


Abstract
A difficult problem concerning the interaction of DNA with amphiphiles of opposite charge above their critical micelle concentration is the propensity for aggregation of the condensed DNA complexes. In this study, this problem was addressed by attenuating amphiphile charge density within a cholate micelle environment. The amphiphile consisted of a cationic peptide, acetyl-CWKKKPKK-amide, conjugated to dilaurylphosphatidylethanolamine. In the presence of cholate, multiple equivalents of cationic charge were required to bring about the completion of DNA condensation. At the end point of condensation, stable, soluble DNA–micelle complexes were formed, which by dynamic light scattering exhibited apparent hydrodynamic diameters between 30 and 60 nm. Aggregation, as measured by static light scattering at 90° and by turbidity, was not observed until further additions of peptide–lipid conjugate were made beyond the end point of DNA condensation. Liposome complexes containing the non-aggregated, compacted DNA were formed by adding dioleoylphosphatidylcholine followed by removing the cholate by dialysis. The resulting complexes were distributed within a narrow density range, the DNA was quantitatively assembled into the liposomes, and liposomes without DNA were not detected. Small particles were formed with a mean hydrodynamic diameter of 77 nm. The liposomal DNA showed complete retention of its supercoiled form and no detectable sensitivity to DNase (25 U/10 µg DNA, 1.5 h, 37°C). The use of an anionic, dialyzable amphiphile to attenuate charge interactions between DNA and cationic amphiphiles is a useful technology for the quantitative assembly of compacted DNA into conventional liposomes, with complete protection against nuclease activity.
INTRODUCTION
The encapsulation of nucleic acids into liposomes has been a subject of investigation since the late 1970s. In the earliest work, DNA or RNA was incorporated into zwitterionic, neutral or anionic liposomes by techniques such as passive entrapment or reverse phase evaporation (reviewed in 1,2). Although liposome-mediated gene transfer to cells was successfully demonstrated, the results were disappointing. A major reason cited for the low levels of gene expression was the inefficiency of the methods used to encapsulate the nucleic acid. Since the original work of Felgner and colleagues (3), formulations containing cationic lipids have proven to be far more effective for gene transfer, in part because of the ability of cationic lipid to complex strongly and completely with nucleic acid by electrostatic interaction. In addition, cationic lipid–nucleic acid complexes (called ‘lipoplexes’; 4) associate avidly with cells and can disrupt endosomal or lysosomal membranes, thus delivering the nucleic acid into the cytoplasm and eventually into the nucleus.
Although cationic lipids continue to be the most common class of molecules for liposome-mediated gene transfer, their complexation with DNA is difficult to control. When DNA and pre-formed cationic liposomes are mixed, strong electrostatic interactions cause the formation of large aggregates of DNA and lipid, with particle sizes generally distributed over a range of several hundred nanometers (5). Although these structures are efficient for DNA delivery to cells in vitro, they diffuse poorly within tissues and are too large to extravasate from blood vessels when introduced by i.v. injection for systemic delivery (6,7). As reported by Liu et al. (8), vascular endothelial cells, monocytes and macrophages are the cell types most commonly transfected following i.v. injection of cationic liposome–DNA complexes. Delivery systems containing smaller, uniform complexes of condensed DNA are needed to significantly enhance gene transfer in vivo.
In this investigation, we report studies of a model system that demonstrates an alternative approach to the compaction of DNA by cationic amphiphiles, in such a way that small, stable particles of condensed DNA are formed. In particular, we have prepared an amphiphile consisting of a cationic peptide, acetyl-CWKKKPKK-amide, conjugated to phosphatidylethanolamine. Using a strategy developed originally by Dubin and co-workers (9–12), we show that mixtures of this cationic peptide–lipid conjugate and anionic detergent form soluble complexes with plasmid DNA. Under appropriate conditions, these complexes are stable in solution, neither aggregate nor precipitate and contain DNA that appears fully condensed and compacted to a small hydrodynamic diameter. We also report that this compacted DNA can be subsequently combined with non-cationic lipid to form small, homogeneous, nuclease-resistant liposome complexes.
MATERIALS AND METHODS
Peptide synthesis
The peptide CWKKKPKK-amide was synthesized by F-MOC chemistry using a FastMoc‘ protocol (13) on an ABI 431A peptide synthesizer. All residues were single coupled to MBHA Rink-amide resin (AnaSpec, San Jose, CA) (500 mg resin, 0.5 meq/g, 0.25 mmol scale synthesis). The N-terminus was acetylated on the resin by reacting twice with 7 ml of acetic anhydride plus 7 ml of pyridine for 2 h. The peptide was cleaved from the resin with 9.5 ml of trifluoroacetic acid, 0.5 ml of water and 0.25 ml each of thioanisole and 1,2-ethanedithiol for 3 h (14), followed by repeated precipitation in cold t-butyl methyl ether. The yield was 150 mg crude product.
The peptide was purified by HPLC by injecting 10 mg, dissolved in 0.5 ml water + 0.1% trifluoroacetic acid, onto a Hamilton PRP-3 reverse phase column and eluting with a 25 min linear gradient (1 ml/min) from 100% water to 40% water/60% acetonitrile (each with 0.1% trifluoroacetic acid). The first 3 ml of the major absorbance peak at 280 nm was collected. Peptide concentration was determined by tryptophan absorption at 280 nm (ɛ = 5600 M–1 cm–1) and confirmed by assay for cysteine (reaction with DTNB, absorbance at 410 nm, ɛ = 13 600 M–1 cm–1).
Conjugation of C12–PE and C18–PE with MBS
The heterobifunctional cross-linking reagent MBS (3-maleimidobenzoic acid n-hydroxysuccinimide ester; Sigma, St Louis, MO) was coupled to either dioleoylphosphatidylethanolamine (di18:1 cis-9) or dilaurylphosphatidylethanolamine (di12:0) as follows. Twenty micromoles of phosphatidylethanolamine were mixed with 35 µmol MBS in 1 ml of chloroform. Ten microliters of triethylamine were added to initiate the reaction. The reaction was judged complete after 1 h by analysis on TLC (solvent system chloroform/acetone/methanol/acetic acid/water, 50:20:10:10:5). The reaction was diluted to 3 ml with chloroform, washed with 3 ml of water + 25 µl of acetic acid, washed twice with 3 ml of water and the chloroform phase evaporated. The residue was dissolved in 3:1 distilled 95% ethanol/chloroform and purified by LH-20 Sephadex chromatography in the same solvent. Concentrations were determined by lipid phosphorus analysis (15).
Conjugation of peptide to PE–MBS
One micromole of dried residue of acetyl-CWKKKPKK-amide peptide was dissolved in 400 µl of dimethylformamide containing 2 µmol of PE–MBS and allowed to react at room temperature. Progress of the reaction was followed by taking 5 µl aliquots and assaying for free cysteine with DTNB (150 µM DTNB, 1% SDS, 1 ml PBS, pH 7.4). If after 1 h free cysteine was detected, an additional 1 µmol of lipid–MBS conjugate was added. The dimethylformamide was evaporated (N2 gas), the residue dissolved in 50 µl of DMSO and the product precipitated three times by addition of 5 ml of t-butyl methyl ether. TLC analysis of the ether supernatants indicated nearly complete separation of product from the PE–MBS reactant (which remains dissolved in the ether) with the first precipitation step.
Peptide–lipid conjugate was stored in DMSO. Concentration was determined by tryptophan fluorescence in dimethylformamide. Tryptophan fluorescence was compared to a standard curve of free peptide (where the concentration of free peptide was previously determined by tryptophan absorbance and DTNB measurements).
DNA condensation measurements
Plasmid DNA used in all experiments was the pGL3 control vector (5256 bp) from Promega (Madison, WI), maintained in Escherichia coli JM109. DNA was prepared using endo-free Qiagen maxi kits (Qiagen, Valencia, CA). Cholic acid (Sigma) was recrystallized twice in ethanol. All fluorescence measurements were carried out on an SLM-Aminco 8100 fluorometer, 450 W Xenon lamp, slit widths 4 nm, with all measurements relative to the fluorescence of a rhodamine dye reference sample. PMT voltages were established by setting the fluorescence of the DNA plus dye sample (described below) equal to 80% of full scale, using the auto-range feature of the instrument.
Condensation of DNA by peptide and peptide–lipid conjugates was measured by fluorescent dye displacement assays (16) using the intercalating dye ethidium bromide (λex = 520 nm, λem = 605 nm) or the minor groove binding dye Hoechst 33258 (λex = 350 nm, λem = 460 nm). Solutions with various concentrations of sodium cholate (10–30 mM) were prepared in 1 mM phosphate buffer, pH 8.0. Fluorescent dye was added to give a final dye to DNA base pair ratio of 1:25. All fluorescence measurements were carried out in 3 ml volumes containing 2–30 µg DNA. DNA condensation was followed by repeated addition of 5 µl aliquots of peptide or peptide–lipid conjugate dissolved in dimethylformamide (typically 0.3 nmol/µl). Fluorescence values were normalized to a range of 0–1 by taking the fluorescence with DNA but without peptide equal to 1.0 (Fmax) and the fluorescence of blank samples (with dye, without DNA, without peptide) equal to 0 (Fblank). Relative fluorescence was therefore equal to (Fobserved – Fblank)/(Fmax – Fblank).
Aggregation of DNA–peptide complexes during and after condensation was followed by static light scattering at a 90° angle (17), by setting the excitation and emission monochrometers of the fluorometer to 500 nm. Turbidity was measured by loss of transmitted light at 500 nm and is reported as 100 minus percent transmitted light, as described by Dubin and co-workers (12).
Dynamic light scattering
Condensed DNA complexes in cholate, and DNA–phosphatidylcholine liposomes, were analyzed by dynamic light scattering in a Microtrac Ultrafine Particle Size Analyzer (Microtrac, Clearwater, FL). Size analysis was performed with the software package supplied with the instrument, with assumptions of spherical and transparent particles. The data channel progression was carried out over the full size range of the instrument, with the format set to a geometrically increasing diameter ranging from ∼3 nm to 6 µm.
Liposome formation
Plasmid DNA (10 µg) was placed in a solution of 3 ml of 30 mM cholate, 1 mM phosphate buffer, pH 8.0. Thirty nanomoles (5× charge equivalents) of peptide–lipid conjugate, dissolved in 250 µl of dimethylformamide, were added to the DNA solution through a capillary pipette while stirring the solution vigorously. Then, 1000 nmol of dioleoylphosphatidylcholine, dissolved in 500 µl of dimethylformamide, were added, also through a capillary pipette while stirring vigorously. The solution was dialyzed three times against 1 l of 1 mM phosphate buffer, with a minimum of 6 h between changes of buffer.
Density gradient analysis was carried out on linear 4 ml gradients of 0–20% sucrose in 1 mM phosphate buffer. Liposomes were prepared as above with the addition of 0.1% 1-(oleoyl)-2-(5,7-dimethylBODIPY-1-hexadecanoyl)-phosphatidylcholine (D-3821; Molecular Probes, Eugene, OR). Samples were centrifuged for 18 h at 150 000 g (SW 50.1 rotor). Fractions of ∼150 µl were collected and diluted to 1 ml with 1 mM phosphate buffer. Hoechst dye was added to a final concentration of 200 pmol/ml. The fluorescence of the BODIPY-labeled lipid was measured at λex = 490 nm, λem = 520 nm. The fluorescence of the Hoechst dye binding to the DNA was measured at λex = 350 nm, λem = 460 nm.
DNase resistance
Liposomes were concentrated on Centriplus concentrators (Amicon, Bedford, MA) and adjusted to a volume of 10 µg DNA/360 µl 1 mM phosphate buffer. To this solution was added 20 µl of 100 mM MgSO4 and 20 µl of DNase (25 U) and the mixture incubated at 37°C for 1.5 h. Control incubations included DNA–phosphatidylcholine liposomes without DNase and 10 µg free DNA with and without DNase. At the end of the incubation, 50 µl of 1 M NaCl and 100 µl of 0.5 M sodium carbonate, pH 11.5, were added. Lipid and peptide–lipid conjugate (with the lysines now neutralized by the sodium carbonate) were removed from the aqueous phase by extracting twice into 500 µl of chloroform. Fifty microliters of the aqueous phase (∼1 µg DNA) were analyzed on a 1% agarose gel containing ethidium bromide.
RESULTS
Condensation of plasmid DNA with free peptide and peptide–lipid conjugate
Figure 1A and B shows the condensation of DNA by peptide–lipid conjugates in the presence of 20 mM cholate, as monitored by fluorescent dye displacement. Two DNA-binding fluorophores were used: (i) ethidium bromide, an intercalating dye; (ii) Hoechst 33258, a minor groove binding dye. DNA (5.3 kb) concentration was 10 µg/3 ml, with a 25:1 (mol/mol) base pair to dye ratio. Free peptide or peptide–lipid conjugate was then added as a concentrated dimethylformamide solution. The data are reported as relative fluorescence (1 unit = fluorescence before addition of peptide; 0 units = fluorescence in the absence of DNA) versus charge equivalents of peptide (lysine positive charge divided by DNA negative charge). Both peptide–lipid conjugates produced an apparent completion of DNA condensation at fewer charge equivalents than free peptide. The C18–PE peptide–lipid conjugate condensed the DNA more effectively (apparent end of condensation ∼1.7 charge equivalents) than the C12–PE conjugate (end of condensation ∼2.0 charge equivalents). However, with the C18–PE peptide–lipid conjugate, solutions became turbid both in the presence and absence of DNA, suggesting that the cholate was unable to completely disperse the C18 conjugate within a mixed micelle. Condensations carried out with the C12–PE conjugate remained optically clear. As a result, all further studies were carried out using the C12–PE conjugate. In addition, the ethidium bromide probe exhibited a sharper indication of the end point of condensation and was used for all subsequent studies.
Figure 1.

DNA condensation by free peptide and peptide–lipid conjugates in anionic detergent. Experimental solutions consisted of 10 µg DNA in 3 ml of 20 mM cholate, 1 mM phosphate buffer, pH 8.0. Solutions of peptide or peptide–lipid conjugate in dimethylformamide were added in 5 µl aliquots (1.5 nmol peptide, 0.25 charge equivalents relative to the DNA). The fluorescence values were normalized to a range of 0–1 as described in Materials and Methods. (A) Ethidium bromide fluorescence, dye:base pair ratio 1:25. Closed triangles, free peptide; open diamonds, C12–PE–peptide; closed squares, C18–PE–peptide. (B) Hoechst dye fluorescence, dye:base pair ratio 1:25. Symbols as in (A). Values are means ± SEM for three separate experiments (for many data points, error bars are eclipsed by the size of the symbol).
Tryptophan fluorescence of the peptide indicated that the peptide–lipid conjugate interacted with the cholate, presumably to form mixed micelles. In the absence of cholate (and absence of DNA), the tryptophan fluorescence exhibited an emission maximum at 352 nm. This maximum was blue-shifted to 343 nm in the presence of either 20 or 30 mM cholate. In contrast, tryptophan fluorescence did not change upon interaction of the peptide–lipid conjugate with DNA, either as a change in fluorescence intensity or by a shift in emission wavelength (data not shown).
Figure 2 shows DNA condensation by the C12–PE peptide–lipid conjugate at various plasmid DNA concentrations in the presence of 20 mM cholate. At low DNA concentrations, slightly more peptide–lipid conjugate (∼0.5 charge equivalents) was required to bring about complete condensation. Interestingly, the extent of dye displacement consistently increased with decreasing DNA concentration. This observation would be consistent with a hypothesis that at lower DNA concentrations, DNA condensation is apparently more effective due to reduced interference from interactions with surrounding DNA molecules.
Figure 2.

DNA condensation by C12–PE peptide–lipid conjugate at different DNA concentrations. Experimental solutions consisted of 20 mM cholate, 1 mM phosphate buffer, pH 8.0, with 2–30 µg DNA in a total volume of 3 ml. The ethidium bromide to DNA base pair ratio was maintained at 1:25 mol/mol regardless of DNA concentration. C12–PE peptide–lipid conjugate in dimethylformamide was added in 5 µl aliquots, 0.25 charge equivalents per aliquot. Observed fluorescence was normalized to a range of 0–1 as described in Materials and Methods. Open diamonds, 2 µg DNA; closed squares, 5 µg DNA; open triangles, 10 µg DNA; closed diamonds, 20 µg DNA; crosses, 30 µg DNA.
Binding of peptide–lipid conjugate is attenuated by cholate
Increasing the amount of negatively charged cholate clearly attenuated the electrostatic interactions between peptide–lipid conjugate and DNA. Figure 3 shows the condensation of plasmid DNA by the C12–PE peptide–lipid conjugate at various concentrations of cholate detergent. In the presence of 10 and 15 mM cholate, condensation takes place quite rapidly, with the DNA fully condensed at ∼1.25 charge equivalents. In 20 mM cholate, condensation was complete at ∼2.0 charge equivalents. In 30 mM cholate, 3.5 charge equivalents of conjugate were required for completion of condensation.
Figure 3.

DNA condensation by C12–PE peptide–lipid conjugate at different concentrations of sodium cholate. Experimental solutions consisted of 1 mM phosphate buffer, pH 8.0, 10 µg DNA, 3 ml total volume, and cholate concentrations of 10–30 mM. C12–PE peptide–lipid conjugate in dimethylformamide was added in 5 µl aliquots, 0.25 charge equivalents per aliquot. Observed fluorescence was normalized to a range of 0–1 as described in Materials and Methods. Values are means ± SEM for three separate experiments (for most data points, error bars are eclipsed by the size of the symbol). Open diamonds, 10 mM cholate; closed squares, 15 mM cholate; closed triangles, 20 mM cholate; open circles, 30 mM cholate.
Dynamic light scattering measurements indicated that the end point of condensation, as determined by ethidium bromide displacement, corresponded to the presence of highly compacted DNA complexes with small hydrodynamic diameters. Diameters increased with increasing DNA concentration (Fig. 4A, 1.7 µg/ml, mean diameter 29 nm; Fig. 4B, 3.3 µg/ml, mean diameter 55 nm), suggesting that individual complexes consisted of more than one DNA molecule. Attempts to find the smallest obtainable complexes were unsuccessful as the dynamic light scattering measurements could not be made at DNA concentrations <1.7 µg/ml DNA. Attempts to define mean diameters at charge equivalencies before the apparent end point of condensation were also unsuccessful, as the instrument reported the presence of multiple species distributed over a wide range.
Figure 4.

Dynamic light scattering of condensed DNA in 20 mM cholate at two different DNA concentrations. (A) Five micrograms plasmid DNA condensed with 2.5 charge equivalents of C12–PE peptide–lipid conjugate, 3 ml total volume. (B) Ten micrograms DNA condensed with 2.0 charge equivalents of peptide–lipid conjugate, 3 ml total volume.
Separation of end point of DNA condensation from onset of aggregation
To determine the separation of the end point of DNA condensation from the onset of aggregation, aggregation was studied by: (i) static light scattering at a 90° angle at 500 nm; (ii) turbidity measured as the loss of transmitted light at 500 nm. We routinely observed an increase in static light scattering at 90° before the onset of turbidity. The light scattering measurement likely detects both unaggregated complexes of compacted DNA as well as aggregation, because compacted DNA scatters light more effectively than DNA in an extended conformation (17). Turbidity is a measure of the molecular weight of the complex, hence it detects aggregation.
Solutions of plasmid DNA were carefully titrated with peptide–lipid conjugate in order to determine the exact charge equivalence where DNA condensation ended (as measured by ethidium bromide dye displacement). Then, the titration was continued to determine the charge equivalence at which the condensed DNA–peptide–lipid complexes began to aggregate. In a solution of 20 mM cholate (Fig. 5A), DNA condensation ended at ∼2.0 charge equivalents and the onset of turbidity occurred at ∼2.5–2.75 charge equivalents. This is seen both by the marked increase in turbidity beginning at 2.5 charge equivalents and also by the ‘plateau’ of ethidium bromide fluorescence between 2.0 and 2.75 charge equivalents. Ethidium bromide fluorescence decreased slightly at higher charge equivalents, due to the loss of excitation and emitted light caused by the appearance of turbidity in the solution. In the plateau region, the solution contained stable, non-aggregating complexes of condensed DNA. In 30 mM cholate (Fig. 5B), the end point of DNA condensation occurred between 4 and 4.25 charge equivalents with the onset of turbidity at ∼5 charge equivalents.
Figure 5.

Determination of the charge equivalence difference between the end point of DNA condensation and the onset of aggregation and turbidity in anionic detergent. Experimental solutions consisted of 1 mM phosphate buffer, pH 8.0, 10 µg DNA, 3 ml total volume. Cholate concentration was 20 (A) or 30 mM (B). DNA condensation (closed diamonds) was measured by ethidium bromide displacement and observed fluorescence was normalized to a range of 0–1 as described in Materials and Methods. Aggregation was measured by static light scattering at 90° (closed squares) or by turbidity (open triangles), as described in Materials and Methods. C12–PE peptide–lipid conjugate in dimethylformamide was added in 5 µl aliquots, 0.25 charge equivalents per aliquot, in the experiment in (A), and 5 µl aliquots, 0.33 charge equivalents per aliquot, for the experiment in (B).
A very different behavior was observed when DNA condensation was carried out in the presence of the neutral detergent octyl glucoside (Fig. 6, 30 mM octyl glucoside). Octyl glucoside did not attenuate the charge interaction between peptide–lipid conjugate and DNA, as ∼1.0 charge equivalent of peptide–lipid conjugate was required for condensation. Changing the octyl glucoside concentration had no effect on the charge equivalency at which DNA condensation occurred (data not shown). Condensation did not complete before the onset of aggregation. Unlike the condensations in 20 and 30 mM cholate, there was no detectable plateau of ethidium bromide fluorescence after the end of condensation and before the onset of turbidity. Turbidity was more severe. At the same DNA concentration, aggregated DNA complexes in octyl glucoside produced a 10% loss of transmitted light as opposed to the 3–5% loss observed when the condensation was carried out in 20 or 30 mM cholate detergent. With octyl glucoside, we were unable to produce solutions of condensed DNA which remained optically clear.
Figure 6.

Determination of the charge equivalence difference between the end point of DNA condensation and the onset of aggregation and turbidity in neutral detergent. Experimental solutions consisted of 1 mM phosphate buffer, pH 8.0, 10 µg DNA, 3 ml total volume. Octyl glucoside concentration was 30 mM. DNA condensation (open diamonds) was measured by ethidium bromide displacement and observed fluorescence was normalized to a range of 0–1 as described in Materials and Methods. Aggregation was measured by static light scattering at 90° (closed squares) or by turbidity (closed triangles), as described in Materials and Methods. C12–PE peptide–lipid conjugate in dimethylformamide was added in 5 µl aliquots, 0.20 charge equivalents per aliquot.
Liposome formation
Liposomes of condensed DNA, peptide–lipid conjugate and phosphatidylcholine were prepared by adding phosphatidylcholine to the cholate solution of condensed DNA, followed by removal of the cholate by dialysis (18). Figure 7 shows a sucrose density gradient analysis of the resulting DNA–phosphatidylcholine complexes. All of the lipid, and almost all of the DNA, banded together in a narrow region towards the center of the 0–20% sucrose gradient. A small fraction of DNA was evident at higher densities. Presumably this was condensed DNA that may have aggregated in solution prior to the addition of phosphatidylcholine and did not form liposome complexes. Otherwise, by this density gradient analysis, the DNA–phosphatidylcholine complexes were homogeneous.
Figure 7.

Density gradient analysis of DNA–phosphatidylcholine liposomes. Liposomes containing 0.1% of the fluorescent lipid 1-(18:1)-2-(C16-BODIPY)-phosphatidylcholine were concentrated to ∼20 µg DNA/ml and a 0.5 ml aliquot placed on a 4 ml 0–20% linear sucrose gradient. Fractions of 150 µl were collected then diluted to 1 ml with 1 mM phosphate buffer, pH 8.0. Lipid content (open squares) was measured by fluorescence (λex 490 nm, λem 520 nm). DNA (closed diamonds) was measured by the addition of Hoechst dye (λex 350 nm, λem 460 nm).
Figure 8 shows the dynamic light scattering analysis of these DNA–phosphatidylcholine complexes, which were distributed over a narrow range of hydrodynamic diameters centered around 77 nm. (This measurement was made several days following the formation of the liposomes, indicating that they were quite stable upon storage.)
Figure 8.

Dynamic light scattering analysis of DNA–phosphatidylcholine liposomes. Liposome composition, method of assembly and instrumental details of the analysis are described in Materials and Methods.
The DNA–phosphatidylcholine liposomes were treated with high levels of DNase (25 U/10 µg DNA) to examine the nuclease resistance of the complexes. In order to visualize the DNA following nuclease treatment, it was necessary to neutralize the lysines of the peptide–lipid conjugate with carbonate buffer, then to extract both the phosphatidylcholine and the neutralized peptide–lipid conjugate into chloroform. The DNA remaining in the aqueous phase could then be analyzed by agarose gel electrophoresis (Fig. 9). As seen in lane 5 (incubation without DNase) versus lane 6 (incubation with DNase), the DNA, when complexed with phosphatidylcholine, was unaffected by nuclease and fully retained its supercoiled form. The remaining lanes are a variety of controls and are described in the figure legend.
Figure 9.
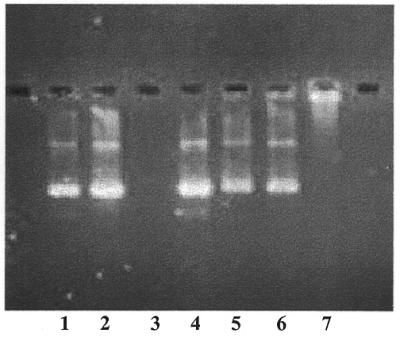
DNase resistance of DNA–phosphatidylcholine liposomes. Liposomes containing 10 µg DNA were treated with 25 U DNase for 1.5 h at 37°C as described in Materials and Methods. Lipid and peptide–lipid conjugate were extracted into chloroform as described and the DNA (which remained in the aqueous phase) was analyzed on a 1% agarose gel containing ethidium bromide (∼1 µg/lane). Lane 1, free DNA; lane 2, free DNA taken through the extraction procedure (no DNase); lane 3, free DNA completely digested by DNase; lane 4, a ‘stop’ control, where free DNA was added after an incubation mixture containing DNase had been extracted with chloroform; lane 5, DNA–phosphatidylcholine liposomes incubated without DNase and carried through the extraction procedure; lane 6, DNA–phosphatidylcholine liposomes incubated with DNase and carried through the extraction procedure; lane 7, DNA–phosphatidylcholine liposomes not treated with DNase and not extracted.
DISCUSSION
Efficient packaging of DNA into lipidic complexes continues to be a major challenge to the preparation of non-viral gene delivery systems. As recently reviewed by Tang and Szoka (19), aggregation of DNA complexes normally occurs when attempting to prepare systems containing condensed DNA, particularly at concentrations and in physiologically compatible buffer systems that are appropriate for in vivo administration. Most preparations of cationic lipid and DNA aggregate in a difficult to control manner during their assembly and the resulting complexes typically show a wide particle size distribution over a range of several hundred nanometers (5). Although these complexes are efficient for in vitro gene transfer, a prevailing view is that smaller and more well-defined particle sizes will be more appropriate to overcome the barriers to diffusion found in vivo, particularly for systemic i.v. administration (5–7)
In this work, we explore a strategy where the cationic amphiphile used to condense DNA is in solution above its critical micelle concentration (c.m.c.). If the cationic amphiphile were the only amphiphile present, this approach would not be feasible. When the concentration of charged amphiphile is above its c.m.c., the electrostatic interactions between polyelectrolyte and micelle of opposite charge are generally so strong that uncontrollable phase separation occurs. The polymer–micelle complex will form either a solid precipitate or a second liquid phase termed a co-acervate, and soluble polymer–micelle complex generally does not co-exist in solution with free micelle. To avoid this problem, Dubin and co-workers (9–12) have shown that by including a second neutral amphiphile in the micelle, the strong electrostatic interactions between polymer and micelle can be attenuated. In the presence of the second amphiphile, which effectively dilutes the surface charge density of the micelle, conditions can be found where soluble, reversible polymer–micelle complexes can be formed.
The work reported here draws heavily upon this experimental strategy developed by Dubin and co-workers. In our experiments, we found that by adding our cationic peptide–lipid conjugate to a solution containing a second amphiphile (cholate), the otherwise strong electrostatic interactions between peptide–lipid conjugate and DNA were attenuated. This resulted in the formation of soluble complexes of compacted DNA which did not aggregate nor precipitate. By increasing or decreasing the concentration of cholate in solution, the charge equivalence of peptide–lipid conjugate required to bring about the condensation of DNA was controllable. It should be recognized that the charge equivalence difference between the end point of DNA condensation and the onset of aggregation was quite narrow. However, our finding of a narrow region between binding of conjugate and the onset of aggregation is similar to the findings of Dubin with a variety of polyelectrolyte–amphiphile systems (9,11,12). These investigators found at most a factor of two difference between the mole fraction of charged amphiphile in the micelle required for the onset of binding of micelle to polyelectrolyte and the mole fraction of charged amphiphile where precipitation of the polymer–micelle complex began. It will, of course, be useful to explore variations of the structure of our peptide–lipid conjugate to see if a wider region of charge equivalence can be found where soluble complexes of condensed DNA can be maintained in solution.
Although the major focus of this investigation was the formation of soluble, stable complexes of condensed DNA, we have also shown that these complexes can then be used for the formation of liposome complexes containing a non-cationic lipid, e.g. phosphatidylcholine (Figs 7–9). In this communication, we report one set of conditions which resulted in the formation of small particles, which were homogeneous in density and nuclease resistant. Further work should be devoted to the study of the many variables involved in this assembly process. These include DNA to peptide charge equivalence, amount of phosphatidylcholine relative to DNA, initial cholate concentration and use of different lipids with different polar head groups and/or hydrocarbon chain lengths, as well as factors such as temperature, dialysis volume and rate of detergent removal.
The assembly of phosphatidylcholine into the condensed DNA complex is driven by the hydrophobic interactions between the phosphatidylcholine hydrocarbon chains and the hydrocarbon chains of the peptide–lipid conjugate. This assembly process is fundamentally different from other methods that have been used to form complexes of DNA associated with or encapsulated into zwitterionic or anionic liposomes. Most of the reported methods for assembling DNA into non-cationic liposomes have relied upon passive entrapment, using the technique of reverse phase evaporation or the technique of dehydration–rehydration (20). Also, Lee and Huang (21) have reported formation of liposome complexes where the DNA is first condensed with a charge excess of polylysine, followed by combination with phosphatidylglycerol. Here the association is driven by electrostatic forces between the positively charged condensed DNA complex and the anionic lipid. All of these previously reported techniques exhibit either a significant percentage of DNA that was not associated with lipid or, in the case of electrostatic association, an excess of free lipid that required removal by density gradient centrifugation.
In contrast, we found that complex formation driven by hydrophobic interactions upon removal of the cholate amphiphile resulted in an efficient assembly process where unincorporated lipid was not detected. This approach should allow for the inclusion of additional functional components, providing they contain a lipophilic region to allow for hydrophobic interactions with the condensed DNA complex during the process of liposome formation. For the field of gene delivery, such functional components could include fusogenic agents (such as fusogenic peptides with hydrophobic regions), targeting ligands coupled to lipid (21) and compounds for steric stabilization, including bioconjugates of polyethyleneglycol and phospholipid, currently in use for systemic drug delivery (22).
REFERENCES
- 1.Fraley R. and Papahadjopoulos,D. (1982) Curr. Top. Microbiol. Immunol., 96, 171–191. [DOI] [PubMed] [Google Scholar]
- 2.Lurquin P.F. (1993) In Gregoriadis,G. (ed.), Liposome Technology, Vol. II, 2nd Edn. CRC Press, Boca Raton, FL, pp. 129–139.
- 3.Felgner P.L., Gadek,T.R., Holm,M., Roman,R., Chan,H.W., Wenz,M., Northrop,J.P., Ringold,G.M. and Danielsen,M. (1987) Proc. Natl Acad. Sci. USA, 84, 7413–7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Felgner P.L., Barenholz,Y., Behr,J.P., Cheng,S.H., Cullis,P., Huang,L., Jessee,J.A., Seymour,L., Szoka,F. and Thierry,A.R. (1997) Hum. Gene Ther., 8, 511–512. [DOI] [PubMed] [Google Scholar]
- 5.Lasic D.D. (1997) Liposomes in Gene Delivery. CRC Press, Boca Raton, FL.
- 6.Allen T.M. and Stuart,D.D. (1999) In Janoff,A.S. (ed.), Liposomes, Rational Design. Marcel Dekker, New York, NY, pp. 63–87.
- 7.Li S. and Huang,L. (1999) In Janoff,A.S. (ed.), Liposomes, Rational Design. Marcel Dekker, New York, NY, pp. 89–124.
- 8.Liu Y., Mounkes,L.C., Liggitt,H.D., Brown,C.S., Solodin,I., Heath,T.D. and Debs,R.J. (1997) Nature Biotechnol., 15, 167–173. [DOI] [PubMed] [Google Scholar]
- 9.Li P., Kichler,A. and Dubin,P.L. (1994) Macromolecules, 27, 7049–7055. [Google Scholar]
- 10.Li Y. and Dubin,P.L. (1994) In Herb,C.A. and Prud’homme,R.K. (eds), Structure and Flow in Surfactant Solutions. American Chemical Society, Washington, DC, pp. 320–336.
- 11.Li Y., Dubin,P.L., Havel,H.A., Edwards,S.L. and Dautzenberg,H. (1995) Langmuir, 11, 2486–2492. [Google Scholar]
- 12.McQuigg D.W., Kaplan,J.I. and Dubin,P.L. (1992) J. Phys. Chem., 96, 1973–1978. [Google Scholar]
- 13.Fields C.G., Lloyd,D.H., Macdonald,R.L., Otteson,K.M. and Noble,R.L. (1991) Peptide Res., 4, 95–101. [PubMed] [Google Scholar]
- 14.Guy C.A. and Fields,G.B. (1997) Methods Enzymol., 289, 67–83. [DOI] [PubMed] [Google Scholar]
- 15.Rouser G., Siakotos,A.N. and Fleischer,S. (1966) Lipids, 1, 85–86. [DOI] [PubMed] [Google Scholar]
- 16.Xu Y. and Szoka,F.C. (1996) Biochemistry, 35, 5616–5623. [DOI] [PubMed] [Google Scholar]
- 17.Tang M.X. and Szoka,F.C. (1998) In Kabanov,A.V., Felgner,P.L. and Seymour,L.W. (eds), Self-assembling Complexes for Gene Delivery: From Laboratory to Clinical Trial. John Wiley & Sons, Chichester, UK, pp. 169–196.
- 18.Allen T.M. (1984) In Gregoriadis,G. (ed.), Liposome Technology, Vol. I. CRC Press, Boca Raton, FL, pp. 109–122.
- 19.Tang M.X. and Szoka,F.C. (1998) In Kabanov,A.V., Felgner,P.L. and Seymour,L.W. (eds), Self-assembling Complexes for Gene Delivery: From Laboratory to Clinical Trial. John Wiley & Sons, Chichester, UK, pp. 26–50.
- 20.Gregoriadis G., Saffie,R. and Hart,S.L. (1996) In Gregoriadis,G. and McCormack,B. (eds), Targeting of Drugs 5: Strategies for Oligonucleotide and Gene Delivery in Therapy. Plenum Press, New York, NY, pp. 143–150.
- 21.Lee R.J. and Huang,L. (1996) J. Biol. Chem., 271, 8481–8487. [DOI] [PubMed] [Google Scholar]
- 22.Papahadjopoulos D. (1999) In Janoff,A.S. (ed.), Liposomes, Rational Design. Marcel Dekker, New York, NY, pp. 1–12.