


Abstract
The synthesis and incorporation into oligodeoxynucleotides of two novel, conformationally restricted abasic (AB) site analogs are described. The stability of oligonucleotide 18mer duplexes containing one such AB site opposite any of the four natural DNA bases was investigated by UV melting curve analysis and compared to that of duplexes containing a conformationally flexible propanediol unit 1 or a tetrahydrofuran unit 2 as an AB site analog. No major differences in the melting temperatures (ΔTm 0–3°C) between the different abasic duplexes were observed. All AB duplexes were found to have Tms that were lower by 9–15°C relative to a fully matched 18mer control duplex, and by 4–10°C relative to the corresponding 19mer duplexes in which the AB site is replaced by a mismatched nucleobase. Thus we conclude that the loss of stability of a duplex that is encountered by removal of a nucleobase from the stack cannot be compensated with conformational restriction of the AB site. From the van’t Hoff transition enthalpies obtained from the melting curves, it appears that melting cooperativity is higher for the duplexes containing the conformationally rigid AB sites. Fluorescence quenching experiments with duplexes containing the fluorescent base 2-aminopurine (2AP) opposite the AB sites showed a weak tendency towards more efficient stacking of this base in duplexes containing the conformationally constrained AB sites. Thus, such AB sites may structurally stabilize the cavity formed by the removal of a base. Potential applications emerging from the properties of such conformationally constrained AB sites in DNA diagnostics are discussed.
INTRODUCTION
Abasic (AB) sites in DNA are deoxyribose residues in their hemiacetal form that arise from hydrolysis of the N-glycosidic bonds of nucleotides (1–3). Under physiological conditions this process occurs spontaneously at a very low rate, preferably at purine nucleotides, and is frequently a consequence of chemical modification (e.g. alkylation) of the nucleobases. AB sites are also selectively produced by N-glycosylases and play a major role in DNA repair processes. Due to the lack of coding information, AB sites are mutagenic.
In view of the biological significance, a considerable body of data describing the structural and thermodynamic impact of AB sites in DNA duplexes is available today. Due to the intrinsic chemical instability of AB DNA, most of these data were obtained from oligonucleotides containing the chemically stable, isostructural tetrahydrofuran analog 2 of an AB site (Fig. 1) (4), or an acyclic analog as e.g. 1 (5). From UV-spectroscopic and calorimetric studies it is known that loss of a base considerably decreases duplex stability due to the interruption of the DNA base-stack (6). The thermodynamic impact is sequence dependent and dominated by the identity of the adjacent base pairs with the cross-strand partner base exerting only a secondary thermodynamic effect (7). Structural investigations by NMR on a series of abasic DNA duplexes were performed. These studies have established that oligonucleotides containing AB sites opposite purine residues conserve a regular right handed DNA geometry in which the base opposite the lesion stacks inside the helix (8–11). On the other hand, oligonucleotides containing AB sites opposite pyrimidine bases display additional forms where the abasic sugar and, sometimes, the pyrimidine residue are extrahelical (12,13). If the AB site is opposite to a thymine base and flanked by two pyrimidine residues, a kink of the helical axis by ∼30° into the major groove results (14).
Figure 1.
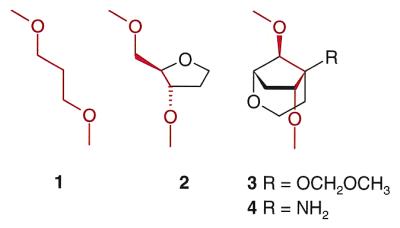
The chemical structures of the four different abasic site analogs used in this study. The respective backbone bonds shared by all analogs are drawn in red.
Fluorescence quenching experiments with oligodeoxynucleotides presenting the base 2-aminopurine (2AP) opposite AB sites showed that this base is predominantly stacked intrahelically and thus supports the conclusions from NMR (15). These experiments also showed that a notable difference in the propensity of stacking of the fluorescent base occurs as a function of the Mg2+ concentration with respect to monovalent Na+. From these structural investigations it becomes clear that AB sites are structurally labile and induce a multitude of different DNA tertiary structures depending on the base sequence and the electrostatic strength of the medium.
While much is known about the role of the excised bases, the structural and energetic impact of the sugar–phosphate backbone at AB sites in DNA duplexes is less well understood. As part of our ongoing program on nucleic acid analogs with conformationally restricted sugar–phosphate backbones (16–18) we set out to explore the effect of conformational restriction of AB sites on the thermal stability and structure of the corresponding duplexes. In this context we have synthesized oligonucleotides containing the two AB site units 3 and 4 (Fig. 1), and compared their thermodynamic and structural properties with duplexes containing the already known AB sites 1 and 2 by UV and fluorescence spectroscopy. While the propanediol unit 1 is completely flexible, the tetrahydrofuran unit 2 exhibits partial restriction around one backbone torsion angle. The new bicyclo-(3.2.1)-units 3 and 4 are fully restricted at two backbone bonds in a geometry that is similar to the corresponding torsion angles in B-DNA duplexes (19). Additionally, 4 contains an amino function that is expected to be protonated at neutral pH. This allows for the assessment of an additional positive charge on the stability of corresponding duplexes.
MATERIALS AND METHODS
Chemical synthesis of the abasic building block 4
Chemicals, solvents and reagents for reactions were generally from Fluka AG (Buchs, Switzerland), and were the highest quality available. Solvents for extraction and chromatography were of technical grade and distilled prior to use. 1H-, 13C-NMR: Bruker AC 300: Δδ in p.p.m. relative to CHCl3 = 7.27 (1H) and 77.0 (13C) as internal standard. Coupling constants J (Hz). High resolution LSIMS-MS: VG Autospec Q, Cs+ ion source.
(1S,5S,6R,8S)-5-(Trifluoroacetamido)-8-{[(tert-butyl)dimethylsilyl]oxy}-2-oxabicyclo(3.2.1)octane-6-ol (6). To a stirred solution of 5 (540 mg, 1.97 mmol) and NEt3 (0.55 ml, 3.95 mmol) in CH2Cl2 (18 ml) at 0°C was added dropwise (CF3CO)2O (0.48 ml, 3.45 mmol). The ice bath was removed, and after 2 h the mixture was extracted with ethyl acetate (AcOEt). The organic phase was washed twice with H2O, dried (MgSO4) and evaporated. The residue was dissolved in MeOH (30 ml) containing NEt3 (0.69 ml, 4.95 mmol) and stirred for 1 h. The solution was diluted with AcOEt, washed twice with H2O, dried (MgSO4) and evaporated. FC (hexane/toluene/AcOEt 4:1:1) afforded 643 mg (88%) of 6 as a white solid.
TLC (hexane/toluene/AcOEt 4:1:1): Rf 0.24. 1H-NMR (300 MHz, CDCl3): 0.13, 0.15 [2s, (CH3)2Si]; 0.89 [s, (CH3)3C]; 1.71 [dt, J = 6.7, 12.1, H-C(4)]; 1.90 [ddd, J = 1.5, 2.9, 15.5, H-C(7)]; 2.54 [dd, J = 3.7, 12.9, H-C(4)]; 2.63 [ddd, J = 5.9, 10.7, 15.8, H-C(7)]; 3.74 [dd, J = 6.6, 11.8, H-C(3)]; 3.87 [m, H-C(8)]; 4.09 [dd, J = 2.2, 5.5, H-C(1)]; 4.18 [dt, J = 4.1, 12.1, H-C(3)]; 4.52 [dd, J = 2.9, 10.3, H-C(6)]; 6.72 (broad s, NH). 13C-NMR (75 MHz, CDCl3): –5.31, –4.42 [(CH3)2Si]; 17.75 [(CH3)3C]; 25.39 [(CH3)3C)]; 31.28 [C(4)]; 35.20 [C(7)]; 59.23 [C(3)]; 66.78 [C(5)]; 76.03 [C(6)]; 78.29 [C(1)]; 81.37 [C(8)]. HR-MS [(M + 1)+]: 370.16599 (calc. 370.16615).
(1S,5S,6R,8S)-6-[(Allyloxycarbonyl)oxy]-5-(trifluoroacetamido)-8-{[(tert-butyl)dimethylsilyl]oxy}-2-oxabicyclo(3.2.1)octane (7). A solution of allyl chloroformate (0.23 ml, 2.16 mmol) in Et2O (4 ml) was added dropwise to a solution of 6 (320 mg, 0.87 mmol) and N,N,N′,N′-tetramethylethylenediamine (0.33 ml, 2.20 mmol) in Et2O (8 ml) at 0°. After 6 h at 0°, AcOEt was added, and the organic phase separated and washed with saturated NH4Cl and H2O (2×), dried (MgSO4) and evaporated. The residue was purified by FC (hexane/AcOEt 12:1 to 10:1) to give 345 mg (87%) of 7 as a colorless oil.
TLC (hexane/AcOEt 10:1): Rf 0.38. 1H-NMR (300 MHz, CDCl3): 0.05, 0.09 [2s, (CH3)2Si]; 0.86 [s, (CH3)3C]; 1.69 [dd, J = 3.7, 12.9, H-C(4)]; 1.98 [ddd, J = 1.8, 3.7, 15.8, H-C(7)]; 2.59–2.71 [m, H-C(4), H-C(7)]; 3.77 [dd, J = 7.0, 12.1, H-C(3)]; 3.90 [dt, J = 4.1, 12.2, H-C(3)]; 4.11 [dd, J = 1.5, 5.9, H-C(1)]; 4.60 [m, H-C(8)]; 4.70 (m, -O-CH2-); 5.24 [dd, J = 3.3, 10.7, H-C(6)]; 5.34 (dq, J = 1.1, 10.3, -CH=CH2, cis); 5.41 (dq, J = 1.5, 17.3, -CH=CH2, trans); 5.95 (m, -CH=CH2); 7.99 (broad s, NH). 13C-NMR (75 MHz, CDCl3): –5.38, –4.81 [(CH3)2Si]; 17.72 [(CH3)3C]; 25.50 [(CH3)3C]; 29.52 [C(4)]; 31.53 [C(7)]; 58.89 [C(3)]; 66.13 [C(5)]; 69.45 (-O-CH2-); 77.92 [C(8)]; 78.37 [C(1)]; 82.35 [C(6)]; 115.61 (q, J = 289, -CF3); 119.96 (-CH=CH2); 130.79 (-CH=CH2); 156.28 (q, J = 37, -COCF3); 156.40 (-OCO2-). HR-MS [(M + 1)+]: 454.18729 (calc. 454.18728).
(1S,5S,6R,8S)-6-[(Allyloxycarbonyl)oxy]-5-(trifluoroacetamido)-2-oxabicyclo(3.2.1)octane-8-ol (8). A solution of 7 (320 mg, 0.71 mmol) and tetrabutylammonium fluoride trihydrate (267 mg, 0.85 mmol) in THF (10 ml) was stirred for 20 min. The mixture was distributed between AcOEt and H2O, and the organic phase was dried (MgSO4) and evaporated. FC (hexane/AcOEt 2.2:1 to 2:1) resulted in 240 mg (100%) of 8 as a colorless oil.
TLC: (hexane/AcOEt 2:1): Rf 0.20. 1H-NMR (300 MHz, CDCl3): 1.68 [dd, J = 3.7, 13.2, H-C(4)]; 1.99 [ddd, J = 1.8, 3.3, 15.8, H-C(7)]; 2.38 [dt, J = 7.0, 12.5, H-C(4)]; 2.73 [ddd, J = 5.5, 11.0, 16.2, H-C(7)]; 3.78 [dd, J = 6.6, 12.1, H-C(3)]; 3.91 [dt, J = 4.1, 12.2, H-C(3)]; 4.27 [dd, J = 1.5, 5.5, H-C(1)]; 4.68 [m, H-C(8)]; 4.70 (m, -O-CH2-); 5.24 [dd, J = 3.3, 11.0, H-C(6)]; 5.35 (dq, J = 1.1, 10.3, -CH=CH2, cis); 5.41 (dq, J = 1.3, 17.3, -CH=CH2, trans); 5.95 (m, -CH=CH2); 8.38 (broad s, NH). 13C-NMR (75 MHz, CDCl3): 31.05 [C(4)]; 31.58 [C(7)]; 58.92 [C(3)]; 66.49 [C(5)]; 69.55 (-O-CH2-); 77.83 [C(1)]; 77.94 [C(8)]; 82.54 [C(6)]; 115.65 (q, J = 288, -CF3); 120.07 (-CH=CH2); 130.67 (-CH=CH2); 156.51 (-OCO2-); 156.64 (q, J = 37, -COCF3). HR-MS [(M + 1)+]: 340.10049 (calc. 340.10080).
(1S,5S,6R,8S)-6-[(Allyloxycarbonyl)oxy]-5-(trifluoroacetamido)-8-[9′-(9′-phenylxanthenyl)oxy]-2-oxabicyclo(3.2.1)octane (9). A solution of 8 (240 mg, 0.71 mmol) and 9-chloro-9-phenylxanthene (410 mg, 1.40 mmol) in dry pyridine (2.5 ml) was stirred at room temperature (RT) in the dark. After 2 days, additional 9-chloro-9-phenylxanthene (310 mg, 1.06 mmol) was added and the suspension stirred for another 2 days. The suspension was partitioned between AcOEt and H2O, the organic phase separated, dried (MgSO4) and evaporated. FC (hexane/AcOEt 7:1 to 5:1 +1% NEt3) afforded 391 mg (92%) of 9 as a slightly yellowish foam.
TLC: (hexane/AcOEt 4:1): Rf 0.35. 1H-NMR (300 MHz, CDCl3): 1.74 [ddd, J = 2.2, 3.7, 15.8, H-C(7)]; 2.08 [dd, J = 4.1, 13.2, H-C(4)]; 2.19 [dt, J = 6.6, 12.9, H-C(4)]; 2.41 [ddd, J = 5.5, 10.7, 15.8, H-C(7)]; 2.98 [d, J = 5.5, H-C(1)]; 3.55 [dd, J = 6.2, 12.1, H-C(3)]; 3.74 [dt, J = 4.4, 12.0, H-C(3)]; 3.76 [m, H-C(8)]; 4.68 (d, J = 5.9, -O-CH2-); 5.32 (dq, J = 1.1, 10.7, -CH=CH2, cis); 5.40 (dq, J = 1.5, 17.3, -CH=CH2, trans); 5.49 [dd, J = 3.7, 11.0, H-C(6)]; 5.96 (m, -CH=CH2); 6.92–7.42 (m, 13 aromatic H). 13C-NMR (75 MHz, CDCl3): 30.04 [C(4)]; 31.88 [C(7)]; 58.59 [C(3)]; 65.20 [C(5)]; 69.28 (-O-CH2-); 75.83 [C(1)]; 80.01 [C(8)]; 81.81 [C(6)]; 116.65 (pixyl); 116.82 (pixyl); 119.65 (-CH=CH2); 123.41; 124.26; 127.04; 127.05; 127.85; 130.09; 130.12; 130.36; 130.56 (all pixyl); 131.07 (-CH=CH2); 155.76 (-OCO2-); 155.98 (q, J = 36, -COCF3).
(1S,5S,6R,8S)-5-(Trifluoroacetamido)-8-[9′-(9′-phenylxanthenyl)oxy]-2-oxabicyclo(3.2.1)octane-6-ol (10). To a solution of 9 (206 mg, 0.35 mmol) and Pd(P(C6H5)3)4 (20 mg, 17 µmol) in THF (18 ml) was added dropwise morpholine (90 µl, 1.03 mmol). After 20 min, the solution was diluted with AcOEt, washed with 10% NaCl, the organic phase separated, dried (MgSO4) and evaporated. The residue was purified by FC (hexane/AcOEt 3:1 to 2.5:1 +1% NEt3) to give 162 mg (90%) of 10 as a white foam.
TLC (hexane/AcOEt 2:1): Rf 0.25. 1H-NMR (300 MHz, CDCl3): 1.34 [dd, J = 7.0, 12.1, H-C(4)]; 1.75 [ddd, J = 1.9, 3.3, 15.5, H-C(7)]; 2.35 [dd, J = 3.7, 12.2, H-C(4)]; 2.62 [ddd, J = 5.5, 10.3, 15.5, H-C(7)]; 2.95 [d, J = 5.5, H-C(1)]; 3.31 [m, H-C(8)]; 3.50 [dd, J = 6.6, 12.1, H-C(3)]; 3.98 [dt, J = 4.1, 12.1, H-C(3)]; 4.24 (broad s, OH); 4.69 [dd, J = 2.9, 10.3, H-C(6)]; 6.32 (broad s, NH); 6.95–7.47 (m, 13 aromatic H). 13C-NMR (75 MHz, CDCl3): 31.84 [C(4)]; 35.14 [C(7)]; 58.70 [C(3)]; 66.16 [C(5)]; 75.96 [C(1)]; 76.64 [C(6)]; 81.69 [C(8)]; 116.90; 117.32; 123.74; 124.72; 127.19; 127.28; 127.97; 128.56; 130.34; 130.37; 130.73 (all pixyl); 156.64 (q, J = 37, -COCF3). HR-MS [(M + 1)+]: 512.16846 (calc. 512.16848).
(1S,5S,6R,8S)-5-(Trifluoroacetamido)-6-{[(2′-cyanoethoxy)(diisopropylamino)phosphino]oxy}-8-[9′-(9′-phenylxanthenyl)oxy]-2-oxabicyclo(3.2.1)octane (11). To a solution of 10 (156 mg, 0.30 mmol) and N,N-diisopropylethylamine (0.31 ml, 1.81 mmol) in MeCN (2 ml) was added dropwise 2-cyanoethyl N,N-diisopropylchlorophosphoramidite (0.20 ml, 0.90 mmol). After 20 min, the solution was diluted with AcOEt and washed with saturated aqueous NaHCO3. The organic phase was separated, dried (MgSO4) and evaporated. The two diastereoisomers could easily be separated by FC (hexane/AcOEt 3.5:1 to 3:1 +1% NEt3) to yield 114 mg (53%) of the apolar diastereoisomer and 72 mg (33%) of the polar diastereoisomer of 11, both as white foams.
Apolar isomer: TLC (hexane/AcOEt 3:1): Rf 0.34. 1H-NMR (300 MHz, CDCl3): 1.20, 1.21, 1.22, 1.23 [4s, 2 (CH3)2CH-N]; 1.60 [ddd, J = 2.2, 3.7, 15.4, H-C(7)]; 2.10 [dd, J = 4.1, 12.9, H-C(4)]; 2.21 [dt, J = 6.6, 12.2, H-C(4)]; 2.30 [ddd, J = 5.9, 11.0, 15.8, H-C(7)]; 2.70 (t, J = 6.3, -CH2-CN); 2.97 [d, J = 5.5, H-C(1)]; 3.54 [dd, J = 6.3, 11.4, H-C(3)]; 3.59–3.65 (m, 2 -CH-N); 3.74 [dt, J = 4.4, 11.8, H-C(3)]; 3.75 [m, H-C(8)]; 3.81–4.02 (m, -CH2-CH2-CN); 4.68 [dt, J = 3.3, 11.6, H-C(6)]; 6.93–7.41 (m, 13 aromatic H). 13C-NMR (75 MHz, CDCl3): 20.18 (d, J = 6.1, -CH2-CN); 24.40, 24.45, 24.51, 24.54 [2 (CH3)2CH-N]; 30.43 [C(4)]; 33.42 [d, J = 2.4, C(7)]; 43.24 (d, J = 12.1, -CH-N); 57.96 (d, J = 17.0, -CH2-CH2-CN); 58.71 [C(3)]; 65.28 [d, J = 2.4, C(5)]; 76.29 [C(1)]; 77.86 [d, J = 14.0, C(6)]; 79.43 [C(8)]; 115.73 (q, J = 288, -CF3); 116.46; 116.69; 123.24; 123.99; 126.86; 127.06; 127.74; 129.87; 129.90; 130.63 (all pixyl); 155.69 (q, J = 36, -COCF3). 31P-NMR (161.9 MHz, CDCl3): 149.56. HR-MS [(M + 1)+]: 712.27563 (calc. 712.27634).
Polar isomer: TLC (hexane/AcOEt 3:1): Rf 0.29. 1H-NMR (300 MHz, CDCl3): 1.18, 1.21 [2s, 2 (CH3)2CH-N]; 1.72 [ddd, J = 2.2, 3.6, 15.8, H-C(7)]; 2.03 [dt, J = 7.4, 12.3, H-C(4)]; 2.36 [m, H-C(4)]; 2.43 [dd, J = 5.9, 11.0, H-C(7)]; 2.68 (t, J = 6.2, -CH2-CN); 2.91 [d, J = 5.1, H-C(1)]; 3.53 [m, H-C(8)]; 3.54–3.65 [m, H-C(3), 2 -CH-N]; 3.71–3.97 [m, H-C(3), -CH2-CH2-CN]; 4.67 [dt, J = 3.3, 10.1, H-C(6)]; 6.48 (broad s, NH); 6.92–7.41 (m, 13 aromatic H). 13C-NMR (75 MHz, CDCl3): 20.46 (d, J = 7.3, -CH2-CN); 24.30, 24.42, 24.50, 24.59 [2 (CH3)2CH-N]; 30.29 [C(4)]; 34.12 [C(7)]; 43.31 (d, J = 12.1, -CH-N); 57.61 (d, J = 18.2, -CH2-CH2-CN); 58.92 [C(3)]; 65.75 [d, J = 6.7, C(5)]; 76.23 [C(1)]; 79.08 [d, J = 15.2, C(6)]; 79.89 [C(8)]; 115.55 (q, J = 288, -CF3); 116.60; 116.76; 123.41; 124.15; 127.00; 127.09; 127.82; 130.01; 130.03; 130.65 (all pixyl); 155.76 (q, J = 36, -COCF3). 31P-NMR (161.9 MHz, CDCl3): 150.59.
Oligodeoxynucleotide synthesis
Oligodeoxynucleotides were synthesized on a Pharmacia Gene Assembler synthesizer using standard phosphoramidite chemistry. The nucleoside phosphoramidites, including those containing the abasic units 1 and 2, were from Glen Research (Sterling, VA) and purchased from Eurogentec (Brussels, Belgium). The synthesis of the phosphoramidite building block for unit 3 will be described elsewhere. The standard synthetic procedure (trityl off mode) was used, and only for the non-natural phosphoramidites the coupling time was extended to 6 min. In the coupling step, tetrazole (0.45 M in CH3CN) was replaced by the more active (S-benzyl)thiotetrazole (0.2 M in CH3CN). Coupling efficiencies for all building blocks, including 3 and 4 were >98%. After standard detachment and deprotection (conc. NH3, 55°C 12–16 h) the crude oligomers were purified by anion exchange HPLC (Macherey-Nagel, nucleogen DEAE 60/7) and desalted over Sep-pak cartridges (Waters, Milford, USA). All oligodeoxynucleotides containing abasic units 1–4 were routinely analyzed by elecrospray mass spectrometry. The masses were found to be within 0.5 of the expected mass.
UV melting curves
UV melting curves were determined at 260 nm on a Varian Cary 3E spectrophotometer that was equipped with a Peltier block using the Varian WinUV software. Complementary oligodeoxynucleotides were mixed to 1:1 stoichiometry and the solutions adjusted to a final duplex concentration of 1.0–1.2 µM in 10 mM NaH2PO4, 150 mM NaCl, pH 7.0. A heating–cooling–heating cycle in the temperature range 20–90°C was applied with a temperature gradient of 0.5°C/min. All ramps were superimposable indicating equilibrium melting processes. Tm values were defined as the maximum of the first derivative of the melting curve. Thermodynamic data were determined directly from the melting curves by a curve fitting procedure that is based on the two-state model for a bimolecular non self-complementary oligonucleotide pair (20).
Steady state fluorescence measurements
Steady state fluorescence emission spectra of the 2AP containing oligodeoxynucleotide samples [c(duplex) = 1 µM, buffer as described for UV measurements] were recorded on a Perkin-Elmer Luminescence Spectrometer LS 50B using a 5 mm square cuvette. The emission spectra were recorded at a scan speed of 60 nm/min over the wavelength range 340–400 nm with an excitation wavelength of 320 nm. The excitation slit was set to 2.5 nm and the emission slit to 10 nm. For each measurement three scans were accumulated.
RESULTS
Synthesis of the building block for the rigid abasic unit 4
In the following we concentrate on the description of the synthesis of the phosphoramidite building block 11 (Scheme 1) which is used in oligonucleotide synthesis as a precursor of the abasic unit 4. The corresponding building block for the abasic unit 3 will be described elsewhere.
The synthesis starts with the bicyclic amine 5 which has been prepared earlier in our laboratory (21). In a first step 5 was N-protected (→ 6) with the trifluoroacetyl group that was shown to be compatible with standard oligonucleotide synthesis. The secondary hydroxyl group in 6 was then transformed into the corresponding allyl carbonate (→ 7) followed by removal of the silyl group (→ 8). The hydroxyl function in 8, structurally corresponding to a 5′-OH function in deoxyribonucleosides, was subsequently converted into the pixyl ether 9. This ether, which can be cleaved under identical conditions as the usual dimethoxytrityl (DMT) group during oligonucleotide synthesis (22,23), was preferred due to the higher reactivity of the reagent pixyl chloride with 8, compared to DMT chloride, and the enhanced deprotection kinetics during acidic removal. Neutral Pd(0)-catalyzed cleavage of the allyl protecting group in 9 yielded 10 which was subsequently converted into the phosphoramidite building block 11 that was directly used in oligonucleotide synthesis. The overall yield in the conversions of 5 to 11 amounts to 54%.
Design of abasic duplexes and UV melting analysis
Throughout the present study we used the following 19mer DNA duplex sequence:
Strand 1: 5′-GATGACXGCTAGCTAGGAC-3′
Strand 2: 3′-CTACTGYCGATCGATCCTG-5′
This sequence conforms in length and base composition with a typical primer sequence. Position X (strand 1) thereby represents the position where the unnatural AB units 1–4 were incorporated, whereas position Y (strand 2) represents all four natural bases opposing the AB site. The flanking, intrastrand nearest neighbor bases to Y remain constant with a 5′-pyrimidine (C) and a 3′-purine (G) base. This sequence context was chosen as to emulate a situation of average structural stability with respect to the intrahelical stacking propensity of Y.
Tm data
The various duplexes were subjected to UV melting curve analyses. The corresponding Tm data (λ = 260 nm) are summarized in Table 1. All melting curves showed typical sigmoidal behavior with only one apparent transition.
Table 1. Tm data (°C) of the duplexes containing the abasic units 1–4. For buffer conditions see Materials and Methods.
| X | ||||||||
|---|---|---|---|---|---|---|---|---|
| Y | 1 | 2 | 3 | 4 | A | G | C | T |
| A | 54 | 53 | 55 | 55 | 57 | 59 | 56 | 64 |
| G | 52 | 55 | 55 | 55 | 59 | 59 | 66 | 60 |
| C | 50 | 52 | 52 | 52 | 56 | 67 | 55 | 56 |
| T | 52 | 50 | 53 | 53 | 64 | 60 | 56 | 59 |
| (-) (-)a | 64.0 |
aDeletion duplex 18mer (X = Y = deleted).
Inspection of the data reveals that all duplexes, whatever the AB unit is and whatever the opposing base to the AB site is, melt with a Tm that is lower by 9–15°C compared to the 18mer deletion duplex in which the positions X and Y are missing. A slight preference for purine bases opposite all AB units 1–4 is observed. This weak stabilizing effect might be due to the higher stacking surface of the purines with respect to the pyrimidines. Comparing the different AB units 1–4, one observes only slightly higher Tms of the duplexes with the conformationally more constrained units 3 and 4 compared to that with the more flexible units 1 and 2. ΔTms are in the range of 0 to +3°C for any given base opposite the AB site (rows 1 to 4 in Table 1). The rigid units 3 and 4 were slightly less discriminating between the nature of the opposite base than the more flexible units 1 and 2 (columns 1 to 4 in Table 1). ΔTm (max.) values amount to 4 and 5°C for 1 and 2, and 3.0°C for 3 and 4, respectively. The introduction of a positive charge in the AB site does not significantly alter the duplex stability. This is evident from a comparison of duplexes with AB units 3 and 4.
In order to get a more complete picture we also measured all 12 possible base-mismatch arrangements (as well as the four matched arrangements) for X and Y being natural DNA bases (Table 1, right half). As expected, decreases in Tm by 4–10°C are observed for the mismatches, the wobble base-pairs (X-Y = G-T or T-G) expectedly giving rise to the most stable non Watson–Crick base-pairs. From a comparison with the Tms of the AB site duplexes it can safely be stated that any base replacing an AB site leads to enhanced duplex stability. In other words, even the conformationally restricted AB sites 3 and 4 destabilize the DNA duplex more than any mismatched natural nucleoside at the same position.
Thermodynamic data
We also determined van’t Hoff enthalpies (ΔHvH) of duplex formation from the UV melting curves by a curve fitting procedure to the normalized α-curves using the two-state model for two interacting, non self-complementary oligonucleotides (20,24). The thus derived ΔHvH values are depicted in Table 2.
Table 2. Thermodynamic data of duplex formation, determined from the UV melting curves.
| –ΔHvH (kcal/mol)a | ||||
|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |
| A | 122 | 131 | 145 | 132 |
| G | 96 | 104 | 131 | 106 |
| C | 108 | 105 | 146 | 115 |
| T | 108 | 109 | 146 | 115 |
| (-)(-) | 111 | |||
aEstimated error arising from baseline correction: ± 5%
By comparison of the data it can roughly be stated that the highest duplex formation enthalpies are observed with adenine being the base opposite to the AB site, regardless of the nature of the AB site. The lowest enthalpies are observed with guanine while the two pyrimidine bases are inbetween (with the exception of AB site 3). Duplexation enthalpies tend to be more negative when a conformationally constrained AB site is present, the unit 3 thereby having the most pronounced effect. By comparison of these data with the enthalpy of the fully matched 18mer duplex it becomes clear that the data are obscured by the fact that most of the duplexes do not melt according to a pure two-state process (see Discussion).
Fluorescence quenching experiments
Fluorescence quenching experiments were performed to obtain information about whether a structurally preorganized AB site promotes base stacking of the base opposing the AB site. For this reason we have prepared an oligonucleotide (strand 2), in which Y corresponds to a deoxynucleoside containing the fluorescent base 2AP. Fluorescence emission spectra were recorded on a series of duplexes containing the 2AP unit opposite to the AB sites 1–4 in the presence and in absence of Mg2+. As controls, the fluorescence spectra of a duplex in which the 2AP unit is opposite to a T (X = T, Y = 2AP, a match situation), of the corresponding single strand containing the 2AP unit alone (strand 2, Y = 2AP), and of a duplex in which the 2AP unit is forced into a single nucleotide bulge (X = deleted, Y = 2AP) were also recorded. Relative fluorescence intensities are tabulated in Table 3, representative emission spectra are depicted in Figure 2.
Table 3. Fluorescent properties of selected 2AP containing oligodeoxynucleotides in the presence and absence of Mg2+.
| F/F0a | F/F0a | |
|---|---|---|
| X-Y | (0.15 M Na+) | (0.15 M Na+ + 15 mM Mg2+) |
| 2AP-1 | 3.4 | 6.0 |
| 2AP-2 | 2.9 | 6.5 |
| 2AP-3 | 2.9 | 7.0 |
| 2AP-4 | 2.4 | 5.4 |
| 2AP-T | 1 | 1 |
| 2AP-(-)b | 12.6 | 15.5 |
| 2APc | 6.6 | 7.1 |
aThe ratio of fluorescence intensities at 370 nm for the 2AP-Y duplexes (F) and the 2AP-T duplex(F0): Buffer cond. c(duplex) = 2 µM, in 10 mM NaH2PO4, pH 7.0.
bDuplex with 2AP residue in a single nucleotide bulge.
c2AP containing single strand.
Figure 2.

Fluorescence emission spectra of selected duplexes (see Table 3) in the absence of Mg2+: black, 2AP-(-); brown, 2AP; green, 2AP-1; blue, 2AP-2; purple, 2AP-3; red, 2AP-4; yellow, 2AP-T.
From the data in Table 3 it can be seen that fluorescence emission from 2AP in the absence of divalent magnesium cations is efficiently quenched when this base is opposite to one of the AB site residues 1–4, indicating a high degree of stacking of 2AP into the helix. Quenching efficiency relative to the control duplex (X-Y = 2AP-T) follows the order 4 > 3 = 2 > 1. In all cases fluorescence quenching is much stronger in the AB duplexes than in the corresponding single strand or in the duplex with the 2AP unit in the bulge. Addition of Mg2+ to the buffer generally leads to less efficient quenching of the 2AP fluorescence in the AB site containing duplexes, indicating a higher population of structures, in which the 2AP base is unstacked as a consequence of the helix compressing power of Mg2+. In these cases again, AB site 4 is more effective in stabilizing the 2AP base in the stack, followed by 1, 2 and 3.
DISCUSSION
AB duplex stability
The principal point of the present investigation was to determine systematically the effect of conformational restriction of an abasic site on duplex stability and structure. Interpretation of the Tm data (Table 1) clearly shows that conformational restriction at the AB site does, if at all, only marginally improve duplex stability. It is also clear that conformational restriction of the AB site does not change stability as a function of the nature of the opposite base. While the AB units 3 and 4 emulate the backbone geometry of a nucleoside in the south conformation (torsion angle δ = 157°) (19) one could argue that this would be different with an abasic nucleoside which is locked in a north- conformation. However, Kvaerno and Wengel have shown that an abasic locked nucleic acid (LNA) unit (a 2′-O,4′-methylene bridged abasic ribonucleoside unit) within a DNA duplex also leads to strong destabilization of the corresponding duplex (25). Thus it emerges that the influence of geometry and backbone flexibility of an abasic site is negligible on duplex stability in comparison to the effect of the missing base. In other words, the destabilization that occurs by perturbation of the base stack, cannot be compensated by a rigid backbone at this position. This view is corroborated by the fact that any natural base within the stack is better than no base (see Tms, Table 1), and by the recent findings that DNA bases can also be replaced by aromatic ring systems devoid of any hydrogen bonding capability without destabilization of the double helix (26,27).
Melting cooperativity
The transition enthalpies (ΔHvH) as determined from the UV melting curves require caution in interpretation. Crude insight into the mechanism of melting of a duplex can be obtained in principle from a comparison of the model-dependent ΔHvH and model independent (e.g. calorimetric) transition enthalpies (ΔH0). Specifically, if ΔHvH < ΔH0, the transition occurs in a non-two-state manner (6). If one compares the experimentally determined ΔHvH value of the fully matched 18mer duplex (X and Y deleted, ΔHvH = –111 kcal/mol, Table 2), with its predicted (28) transition enthalpy under standard conditions (ΔH0 = –132.5 kcal/mol) it becomes clear that most of the duplexes investigated seem not to melt according to the two-state model. It is therefore likely that the differences in the measured enthalpies reflect differences in transition cooperativity rather than real enthalpic differences. Thus, AB duplexes with the conformationally restricted AB sites 3 and 4 seem to have a higher tendency to melt as a single cooperative unit than those with the AB sites 1 and 2, which essentially do not alter the melting cooperativity (6). Thus conformational restriction of AB sites seems to promote the melting cooperativity but not the duplex stability.
Structure at the AB sites
The fluorescence spectroscopic investigation revealed that in the absence of divalent cations, the fluorescence signal of the 2AP residue opposite the AB site is more efficiently reduced if the AB site is conformationally restricted (3 and 4) relative to the arrangements where the conformationally more flexible AB site 1 is opposite the 2AP base. Qualitatively this can be interpreted in the way that the base 2AP has a higher tendency to be intrahelically stacked in the former case (15,29,30). The conformationally more rigid AB sites thus seem to preorganize the cavity formed by the missing base more efficiently, reducing the number of possible conformational states of an abasic duplex.
It was recently shown by Stivers (15) that divalent metals, as Mg2+, have a dramatic effect on AB duplex structure. Due to the helix compressing power of Mg2+, a base opposite to an AB site has a higher tendency to be unstacked. In our experiments we observed that this is also true for duplexes with conformationally constrained AB sites. The fluorescence quenching efficiency of the AB sites increases in the order 4 > 1 > 2 > 3. The observed differences for the two conformationally restricted AB units 3 and 4 seems to arise from the different nature of the functional groups at the bridgehead position (-NH3+ versus -OCH2OMe).
Effect of a positive charge at the AB site
With the AB unit 4, we investigated the effect of a structurally well positioned positive charge (replacement of O-4′, of an abasic nucleoside by a protonated amino function). As a general picture it emerges that this positive charge is moderately beneficial for both, the energy of duplex formation as well as the structural preorganization of the cavity around the AB site. Especially in the case of Mg2+ containing media, the presence of this charge seems to counterbalance the helix compressing activity of the metal ion.
CONCLUSIONS
The following conclusions can be drawn in view of potential applications of conformationally restricted AB site analogs. With respect to the use of AB sites as universal bases it can be stated that all AB site analogs 1–4 do fulfil the criteria of pairing ambiguity with respect to an opposite nucleobase in a duplex, however, none of these analogs fulfil the criteria of high thermal stability of the corresponding duplexes. While this has been recognized earlier for the case of AB unit 2 (4), these findings can now be extended to conformationally restricted AB units, such as 3 and 4. At low salt conditions the conformationally constrained AB units 3 and 4 show a tendency to stabilize the cavity formed by the missing base in an AB duplex more efficiently than the more flexible units 1 and 2. This can be of interest e.g. for the introduction of preferred intercalator binding sites in DNA duplexes. Furthermore such units could act as tools to investigate charge or electron transport phenomena through the π-stack of DNA (31). Given the reduced degree of conformational freedom, the units 3 or 4 may be useful for investigating the contribution of the sugar–phosphate backbone to the cooperativity of the DNA melting process.

Scheme 1. (a) (CF3CO)2O, CH2Cl2, 0°C→RT, 2 h, 88%; (b) allyl chloroformate, Et2O, TMEDA, 0°C, 6 h, 87%; (c) n-Bu4NF, THF, RT, 20 min, 100%; (d) 9-chloro-9-phenylxanthene, pyridine, RT, 4 days, 92%; (e) Pd[P(C6H5)3]4, morpholine, THF, RT, 20 min, 90%; (f) Cl-P(OCH2CH2CN)N(iPr)2, EtN(iPr)2, CH3CN, RT, 20 min, 86%.
Acknowledgments
ACKNOWLEDGEMENTS
Financial support from the Swiss National Science Foundation is gratefully acknowledged. A.H. thanks the Novartis Stpendienfonds, Basel, for a Ph.D. grant. We also thank Roche Diagnostics (Penzberg, Germany), for providing us with some of the unmodified oligodeoxynucleotides.
References
- 1.Lindahl T. and Nyberg,B. (1972) Biochemistry, 11, 3610–3618. [DOI] [PubMed] [Google Scholar]
- 2.Loeb L.A. and Preston,B.D. (1986) Annu. Rev. Genet., 20, 201–230. [DOI] [PubMed] [Google Scholar]
- 3.Weiss B. and Grossman,L. (1987) Adv. Enzymol. Relat. Areas Mol. Biol., 60, 1–34. [DOI] [PubMed] [Google Scholar]
- 4.Millican T.A., Mock,G.A., Chauncey,M.A., Patel,T.P., Eaton,M.A.W., Gunning,J., Cutbush,S.D., Neidle,S. and Mann,J. (1984) Nucleic Acids Res., 12, 7435–7453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seela F. and Kaiser,K. (1987) Nucleic Acids Res., 15, 3113–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vesnaver G., Chang,C.-N., Eisenberg,M., Grollman,A.P. and Breslauer,K.J. (1989) Proc. Natl Acad. Sci. USA, 86, 3614–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gelfand C.A., Plum,G.E., Grollman,A.P., Johnson,F. and Breslauer,K.J. (1998) Biochemistry, 37, 7321–7327. [DOI] [PubMed] [Google Scholar]
- 8.Cuniasse P., Sowers,L.C., Eritja,R., Kaplan,B., Goodman,M.F., Cognet,J.A.H., LeBret,M., Guschlbauer,W. and Fazakerley,G.V. (1987) Nucleic Acids Res., 15, 8003–8022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goljer I., Kumar,S. and Bolton,P.H. (1995) J. Biol. Chem., 270, 22980–22987. [DOI] [PubMed] [Google Scholar]
- 10.Kalnik M.W., Chang,C.-N., Grollman,A.P. and Patel,D.J. (1988) Biochemistry, 27, 924–931. [DOI] [PubMed] [Google Scholar]
- 11.Withka J.M., Wilde,J.A. and Bolton,P.H. (1991) Biochemistry, 30, 9931–9940. [DOI] [PubMed] [Google Scholar]
- 12.Cuniasse P., Fazakerley,G.V., Guschlbauer,W., Kaplan,B. and Sowers,L.C. (1990) J. Mol. Biol., 213, 303–314. [DOI] [PubMed] [Google Scholar]
- 13.Singh M.P., Hill,G.C., Péoc’h,D., Rayner,B., Imbach,J.-L. and Lown,J.W. (1994) Biochemistry, 33, 10271–10285. [DOI] [PubMed] [Google Scholar]
- 14.Coppel Y., Berthet,N., Coulombeau,C., Coulombeau,C., Garcia,J. and Lhomme,J. (1997) Biochemistry, 36, 4817–4830. [DOI] [PubMed] [Google Scholar]
- 15.Stivers J.T. (1998) Nucleic Acids Res., 26, 3837–3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bolli M., Trafelet,H.U. and Leumann,C. (1996) Nucleic Acids Res., 24, 4660–4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Epple C. and Leumann,C. (1998) Chem. Biol., 5, 209–216. [DOI] [PubMed] [Google Scholar]
- 18.Steffens R. and Leumann,C.J. (1999) J. Am. Chem. Soc., 121, 3249–3255. [Google Scholar]
- 19.Epple C., Leumann,C. and Stoeckli-Evans,H. (1998) Acta Crystallogr. C, 54, 1141–1143. [Google Scholar]
- 20.Marky L.A. and Breslauer,K.J. (1987) Biopolymers, 26, 1601–1620. [DOI] [PubMed] [Google Scholar]
- 21.Egger A., Hunziker,J., Rihs,G. and Leumann,C. (1998) Helv. Chim. Acta, 81, 734–743. [Google Scholar]
- 22.Chattopadhyaya J.B. and Reese,C.B. (1978) J. Chem. Soc. Chem. Commun., 639–640. [Google Scholar]
- 23.Sproat B.S., Beijer,B., Grotli,M., Ryder,U., Morand,K.L. and Lamond,A.I. (1994) J. Chem. Soc. [Perkin], 1, 419–431. [Google Scholar]
- 24.Breslauer K.J. (1986) In Hinz,H.-J. (ed.), Thermodynamic Data for Biochemistry and Biotechnology. Springer-Verlag, Berlin, pp. 402–427.
- 25.Kvaerno L. and Wengel,J. (1999) J. Chem. Soc. Chem. Commun., 657–658. [Google Scholar]
- 26.McMinn D.L., Ogawa,A.K., Wu,Y., Liu,J., Schultz,P.G. and Romesberg,F.E. (1999) J. Am. Chem. Soc., 121, 11585–11586. [Google Scholar]
- 27.Kool E.T., Morales,J.C. and Guckian,K.M. (2000) Angew. Chem. Intl. Ed. Engl., 39, 990–1009. [DOI] [PubMed] [Google Scholar]
- 28.Breslauer K.J., Frank,R., Blöcker,H. and Marky,L.A. (1986) Proc. Natl Acad. Sci. USA, 83, 3746–3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Law S., Eritja,R., Goodman,M.F. and Breslauer,K.J. (1996) Biochemistry, 35, 12329–12337. [DOI] [PubMed] [Google Scholar]
- 30.Holz B., Klimasauskas,S., Serva,S. and Weinhold,E. (1998) Nucleic Acids Res., 26, 1076–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meggers E., Michel-Beyerle,M.E. and Giese,B. (1998) J. Am. Chem. Soc., 120, 12950–12955. [Google Scholar]