

Abstract
Anelloviruses are highly prevalent in diverse mammals, including humans, but so far have not been linked to any disease and are considered to be part of the ‘healthy virome’. These viruses have small circular single-stranded DNA (ssDNA) genomes and encode several proteins with no detectable sequence similarity to proteins of other known viruses. Thus, anelloviruses are the only family of eukaryotic ssDNA viruses currently not included in the realm Monodnaviria. To gain insights into the provenance of these enigmatic viruses, we sequenced more than 250 complete genomes of anelloviruses from nasal and vaginal swab samples of Weddell seal (Leptonychotes weddellii) from Antarctica and a fecal sample of grizzly bear (Ursus arctos horribilis) from the USA and performed a comprehensive family-wide analysis of the signature anellovirus protein ORF1. Using state-of-the-art remote sequence similarity detection approaches and structural modeling with AlphaFold2, we show that ORF1 orthologs from all Anelloviridae genera adopt a jelly-roll fold typical of viral capsid proteins (CPs), establishing an evolutionary link to other eukaryotic ssDNA viruses, specifically, circoviruses. However, unlike CPs of other ssDNA viruses, ORF1 encoded by anelloviruses from different genera display remarkable variation in size, due to insertions into the jelly-roll domain. In particular, the insertion between β-strands H and I forms a projection domain predicted to face away from the capsid surface and function at the interface of virus–host interactions. Consistent with this prediction and supported by recent experimental evidence, the outermost region of the projection domain is a mutational hotspot, where rapid evolution was likely precipitated by the host immune system. Collectively, our findings further expand the known diversity of anelloviruses and explain how anellovirus ORF1 proteins likely diverged from canonical jelly-roll CPs through gradual augmentation of the projection domain. We suggest assigning Anelloviridae to a new phylum, ‘Commensaviricota’, and including it into the kingdom Shotokuvirae (realm Monodnaviria), alongside Cressdnaviricota and Cossaviricota.
Keywords: Anellovirus, structural modelling, capsid proteins, jelly-roll fold, virus evolution, taxonomy and classification, Commensaviricota
Introduction
Anelloviruses (family Anelloviridae) are one of the most enigmatic components of the human virome, in terms of both their biology and provenance. A large proportion of the human population appears to be infected with anelloviruses, yet no disease has been unequivocally linked to anellovirus infections (Kaczorowska and van der Hoek 2020; Webb, Rakibuzzaman, and Ramamoorthy 2020). The infections in humans have been shown to occur at an early age (Kaczorowska et al. 2022a), with high virus loads detected throughout the lifetime, and have been identified in nearly every human tissue (Leppik et al. 2007; Focosi et al. 2016; Blatter et al. 2018) including the brain and in fluid samples (Gerner et al. 2000; Goto et al. 2000; Inami et al. 2000; Itoh et al. 2000; Maggi et al. 2001; Naganuma et al. 2008; Arze et al. 2021; Kaczorowska et al. 2022b, 2023), consistent with the lymphocytes being the primary site of anellovirus replication (Leppik et al. 2007). The virus load appears to be controlled by the immune system because virus levels increase with the level of host immunosuppression (Focosi et al. 2016; Blatter et al. 2018). Although various studies have indicated a possible association between the presence of anelloviruses and certain medical conditions, such as diabetes, fever, cancer, liver cirrhosis, acquired immunodeficiency syndrome, and schizophrenia (Camci et al. 2002; Madsen. et al. 2002; Thom and Petrik 2007,McElvania TeKippe et al. 2012; Canuti et al. 2015; Kazemi et al. 2015; Hettmann et al. 2016; Spandole-Dinu et al. 2018), no significant link was identified with any of these. Furthermore, it has been suggested that anelloviruses positively affect human health by shaping immunity during early development (Kaczorowska and van der Hoek 2020). Thus, anelloviruses are considered to be part of the ‘healthy human virome’ (Arze et al. 2021; Koonin, Dolja, and Krupovic 2021). Apparently, asymptomatic anellovirus infections are also common in other mammals (Fahsbender et al. 2017; Crane et al. 2018; de Souza Wm et al. 2018; Webb, Rakibuzzaman, and Ramamoorthy 2020), suggesting extensive coevolution of anelloviruses with mammalian hosts. Notably, outside of mammals, anelloviruses have been characterized in chickens and several other avian species, and with members of one species, Gyroviruschickenanemia, having been shown to cause anemia, intramuscular hemorrhage, lymphoid atrophy, and bone marrow aplasia (Fatoba and Adeleke 2019; Kraberger et al. 2021).
With icosahedral capsids of ∼30 nm in diameter and circular single-stranded (ss) negative-sense DNA genomes of 1.6–3.9 kb (Miyata et al. 1999; Biagini 2009; Biagini et al. 2012), anelloviruses are among the smallest animal DNA viruses. Their genomes include one large open reading frame (ORF1) and several additional short ORFs, typically overlapping with ORF1. The product of ORF1 (∼350–710 amino acids (aa)) has been suggested to function as both the capsid protein (CP) and the replication initiation protein (Rep) (Erker et al. 1999; Mushahwar et al. 1999; Kamahora, Hino, and Miyata 2000; Bendinelli et al. 2001). The N-terminal region of the ORF1 product contains a stretch of positively charged residues, a feature typical of CPs of small RNA and DNA viruses (Rosario, Duffy, and Breitbart 2012; Requião et al. 2020). This protein also has been claimed to contain sequence motifs that are diagnostic of the rolling-circle replication initiation endonucleases of eukaryotic ssDNA viruses (Biagini et al. 2001; Kazlauskas et al. 2019). Recent cryo-electron microscopy analysis has shown that ORF1 of torque teno mini virus (Betatorquevirus) contains a jelly-roll domain and indeed forms an icosahedral capsid (Liou et al. 2022). However, the conservation of the jelly-roll domain across the family Anelloviridae and the presence or absence of the Rep domain in ORF1 were not investigated. ORF2 (∼100–120 aa), with a conserved N-terminal W-x7-H-x3-C-x-C-x5-H motif (x—any amino acid), has been suggested to function as a phosphatase and a suppressor of the nuclear factor-kappa B pathway (Hijikata, Takahashi, and Mishiro 1999; Biagini et, al. 2000; Okamoto et al. 2000; Peters et al. 2002; Zheng et al. 2007; Kakkola et al. 2009). Finally, ORF3 (∼70–200 aa) contains a C-terminal serine-rich domain, which has been suggested to be a target for phosphorylation and to play a role in anellovirus infection persistence and possibly in the inhibition of apoptosis (Kamahora, Hino, and Miyata 2000; Asabe et al. 2001; Kooistra et al. 2004; Qiu et al. 2005; Prasetyo et al. 2009; Singh and Ramamoorthy 2016).
None of the anellovirus ORFs bear detectable sequence similarity to proteins of other viruses or any other available proteins. In the absence of an identifiable Rep or a CP typical of other eukaryotic ssDNA viruses, Anelloviridae was the only family of eukaryote-infecting viruses with circular ssDNA genomes not included in the phylum Cressdnaviricota of the realm Monodnaviria (Krupovic et al. 2020). More generally, given the current absence of detected anelloviruses outside of vertebrate hosts and the lack of detectable homology to other known viruses or cellular genes, the evolutionary origins of this group of viruses remain enigmatic.
Based on comparisons of ORF1 protein sequences, known anelloviruses have been classified into 156 species, which are distributed across 30 genera (Biagini 2009; Biagini et al. 2012; Kraberger et al. 2021; Varsani et al. 2021), with 69 percent pairwise nucleotide (nt) identity between ORF1 genes chosen as a species demarcation threshold and genera defined based on the topology of the ORF1 protein phylogenetic tree (Varsani et al. 2021). The most populous genera are Alphatorquevirus, Betatorquevirus, and Gammatorquevirus, which include viruses that infect exclusively primates including humans (Okamoto 2009; Varsani et al. 2021).
Here, we further expand the breadth of known anellovirus diversity by sequencing 256 new complete genomes of anelloviruses from Weddell seals (Leptonychotes weddellii) and a grizzly bear (Ursus arctos horribilis). A comprehensive family-wide analysis of sequences and the predicted structures of the signature anellovirus protein ORF1 confirmed that they all adopt a jelly-roll fold typical of viral CPs, establishing an evolutionary link to other eukaryotic ssDNA viruses. However, the results of our analysis are inconsistent with ORF1 having any relationship with viral rolling-circle Reps. A comparison of the modeled structures of anellovirus CPs allowed us to reconstruct their evolution through the acquisition and gradual augmentation of a ‘projection domain’ within the ORF1 product, which appears to be at the forefront of virus–host interactions. Collectively, our findings suggest that, whereas the anellovirus ORF1 likely evolved from a canonical single jelly-roll (SJR) CP gene typical of eukaryote-infecting viruses in the phylum Cressdnaviricota (Krupovic et al. 2020), the most recent common ancestor of all known anelloviruses apparently lost the Rep-encoding gene.
Materials and methods
Sampling, identification, and sequencing of the anellovirus genomes
A single fecal sample from a grizzly bear (U. arctos horribilis) was collected in 2016 in Wyoming state (USA). Approximately 5 g of the sample was resuspended in 20 ml of SM buffer (G-Biosciences, USA) (0.1 M NaCl, 50 mM Tris/HCl—pH 7.4, and 10 mM MgSO4) buffer and centrifuged at 10,000 × g for 10 min. The supernatant was sequentially filtered through 0.45- and 0.2-µm syringe filters, and viral particles in the filtrate were precipitated with 15 per cent w/v PEG 8000 (Sigma, USA). The resulting suspension was incubated overnight at 4°C to precipitate virions and then centrifuged at 6000 × g for 20 min. The supernatant was discarded, and the pellet was resuspended in 2 ml of SM buffer. From this suspension, 200 µl was used for viral DNA extraction using the High Pure Viral Nucleic Acid Kit (Roche Diagnostics, USA), and circular DNA molecules in the DNA extract were amplified using rolling-circle amplification (RCA) using the TempliPhi™ kit (GE Healthcare, USA). The RCA product was used to generate a library (TruSeq Nano DNA kit) for paired-end (2 × 100 base pair (bp)) sequencing on an Illumina 4000 sequencer at Macrogen Inc. (Korea).
Nasal and vaginal swabs from adult female Weddell seals (L. weddellii) were taken from sedated individuals during the Austral summer seasons of 2015/2016 (n = 39) and 2016/2017 (n = 40). These samples were collected under National Marine Fisheries Service Marine Mammal permit #17411, Antarctic Conservation Act permit #2014-003, and the University of Alaska Anchorage and the University of Alaska Fairbank’s Institutional Animal Care and Use Committee approvals #419971 and #854089. Following sampling, the vaginal and nasal swabs were stored at 4°C in UTM™ Viral Transport Media (Copan, USA). An aliquot of 1 ml of the transport media was filtered through a 0.2-µm syringe filter, and 200 µl of the filtrate was used for viral DNA extraction using the High Pure Viral Nucleic Acid Kit (Roche Diagnostics). Circular DNA molecules in the extract were amplified using RCA using the TempliPhi™ kit (GE Healthcare). For each of the two seasons, a 5 µl aliquot of the RCA product was pooled for nasal and vaginal swabs and used to generate four libraries (TruSeq Nano DNA kit) for paired-end (2 × 100 bp) sequencing on an Illumina 4000 sequencer at Macrogen Inc. (Korea).
The 2 × 100 bp raw reads from the five libraries were trimmed with Trimmomatic v0.39 (Bolger, Lohse, and Usadel 2014) and then de novo assembled using metaSPades v3.14 (Bankevich et al. 2012). The de novo assembled contigs (>1,000 nts) were checked for terminal redundancy to determine if they represent circular molecules. All contigs >1,000 nts were analyzed using BLASTx (Altschul et al., 1990) against a RefSeq viral protein database (RefSeq release 205). For all contigs that we identified as anellovirus-like, we assembled a dataset and designed sets of abutting primers (Supplementary Table S1) to recover complete genomes from each of the individual samples of the Weddell seal nasal and vaginal swabs and the grizzly bear fecal sample. The RCA products were used as templates with specific primer pairs to amplify the anellovirus genomes using Kapa HiFi Hotstart DNA polymerase with the following thermal cycling conditions: 95°C for 3 min; 25 cycles of 98°C for 20 s, 60°C for 15 s, 72°C for 2 min, and a final extension of 72°C for 3 min. The amplicons were resolved on a 0.7 per cent agarose gel stained with SYBR Safe (ThermoFisher, USA), and ∼2–2.5 kb size fragments were excised, gel purified, and cloned into pJET1.2 plasmid vector (ThermoFisher) and transformed into Escherichia coli XL1-blue competent cells. The resulting recombinant plasmids were Sanger sequenced by primer walking at Macrogen Inc. (South Korea).
ORFs in the cloned and Sanger sequenced genomes were identified using ORF finder tool in Geneious Prime 2022.1.1 (Biomatters Ltd, New Zealand) and manually checked by global alignments. The sequences have been deposited in GenBank under accession numbers OP629190–OP629445.
Pairwise identity analysis to determine species classification was performed using SDT v 1.2 (Muhire, Varsani, and Martin 2014).
Recombination analyses
The 246 sequences of the anelloviruses identified from Weddell seal samples in this study represent two anellovirus species Torque teno pinniped virus 8 (n = 160) and Torque teno pinniped virus 9 (n = 86), and thus, these were aligned together with other members of these two species using Multiple Alignment using Fast Fourier Transform (MAFFT) with auto option (Katoh and Standley 2013). Recombination in the aligned datasets was inferred using Recombination Detection Program 5 (RDP5) (Martin et al. 2021) with default settings. Sequences were auto-masked for optimal recombination detection, and only events detected with more than three different methods implemented in RDP5 coupled with phylogenetic support for recombination and a P-value of <0.05 were considered credible.
The ten anelloviruses derived from the grizzly bear feces are diverse (each representing a new species sharing <69 per cent ORF1 identity) and thus could not be reliably aligned and analyzed for recombination with other closely related anelloviruses.
Dataset of anellovirus proteins
Sequences representing all anellovirus species (n = 153) listed in the Virus Metadata Resource provided by the International Committee on Taxonomy of Viruses (ICTV) were downloaded from the National Center for Biotechnology Information. This reference dataset was supplemented with the newly sequenced anelloviruses from Weddell seals (n = 246) and grizzly bears (n = 10) (Supplementary Tables S2 and 3). Three main ORFs, ORF1-3, and hypothetical ORFs from Weddell seal anelloviruses (WSAs) were extracted from the annotated sequence files in Geneious Prime 2022.1.1 (Biomatters Ltd) and used in the subsequent analyses.
Phylogenetic analysis
For taxonomic and phylogenetic analyses, the ORF1 protein sequences of representative anelloviruses (one per species) together with the 256 from this study were clustered to 80 per cent identity with CD-HIT (Li and Godzik 2006) and aligned using MAFFT with G-INS-i option (Katoh and Standley 2013). The alignment was trimmed with TrimAL (Capella-Gutierrez, Silla-Martinez, and Gabaldon 2009) with gap = 0.2 option, and the resulting alignment was used to infer a maximum-likelihood phylogenetic tree with PhyML 3.0 (Guindon et al. 2010) with VT+G+ F as the amino acid substitution model determined using ProtTest (Darriba et al. 2011) and SH-like branch support. The tree was annotated and visualized in iTOL (Letunic and Bork 2021).
The sizes of anellovirus genomes and projection domains were mapped onto a tree of their respective host species obtained from TimeTree of Life (Kumar et al. 2022).
Structural modeling and similarity search
For representatives of each anellovirus genus, including the new WSA and grizzly bear anellovirus (GBA), protein sequences of ORFs 1, 2, or 3 were used as inputs for AlphaFold2 (version 2.1.1; Jumper et al. 2021) structural prediction. The quality of generated structural models was assessed using the local distance difference test (Mariani et al. 2013) (Supplementary Fig. 1). All ORF1 models generated in this study are provided in Supplementary Data File 1.
The predicted structures of ORFs 1, 2, and 3 were used as inputs for the DALI (Holm 2020) server, and the structural similarity was evaluated based on the DALI Z score, where scores above 2 are considered potentially significant (Holm 2020). For ORF1 homologs with large spike insertions, the jelly-roll and spike domains were split, and DALI searches were run separately. Only the top 20 hits against the Protein Data Bank (PDB50) database were extracted. To build the structure-based dendrogram, structures of ORF1 and their top DALI hits were used in an all-against-all comparison, and an average linkage clustering was performed via DALI.
The protein sequences representing each anellovirus genus were used as queries in profile–profile comparisons with HHsearch (Steinegger et al. 2019) against the PDB70 and viral protein (UniProt-SwissProt-viral70_3_Nov_2021) profile databases (Gabler et al. 2020).
Search for Rep protein motifs
To search for the potential anellovirus Rep homologs, all six possible frames were in silico translated from representative anellovirus genomes using Prodigal (Hyatt et al. 2010) in meta mode and five different translation tables: standard code; vertebrate mitochondrial code; yeast mitochondrial code; invertebrate mitochondrial code, and bacterial, archaeal, and plant plastid code. These resulting amino acid sequences were used as input for InterProScan (Jones et al. 2014) against all available databases with default settings.
Results and discussion
New anelloviruses from Weddell seals and grizzly bears
The newly sequenced genomes of WSAs and GBAs range from ∼2,000 to 2,600 nts in length and are on the smaller side of the anellovirus genome size distribution (Fig. 1A). The WSA genomes display uniform sizes similar to those of viruses in the genus Lambdatorquevirus (∼2,150 nts; Fig. 1A), with the exception of WSA isolate N1637_a (OP629327), which has a considerably shorter genome of 1,976 nts, with a 210-nt region in the ORF1 (5′ region) missing. The latter sequence might therefore be a subgenomic molecule. By contrast, the GBA genomes are more variable, ranging from 1,965 to 2,557 nts (Fig. 1A, B). The WSA and GBA genomes display an organization similar to that of other anelloviruses, with one large (∼1,200 nts) and several shorter (∼200 nts) ORFs, three ORFs in the WSA genome sequences, and two in the GBA genome sequences (Fig. 1B). Similar to other anelloviruses, the WSA and GBA genomes contain a GC-rich sequence in the intergenic region (Fig. 1B) that likely plays a role in genome replication.
Figure 1.
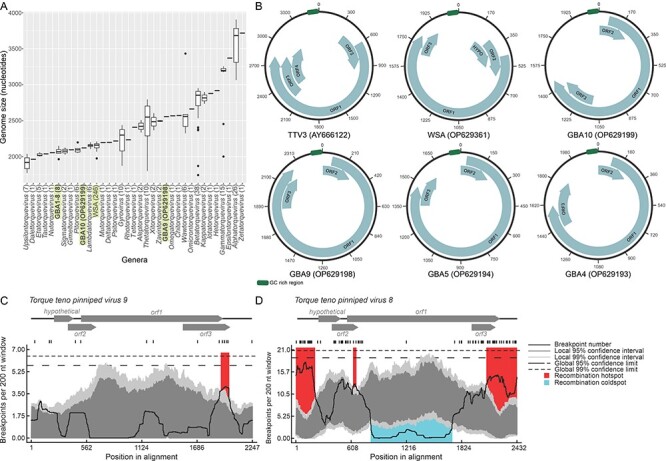
Genomic characteristics of new anelloviruses. (A) A barplot of genome sizes per anellovirus genera. The number of genomes in each category is provided in parentheses. (B) Genome maps of a prototypical anellovirus, TTV3, as well as a representative WSA and four grizzly bear anelloviruses. (C–D) The recombination analysis within the anellovirus species Torque teno pinniped virus 9 (C) and Torque teno pinniped virus 8 (D), with breakpoint hotspots shown in red and cold spots in blue. The dark gray and light gray areas of the plots indicate 95 and 99 per cent confidence intervals, respectively. Detectable breakpoint positions are shown with vertical lines above the graphs. The thick black line is the plot of the number of breakpoints detected within the 200-nt window region, and the window was moved along each of the represented alignments 1 nt at a time.
Maximum-likelihood phylogenetic analysis based on the ORF1 amino acid sequences from the anelloviruses representing each of the established species from the 31 genera and the new anelloviruses sequenced in this study (n = 409; Supplementary Files 1 and 2) largely recapitulated the established ICTV taxonomy (Fig. 2). Based on ORF1 nt pairwise identities and phylogenetic analysis, the WSA sequences belong to two species Torque teno pinniped virus 8 (n = 160) and Torque teno pinniped virus 9 (n = 86) in the genus Lambdatorquevirus. Both these species include anelloviruses that have been previously sampled only from Weddell seals (Fahsbender et al. 2017). By contrast, the GBA sequences belong to the Pitorquevirus (n = 8) and Omicrontorquevirus (n = 1) genera, and one is likely a member of a new genus. In particular, seven genomes, GBA-1, -2, -3, -4, -6, -7, and -8 (OP629190, OP629191, OP629192, OP629193, OP629195, OP629196, and OP629197), form a clade that is a sister group to other classified pitorqueviruses, all of which were sampled from giant pandas (Ailuropoda melanoleuca) (Zhang et al. 2017). The GBA-10 sequence (OP629199) branches separately from viruses in the established genera and hence can be considered a founding member of a new genus, which we propose to name ‘Ayintorquevirus’ (Fig. 2). Based on the pairwise identities of the ORF1s of the GBAs, which share <69 per cent identity with those of other anelloviruses, each of these represents a new species.
Figure 2.

The maximum-likelihood phylogenetic tree of ORF1 protein sequences of anelloviruses representing each of the established anellovirus species and those from the newly sequenced Weddell seal and grizzly bear anelloviruses. Branches representing species of particular genera to which the ORF1s of the anelloviruses identified in this study are not part of have been collapsed. In cases where there is only one species in a genus, the genus and the species names (with exemplar sequence accession number) are provided. The bold taxa names represent the ORF1s of the anelloviruses from this study, and the genera they belong to have been color coded. Branches with >0.8 SH-like support are shown.
In viruses of the species Torque teno pinniped virus 9, there is a recombination hotspot in the 3′ region of the orf1 and orf3 genes, which is similar to that reported previously (Fahsbender et al. 2017) (Fig. 1C). Notably, an equivalent hotspot is also present in viruses of the species Torque teno pinniped virus 8, where it not only encompasses the 3′ of the orf1 and orf3 genes but extends to the entire non-coding intergenic region. An additional small hotspot was detected at the 3ʹ end of orf2 and 5ʹend of orf1 genes (Fig. 1D, Supplementary Table 4). Conversely, the middle of the orf1 gene, spanning ∼800–1,700 nts of the genome alignment, is a recombination cold spot (Fig. 1D). These observations strongly suggest that the termini of orf1 as well as the non-coding region can be exchanged through recombination between anellovirus genomes, whereas the middle region is protected from such exchanges, possibly, due to strong purifying selection on the protein structure.
Anellovirus ORF1 is a homolog of SJR CPs
To analyze the relationship between anelloviruses and other viruses, we performed BLASTp searches against the viral RefSeq database queried with ORF1 protein sequences representing each of the Anelloviridae genera. No significant matches were obtained outside of the Anelloviridae. Thus, we sought to gain insights into the provenance of anelloviruses through more sensitive profile–profile comparisons. Searches queried with the ORF1 sequence of torque teno virus 1 (TTV1; NP_817122) against the PDB and viral protein profile databases using HHsearch yielded a highly significant hit (probability = 95.8) to the CP of a bat circovirus (PDB id: 6RPO; Supplementary Table S5). Importantly, the aligned regions encompassed nearly the entirety of the circovirus CP but only ∼30 per cent of the anellovirus protein, highlighting the disparity in their respective sizes (233 versus 770 aa). Significant hits were also obtained with ORF1 sequences from viruses of other anellovirus genera (Supplementary Table S5). Notably, however, not all anellovirus ORF1 sequences yielded global alignments. For instance, a search queried with the more compact ORF1 of chicken anemia virus (449 aa) retrieved only a match to the N-terminal half of the circovirus CP, indicating a higher divergence between these proteins.
Circovirus CPs contain an N-terminal arginine-rich region involved in DNA binding (Erker et al. 1999; Sarker et al. 2016) and a jelly-roll domain comprising eight antiparallel β-strands (B through I), which form two juxtaposed β-sheets, BIDG and CHEF (Fig. 3A) (Sarker et al. 2016; Khayat et al. 2019). Structurally similar CPs with the SJR fold are encoded by a vast diversity of eukaryote-infecting ssDNA and ssRNA viruses with icosahedral capsids (Rossmann and Johnson 1989; Krupovic and Koonin 2017). Notably, circovirus CPs have one of the most compact SJR cores, with no structural elaborations, such as long loops connecting the β-strands, additional secondary structure elements, or even distinct domains that are occasionally found to be inserted into CPs of other viruses (Wery et al. 1994; Wada et al. 2008; Kazlauskas et al. 2017; Munke et al. 2022).
Figure 3.

The structure of the circovirus jelly-roll CP and structural model of anellovirus ORF1. (A) CP of bat circovirus (BCV, family Circoviridae; PDB id: 6RPK) colored using the rainbow scheme from blue N-terminus to red C-terminus. The insert shows the schematic topology of the jelly-roll fold. The eight β-strands are labeled from B to I and form two antiparallel β-sheets BIDG and CHEF. The location of the projection (P)-domain is shown as a gray rectangle. (B) The structural model of ORF1 from TTV 21 (genus Alphatorquevirus; GenBank accession number: AAK27697). The jelly-roll domain shared with circoviruses is colored using the rainbow scheme, whereas the P-domain is shown in gray. (C) The superposed structures of the BCV (green) and TTV21 (gray) structures.
The anellovirus ORF1 protein also contains an arginine-rich N-terminal region similar to that of circovirus CPs. To verify to what extent the structural similarity extends to the SJR domain, representative ORF1 protein sequences from each of the established Anelloviridae genera as well as those of the newly sequenced WSA and GBA genomes were modeled with AlphaFold2 (Jumper et al. 2021). Inspection of the generated models confirmed that all ORF1 protein homologs contained the conserved SJR domain (Fig. 3B, C, Supplementary Fig. 2). Indeed, DALI (Holm 2020) searches against the PDB database queried with the predicted jelly-roll domains of different ORF1 protein orthologs consistently yielded best hits, with significant Z-scores (Z = 6.0–9.6), to the CPs of eukaryotic ssDNA and ssRNA viruses from the families Circoviridae, Hepeviridae, Astroviridae, Marnaviridae, Nodaviridae, and Dicistroviridae, and the genus Papanivirus (Supplementary Table S6). Collectively, the results of profile–profile and structure comparisons indicate that the anellovirus ORF1 protein is homologous to the jelly-roll CPs of other eukaryote-infecting ssDNA viruses and, in particular, to the CPs of circoviruses.
We also modeled the structures of the anellovirus ORF2 and ORF3 as well as hypothetical proteins. However, in all cases, these short proteins yielded structures with generic, simple folds, such as helix-turn-helix, which gave no useful information on their provenance (Supplementary Fig. S3).
Structural dissection of the anellovirus ORF1 proteins
Anellovirus ORF1 proteins display a remarkable variation in length and sequence. To verify whether these proteins are orthologous, we performed an all-against-all structural comparison of the ORF1 homologs from different Anelloviridae genera and CPs of other viruses identified as the top hits in the DALI searches.
This analysis showed that all anelloviruses form a compact, arguably, monophyletic group (Fig. 4A), suggesting that the dramatic evolution observed among the ORF1 sequences took place following their divergence from a common ancestor. To reconstruct the gradual evolution of the ORF1 structure, we superposed the structures of ORF1 homologs from different genera with the compact CP of a circovirus (PDB id: 6RPK; Fig. 4B). This comparison allowed further delineating the borders of the anellovirus jelly-roll domain and exposed the differences compared to the SJR CP of circoviruses. In particular, most ORF1 proteins contain two prominent insertions. One is located between β-strands B and C (Fig. 4B) and, depending on the virus, can adopt an extended loop, α-helix or β-hairpin conformation. The other insertion is located within a loop connecting the β-strands H and I. The H-I insertion varies substantially in both size and predicted structure, from an extended loop to an elaborate α + β domain (Fig. 4B, C). Structural searches queried with the latter domain did not produce significant matches against the PDB database, suggesting that this fold is so far unique to anelloviruses. Notably, the relative orientation of the α + β domain with respect to the jelly-roll domain is similar to the ‘projection’ (P) domains present in the CPs of a variety of ssDNA and ssRNA viruses, such as bacilladnaviruses, tombusviruses, nodaviruses, caliciviruses, and several other groups of viruses (Wery et al. 1994; Wada et al. 2008; Kazlauskas et al. 2017; Munke et al. 2022). In all these viruses, the projection domains face away from the capsid surface and, at least in some cases, are implicated in host recognition and binding (Tan, Hegde, and Jiang 2004). This prediction is confirmed by the recent cryogenic electron microscopy structure of the betatorquevirus LY1 (Liou et al. 2022). It is plausible, therefore, that the P-domains of anellovirus ORF1 proteins play a similar role, despite the absence of sequence or structural similarity to the equivalent domains found in the CPs of other viruses. Consistent with the role of the P-domain in host recognition/interaction, mapping of the sequence conservation (Shannon diversity) on the ORF1 structure revealed a high variability in the outermost region of the P-domain. Thus, based on sequence divergence, the P-domain can be subdivided into the SJR-proximal P1 and the SJR-distal, highly divergent P2 subdomains. The latter region has been previously noted to be hypervariable in anelloviruses (Nishizawa et al. 1999; Jelcic et al. 2004; Arze et al. 2021; Kaczorowska et al. 2023), and our findings provide a biological rationale for its high evolution rate. Notably, the same region was identified in our analysis as a recombinational cold spot in Torque teno pinniped virus 8 (Fig. 1D), suggesting that the P-domain primarily evolves through substitutions rather than recombination, which is likely to be suppressed by natural selection, conceivably, due to the selection for the preservation of structurally important amino acid interactions in this domain. Unlike the P-domain, and especially, the P2 subdomain, which appears to co-evolve with the host factors following Red Queen dynamics, the evolution of the SJR domain is constrained by its likely role in capsid shell formation and is accordingly considerably slower (Fig. 5A, B). A similar distribution of conservation across the SJR and P-domains has been also observed for other ssDNA viruses, e.g. ‘cruciviruses’ (Roux et al. 2013; Quaiser et al. 2016; de la Higuera I et al. 2020).
Figure 4.

The evolution of the anellovirus CP by the projection domain augmentation. (A) The average linkage dendrogram of anellovirus ORF1 structures and SJR CP homologs obtained from the DALI server. The homologous CP branches are marked in blue, and the anellovirus ORF1 structures are marked in green. (B) Examples of ORF1 structures with the circled projection domains and an arrow indicating the structure between strands B and C are also shown. The stars mark each structure from the newly sequenced anelloviruses in the dendrogram. The models are colored using the rainbow scheme, from blue N-terminus to red C-terminus. (C) The plot of spike domain sizes from each anellovirus genus. The colors mark the host of the viruses.
Figure 5.

The conservation of the jelly-roll and extensive divergence of the projection domain in the anellovirus CPs. (A) The structural model of TTV 21 ORF1 (AAK27697). Different regions of ORF1 are highlighted using different colors. (B) Shannon conservation index plotted for each amino acid position in the ORF1 alignment. High lines mark high conservation and low lines mark low conservation. Domains of the ORF1 structure as in A are marked. (c) Correlation plots of genome sizes, ORF1 sizes, and P-domain sizes. The black dots correspond to median values per genus.
Evolution of ORF1 through gradual augmentation of the P-domain
There is a notable correlation between the anellovirus genome size, ORF1 length, and the size of the P-domain (R2 = 0.92 for ORF1-P-domain size and >0.5 for the rest of the combinations; P-value <2.2e-16 for ORF1-P-domain size and <1e-06 for the rest of the combinations; and Spearman rank correlation coefficient = 0.97 for ORF1-P-domain size and >0.72 for the rest of the combinations). These correlations indicate that the increase in the genome size is almost exclusively attributable to elaborations within the P-domain (Fig. 5C). Based on the size of the P-domain, anelloviruses can be classified into seven groups (Fig. 4C). Group 1 includes anelloviruses with the most compact ORF1 proteins, among which members of the genus Gyrovirus, the only group of anelloviruses known to infect birds, have the smallest ORF1. The ORF1 proteins of gyroviruses, such as chicken anemia virus, lack the P-domain, and instead, the β-strands of the SJR core are connected through extended loops, which occasionally contain short α-helices. In the structure-based tree, gyrovirus ORF1 forms a sister group to mammalian anelloviruses (Fig. 4A), suggesting an early divergence. Among the mammalian anelloviruses, one of the newly sequenced GBA, GBA-10 (OP629199), and members of the genus Etatorquevirus, which are most similar to members of the genus Gyrovirus in Anelloviridae family, also lack an elaborate P-domain, and instead, β-strands H and I are connected through an extended loop with a short α-helix. Extensive loops connecting the β-strands of the SJR domain are also typical of viruses with linear ssDNA genome in the family Parvoviridae, such as adeno-associated virus and canine parvovirus (de Villiers and Hausen 2009; Grekova et al. 2012). However, unlike in anelloviruses, the extended loops in parvovirus CPs are located between β-strands G and H.
The predicted ORF1 structures in the six other anellovirus groups display a gradual complexification of the P-domain through (1) increase in the α-helical content within the H-I loop in Groups 2 and 3, which can be considered as the emergence of the P1 subdomain; (2) appearance of a four-stranded β-sheet in Group 4; (3) acquisition of additional β-sheets in Groups 5 and 6; (4) further proliferation of the unstructured loops and secondary structure elements in the SJR-distal region of the P1 subdomain in Group 6, signifying the emergence of the P2 subdomain; and (5) in Group 7, the P2 subdomain reaches the peak of its complexity, becoming more structured, with an increased α-helical content. The existence of such a broad range of structural complexity in ORF1 proteins of contemporary anelloviruses appears surprising, and its biological implications, in particular, for virus–host interactions, remain to be explored. Furthermore, it would be interesting to determine whether the increasingly augmented P-domains represent intermediate evolutionary steps, or these elaborations evolved independently in different lineages.
Seven of the ten anellovirus genera with the largest P-domains (ORF1 Groups 6 and 7) include viruses infecting primates (Alpha-, Beta-, Gamma-, Epsilon-, Zeta-, Omega-, and Hetorquevirus genera), whereas the remaining three genera, all in Group 6, are associated with bears and pigs (Omicron- and Kappatorquevirus, respectively) (Fig. 4C). Viruses from the remaining 20 Anelloviridae genera showed no recognizable patterns of clustering based on the P-domain size and the host. To further explore this relationship, we mapped the anellovirus genome and P-domain sizes onto a phylogenetic tree of their respective hosts derived from the TimeTree of Life website (http://www.timetree.org/). This analysis further highlighted that primate-infecting anelloviruses have considerably larger genomes and P-domains compared to any other group of hosts (Supplementary Fig. S4). A distinguishing feature of ORF1 proteins in Groups 6 and 7 is the presence of the P2 subdomain, suggesting that it might play an important role in the evasion of the primate immune response and/or the recognition of primate-specific host receptors. What differences between the immune systems of primates and those of other mammals might be driving this difference remains unclear.
Outstanding questions about the anellovirus genome replication
As mentioned earlier, it has been previously reported that the ORF1 protein of betatorqueviruses contains conserved motifs indicative of it potentially being related to the rolling-circle replication initiation endonucleases of the HUH superfamily (Biagini et al. 2001; Umemura et al. 2002; Huang et al. 2010). However, these motifs were subsequently not found to be conserved throughout the family (Rosario, Duffy, and Breitbart 2012). The structural model of ORF1 from torque teno mini virus 1 (Betatorquevirus) allows us to reevaluate the relevance and validity of the ‘Rep motifs’ in the structural context. The HUH superfamily Rep proteins have the RNA-recognition-motif (RRM) fold, similar to the Palm domain of RNA and DNA polymerases (Umemura et al. 2002), with the conserved Motifs I and II being located within Strands β1 and β3, respectively, and the invariable catalytic tyrosine residue of Motif III located within an α-helix (Krupovic, Dolja, and Koonin 2019; Tarasova and Khayat 2021). It is obvious from our structural model that no part of the anellovirus ORF1 adopts the RRM fold. Furthermore, the putative Rep motifs are located within the P-domain, with ‘Motifs I and II’ mapping to spatially separated loops and ‘Motif III’ residing within a β-strand (Supplementary Fig. S5). Thus, beyond any reasonable doubt, the ‘RCR motifs’ in ORF1 do not signify ORF1 homology to HUH Reps of other ssDNA viruses and therefore cannot play equivalent roles to those of the HUH Reps during viral genome replication. Other proteins encoded by anelloviruses, even when alternative coding tables are used for in silico protein translation, did not show any similarity to known replication proteins either.
Thus, the question of how anelloviruses replicate their genomes remains open and enigmatic. In particular, it is unclear which viral and/or cellular factors are involved in the initiation of anellovirus replication. One possibility is that, similar to satellite viruses (Krupovic, Kuhn, and Fischer 2016), anelloviruses depend for replication on a coinfecting virus, which would provide the Rep protein in trans. Indeed, it has been shown that TTV-HD14 genome replication was enhanced during coinfection with Epstein–Barr virus (Othoherpesviridae) or in a cell line expressing the large T-antigen of simian virus 40 (Polyomaviridae) (de Villiers Em et al. 2011; Borkosky et al. 2012). However, neither orthoherpesviruses nor polyomaviruses produce ss replication intermediates, suggesting that the positive effect of these viruses on anellovirus replication was indirect. Furthermore, the importance of a helper virus appears unlikely, given the recent breakthrough in achieving anellovirus replication and virion formation in vitro in the T cell–derived human cell line MOLT-4 (Nawandar et al. 2022). Transfection of the MOLT-4 cells with a plasmid comprising a tandem anellovirus genome yielded unit-length, replicated anellovirus genomes, which were packaged into isometric virus particles. This result indicates that all factors required for genome replication, its conversion into an ssDNA form, and packaging are provided by the virus and/or the host. Alternatively, the Rep function could be provided in trans by the rolling-circle transposons, such as Helitrons (Kapitonov and Jurka 2001) or their fish relatives Heletrons (Poulter, Goodwin, and Butler 2003). However, although functional helitrons are found in bats (Kosek et al. 2021), no active copies have been described in any primates. Previous studies have documented the accumulation of subgenomic anellovirus DNA molecules smaller than the size of the wild-type virus genome, which could be by-products of recombination-dependent replication (Leppik et al. 2007; de Villiers Em et al. 2011), a replication mechanism alternative to RCR that has been described in circular ssDNA viruses in the family Geminiviridae (Jeske 2001; Preiss and Jeske 2003; Alberter, Ali Rezaian, and Jeske 2005; Jovel, Preiß, and Jeske 2007; Erdmann et al. 2010). In this mode of replication, the host DNA polymerase produces a covalently closed double-stranded circular form of the viral genome and then generates long concatemers using this dsDNA as a template. The linear genome concatemers produced from the template are then resolved as double-stranded circular molecules by homologous recombination. Thus, conceivably, anelloviruses could rely exclusively on recombination-dependent replication. However, how the dsDNA intermediates would be converted into the ssDNA mature genomes that are eventually packaged is unclear. The availability of the transfection system for anelloviruses will be instrumental in elucidating the mechanisms of anellovirus genome replication.
Concluding remarks
Here, we explored the evolution of anelloviruses through a comparative structural analysis of their ORF1 proteins. We showed that ORF1, the signature protein of anelloviruses, has the jelly-roll fold, the most common fold in the CPs of viruses with small icosahedral capsids. While this manuscript was in preparation, a preprint describing the structure of ORF1 from the human betatorquevirus LY1 was posted online (Liou et al. 2022). Although the structural model was not publicly available at the time of this writing, precluding direct comparison, the experimentally determined structure appears to be closely similar to the betatorquevirus ORF1 models generated herein using AlphaFold2. There is extensive variability in the overall size of ORF1 protein of anelloviruses belonging to different genera, with the jelly-roll domain being highly conserved, consistent with its key role in virus particle formation, whereas the P-domain is highly variable. Systematic comparison of ORF1 protein structures across the family Anelloviridae suggests that anellovirus ORF1 protein evolved from an ancestral virus with a more compact CP, through incremental augmentation and complexification of the ORF1 protein structure. Sensitive profile–profile comparisons have shown that anellovirus ORF1 proteins are homologous to the CPs of circoviruses, currently the only other known group of viruses with small circular ssDNA genomes that infect mammals (Krupovic et al. 2020, 2020). Given the compact structure of the circovirus CPs, the canonical circovirus genome organization, with the cp and rep genes, similar to that of other ssDNA viruses, it appears likely that anelloviruses evolved in the vertebrate hosts from a circovirus-like ancestor. However, whether this virus was a member of the extant family Circoviridae or was ancestral to both circoviruses and anelloviruses remains unclear due to the high sequence divergence. Under this scenario, the emergence of the anellovirus ancestor would entail a dramatic change in the genome replication strategy, accompanied by the loss of the rep gene, a feature that sets anelloviruses apart from all other viruses in the phylum Cressdnaviricota (Krupovic et al. 2020). Notably, bidnaviruses have apparently evolved along a similar evolutionary path from parvoviruses (both classified in the phylum Cossaviricota) by replacing the HUH superfamily Rep with a protein-primed family B DNA polymerase (Krupovic and Koonin 2014). An alternative scenario that could be consistent with the available data involves the transfer of the capsid gene from a circovirus-like ancestor to a distinct replicon that replicated without the involvement of an HUH-Rep. However, such non-viral replicons related to anelloviruses are not known, so that, in the absence of understanding of the anellovirus genome replication mechanism as well as cellular or viral replication determinants, this scenario cannot be currently substantiated.
Due to the lack of identifiable relationship to other known ssDNA viruses, anelloviruses so far have not been considered as belonging to the realm Monodnaviria (Koonin et al. 2020). However, in light of our current results, anelloviruses arguably should be included in this realm, probably, as a separate new phylum within the kingdom Shotokuvirae, alongside Cressdnaviricota and Cossaviricota. Thus, we propose assigning Anelloviridae to the order ‘Sanitavirales’ (after Latin sanitas for health, referring to anelloviruses being part of the healthy human virome), within the class ‘Cardeaviricetes’ (after Cardea, the Roman goddess of health) and phylum Commensaviricota, referring to the commensal lifestyle of anelloviruses.
Supplementary Material
Funding
A.B. was supported by a post-doctoral fellowship from Foundation pour la Recherche Mèdicale (grant number SPF202110014092). M.K. was supported by a grant from the l’Agence Nationale de la Recherche (ANR-20-CE20-0009-02). A.L.K. and R.S.B. were supported by Institutional Development Awards (IDeA) Networks of Biomedical Research Excellence Assistantships (grant number P20GM103395) from the National Institute of General Medical Sciences of the National Institutes of Health (NIH). E.V.K. was supported by the Intramural Research Program of the NIH (National Library of Medicine).
Weddell seal samples were collected under National Marine Fisheries Service Marine Mammal permit #17411, Antarctic Conservation Act permit #2014-003, and the University of Alaska Anchorage and University of Alaska Fairbanks’s Institutional Animal Care and Use Committee approvals #419971 and #854089, with funding from the National Science Foundation grant ANT-1246463 to J.M.B.
The molecular work described in this study is supported by the Center of Evolution and Medicine Venture Fund (Center of Evolution and Medicine, Arizona State University, USA) grant awarded to A.V.
The content is solely the responsibility of the authors and does not necessarily reflect the official views of the NIH. J.M.B. contributed to this work while serving at the National Science Foundation. Any opinion, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation. The findings and conclusions in this article are those of the authors and do not necessarily represent the views of the US Fish and Wildlife Service.
Collection of grizzly bear fecal samples was undertaken by Daniel Thompson and Kent Schmidlin at the Wyoming Game and Fish Department (Lander, Wyoming, USA) and the team at Trophy Game (USA).
Contributor Information
Anamarija Butkovic, Institut Pasteur, Université Paris Cité, CNRS UMR6047, Archaeal Virology Unit, 25 rue du Dr Roux, Paris 75015, France.
Simona Kraberger, The Biodesign Center for Fundamental and Applied Microbiomics, Center for Evolution and Medicine, School of Life Sciences, Arizona State University, 1001 S. McAllister Ave, Tempe, AZ 85287, USA.
Zoe Smeele, The Biodesign Center for Fundamental and Applied Microbiomics, Center for Evolution and Medicine, School of Life Sciences, Arizona State University, 1001 S. McAllister Ave, Tempe, AZ 85287, USA.
Darren P Martin, The Biodesign Center for Fundamental and Applied Microbiomics, Center for Evolution and Medicine, School of Life Sciences, Arizona State University, 1001 S. McAllister Ave, Tempe, AZ 85287, USA.
Kara Schmidlin, The Biodesign Center for Fundamental and Applied Microbiomics, Center for Evolution and Medicine, School of Life Sciences, Arizona State University, 1001 S. McAllister Ave, Tempe, AZ 85287, USA.
Rafaela S Fontenele, The Biodesign Center for Fundamental and Applied Microbiomics, Center for Evolution and Medicine, School of Life Sciences, Arizona State University, 1001 S. McAllister Ave, Tempe, AZ 85287, USA.
Michelle R Shero, Biology Department, Woods Hole Oceanographic Institution, 266 Woods Hole Rd, Woods Hole, MA 02543, USA.
Roxanne S Beltran, Department of Ecology and Evolutionary Biology, University of California Santa Cruz, 130 McAllister Way, Santa Cruz, CA 95060, USA.
Amy L Kirkham, U.S. Fish and Wildlife Service, Marine Mammals Management, 1011 E, Tudor Road, Anchorage, AK 99503, USA.
Maketalena Aleamotu’a, School of Environmental and Life Sciences, The University of Newcastle, University Drive, Callaghan, NSW 2308, Australia.
Jennifer M Burns, Department of Biological Sciences, Texas Tech University, 2500 Broadway, Lubbock, TX 79409, USA.
Eugene V Koonin, National Center for Biotechnology Information, National Library of Medicine, 8600 Rockville Pike, Bethesda, MD 20894, USA.
Arvind Varsani, The Biodesign Center for Fundamental and Applied Microbiomics, Center for Evolution and Medicine, School of Life Sciences, Arizona State University, 1001 S. McAllister Ave, Tempe, AZ 85287, USA; Computational Biology Division, Department of Integrative Biomedical Sciences, Institute of Infectious Diseases and Molecular Medicine, University of Cape Town, Observatory, 1 Anzio Road, Cape Town 7925, South Africa.
Mart Krupovic, Institut Pasteur, Université Paris Cité, CNRS UMR6047, Archaeal Virology Unit, 25 rue du Dr Roux, Paris 75015, France.
Supplementary data
Supplementary data are available at Virus Evolution online.
Conflict of interest:
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Alberter B., Ali Rezaian M., and Jeske H. (2005) ‘Replicative Intermediates of Tomato Leaf Curl Virus and Its Satellite DNAs’, Virology, 331: 441–8. [DOI] [PubMed] [Google Scholar]
- Altschul S. F. et al. (1990) ‘Basic Local Alignment Search Tool’, Journal of Molecular Biology, 215: 403–10. [DOI] [PubMed] [Google Scholar]
- Arze C. A. et al. (2021) ‘Global Genome Analysis Reveals a Vast and Dynamic Anellovirus Landscape within the Human Virome’, Cell Host & Microbe, 29: 1305–1315.e6. [DOI] [PubMed] [Google Scholar]
- Asabe S.-I. et al. (2001) ‘Phosphorylation of Serine-Rich Protein Encoded by Open Reading Frame 3 of the TT Virus Genome’, Biochemical and Biophysical Research Communications, 286: 298–304. [DOI] [PubMed] [Google Scholar]
- Bankevich A. et al. (2012) ‘SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing’, Journal of Computational Biology, 19: 455–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendinelli M. et al. (2001) ‘Molecular Properties, Biology, and Clinical Implications of TT Virus, a Recently Identified Widespread Infectious Agent of Humans’, Clinical Microbiology Reviews, 14: 98–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biagini P. (2009) ‘Classification of TTV and Related Viruses (Anelloviruses)’. In: de Villiers, E. M., and Hausen, H. Z. (eds.) TT Viruses, pp. 21–33. Berlin Heidelberg: Springer Berlin, Heidelberg. [DOI] [PubMed] [Google Scholar]
- ——— et al. (2012) ‘Family Anelloviridae’, In: King Andrew M. Q., Adams M. J., Carstens E. B. and Lefkowitz E. J. (eds.) Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses, pp. 331–41. Elsevier. [Google Scholar]
- ——— et al. (2001) ‘Genetic Analysis of Full-Length Genomes and Subgenomic Sequences of TT Virus-Like Mini Virus Human Isolates’, Journal of General Virology, 82: 379–83. [DOI] [PubMed] [Google Scholar]
- Biagini P. et al. (2000) ‘Complete Sequences of Two Highly Divergent European Isolates of TT Virus’, Biochemical and Biophysical Research Communications, 271: 837–41. [DOI] [PubMed] [Google Scholar]
- Blatter J. A. et al. (2018) ‘Anellovirus Loads Are Associated with Outcomes in Pediatric Lung Transplantation’, Pediatric Transplantation, 22: e13069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger A. M., Lohse M., and Usadel B. (2014) ‘Trimmomatic: A Flexible Trimmer for Illumina Sequence Data’, Bioinformatics, 30: 2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkosky S. S. et al. (2012) ‘Epstein-Barr Virus Stimulates Torque Teno Virus Replication: A Possible Relationship to Multiple Sclerosis’, PLoS One, 7: e32160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camci C. et al. (2002) ‘The Prevalence of TT Virus in Cancer Patients.’, The New Microbiologica, 25: 463–8. [PubMed] [Google Scholar]
- Canuti M. et al. (2015) ‘Reduced Maternal Levels of Common Viruses during Pregnancy Predict Offspring Psychosis: Potential Role of Enhanced Maternal Immune Activity?’, Schizophrenia Research, 166: 248–54. [DOI] [PubMed] [Google Scholar]
- Capella-Gutierrez S., Silla-Martinez J. M., and Gabaldon T. (2009) ‘trimAl: A Tool for Automated Alignment Trimming in Large-Scale Phylogenetic Analyses’, Bioinformatics, 25: 1972–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane A. et al. (2018) ‘Novel Anelloviruses Identified in Buccal Swabs of Antarctic Fur Seals’, Virus Genes, 54: 719–23. [DOI] [PubMed] [Google Scholar]
- Darriba D. et al. (2011) ‘ProtTest 3: Fast Selection of Best-Fit Models of Protein Evolution’, Bioinformatics, 27: 1164–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Higuera I. et al. (2020) ‘Unveiling Crucivirus Diversity by Mining Metagenomic Data’, mBio, 11: e01410–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza W. M. et al. (2018) ‘Discovery of Novel Anelloviruses in Small Mammals Expands the Host Range and Diversity of the Anelloviridae’, Virology, 514: 9–17. [DOI] [PubMed] [Google Scholar]
- de Villiers E.M. et al. (2011) ‘The Diversity of Torque Teno Viruses: In Vitro Replication Leads to the Formation of Additional Replication-Competent Subviral Molecules’, Journal of Virology, 85: 7284–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Villiers, E. M., and Hausen, H. Z. (ed.) (2009) TT Viruses: The Still Elusive Human Pathogens. Berlin, Heidelberg: Springer Berlin Heidelberg. [Google Scholar]
- Erdmann J. B. et al. (2010) ‘Replicative Intermediates of Maize Streak Virus Found during Leaf Development’, Journal of General Virology, 91: 1077–81. [DOI] [PubMed] [Google Scholar]
- Erker J. C. et al. (1999) ‘Analyses of TT Virus Full-length Genomic Sequences.’, Journal of General Virology, 80: 1743–50. [DOI] [PubMed] [Google Scholar]
- Fahsbender E. et al. (2017) ‘Diverse and Highly Recombinant Anelloviruses Associated with Weddell Seals in Antarctica’, Virus Evolution, 3: vex017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatoba A. J., and Adeleke M. A. (2019) ‘Chicken Anemia Virus: A Deadly Pathogen of Poultry’, Acta Virologica, 63: 19–25. [DOI] [PubMed] [Google Scholar]
- Focosi D. et al. (2016) ‘Torquetenovirus: The Human Virome from Bench to Bedside’, Clinical Microbiology and Infection, 22: 589–93. [DOI] [PubMed] [Google Scholar]
- Gabler F. et al. (2020) ‘Protein Sequence Analysis Using the MPI Bioinformatics Toolkit’, Current Protocols in Bioinformatics, 72: e108. [DOI] [PubMed] [Google Scholar]
- Gerner P. et al. (2000) ‘Mother-to-infant Transmission of TT Virus: Prevalence, Extent and Mechanism of Vertical Transmission’, The Pediatric Infectious Disease Journal, 19: 1074–8. [DOI] [PubMed] [Google Scholar]
- Goto K. et al. (2000) ‘Detection Rates of TT Virus DNA in Serum of Umbilical Cord Blood, Breast Milk and Saliva’, The Tohoku Journal of Experimental Medicine, 191: 203–7. [DOI] [PubMed] [Google Scholar]
- Grekova S. P. et al. (2012) ‘Activation of a Glioma-Specific Immune Response by Oncolytic Parvovirus Minute Virus of Mice Infection’, Cancer Gene Therapy, 19: 468–75. [DOI] [PubMed] [Google Scholar]
- Guindon S. et al. (2010) ‘New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0’, Systematic Biology, 59: 307–21. [DOI] [PubMed] [Google Scholar]
- Hettmann A. et al. (2016) ‘Detection and Phylogenetic Analysis of Torque Teno Virus in Salivary and Tumor Biopsy Samples from Head and Neck Carcinoma Patients’, Intervirology, 59: 123–9. [DOI] [PubMed] [Google Scholar]
- Hijikata M., Takahashi K., and Mishiro S. (1999) ‘Complete Circular DNA Genome of a TT Virus Variant (Isolate Name SANBAN) and 44 Partial ORF2 Sequences Implicating a Great Degree of Diversity beyond Genotypes’, Virology, 260: 17–22. [DOI] [PubMed] [Google Scholar]
- Holm L. (2020) ‘Using Dali for Protein Structure Comparison’. In: Gáspári, Z. (ed.). Structural Bioinformatics, pp. 29–42, Methods in Molecular Biology, Vol. 2112. Springer US: New York, NY. [DOI] [PubMed] [Google Scholar]
- Huang Y. W. et al. (2010) ‘Multiple Infection of Porcine Torque Teno Virus in a Single Pig and Characterization of the Full-Length Genomic Sequences of Four U.S. Prototype PTTV Strains: Implication for Genotyping of PTTV’, Virology, 396: 289–97. [DOI] [PubMed] [Google Scholar]
- Hyatt D. et al. (2010) ‘Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification’, BMC Bioinformatics, 11: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inami T. et al. (2000) ‘High Prevalence of TT Virus DNA in Human Saliva and Semen’, Journal of Clinical Microbiology, 38: 2407–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y. et al. (2000) ‘Visualization of TT Virus Particles Recovered from the Sera and Feces of Infected Humans’, Biochemical and Biophysical Research Communications, 279: 718–24. [DOI] [PubMed] [Google Scholar]
- Jelcic I. et al. (2004) ‘Isolation of Multiple TT Virus Genotypes from Spleen Biopsy Tissue from a Hodgkin’s Disease Patient: Genome Reorganization and Diversity in the Hypervariable Region’, Journal of Virology, 78: 7498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeske H. (2001) ‘DNA Forms Indicate Rolling Circle and Recombination-Dependent Replication of Abutilon Mosaic Virus’, The EMBO Journal, 20: 6158–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P. et al. (2014) ‘InterProScan 5: Genome-Scale Protein Function Classification’, Bioinformatics, 30: 1236–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovel J., Preiß W., and Jeske H. (2007) ‘Characterization of DNA Intermediates of an Arising Geminivirus’, Virus Research, 130: 63–70. [DOI] [PubMed] [Google Scholar]
- Jumper J. et al. (2021) ‘Highly Accurate Protein Structure Prediction with AlphaFold’, Nature, 596: 583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczorowska J. et al. (2022a) ‘Early-Life Colonization by Anelloviruses in Infants’, Viruses, 14: 865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2022b) ‘Diversity and Long-Term Dynamics of Human Blood Anelloviruses’, Journal of Virology, 96: e00109–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2023) ‘Anellovirus Evolution during Long-Term Chronic Infection’, Virus Evolution, 9: vead001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczorowska J., and van der Hoek L. (2020) ‘Human Anelloviruses: Diverse, Omnipresent and Commensal Members of the Virome’, FEMS Microbiology Reviews, 44: 305–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakkola L. et al. (2009) ‘Replication of and Protein Synthesis by TT Viruses’. In: de Villiers, E.M., and Zur, H. H. (eds.) TT Viruses. Berlin, Heidelberg: Springer Berlin Heidelberg. pp. 53–64. [DOI] [PubMed] [Google Scholar]
- Kamahora T., Hino S., and Miyata H. (2000) ‘Three Spliced mRNAs of TT Virus Transcribed from a Plasmid Containing the Entire Genome in COS1 Cells’, Journal of Virology, 74: 9980–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitonov V. V., and Jurka J. (2001) ‘Rolling-Circle Transposons in Eukaryotes’, Proceedings of the National Academy of Sciences of the United States of America, 98: 8714–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., and Standley D. M. (2013) ‘MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability’, Molecular Biology and Evolution, 30: 772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazemi M. J. et al. (2015) ‘Association Between TT Virus Infection and Cirrhosis in Liver Transplant Patients’, Hepatitis Monthly, 15: e28370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskas D. et al. (2017) ‘Evolutionary History of ssDNA Bacilladnaviruses Features Horizontal Acquisition of the Capsid Gene from ssRNA Nodaviruses’, Virology, 504: 114–21. [DOI] [PubMed] [Google Scholar]
- ——— et al. (2019) ‘Multiple Origins of Prokaryotic and Eukaryotic Single-Stranded DNA Viruses from Bacterial and Archaeal Plasmids’, Nature Communications, 10: 3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khayat R. et al. (2019) ‘Structural Characterization of the PCV2d Virus-Like Particle at 3.3 Å Resolution Reveals Differences to PCV2a and PCV2b Capsids, a Tetranucleotide, and an N-terminus near the Icosahedral 3-Fold Axes’, Virology, 537: 186–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooistra K. et al. (2004) ‘TT Virus-Derived Apoptosis-Inducing Protein Induces Apoptosis Preferentially in Hepatocellular Carcinoma-Derived Cells’, Journal of General Virology, 85: 1445–50. [DOI] [PubMed] [Google Scholar]
- Koonin E. V. et al. (2020) ‘Global Organization and Proposed Megataxonomy of the Virus World’, Microbiology and Molecular Biology Reviews, 84: e00061–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin E. V., Dolja V. V., and Krupovic M. (2021) ‘The Healthy Human Virome: From Virus–Host Symbiosis to Disease’, Current Opinion in Virology, 47: 86–94. [DOI] [PubMed] [Google Scholar]
- Kosek D. et al. (2021) ‘The Large Bat Helitron DNA Transposase Forms a Compact Monomeric Assembly that Buries and Protects Its Covalently Bound 5′-Transposon End’, Molecular Cell, 81: 4271–4286.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraberger S. et al. (2021) ‘Taxonomic Updates for the Genus Gyrovirus (Family Anelloviridae): Recognition of Several New Members and Establishment of Species Demarcation Criteria’, Archives of Virology, 166: 2937–42. [DOI] [PubMed] [Google Scholar]
- Krupovic M. et al. (2020) ‘Cressdnaviricota : A Virus Phylum Unifying Seven Families of Rep-Encoding Viruses with Single-Stranded, Circular DNA Genomes’, Journal of Virology, 94: e00582–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupovic M., Dolja V. V., and Koonin E. V. (2019) ‘Origin of Viruses: Primordial Replicators Recruiting Capsids from Hosts’, Nature Reviews. Microbiology, 17: 449–58. [DOI] [PubMed] [Google Scholar]
- Krupovic M., and Koonin E. V. (2014) ‘Evolution of Eukaryotic Single-Stranded DNA Viruses of the Bidnaviridae Family from Genes of Four Other Groups of Widely Different Viruses’, Scientific Reports, 4: 5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— (2017) ‘Multiple Origins of Viral Capsid Proteins from Cellular Ancestors’, Proceedings of the National Academy of Sciences, 114: E2401–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupovic M., Kuhn J. H., and Fischer M. G. (2016) ‘A Classification System for Virophages and Satellite Viruses’, Archives of Virology, 161: 233–47. [DOI] [PubMed] [Google Scholar]
- Kumar S. et al. (2022) ‘TimeTree 5: An Expanded Resource for Species Divergence Times’, Molecular Biology and Evolution, 39: msac174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppik L. et al. (2007) ‘In Vivo and In Vitro Intragenomic Rearrangement of TT Viruses’, Journal of Virology, 81: 9346–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I., and Bork P. (2021) ‘Interactive Tree of Life (Itol) V5: An Online Tool for Phylogenetic Tree Display and Annotation’, Nucleic Acids Research, 49: W293–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., and Godzik A. (2006) ‘Cd-hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences’, Bioinformatics, 22: 1658–9. [DOI] [PubMed] [Google Scholar]
- Liou S. H. et al. (2022) ‘Anellovirus Structure Reveals a Mechanism for Immune Evasion’, bioRxiv <http://biorxiv.org/lookup/doi/10.1101/2022.07.01.498313>. [Google Scholar]
- Madsen C.D. et al. (2002) ‘TTV Viral Load as a Marker for Immune Reconstitution after Initiation of HAART in HIV-Infected Patients’, HIV Clinical Trials, 3: 287–95. [DOI] [PubMed] [Google Scholar]
- Maggi F. et al. (2001) ‘Dynamics of Persistent TT Virus Infection, as Determined in Patients Treated with Alpha Interferon for Concomitant Hepatitis C Virus Infection’, Journal of Virology, 75: 11999–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani V. et al. (2013) ‘lDDT: A Local Superposition-Free Score for Comparing Protein Structures and Models Using Distance Difference Tests’, Bioinformatics, 29: 2722–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D. P. et al. (2021) ‘RDP5: A Computer Program for Analyzing Recombination in, and Removing Signals of Recombination From, Nucleotide Sequence Datasets’, Virus Evolution, 7: veaa087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElvania TeKippe E. et al. (2012) ‘Increased Prevalence of Anellovirus in Pediatric Patients with Fever’’, PLoS One, 7: e50937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata H. et al. (1999) ‘Identification of a Novel GC-Rich 113-Nucleotide Region to Complete the Circular, Single-Stranded DNA Genome of TT Virus, the First Human Circovirus’, Journal of Virology, 73: 3582–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhire B. M., Varsani A., and Martin D. P. (2014) ‘SDT: A Virus Classification Tool Based on Pairwise Sequence Alignment and Identity Calculation’, PLoS One, 9: e108277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munke A. et al. (2022) ‘Primordial Capsid and Spooled ssDNA Genome Structures Unravel Ancestral Events of Eukaryotic Viruses’, mBio, 13: e0015622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mushahwar I. K. et al. (1999) ‘Molecular and Biophysical Characterization of TT Virus: Evidence for a New Virus Family Infecting Humans’, Proceedings of the National Academy of Sciences, 96: 3177–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naganuma M. et al. (2008) ‘TT Virus Prevalence, Viral Loads and Genotypic Variability in Saliva from Healthy Japanese Children’, Acta Paediatrica, 97: 1686–90. [DOI] [PubMed] [Google Scholar]
- Nawandar D. M. et al. (2022) ‘Human Anelloviruses Produced by Recombinant Expression of Synthetic Genomes’, bioRxiv <http://biorxiv.org/lookup/doi/10.1101/2022.04.28.489885>. [Google Scholar]
- Nishizawa T. et al. (1999) ‘Quasispecies of TT Virus (TTV) with Sequence Divergence in Hypervariable Regions of the Capsid Protein in Chronic TTV Infection’, Journal of Virology, 73: 9604–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto H. (2009) ‘TT Viruses in Animals’. In: de Villiers, E. M., and Hausen, H. Z. (eds). TT Viruses, pp. 35–52, Springer Berlin Heidelberg: Berlin, Heidelberg. [Google Scholar]
- ——— et al. (2000) ‘TT Virus mRNAs Detected in the Bone Marrow Cells from an Infected Individual’, Biochemical and Biophysical Research Communications, 279: 700–7. [DOI] [PubMed] [Google Scholar]
- Peters M. A. et al. (2002) ‘Chicken Anemia Virus VP2 Is a Novel Dual Specificity Protein Phosphatase’, Journal of Biological Chemistry, 277: 39566–73. [DOI] [PubMed] [Google Scholar]
- Poulter R. T. M., Goodwin T. J. D., and Butler M. I. (2003) ‘Vertebrate Helentrons and Other Novel Helitrons’, Gene, 313: 201–12. [DOI] [PubMed] [Google Scholar]
- Prasetyo A. A. et al. (2009) ‘Replication of Chicken Anemia Virus (CAV) Requires Apoptin and Is Complemented by VP3 of Human Torque Teno Virus (TTV)’, Virology, 385: 85–92. [DOI] [PubMed] [Google Scholar]
- Preiss W., and Jeske H. (2003) ‘Multitasking in Replication Is Common among Geminiviruses’, Journal of Virology, 77: 2972–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J. et al. (2005) ‘Human Circovirus TT Virus Genotype 6 Expresses Six Proteins following Transfection of a Full-Length Clone’, Journal of Virology, 79: 6505–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaiser A. et al. (2016) ‘Diversity and Comparative Genomics of Chimeric Viruses in Sphagnum- Dominated Peatlands’, Virus Evolution, 2: vew025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Requião R. D. et al. (2020) ‘Viruses with Different Genome Types Adopt a Similar Strategy to Pack Nucleic Acids Based on Positively Charged Protein Domains’, Scientific Reports, 10: 5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario K., Duffy S., and Breitbart M. (2012) ‘A Field Guide to Eukaryotic Circular Single-Stranded DNA Viruses: Insights Gained from Metagenomics’, Archives of Virology, 157: 1851–71. [DOI] [PubMed] [Google Scholar]
- Rossmann M. G., and Johnson J. E. (1989) ‘Icosahedral RNA Virus Structure’, Annual Review of Biochemistry, 58: 533–73. [DOI] [PubMed] [Google Scholar]
- Roux S. et al. (2013) ‘Chimeric Viruses Blur the Borders between the Major Groups of Eukaryotic Single-Stranded DNA Viruses’, Nature Communications, 4: 2700. [DOI] [PubMed] [Google Scholar]
- Sarker S. et al. (2016) ‘Structural Insights into the Assembly and Regulation of Distinct Viral Capsid Complexes’, Nature Communications, 7: 13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P., and Ramamoorthy S. (2016) ‘Lack of Strong Anti-Viral Immune Gene Stimulation in Torque Teno Sus Virus1 Infected Macrophage Cells’, Virology, 495: 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spandole-Dinu S. et al. (2018) ‘Prevalence of Human Anelloviruses in Romanian Healthy Subjects and Patients with Common Pathologies’, BMC Infectious Diseases, 18: 334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinegger M. et al. (2019) ‘HH-suite3 for Fast Remote Homology Detection and Deep Protein Annotation’, BMC Bioinformatics, 20: 473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M., Hegde R. S., and Jiang X. (2004) ‘The P Domain of Norovirus Capsid Protein Forms Dimer and Binds to Histo-Blood Group Antigen Receptors’, Journal of Virology, 78: 6233–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasova E., and Khayat R. (2021) ‘A Structural Perspective of Reps from CRESS-DNA Viruses and Their Bacterial Plasmid Homologues’, Viruses, 14: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thom K., and Petrik J. (2007) ‘Progression Towards AIDS Leads to Increased Torque Teno Virus and Torque Teno Minivirus Titers in Tissues of HIV Infected Individuals’, Journal of Medical Virology, 79: 1–7. [DOI] [PubMed] [Google Scholar]
- Umemura T. et al. (2002) ‘Observation of Positive Selection within Hypervariable Regions of a Newly Identified DNA Virus (SEN Virus) 1’, FEBS Letters, 510: 171–4. [DOI] [PubMed] [Google Scholar]
- Varsani A. et al. (2021) ‘Taxonomic Update for Mammalian Anelloviruses (Family Anelloviridae)’, Archives of Virology, 166: 2943–53. [DOI] [PubMed] [Google Scholar]
- Wada Y. et al. (2008) ‘The Structure of Melon Necrotic Spot Virus Determined at 2.8 Å Resolution’, Acta Crystallographica. Section F, Structural Biology and Crystallization Communications, 64: 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb B., Rakibuzzaman A., and Ramamoorthy S. (2020) ‘Torque Teno Viruses in Health and Disease’, Virus Research, 285: 198013. [DOI] [PubMed] [Google Scholar]
- Wery J.-P. et al. (1994) ‘The Refined Three-Dimensional Structure of an Insect Virus at 2·8 Å Resolution’, Journal of Molecular Biology, 235: 565–86. [DOI] [PubMed] [Google Scholar]
- Zhang W. et al. (2017) ‘Virome Comparisons in Wild-Diseased and Healthy Captive Giant Pandas’, Microbiome, 5: 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H. et al. (2007) ‘Torque Teno Virus (SANBAN Isolate) ORF2 Protein Suppresses NF-κB Pathways via Interaction with IκB Kinases’, Journal of Virology, 81: 11917–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.