy barrier representations to this description. Finally, we note that NMR initiatives such as those described above may help to identify the appropriate adjuvants necessary to stabilize given functional states of interest as a prelude to cryo-EM, X-ray crystallography, and the production of state-specific GPCR antibodies.
Limitations of study
The mechanistic interpretations presented in the current study are based on the observation of a single probe on TM6. While the V229C location is excellent for probing conformational changes of TM6, the study is limited in terms of addressing co-operativity between different domains of the receptor such as the extracellular and intracellular loops as well as other transmembrane helices. Additionally, while the methods used here are applicable to the study of receptors at large, our results are specific to A2AR and may not generalize to all GPCRs.
METHOD DETAILS
A2AR expression, purification, and labeling:
Plasmid construction, transformation, and colony screening has been described previously (Ye et al., 2016). Briefly, the plasmid pPIC9K_Fa-Factor–Flag–TEV-A2aARTr316-H10_V229C containing the human A2AR gene with a truncated C-terminal tail and the V229C mutation was transformed into Pichia pastoris (P. pastoris) SMD 1163 (Δhis4 Δpep4 Δprb1). A single colony of P. pastoris containing high copy number of the gene encoding A2AR-V229C was inoculated into 200 mL of YPD medium (1% (w/v) yeast extract, 2% (w/v) peptone, 2% (w/v) glucose, and 0.2 mg/mL G418) and grown at 30 ° C for 24–36 h. This starter culture was inoculated into 2 L of BMGY medium (1% (w/v) yeast extract, 2% (w/v) peptone, 1.34% (w/v) yeast nitrogenase base (YNB) without amino acids, 100 mM sodium phosphate (pH 6.5), 0.4 mg/L biotin, 1% (v/v) glycerol, and 100 mg/mL ampicillin), grown at 30 ° C for 24 h, then transferred to a bioreactor (Genesis, Solaris Biotechnology) containing 12 L of BMMY medium without methanol (1% (w/v) yeast extract, 2% (w/v) peptone, 1.34% (w/v) YNB without amino acids, 0.4 mg/L biotin, 0.4 g/L histidine, 2% (v/v) DMSO, 5 mM theophylline, 100 mg/mL ampicillin, and 0.1 mL/L antifoam A. pH was maintained at 6.5 and dissolved oxygen was maintained at 30%). The culture was grown for 3 h at 19° C, then induced with 0.5% (v/v) methanol every 12 h for a total of 72 h. Cells were harvested by centrifugation at 4,000 g for 30 min, flash-frozen with liquid N2, and stored at 80° C.
Cell pellets were resuspended in ice-cold lysis buffer (50 mM HEPES, pH 7.4, 300 mM NaCl, 4 mM theophylline, 5 mM 6-amino-caproic acid, 5 mM benzamidine, and 10% glycerol) and lysed using a microfluidizer (LM 20, ATS Scientific) at 20,000 psi in 2 passes. The lysate was centrifuged at 8,000 g for 30 min. The supernatant was collected and centrifuged at 100,000 g for 90 min, and the resulting pellet was resuspended in membrane solubilization buffer (50 mM HEPES, pH 7.4, 300 mM NaCl, 4 mM theophylline, 1 mM 6-aminocaproic acid, 1 mM benzamidine, 0.75% lauryl maltose neopentyl glycol (LMNG), and 0.05% cholesteryl hemisuccinate (CHS)) at 4 ° C overnight with gentle stirring. The solubilized membrane was centrifuged at 10,000 g for 1 h. Supernatant was collected and incubated with TALON resin (Takara) at 4 ° C for 12–24 h with gentle mixing.
The A2AR-bound TALON resin was washed with 5 bed-volumes of labeling buffer (50 mM HEPES, pH 7.4, 300 mM NaCl, 0.05% LMNG, and 0.005% CHS), then incubated with 2 bed-volumes of degassed labeling buffer containing 200 mM 2-bromo-N-(4-(trifluoromethyl)phenyl)acetamide (BTFMA) at 4 ° C overnight with gentle agitation. A second aliquot of BTFMA was added and incubated for 6 h before the resin was loaded onto a gravity column. The resin was washed with 4 bed-volumes of wash buffer (50 mM HEPES, pH 7.4, 300 mM NaCl, 0.05% LMNG, and 20 mM imidazole), and A2AR was eluted with 50 mM HEPES, pH 7.4, 300 mM NaCl, 0.05% LMNG, and 250 mM imidazole. Eluted receptors were exchanged to LMNG storage buffer (50 mM HEPES, pH 7.4, 300 mM NaCl, 0.05% LMNG), then incubated with XAC-agarose gel (Affi-Gel 10 conjugated with A2AR antagonist xanthine amine congener) at 4° C overnight with gentle agitation (Weiss and Grisshammer, 2002).
A2AR-bound XAC resin was loaded onto a gravity column, washed with 2 bed-volumes of LMNG storage buffer (50 mM HEPES, pH 7.4, 300 mM NaCl, 0.05% LMNG) followed by 1 bed-volume of cholate buffer (50 mM HEPES, pH 7.4, 100 mM NaCl, 15 mM sodium cholate). A2AR was eluted with 50 mM HEPES, pH 7.4, 100 mM NaCl, 15 mM sodium cholate, and 30 mM theophylline, then exchanged to cholate buffer prior to nanodisc assembly.
Nanodisc assembly and purification:
Cholate-solubilized lipid, A2AR, and MSPDH5 (Hagn et al., 2013) (produced in-house) were mixed in HNE buffer (20 mM HEPES, pH 8.0, 100 mM NaCl, 0.5 mM EDTA) with a final concentration of 15 mM cholate, 3.5 mM lipid, and 100 mM MSPDH5. The lipid mixture contained a 3:2 ratio of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) to 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (POPG). A minimum 10-to-1 MSPDH5: A2AR ratio was employed to ensure each nanodisc contains only one receptor. Empty nanodiscs were prepared using the identical procedure without including A2AR. The mixture was incubated on ice for 1 h, followed by the addition of 0.6 g/mL Bio-Beads SM-2 resin (BioRad) and an additional 5 h incubation at 4 ° C with gentle agitation. Bio-Beads were removed using a gravity column, and the assembled nanodiscs were purified on a HiLoad 16/600 Superdex 200 prep grade size exclusion column equilibrated with nanodisc storage buffer (50 mM HEPES, pH 7.4, 100 mM NaCl) at a flow rate of 1 mL/min. Monomeric nanodiscs eluted at around 65 mL (Figure S1). The peak was pooled, incubated with TALON resin for 2 h at 4 ° C, then washed extensively with nanodisc storage buffer to remove empty nanodiscs. A2AR-containing nanodiscs were eluted with 50 mM HEPES, pH 7.4, 100 mM NaCl, and 250 mM imidazole, then concentrated and buffer-exchanged to nanodisc storage buffer for downstream experiments.
Gsa expression and purification:
A pET15b plasmid containing a modified GNAS2 gene immediately downstream of an open reading frame for His6-tagged maltose binding protein (MBP) and TEV cleavage site was the generous gift from Dr. Oliver Ernst at the University of Toronto. This short isoform of human Gsα was originally designed for the purpose of chemoselective labeling and harbors the Y358C mutation along with all other solvent-exposed cysteines replaced (C3S, C200T, C237S, C359I, C365A, C379V). Analogous mutants have previously been made for Giα, and have been shown to retained native-like fold and function (Kaya et al., 2014; Medkova et al., 2002). The plasmid was transformed into Escherichia coli (E. coli) BL21 (DE3) cells by heat shock and screened on agar plates containing 100 mg/mL ampicillin. A single colony was grown in 10 mL of LB medium (0.5% yeast extract, 1% tryptone, 1% NaCl, 100 mg/mL ampicillin) overnight at 37 ° C with shaking. This starter culture was inoculated into 1 L of LB medium with ampicillin and grown at 25 ° C with shaking to an optical density at 600 nm (OD600) of 0.3. Cells were grown for an addition hour at 19 ° C, then induced with isopropyl β-d-1-thio-galactopyranoside (IPTG) to a final concentration of 50 mM. After overnight induction, cells were harvested by centrifugation at 6,000 g for 10 min.
Cell pellets were resuspended in ice-cold lysis buffer (50 mM sodium phosphate, pH 8.0, 300 mM NaCl, 10 mM imidazole, 50 mM GDP, 2 mM MgCl2, 0.5 mM TCEP, 5 mM 6-aminocaproic acid, 5 mM benzamidine, 0.4 mg/mL lysozyme, 2 mg/mL DNase I, 10% glycerol) and lysed by sonication. The lysate was centrifuged at 8,000 g for 2 h, and the resulting supernatant was incubated with Ni-NTA resin for 3 h at 4 ° C with gentle mixing. The resin was washed with 4 bed-volumes of lysis buffer containing 20 mM imidazole, then eluted with 50 mM sodium phosphate (pH 8.0), 100 mM NaCl, 50 mM GDP, 2 mM MgCl2, 0.5 mM TCEP, 10% glycerol, and 250 mM imidazole. The eluted MBP-Gsα fusion protein was buffer exchanged to remove imidazole, then incubated with 10 mg/mL of TEV protease (produced in-house) overnight at 4 ° C without agitation. The sample was loaded onto a Ni-NTA column, and the flow-through containing Gsα without an N-terminal MBP was purified on a HiLoad 16/600 Superdex 200 prep grade size exclusion column equilibrated with 50 mM HEPES, pH 7.4, 100 mM NaCl, 2 mM MgCl2, 5 mM GDP, and 10% glycerol at a flow rate of 1 mL/min. Gαs eluted at around 80 mL (Figure S2). The peak was pooled, concentrated, and GDP was added to a final concentration of 100 mM for downstream experiments.
Mini-Gsα expression and purification:
A pET15b plasmid containing a modified mini-Gsα gene (Carpenter et al., 2016) immediately downstream of an open reading frame for a His6 tag and TEV cleavage site was gifted by Dr. Oliver Ernst at the University of Toronto. Like Gsα above, this protein was originally designed for the purpose of chemoselective labeling and harbors the Y358C mutation along with all other solvent-exposed cysteines replaced (C237S, C359I, C365V, C379V; amino acid number aligned with that of Gsα). The plasmid was transformed into E. coli BL21 (DE3) cells by heat shock and screened on agar plates containing 100 mg/mL ampicillin. A single colony was grown in 10 mL of LB medium (0.5% yeast extract, 1% tryptone, 1% NaCl, 100 mg/mL ampicillin) overnight at 37 ° C with shaking. This starter culture was inoculated into 1 L of LB medium with ampicillin and grown at 37 ° C until an OD600 of 0.6. Cells were induced overnight at 25 ° C with 100 mM IPTG and harvested by centrifugation at 6,000 g for 10 min. Mini-Gsα was purified using identical protocol as Gsα, with the exception that the final size-exclusion chromatography step was omitted. The purified protein was buffer exchanged to 50 mM HEPES, pH 7.4, 100 mM NaCl, 2 mM MgCl2, 100 mM GDP, and 10% glycerol for downstream experiments.
Gβγ expression and purification:
Spodoptera frugiperda Sf9 cells were grown in I-Max insect cell culture medium (Wisent Inc.) to a density of 2 million cells/ml (≥98% viability) and then co-infected with two separate Autographa californica nuclear polyhedrosis viruses, one harboring the gene for His6-human Gβ1 (GNB1) and human Gγ2 (GNG2), and the other harboring the gene for human Giα, to produce Gβγ complex for experiments. Multiplicity of infection (MOI) of 0.75 (Gβγ) and 1.0 (Giα) were used for viral infection. The infected cells were grown at 27 ° C for 48–58 h before harvesting at 1000 g for 20 mins.
Cell pellets were resuspended in ice-cold lysis buffer (50 mM HEPES, pH 8.0, 65 mM NaCl, 2 mM MgCl2, 20 mM GDP, 5 mM 6-aminocaproic acid, 5 mM benzamidine, 5 mM β-mercaptoethanol, 1 mg/mL DNase I) and lysed using a microfluidizer at 2,000 psi in 1 pass. The lysate was centrifuged at 1,000 g for 10 min and the resulting supernatant was centrifuged at 100,000 g for 90 min. Pellets from the ultracentrifugation was resuspended in membrane wash buffer (50 mM HEPES, pH 8.0, 50 mM NaCl, 5 mM MgCl2, 20 mM GDP, 5 mM 6-aminocaproic acid, 5 mM benzamidine, 5 mM β-mercaptoethanol), centrifuged again at 100,000 g for 90 min, then resuspended in membrane wash buffer containing 2% sodium cholate. The resuspended membrane was solubilized for 1 h at 4 ° C prior to centrifugation at 100,000 g for 30 min. The resulting supernatant was collected, and detergent was exchanged by slowly adding a five-fold volume of dilution buffer (20 mM HEPES, pH 8.0, 200 mM NaCl, 5 mM MgCl2, 10 mM imidazole, 0.05% n-dodecyl β-D-maltoside (DDM), 30 mM AlCl3, 5 mM NaF, 20 mM GDP, 1 mM DTT) over a 1 h period. The sample was incubated with Ni-NTA resin for 2 h at 4 ° C with gentle mixing. The resin was washed extensively with wash buffer (20 mM HEPES, pH 8.0, 40 mM NaCl, 5 mM MgCl2, 10 mM imidazole, 30 mM AlCl3, 5 mM NaF, 0.05% DDM, 20 mM GDP, 5 mM β−mercaptoethanol) prior to elution with 250 mM imidazole. The eluted protein was loaded onto a Macro-Prep High Q anion exchange column (Bio-Rad) equilibrated with buffer A (20 mM HEPES, pH 8.0, 40 mM NaCl, 2 mM MgCl2, 0.025% DDM, 20 mM GDP, 200 mM TCEP), washed extensively with 5% buffer B (buffer A with 1 M NaCl), then eluted with a gradient of 6%–40% buffer B. Fractions containing Gβγ (~24% buffer B, Figure S2) were pooled and exchanged to final storage buffer (50 mM HEPES, pH 7.4, 100 mM NaCl, 2 mM MgCl2, 0.0125% DDM, 100 mM TCEP, 100 mM GDP, 10% glycerol) for downstream experiments.
NMR experiments:
NMR samples were prepared in either nanodisc storage buffer or G protein storage buffer (when Gα or Gαβγ were present) with 20–100 mM BTFMA-labeled A2AR-V229C, 20 mM sodium trifluoroacetate (TFA) as the 19F chemical shift reference, and 10% D2O. When applicable, samples also contained a 5-fold excess of Gα (relative to A2AR) or 1.2-fold excess of Gαβγ, and saturating concentrations of A2AR ligands (2 mM NECA, 500 mM LUF5834, or 500 mM ZM241385). All samples were sterile-filtered and prepared in sterile Shigemi tubes to prevent microbial contamination. NMR experiments were acquired at 20 ° C on a 600 MHz Varian Inova spectrometer equipped with a triple-resonance cryoprobe tunable to 19F. A typical fluorine NMR experiment included a 300 ms recycle delay, a 7 ms (45 °) excitation pulse, and a 600 ms acquisition time. Spectra were acquired using 50,000–200,000 scans, yielding a S/N of approximately 50–100. Spectra were processed using MestReNova (Mestrelab Research S.L.) employing chemical shift referencing (75.6 ppm for TFA), baseline correction, zero filling, and exponential apodization equivalent to a 20 Hz line broadening. The transverse relaxation time (T2) of 19F -labeled A2AR-V229C saturated with agonist (NECA) was measured using a CPMG T2 pulse sequence, using a refocusing period of 120 ms and total transverse magnetization evolution times of 0.48, 0.96, 1.44, 1.92, 2.4, 2.88, 3.36, 3.84, 4.32, 4.8 and 6.24 ms. Peak intensities from spectra in the T2 series were fit to exponential decay functions () (Figure S3). To the extent that the resonances could be resolved, T2 relaxation measurements provided an estimate of the homogeneous contribution to linewidth (Δvhomo1/2=1/πT2).
NMR spectral deconvolution:
Spectral deconvolutions were performed using MestReNova assuming generalized Lorentzian line shapes, characterized by a frequency, νi, intensity, Ii, width at half-height, Δν1/2,ι, and a kurtosis parameter in the range of 1 to 1. As discussed below, it was possible to define the activation ensemble in terms of three resonances, A1-A3, whose frequencies (ν1-ν3) could be globally fitted from the entire spectral series as a function of ligand, GDP-bound G protein, and nucleotide-free G protein. In some cases, exchange broadening and/or coalescence suggested very slight deviations in these resonances (e.g. a slight coalescence of A1 and A2 in the case of the NECA + G protein spectra). In general, chemical shift values for A1, A2, and A3 were allowed to vary by no more than 0.03 ppm. To assess the uncertainty in peak fitting and to ensure deconvolution is robust against spectral noise, three additional spectra were acquired under identical conditions but in the absence of A2AR. The noise spectra were then added to the nine parent spectra shown in Figure 3, generating three additional noise-added spectra for each condition. Deconvolutions were performed again for each of the noise-added spectra while holding the chemical shifts and kurtosis factors constant to determine variations in intensity of the deconvolved peaks and sensitivity of deconvolved peak intensities to noise. Peaks from all four spectral fittings (parent + three noise-added) were used to calculate the mean and standard deviation of the percent populations presented in Figures S4 and S7.
NMR spectral assignments and validation of chemical shifts:
Building on our earlier 19F NMR study of A2AR-V229C in detergent micelles (Ye et al., 2016), the 19F NMR spectra in nanodiscs exhibit a clear downfield resonance whose intensity decreases with the addition of agonist and/or G protein. The chemical shift of this inactive state, designated as S1-2, was also observed to vary in accordance with the hypothesis that there is an equilibrium between two unique inactive states (i.e. ionic lock “on” or S1 and ionic lock “off” or S2) which undergo mutual millisecond timescale exchange, resulting in some weighted average of the two (S1-2). In nanodiscs, the spectral series suggests that S1 and S2 states exchange on an intermediate or slow timescale, relative to that observed earlier in micelles. Upon saturation of the receptor by agonist the intensity of the inactive signature decreases while the inactive state resonance is observed to shift downfield, suggesting the equilibrium has shifted to the ionic lock “off” state for the inactive conformers remaining in the ensemble. In addition to the above inactive state resonance, all of the 19F NMR spectra (i.e. as a function of ligand, G protein, and nucleotide) exhibit multiple upfield resonances characteristic of the active ensemble states. Three active states (A1, A2, A3) were assigned from a global analysis of all nine spectra in Figures 3B–3D by first identifying common local maxima and inflection points. The location of the A1 resonance was identified in the receptor + full agonist spectrum (without G protein, dark green trace in Figure 3D). This state is also partially resolved in the receptor + partial agonist + nucleotide-free G protein spectrum (light blue trace in Figure 3C) and appears as a shoulder in the spectra of the apo receptor + G protein both with and without GDP (dark gray and light gray traces in Figure 3B). A2 is assigned from the fully resolved peak in the receptor + partial agonist + nucleotide-free G protein spectrum (light blue trace in Figure 3C), as well as the apo receptor spectrum in the presence of G protein (dark gray trace in Figure 3B). Similarly, A3 was assigned from the maxima observed in the dark gray trace in Figure 3B, the shoulder in the light blue trace in Figure 3C, and the shoulder in the middle green trace in Figure 3D.
To further validate the assigned chemical shifts, we performed non-linear least-squares curve fitting for each spectrum (from 59 ppm to 64 ppm) using the MATLAB built-in function lsqcurvefit. Here, each NMR profile was modeled as a sum of four Lorentzian lines (one corresponding to the inactive resonance and three corresponding to the active resonances). An initial guess for each parameter (νi, Ii, and Δν1=2;i), based on spectral deconvolutions performed in MestReNova (described above), was supplied and when applicable, boundaries for line widths and intensities were specified. After fixing the parameters associated with the inactive resonance, lsqcurvefit was tasked to find the optimal frequency, intensity, and width associated with each active resonance while minimizing the sum of squared residuals. A summary of the analysis is provided below.
Beginning with the spectra of A2AR with saturating concentrations of agonist, NECA (Figure S5A), it is clearly possible in this case to fit the active state resonances to two Lorentzian lines as shown. However, as discussed in Figure S3 and the main text, the deconvolved line widths associated with the A2 resonance disagree with that estimated from T2 measurements by a factor of ~2.5. After imposing an upper limit on the line widths of 140–170 Hz (accounting for line broadening from exponential apodization) for A1 and A2, the resulting fit recapitulates the two maxima but does not capture the upfield signal in the deconvolution. In comparison, a three-state activation ensemble, being the next simplest possible model, produces a better fit. The third Lorentzian line, A3, was found to be a broad resonance in the absence of G protein.
We next consider the apo receptor spectra as shown in Figure S5B, where all three spectra are deconvolved assuming the active ensemble is represented by the identical three states, A1, A2, and A3, defined above. Beginning with the spectrum associated with GDP-loaded G protein bound to the apo receptor, we can distinguish all three active state resonances as local maxima, without any adjustment to the fitted frequencies, ν1, ν2, or ν3. Upon removal of GDP, these same frequencies appear to persist in the deconvolution in the topmost spectrum. In the absence of either G protein or ligand, however, the active ensemble is characterized by significant exchange broadening and we assume that the identical resonances are present, albeit broadened.
The NECA (full agonist) series of spectra, shown in Figure S5C, reveal a similar story. Again, the addition of GDP-bound G protein results in a well-defined A3 resonance, although A1 and A2 cannot be deconvolved with confidence due to apparent coalescence of the two resonances and exchange broadening. The identical frequencies, ν1, ν2, and ν3. used in the global analysis, are nevertheless shown as a reference. Finally, the partial agonist series of spectra, shown in Figure S5D, suggest the identical 3 resonances are again present in the nucleotide-free spectrum (topmost) while excessive exchange broadening and coalescence in the other two spectra make them difficult to be definitively deconvolved.
Deconvolutions of all nine spectra reveal some interesting observations: 1) a three-state active ensemble model (A1, A2, A3) is necessary. Following the principle of Occam’s razor, this is the simplest model if we are to take into account T2 relaxation measurements, 2) in the absence of G protein, the A3 resonance is characteristically broad for apo, NECA- and LUF-bound receptor, in keeping with the idea that the precoupled state is dynamic and samples a broad range of local sub-states, perhaps to facilitate binding to G protein, and 3) while there is a modest degree of exchange broadening and coalescence in a few of the spectra, the global assignment of the three frequencies, ν1, ν2, or ν3, agrees well with the spectra without the need for any adjustments in the frequencies.
GTP hydrolysis experiments:
The GTP hydrolysis experiments were performed using the Promega GTPase-Glo™ assay kit following the manufacturer’s protocol (Mondal et al., 2015). Briefly, Gα or Gαβγ were incubated with A2AR or empty nanodiscs at room temperature in G protein storage buffer containing 1 mM GDP, 4 mM GTP, and 25 mM A2AR ligand (when applicable), at a final concentration of 250 nM G protein, 250 nM, 500 nM, or 1000 nM A2AR, and 250 nM empty nanodiscs. Control reactions consist of identical buffer solutions with GTP but in the absence of either A2AR, G protein, or both. After 90 min, unreacted GTP was converted to ATP through the addition of a GTPase-Glo reagent. Subsequently, detection reagent containing luciferase was added and the resulting luminescence was measured using a TECAN Spark multi-mode plate reader with an integration time of 1 min. The luminescence signal intensity is directly proportional to the amount of unreacted GTP. Therefore, GTP hydrolysis can be calculated for the following:
In the absence of A2AR: ΔLumG = Lum(buffer only) -Lum(G protein only)
In the presence of A2AR: ΔLumG+R=Lum(buffer with A2AR but no G protein) – Lum(G protein + A2AR)
The increase in GTP hydrolysis as a result of increased nucleotide exchange mediated by A2AR (Figure 4 in the main text) was calculated as follows: %Increase GTP hydrolysis=(ΔLumG+R-ΔLumG)/(ΔLumG)*100
where Lum = luminescence signal intensity
Native-PAGE;
A2AR (20 mM) was combined with equimolar Gα or Gαβγ in G protein storage buffer containing 500 mM A2AR ligand (when applicable). Loading buffer was added (final concentration of 50 mM Tris-HCl, pH 7.0, 20% glycerol, and 0.01% bromophenol blue) and the samples were resolved on a 4%–15% tris-glycine polyacrylamide gel (BioRad) at 100 V for 2 h. The gel was stained with Coomassie brilliant blue.
SPR experiments:
SPR experiments were performed in triplicates using a benchtop OpenSPR instrument (Nicoya) equipped with a gold nanoparticle sensor chip conjugated with Ni-NTA. All experiments were performed under constant flow (40 mL/min G protein storage buffer containing 20 mM GDP and 100 mM NECA or LUF5834) over a sensor chip saturated with nanodisc-stabilized A2AR, which was immobilized to the chip via the C-terminal histidine tag. After a stable baseline was reached, Gαβγ was injected at three different concentrations (30 nM, 90 nM, and 270 nM) and allowed to interact with the A2AR-bound sensor chip until maximum dissociation was observed. Binding curves obtained at the three concentrations were simultaneously fit to a one-to-one binding model using Trace-Drawer (Ridgeview Instruments). We note that in our system, incomplete dissociation was observed which was likely a result of a small fraction of Gβγ inserting into the nanodiscs (due to the isoprenyl lipid anchor on the G subunit). Therefore, the last segment of the dissociation phase was excluded in the analysis of the binding curves. This was possible since the minimum amount of dissociation needed for an accurate off rate calculation is 5%.
A2AR-Gαβγ modeling:
The wild-type A2AR bound to NECA and mini-Gsα (based on the PDB structure 5G53 (Carpenter et al., 2016)) was first relaxed by MD simulations in 4:1 POPC:cholesterol (CHL) membranes. The relaxed structure was used as starting point to construct the fully active state of A2AR in complex with wild-type Gsαβγ. The relaxation procedure of the A2AR/NECA/mini-G complex was as follows: The missing intracellular loop 3 connecting TM5 and TM6 was modeled using the MODELER package. After energy minimization and equilibration, 3 successive iterations of 1 ms leap-frog integration MD simulations were conducted, assuming an isothermal-isobaric ensemble using a Nose-Hoover thermostat (Hoover, 1985; Nosè, 1984) and a Parrinello-Rahman barostat (Parrinello and Rahman, 1981) to mimic 300 K and 1 bar conditions. We carried out carbon-α clustering for the last 500 ns of simulation using a GROMOS clustering algorithm (Daura et al., 1999) to obtain the highest populated structure before proceeding to the next step. The construction of the A2AR/NECA/Gs complex was as follows: The Gα subunit was homology modeled using the mini-G structure while the missing regions in the nucleotide binding pocket and the remainder of Gs were modeled based on the structure of the GDP-bound Gs heterotrimer (PDB: 6EG8 (Liu et al., 2019)). The A2AR-Gsαβγ complex was then inserted into the POPC-4:1-CHL extended membrane, equilibrated and relaxed in a 1 ms simulation. An AMBER 14SB forcefield (Maier et al., 2015) was used to represent the intramolecular interactions of protein, and a GAFF forcefield (Wang et al., 2004) was introduced for NECA and GDP. All the simulation boxes were solvated using a TIP3P water model (Jorgensen et al., 1983), while maintaining a 0.15 M NaCl concentration.
Allostery computations using rigidity transmission theory:
To probe allosteric communication in the A2AR-heterotrimeric G protein complex, we utilized the rigidity-transmission allostery (RTA) algorithm, whose details have been previously described (Ye et al., 2018). The RTA algorithm is a computational method based on mathematical rigidity theory (Whiteley, 2005), which predicts how perturbations of conformational rigidity and flexibility (conformational degrees of freedom) at one site transmit across a protein structure to modify degrees of freedom at other distant sites. Here, RTA was applied to examine the allosteric pathways between the orthosteric pocket of the receptor and distal regions in the A2AR-Gαβγ complex with a focus on the GDP pocket and Gβγ.
Starting with the model of NECA-bound A2AR complexed with GDP-bound heterotrimer, a constrained network representation of protein structure was generated with the method FIRST (Floppy Inclusions and Rigid Substructure Topography (Jacobs et al., 2001))- an initial step in the RTA method. The constrained network consists of nodes (atoms) and edges representing covalent and non-covalent interactions (i.e. covalent bonds, hydrogen bonds, hydrophobic interactions etc.). Hydrogen bonds are ranked in terms of overall strength, whereupon a hydrogen bond energy cut-off value is selected such that all bonds weaker than this cut-off are ignored. The pebble game algorithm (a component of FIRST software) was used to quantify rigidity and flexibility and evaluate available conformational degrees of freedom throughout the constrained network. We then applied the RTA algorithm to quantify the available conformational degrees of freedom at every window of three consecutive residues (i.e. a sliding window filter along the length of the receptor) before and after perturbation of rigidity at the orthosteric pocket (NECA). The extent of the “degree of freedom transmission” was then extracted for each residue. This process was repeated by successively ignoring weak hydrogen bonds in small steps of 0.01 kcal/mol. Each residue was then assigned the allosteric intensity response by averaging the total degree of freedom transmission as a function of energy cut-off using three neighboring windows containing that residue. Residues with high allosteric intensity transmission define the allosteric pathway connecting the orthosteric pocket and distant residues in the receptor and Gsαβγ. To visualize results, residues are colored on the structure based on the amount of allosteric transmission.
QUANTIFICATION AND STATISTICAL ANALYSIS
Each NMR spectrum was acquired using 1–2 freshly prepared sample(s) and a cumulative 50,000 to 200,000 scans until sufficient S/N was achieved. Experiments were monitored to ensure that the peak shapes do not change over time and that the sample remains stable throughout the experiment. Each experimental series were performed on the same batch of receptors (i.e., all spectra in Figure 1 were acquired using receptors from the same cell pellet, purified as a single batch. Same goes for Figure 3). Statistical analysis was performed in either Microsoft Excel (Figures 5, S3, and S7) or GraphPad Prism 8.4.2 (Figure 4). Statistical details can be found in figure legends where applicable. Briefly, GTP hydrolysis data were presented as mean ± SEM (n=3). In Figure 4A, statistical significance was determined by either a two-way ANOVA followed by the Bonferroni (comparison of Gα and Gαβγ for each ligand) or the Tukey test (comparison of each ligand condition to each other). In Figure 4B, statistical significance was determined a multiple t test using the Holm-Sidak method. In all cases, * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001. Kinetic parameters derived from SPR data represent mean ± SD (n = 3, Figure 5). Given that the uncertainties between trials are much larger than the fitting error of the binding curves within each trial, the former is presented. In estimating percent population of states from deconvolution of NMR spectra (Figure S7), data represent mean ± SD from four individual fits. In each case, three noise spectra (of equivalent noise amplitude to that of the baseline in the original spectrum) were added to the original spectrum, generating three new daughter spectra. All 4 spectra were then deconvolved and the intensities were allowed to vary in each fit. For additional details see the STAR Methods section.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Canadian Institutes of Health Research (CIHR) Operating Grant MOP-43998 to R.S.P., the National Institute of General Medical Sciences grants GM106990 and GM083118 to R.K.S., and the MEXT/JSPS KAKENHI no. JP19K23721 to D.P.T. and no. JP19H03191 to A.K. S.K.H. is supported by Alexander Graham Bell Canada Graduate Scholarship-Doctoral from NSERC. A.S. was supported by CREST, Japan Science and Technology Agency (JST), Japan, JPMJCR1402. We thank Dr. Oliver Ernst and Dr. Ned Van Eps for helpful discussions and for providing plasmids with the Gsa and the mini-Gsa sequence. Special thanks to Dr. Marko Jovic from Nicoya Lifesciences for help with SPR data analysis, Dr. Voula Kanelis for access to her lab equipment, Dmitry Pichugin for NMR spectrometer maintenance, and Zixin Chen for artistic input. In memory of our cherished colleague and friend, Prof. Éric Marsault, at the University of Sherbrooke.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ballesteros JA, and Weinstein H (1995). Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 25, 366–428. [Google Scholar]
- Ballesteros JA, Jensen AD, Liapakis G, Rasmussen SGF, Shi L, Gether U, and Javitch JA (2001). Activation of the b 2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J. Biol. Chem 276, 29171–29177. [DOI] [PubMed] [Google Scholar]
- Bockenhauer S, Furstenberg A, Yao XJ, Kobilka BK, and Moerner WE (2011). Conformational dynamics of single G protein-coupled receptors in solution. J. Phys. Chem. B 115, 13328–13338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford W, Buckholz A, Morton J, Price C, Jones AM, and Urano D (2013). Eukaryotic G protein signaling evolved to require G protein-coupled receptors for activation. Sci. Signal 6, ra37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun S, and Levitzki A (1979). Adenosine receptor permanently coupled to turkey erythrocyte adenylate cyclase. Biochemistry 18, 2134–2138. [DOI] [PubMed] [Google Scholar]
- Carpenter B, and Tate CG (2016). Engineering a minimal G protein to facilitate crystallisation of G protein-coupled receptors in their active conformation. Protein Eng. Des. Sel 29, 583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter B, Nehmè R, Warne T, Leslie AGW, and Tate CG (2016). Erratum: Structure of the adenosine A2A receptor bound to an engineered G protein. Nature 538, 542. [DOI] [PubMed] [Google Scholar]
- Casiraghi M, Damian M, Lescop E, Point E, Moncoq K, Morellet N, Levy D, Marie J, Guittet E, Banères JL, and Catoire LJ (2016). Functional modulation of a G protein-coupled receptor conformational landscape in a lipid bilayer. J. Am. Chem. Soc 138, 11170–11175. [DOI] [PubMed] [Google Scholar]
- Cavanagh J, Skelton NJ, Fairbrother WJ, Rance M, and Palmer AG III. (2007). Protein NMR Spectroscopy (Academic Press; ). [Google Scholar]
- Clark LD, Dikiy I, Chapman K, Rödström KEJ, Aramini J, LeVine MV, Khelashvili G, Rasmussen SGF, Gardner KH, and Rosenbaum DM (2017). Ligand modulation of sidechain dynamics in a wild-type human GPCR. eLife 6, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congreve M, de Graaf C, Swain NA, and Tate CG (2020). Impact of GPCR Structures on Drug Discovery. Cell 181, 81–91. [DOI] [PubMed] [Google Scholar]
- Daura X, Gademann K, Jaun B, Seebach D, van Gunsteren WF, and Mark AE (1999). Peptide Folding: When Simulation Meets Experiment. Angew. Chemie Int 38, 236–240. [Google Scholar]
- de Lera Ruiz M, Lim YH, and Zheng J (2014). Adenosine A2A receptor as a drug discovery target. J. Med. Chem 57, 3623–3650. [DOI] [PubMed] [Google Scholar]
- Dorè AS, Robertson N, Errey JC, Ng I, Hollenstein K, Tehan B, Hurrell E, Bennett K, Congreve M, Magnani F, et al. (2011). Structure of the adenosine A(2A) receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure 19, 1283–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror RO, Arlow DH, Maragakis P, Mildorf TJ, Pan AC, Xu H, Borhani DW, and Shaw DE (2011). Activation mechanism of the b2-adrenergic receptor. Proc. Natl. Acad. Sci. USA 108, 18684–18689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Duc NM, Rasmussen SGF, Hilger D, Kubiak X, Wang L, Bohon J, Kim HR, Wegrecki M, Asuru A, et al. (2019). Assembly of a GPCR-G protein complex. Cell 177, 1232–1242.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy MT, Lee MY, Gao ZG, White KL, Didenko T, Horst R, Audet M, Stanczak P, McClary KM, Han GW, et al. (2018). Allosteric coupling of drug binding and intracellular signaling in the A2A adenosine receptor. Cell 172, 68–80.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Effendi WI, Nagano T, Kobayashi K, and Nishimura Y (2020). Focusing on Adenosine Receptors as a Potential Targeted Therapy in Human Diseases. Cells 9, 785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanelli F, Felline A, Raimondi F, and Seeber M (2016). Structure network analysis to gain insights into GPCR function. Biochem. Soc. Trans 44, 613–618. [DOI] [PubMed] [Google Scholar]
- Fredriksson R, Lagerström MC, Lundin LG, and Schiöth HB (2003). The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol 63, 1256–1272. [DOI] [PubMed] [Google Scholar]
- Frei JN, Broadhurst RW, Bostock MJ, Solt A, Jones AJY, Gabriel F, Tandale A, Shrestha B, and Nietlispach D (2020). Conformational plasticity of ligand-bound and ternary GPCR complexes studied by 19F NMR of the b1-adrenergic receptor. Nat. Commun 11, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galès C, Van Durm JJJ, Schaak S, Pontier S, Percherancier Y, Audet M, Paris H, and Bouvier M (2006). Probing the activation-promoted structural rearrangements in preassembled receptor-G protein complexes. Nat. Struct. Mol. Biol 13, 778–786. [DOI] [PubMed] [Google Scholar]
- García-Nafría J, Lee Y, Bai X, Carpenter B, and Tate CG (2018). Cryo-EM structure of the adenosine A2A receptor coupled to an engineered heterotrimeric G protein. eLife 7, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorio GG, Masureel M, Hilger D, Terry DS, Juette M, Zhao H, Zhou Z, Perez-Aguilar JM, Hauge M, Mathiasen S, et al. (2017). Single molecule analysis of ligand efficacy in b2AR-G-protein activation. Nature 547, 68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero A (2018). A2A Adenosine Receptor Agonists and their Potential Therapeutic Applications. An Update. Curr. Med. Chem 25, 3597–3612. [DOI] [PubMed] [Google Scholar]
- Hagn F, Etzkorn M, Raschle T, and Wagner G (2013). Optimized phospholipid bilayer nanodiscs facilitate high-resolution structure determination of membrane proteins. J. Am. Chem. Soc 135, 1919–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser AS, Attwood MM, Rask-Andersen M, Schioth HB, and Gloriam DE (2017). Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov 16, 829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover WG (1985). Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A Gen. Phys 31, 1695–1697. [DOI] [PubMed] [Google Scholar]
- Isogai S, Deupi X, Opitz C, Heydenreich FM, Tsai CJ, Brueckner F, Schertler GFX, Veprintsev DB, and Grzesiek S (2016). Backbone NMR reveals allosteric signal transduction networks in the b1-adrenergic receptor. Nature 530, 237–241. [DOI] [PubMed] [Google Scholar]
- Jacobs DJ, Rader AJ, Kuhn LA, and Thorpe MF (2001). Protein flexibility predictions using graph theory. Proteins 44, 150–165. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, and Klein ML (1983). Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 79, 926–935. [Google Scholar]
- Kaya AI, Lokits AD, Gilbert JA, Iverson TM, Meiler J, and Hamm HE (2014). A conserved phenylalanine as a relay between the a5 helix and the GDP binding region of heterotrimeric Gi protein a subunit. J. Biol. Chem 289, 24475–24487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuku Y, Ueda T, Okude J, Shiraishi Y, Kondo K, Mizumura T, Suzuki S, and Shimada I (2014). Functional dynamics of deuterated β2-adrenergic receptor in lipid bilayers revealed by NMR spectroscopy. Angew. Chem. Int. Ed. Engl 53, 13376–13379. [DOI] [PubMed] [Google Scholar]
- Lee S, Nivedha AK, Tate CG, and Vaidehi N (2019). Dynamic role of the G protein in stabilizing the active state of the adenosine A2A receptor. Structure 27, 703–712.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Chun E, Thompson AA, Chubukov P, Xu F, Katritch V, Han GW, Roth CB, Heitman LH, IJzerman AP, et al. (2012). Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Xu X, Hilger D, Aschauer P, Tiemann JKS, Du Y, Liu H, Hirata K, Sun X, Guixa -Gonzalez R, et al. (2019). Structural Insights into the Process of GPCR-G Protein Complex Formation. Cell 177, 1243–1251.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, and Simmerling C (2015). ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Kim TH, Masureel M, Altenbach C, Yang Z, Hilger D, Lerch MT, Kobilka TS, Thian FS, Hubbell WL, et al. (2015). Structural insights into the dynamic process of β2-adrenergic receptor signaling. Cell 161, 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medkova M, Preininger AM, Yu NJ, Hubbell WL, and Hamm HE (2002). Conformational changes in the amino-terminal helix of the G protein α(i1) following dissociation from Gbetagamma subunit and activation. Biochemistry 41, 9962–9972. [DOI] [PubMed] [Google Scholar]
- Mondal S, Hsiao K, and Goueli SA (2015). A homogenous bioluminescent system for measuring GTPase, GTPase activating protein, and guanine nucleotide exchange factor activities. Assay Drug Dev. Technol 13, 444–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanoff C, and Stiles GL (1993). Solubilization and characterization of the A2-adenosine receptor. J. Recept. Res 13, 961–973. [DOI] [PubMed] [Google Scholar]
- Neubig RR (1994). Membrane organization in G-protein mechanisms. FASEB J. 8, 939–946. [DOI] [PubMed] [Google Scholar]
- Nobles M, Benians A, and Tinker A (2005). Heterotrimeric G proteins pre-couple with G protein-coupled receptors in living cells. Proc. Natl. Acad. Sci. USA 102, 18706–18711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosé S (1984). A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys 81, 511–519. [Google Scholar]
- Nygaard R, Zou Y, Dror RO, Mildorf TJ, Arlow DH, Manglik A, Pan AC, Liu CW, Fung JJ, Bokoch MP, et al. (2013). The dynamic process of β(2)-adrenergic receptor activation. Cell 152, 532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham WM, and Hamm HE (2008). Heterotrimeric G protein activation by G-protein-coupled receptors. Nat. Rev. Mol. Cell Biol 9, 60–71. [DOI] [PubMed] [Google Scholar]
- Park PS-H, Lodowski DT, and Palczewski K (2008). Activation of G protein-coupled receptors: beyond two-state models and tertiary conformational changes. Annu. Rev. Pharmacol. Toxicol 48, 107–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrinello M, and Rahman A (1981). Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Physiol 52, 7182–7190. [Google Scholar]
- Provasi D, Artacho MC, Negri A, Mobarec JC, and Filizola M (2011). Ligand-induced modulation of the free-energy landscape of G protein-coupled receptors explored by adaptive biasing techniques. PLoS Comput. Biol 7, e1002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebois RV, and Hébert TE (2003). Protein complexes involved in heptahelical receptor-mediated signal transduction. Receptors Channels 9, 169–194. [PubMed] [Google Scholar]
- Sljoka A (2021). Probing Allosteric Mechanism with Long-Range Rigidity Transmission Across Protein Networks. Methods Mol. Biol 2253, 61–75. [DOI] [PubMed] [Google Scholar]
- Smrcka AV, and Fisher I (2019). G-protein βγ subunits as multi-functional scaffolds and transducers in G protein-coupled receptor signaling. Cell. Mol. Life Sci 76, 4447–4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solt AS, Bostock MJ, Shrestha B, Kumar P, Warne T, Tate CG, and Nietlispach D (2017). Insight into partial agonism by observing multiple equilibria for ligand-bound and Gs-mimetic nanobody-bound β1-adrenergic receptor. Nat. Commun 8, 1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sounier R, Mas C, Steyaert J, Laeremans T, Manglik A, Huang W, Kobilka BK, Demene H, and Granier S (2015). Propagation of conformational changes during μ-opioid receptor activation. Nature 524, 375–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staus DP, Strachan RT, Manglik A, Pani B, Kahsai AW, Kim TH, Wingler LM, Ahn S, Chatterjee A, Masoudi A, et al. (2016). Allosteric nanobodies reveal the dynamic range and diverse mechanisms of G-protein-coupled receptor activation. Nature 535, 448–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susac L, Eddy MT, Didenko T, Stevens RC, and Wüthrich K (2018). A2A adenosine receptor functional states characterized by 19F-NMR. Proc. Natl. Acad. Sci. USA 115, 12733–12738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eps N, Caro LN, Morizumi T, Kusnetzow AK, Szczepek M, Hofmann KP, Bayburt TH, Sligar SG, Ernst OP, and Hubbell WL (2017). Conformational equilibria of light-activated rhodopsin in nanodiscs. Proc. Natl. Acad. Sci. USA 114, E3268–E3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel R, Mahalingam M, Lüdeke S, Huber T, Siebert F, and Sakmar TP (2008). Functional role of the “ionic lock”–an interhelical hydrogen-bond network in family A heptahelical receptors. J. Mol. Biol 380, 648–655. [DOI] [PubMed] [Google Scholar]
- Wang J, Wolf RM, Caldwell JW, Kollman PA, and Case DA (2004). Development and testing of a general amber force field. J. Comput. Chem 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- Weis WI, and Kobilka BK (2018). The molecular basis of G protein–coupled receptor activation. Annu. Rev. Biochem 87, 897–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss HM, and Grisshammer R (2002). Purification and characterization of the human adenosine A(2a) receptor functionally expressed in Escherichia coli. Eur. J. Biochem 269, 82–92. [DOI] [PubMed] [Google Scholar]
- Weiss JM, Morgan PH, Lutz MW, and Kenakin TP (1996). The cubic ternary complex receptor-occupancy model I. Model description. J. Theor. Biol 178, 151–167. [DOI] [PubMed] [Google Scholar]
- Whiteley W (2005). Counting out to the flexibility of molecules. Phys. Biol 2, S116–S126. [DOI] [PubMed] [Google Scholar]
- Wu F, Williams LM, Abdul-ridha A, Gunatilaka A, Vaid TM, Kocan M, Whitehead AR, Griffin MDW, Bathgate RAD, Scott DJ, et al. (2020). Probing the correlation between ligand efficacy and conformational diversity at the a1A-adrenoreceptor reveals allosteric coupling of its microswitches. J. Biol. Chem 295, 7404–7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L, Larda ST, Frank Li YF, Manglik A, and Prosser RS (2015). A comparison of chemical shift sensitivity of trifluoromethyl tags: optimizing resolution in 19F NMR studies of proteins. J. Biomol. NMR 62, 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L, Van Eps N, Zimmer M, Ernst OP, and Prosser RS (2016). Activation of the A2A adenosine G-protein-coupled receptor by conformational selection. Nature 533, 265–268. [DOI] [PubMed] [Google Scholar]
- Ye L, Neale C, Sljoka A, Lyda B, Pichugin D, Tsuchimura N, Larda ST, Pomes R, Garcıa AE, Ernst OP, et al. (2018). Mechanistic insights into allosteric regulation of the A2A adenosine G protein-coupled receptor by physiological cations. Nat. Commun 9, 1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J, Chen KM, Clark MJ, Hijazi M, Kumari P, Bai XC, Sunahara RK, Barth P, and Rosenbaum DM (2020). Structure of a D2 dopamine receptor-G-protein complex in a lipid membrane. Nature 584, 125–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Zhu C, Xie Q, and Wang Y (2020). Adenosine A2A Receptor Antagonists for Cancer Immunotherapy. J. Med. Chem 63, 12196–12212. [DOI] [PubMed] [Google Scholar]
- Zheng J, Zhang X, and Zhen X (2019). Development of Adenosine A2A Receptor Antagonists for the Treatment of Parkinson’s Disease: A Recent Update and Challenge. ACS Chem. Neurosci 10, 783–791. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.