

Abstract
Objectives
Molecular structures, spectroscopic properties, charge distributions, frontier orbital energies, nonlinear optical (NLO) properties and molecular docking simulations were analyzed to examine the bio-usefulness of a series of (4-fluorophenyl)[5-(4-nitrophenyl)-3-phenyl-4,5-dihydro-1H-pyrazol-1-yl]methanone derivatives.
Methods
The compounds were studied through computational methods. Equilibrium optimization of the compounds was performed at the B3LYP/6-31G(d,p) level of theory, and geometric parameters, frequency vibration, UV–vis spectroscopy and reactivity properties were predicted on the basis of density functional theory (DFT) calculations.
Results
The energy gap (ΔEg), electron donating/accepting power (ω−/ω+) and electron density response toward electrophiles/nucleophiles calculated for M1 and M2 revealed the importance of substituent positioning on compound chemical behavior. In addition, ω−/ω+ and ΔEn/ΔEe indicated that M6 is more electrophilic because of the presence of two NO2 groups, which enhanced its NLO properties. The hyperpolarizability (β0) of the compounds ranged from 5.21 × 10−30 to 7.26 × 10−30 esu and was greater than that of urea; thus, M1–M6 were considered possible candidates for NLO applications. Docking simulation was also performed on the studied compounds and targets (PDB ID: 5ADH and 1RO6), and the calculated binding affinity and non-bonding interactions are reported.
Conclusion
The calculated ω− and ω+ indicated the electrophilic nature of the compounds; M6, a compound with two NO2 groups, showed enhanced effects. Molecular electrostatic potential (MEP) analysis indicated that amide and nitro groups on the compounds were centers of electrophilic attacks. The magnitude of the molecular hyperpolarizability suggested that the entire compound had good NLO properties and therefore could be explored as a candidate NLO material. The docking results indicated that these compounds have excellent antioxidant and anti-inflammatory properties.
Keywords: DFT; Molecular docking; [5-(4-nitrophenyl)-3-phenyl-4,5-dihydro-1H-pyrazol-1-yl]methanone
المخلص
تمت دراسة سلسلة من مشتقات الميثانون (4-فلوروفينيل) [5- (4-نيتروفينيل) -3-فينيل-4،5-ثنائي هيدرو-1اتش-بيرازول-1-يل] باستخدام الطرق الحسابية. تم إجراء تحسين التوازن لهذه المركبات عند مستوى "ب3ليب/6-31ج∗∗" من الناحية النظرية، وتم التنبؤ بالمعلمات الهندسية، والاهتزاز الترددي، وخصائص الأشعة المرئية وفوق البنفسجية على أساس حسابات نظرية الكثافة الوظيفية. كشفت فجوة الطاقة، والتبرع بالإلكترون / قبول الطاقة واستجابة كثافة الإلكترون تجاه الإلكتروفيل / النوكليوفيل المحسوبة لـ إم1 و إم2، عن أهمية وضع البديل على السلوك الكيميائي للمركب. أيضا، أظهر التبرع بالإلكترون / قبول الطاقة و فجوة الطاقة أن إم6 أكثر إلكتروفيلا بسبب وجود مجموعتين من "إن أو 2"، مما يعزز خصائص "إن إل أو" الخاصة به. تراوحت قابلية الاستقطاب المفرط للمركبات 5.21-7.26 × 10-30 وحدة كهرباء وكانت أكبر من مادة اليوريا القياسية "إن إل أو"؛ وبالتالي جعل إم1-إم6 مرشحين محتملين لتطبيقات "إن إل أو". كشفت عمليات محاكاة الإرساء أن التقاربات الملزمة للمركبات الستة تراوحت من -8.8 إلى -9.3 كيلو كالوري / مول لـ "أ ب أو"-نازعة هيدروجين الكبد (عنوان بي دي بي: 5 أ دي إتش) و -8.5 إلى -9.7 كيلو كالوري / مول لـ هيدرولاز البروتين المضاد للالتهابات (عنوان بي دي بي: 1 آر أو 6)؛ وبالتالي يمكن أن تمتلك خصائص جيدة مضادة للأكسدة ومضادة للالتهابات.
الكلمات المفتاحية: ميثانون (4-فلوروفينيل) [5- (4-نيتروفينيل) -3-فينيل-4،5-ثنائي هيدرو-1اتش-بيرازول-1-يل], نظرية الكثافة الوظيفية, إرساء جزيئي
Introduction
Pyrazole derivatives are heterocyclic compounds recognized to possess a wide range of biological and pharmacological activities.1,2 The unique properties of pyrazoles have been attributed to the electrophilic substitution reactions that occur specially at position 4, and nucleophilic attacks that occur at positions 3 and 5, thus leading to diverse pyrazole structures with broad potential applications in areas such as medicine, agriculture and technology.3, 4, 5 In addition, compounds containing a pyrazole nucleus have been reported to be anti-inflammatory, antiviral, anticancer, antiparasitic, antibacterial, antirheumatoid, antidepressant, analgesic, antinociceptive, antihypertensive, antipyretic and antifungal agents.6, 7, 8, 9, 10, 11, 12 Beyond these biological activities, this class of compounds displays substantial nonlinear optical (NLO) properties,13, 14, 15, 16, 17 electroluminescent properties due to photo-induced-electron transfer,18 and light amplification properties due to stimulated emission or lasing/random lasing action.14,15
Several pyrazole derivatives, such as 3-(1,1-dicyanoethenyl)-1-phenyl-4,5-dihydro-1H-pyrazole, have been investigated for their NLO properties. The size of the nano-crystals of the compound has been suggested to play a major role in the excitation or emission efficiency.19,20 Thus, these compounds can be used for ultrafast optics.21 The optical nonlinearity of a series of N-substituted-5-phenyl-1H-pyrazole-4-ethyl carboxylates of compounds in chloroform solution has been assessed, and these compounds have been found to be good candidates for NLO applications.22 In addition, a series of (Z)-2-(4-nitrophenyl)-3-(1-phenyl-4,5-dihydro-1H-pyrazol-3-yl)acrylonitrile and (E)-3-(4-nitrostyryl)-1-phenyl-4,5-dihydro-1H-pyrazole compounds have been found to have several electron accepting groups attached and to show high NLO responses dependent on functionalization of the pyrazoline derivatives.23 The compounds 1-N-phenyl-3(3,4-dichlorophenyl)-5-phenyl-2-pyrazoline,24 diethyl-1H-pyrazole-3,5-dicarboxylate and 4-(4-bromophenyl)-1-tert-butyl-3-methyl-1H-pyrazol-5-amine25 have been studied with experimental and density functional theory (DFT) methods and found to have promising NLO properties.
More recently, DFT has been used in conformational and NBO analysis of (4-chloro-3,5-dimethyl-1H-pyrazol-1-yl) (p-tolyl)methanone, and the results have shown excellent agreement with experimental data.26 Likewise, 2-bromo-N-(2,3-dihydro-1,5-dimethyl-3-oxo-2-phenyl-1H-pyrazol-4-yl)benzamide and 2-chloro-N-(2,3-dihydro-1,5-dimethyl-3-oxo-2-phenyl-1H-pyrazol-4-yl)benzamide have been studied with both experimental and DFT methods; the theoretical results have revealed that both compounds display energetic hydrogen bonding interactions and are stabilized by electrostatic energy contribution, in line with experimental observations.27 Santhi and Bharathi have reported the synthesis and molecular structural elucidation of a series of 4-(3-(2-amino-3,5-dibromophenyl)-1-(benzoyl)-4,5-dihydro-1H-pyrazol-5-yl)benzonitriles through spectroscopic and DFT methods. The DFT method has been used to analyze the studied molecules’ molecular electrostatic potential, natural bonding orbitals, Mulliken charges, frontier molecular orbital energies and NLO properties. The results have indicated inter- and intra-molecular delocalization and acceptor–donor interactions based on second-order perturbation interactions; moreover, polarizability and hyperpolarizability calculations have indicated that the compounds possess good NLO properties.28
In this work, DFT calculations were performed on 4-(3-(2-amino-3,5-dibromophenyl)-1-(benzoyl)-4,5-dihydro-1H-pyrazol-5-yl)benzonitriles reported by Santhi and Bharathi.28 Structural modifications were used to design six new compounds, as shown in Table 1. The molecular structures, spectroscopic properties, charge distributions, frontier orbital energies, NLO properties and molecular docking simulations were analyzed to examine the bio-usefulness of the studied compounds.
Table 1.
Schematic structures of the modeled compounds; Ma∗ = compound from.28
 | |||
|---|---|---|---|
| Compound | R1 | R2 | Name |
| M1 | F | NO2 | [3-(2-aminophenyl)-5-(4-nitrophenyl)-4,5-dihydro-1H-pyrazol-1-yl](4-fluorophenyl)methanone |
| M2 | NO2 | F | [3-(2-aminophenyl)-5-(4-fluorophenyl)-4,5-dihydro-1H-pyrazol-1-yl](4-nitrophenyl)methanone |
| M3 | CH3 | NO2 | [3-(2-aminophenyl)-5-(4-nitrophenyl)-4,5-dihydro-1H-pyrazol-1-yl](4-methylphenyl)methanone |
| M4 | OH | NO2 | [3-(2-aminophenyl)-5-(4-nitrophenyl)-4,5-dihydro-1H-pyrazol-1-yl](4-hydroxyphenyl)methanone |
| M5 | OCH3 | NO2 | [3-(2-aminophenyl)-5-(4-nitrophenyl)-4,5-dihydro-1H-pyrazol-1-yl](4-methoxyphenyl)methanone |
| M6 | NO2 | NO2 | [3-(2-aminophenyl)-5-(4-nitrophenyl)-4,5-dihydro-1H-pyrazol-1-yl](4-nitrophenyl)methanone |
| Ma | F | CN | 4-[3-(2-amino-3,5-dibromophenyl)-1-(4-fluorobenzoyl)-4,5-dihydro-1H-pyrazol-5-yl]benzonitrile |
Theoretical details
Before DFT calculations, an equilibrium conformer search was performed on all compounds with a semi-empirical AM1 method to identify the lowest conformer for each compound, which was used for further DFT calculations.29 All calculations were performed on these compounds with Becke's three parameter hybrid functional DFT, with Lee, Yang and Parr correlation,30 and optimized at B3LYP/6-31G(d,p) level of theory in gas. Frequency calculation was also performed by using the same basis set to confirm that the optimized molecules were minima, as characterized by positive harmonic frequencies31,32 in Spartan 14.33 The DFT hybrid B3LYP functional has been reported to overestimate the fundamental modes; however, this overestimation can be addressed by calculating harmonic frequencies with a scaling factor of 0.9619 to yield frequencies consistent with experimental data.34 The molecular descriptors calculated from conceptual DFT were the ionization potential (I = −HOMO), electron affinity (A = −LUMO), chemical hardness (η), chemical potential (μ), global electrophilicity (ω), electron donating power , electron accepting power (, nucleofugality (ΔEn) and electrofugality (ΔEe) (equations 1, 2, 3, 4, 5, 6, 7)).35, 36, 37, 38, 39
| 1 |
| 2 |
| 3 |
| 4 |
| 5 |
| 6 |
| 7 |
Results and discussion
Geometry parameters
The geometry parameters extracted from the equilibrium structures optimized at the B3LYP/6-31G(d,p) level for the six compounds M1–M6 are displayed in Tables 2 and 3. The results were compared with the geometries of 4-(3-(2-amino-3,5-dibromophenyl)-1-(4-nitrobenzoyl)-4,5-dihydro-1H-pyrazol-5-yl)benzonitrile predicted at the same level of theory.28 The calculated C3–N7 bonds were 1.299, 1.298, 1.300, 1.299, 1.299 and 1.298 Å for M1–M6, respectively, in agreement with the 1.299 Å calculated for Ma. This type of bond has been experimentally observed to be 1.275 Å and calculated to be 1.287 Å for bis-spiropipridinon/pyrazole derivatives.40 The C1–N8 bond in the pyrazole ring displayed a typical single bond character with a bond length of 1.482 Å for M1, 1.490 Å for M2, 1.490 Å for M3, 1.479 Å for M4, 1.481 Å for M5, 1.486 Å for M6 and 1.487 Å for Ma. The N–N bond length in the pyrazole ring was calculated to be 1.376 Å for M1, 1.378 Å for M2, 1.372 Å for M3, 1.376 Å for M4, 1.375 Å for M5, 1.381 Å for M6 and 1.374 Å for Ma; these values have been experimentally found to be 1.385 and 1.369 Å,40 and 1.3827 Å27 for similar compounds. In addition, The N8–C34 bond length value was 1.383 Å for M1, 1.378 Å for M2, 1.381 Å for M3, 1.360 Å for M4, 1.380 Å for M5, 1.380 Å for M6 and 1.374 Å for Ma.
Table 2.
Optimized structures of M1–M6 in 2-D and 3-D.
| Compound | 2-D structure | 3-D structure |
|---|---|---|
| M1 | 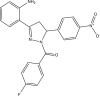 |
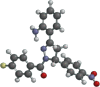 |
| M2 | 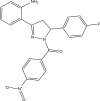 |
 |
| M3 |  |
 |
| M4 |  |
 |
| M5 |  |
 |
| M6 |  |
 |
Table 3.
Selected bond length, bond angle and dihedral angle for the studied compounds.
| Bond length(Å) | M1 | M2 | M3 | M4 | M5 | M6 | Ma |
|---|---|---|---|---|---|---|---|
| C1–C2 | 1.552 | 1.550 | 1.547 | 1.551 | 1.552 | 1.550 | 1.546 |
| C1–N8 | 1.482 | 1.490 | 1.490 | 1.479 | 1.481 | 1.486 | 1.487 |
| C2–C3 | 1.523 | 1.520 | 1.522 | 1.522 | 1.523 | 1.522 | 1.522 |
| C3–N7 | 1.299 | 1.298 | 1.300 | 1.299 | 1.299 | 1.298 | 1.295 |
| N7–N8 | 1.376 | 1.378 | 1.372 | 1.376 | 1.375 | 1.381 | 1.374 |
| C3–C9 | 1.457 | 1.458 | 1.457 | 1.450 | 1.457 | 1.457 | 1.457 |
| N8–C34 | 1.383 | 1.378 | 1.381 | 1.360 | 1.386 | 1.380 | 1.381 |
| C34–O35 | 1.228 | 1.228 | 1.235 | 1.221 | 1.229 | 1.227 | 1.223 |
| C11–N17 | 1.358 | 1.357 | 1.36 | 1.359 | 1.357 | 1.357 | 1.358 |
| C14–Br20 | 1.92 | 1.919 | 1.92 | 1.924 | 1.921 | 1.919 | 1.921 |
| C12–Br21 | 1.916 | 1.917 | 1.914 | 1.914 | 1.918 | 1.913 | 1.914 |
| C29–C32 | 1.471 | 1.348 | 1.466 | 1.471 | 1.471 | 1.472 | 1.430 |
| C32–N33 | 1.231 | – | 1.231 | 1.231 | 1.231 | 1.230 | 1.155 |
| C43-X46 | 1.346 | 1.474 | 1.498 | 1.361 | 1.359 | 1.474 | 1.389 |
| Bond angle (°) | |||||||
| C1–C2–C3 | 102.65 | 103.26 | 102.71 | 102.41 | 102.56 | 103.11 | 102.80 |
| C2–C3–N7 | 112.22 | 112.6 | 112.23 | 112.17 | 112.15 | 112.63 | 112.15 |
| C3–N7–N8 | 109.66 | 109.65 | 109.48 | 109.55 | 109.74 | 109.67 | 109.73 |
| N8–C34–C36 | 119.93 | 119.14 | 102.67 | 120.37 | 119.1 | 119.2 | 120.22 |
| Dihedral angle (°) | |||||||
| C11–C9–C3–N7 | −0.64 | 0.30 | −2.42 | −3.53 | −0.72 | 1.85 | −2.94 |
| C10–C9–C3–C2 | 0.30 | 0.35 | −0.77 | −2.30 | 0.26 | 1.91 | −2.09 |
| C2–C1–C22–C24 | 111.01 | −52.34 | −41.6 | −71.79 | −110.87 | −57.52 | −73.11 |
| N8–C1–C22–C23 | 47.07 | 62.37 | −92.22 | −140.77 | 46.92 | −123.25 | −112.47 |
| C1–N8–C34–035 | −1.37 | 1.16 | −0.61 | −3.23 | −1.30 | −0.47 | −2.35 |
| C38–C36–C34–O35 | −30.08 | 30.81 | −10.79 | −25.60 | −29.55 | 29.72 | −29.56 |
Ma = Experimental data of the compound from Ref.28.
The C2–C3–N7 bond angle for the six compounds was calculated to be 112.22°, 112.6°, 112.23°, 112.17°, 112.15° and 112.63° for M1–M6, respectively. However, N7–N8–C3 was 109.66° for M1, 109.65° for M2, 109.48° for C3, 109.55° for M4, 109.74° for M5 and 109.67° for M6. These two bond angles indicated that the pyrazole ring was distorted from planarity by the aryl and benzoyl rings, as reflected in the dihedral angles (Table 3), thus revealing that aryl and benzoyl groups have more profound effects on the pyrazole ring than the extended R1 and R2 substituents.
Frontier molecular orbital and UV–vis absorption properties
The frontier orbital energies, such as the highest occupied molecular orbital energy (HOMO), lowest unoccupied molecular orbital (LUMO) and band gaps, are critical parameters for kinetic and thermodynamic stability studies, and prediction of reactivity and photochemical properties.41, 42, 43, 44, 45, 46 The frontier orbital molecular overlay revealed that the HOMO overlay was essentially on ring, extending over the two nitrogen atoms of the pyrazole ring, whereas the LUMO overlay was on the phenyl ring for M1, M3, M4 and M5, and was on the benzoyl ring for M1 and M6 (Figure 1). The calculated frontier molecular orbitals LUMO, HOMO and HOMO–LUMO (ΔEg) were, respectively, −2.50, −5.92 and 3.42 eV for M1; −2.77, −5.91 and 3.14 eV for M2; −2.85, −5.87 and 3.02 eV for M3; −2.43, −5.86 and 3.43 eV for M4; −2.42, −5.86 and 3.37 eV for M5; −2.90, −6.06 and 3.16 eV for M6; and −2.26, −6.10 and 3.83 for Ma. The replacement of a cyano group in Ma with NO2 as in M1 led to a decrease in LUMO energy with an increase in HOMO energy, thus resulting in a decrease in ΔEg by 0.41 eV and profound effects on the electron withdrawing capacity of NO2 compared with the CN group. The interchanged M1 F and NO2 positions in M2 further decreased the ΔEg by 0.28 eV with respect to M1, thus indicating that the amide group enhanced the electron withdrawing ability of NO2. M3, M4, M5 and M6 were modeled by the replacement of fluorine in M1 with CH3, OH, OCH3 and NO2, respectively. M3, M5 and M6 presented ΔEg than M1; however, M6 showed the lowest ΔEg because of lowering of the LUMO energy (−2.90 eV) and stabilization of the HOMO, thereby decreasing the π-electron density of the aromatic rings. Thus, M6 was expected to be relatively more reactive toward nucleophiles (Table 2).
Figure 1.

Frontier molecular orbitals for compounds M1–M6.
Other calculated reactivity descriptors such as chemical hardness (η), chemical potential (μ) and global electrophilicity (ω) were, respectively, 1.71, −4.21 and 5.182 eV for M1; 1.57, −4.34 and 5.998 eV for M2; 1.51, −4.36 and 6.295 eV for M3; 1.72, −4.15, and 5.009 eV for M4;1.69, −4.12 and 5.00 eV for M5; and 1.58, −4.48 and 6.351 eV for M6. The μ and ω values for M6 further indicated that the compound would be a good electron acceptor with strong electron pulling effects toward the two NO2 groups. This finding was in agreement with the effect of the NO2 group observed on 4-(3-(2-amino-3,5-dibromophenyl)-1-(4-nitrobenzoyl)-4,5-dihydro-1H-pyrazol-5-yl)benzonitrile by Santhi and Bharathi.28 The electron donating power (ω−) and electron accepting power (ω+) describe the tendency of a molecule to release electrons and to accept electrons, respectively; a smaller ω− indicates a better donor of electron density, and a greater ω+ indicates better accepting electron capacity. The values for ω− demonstrated that M1, M4 and M5 were good electron donors, whereas ω+ indicated the tendency of M2, M3 and M5 to be good electron acceptors, in line with the μ and ω values (Table 4).
Table 4.
Molecular orbitals and reactivity indices.
| Parameter | M1 | M2 | M3 | M4 | M5 | M6 | Ma |
|---|---|---|---|---|---|---|---|
| HOMO (eV) | −5.92 | −5.91 | −5.87 | −5.86 | −5.79 | −6.06 | −6.10 |
| LUMO (eV) | −2.50 | −2.77 | −2.85 | −2.43 | −2.42 | −2.90 | −2.26 |
| HOMO−1 (eV) | −6.71 | −6.62 | −6.10 | −6.41 | −6.32 | −6.90 | −6.79 |
| HOMO−2 (eV) | −7.02 | −6.91 | −6,32 | −6.61 | −6.51 | −7.60 | −7.21 |
| LUMO+1 (eV) | −2.01 | −2.02 | 2.41 | −2.03 | −1.92 | −2.61 | −1.78 |
| ΔEg (eV) | 3.42 | 3.14 | 3.02 | 3.43 | 3.37 | 3.16 | 3.83 |
| η (eV) | 1.71 | 1.57 | 1.51 | 1.715 | 1.685 | 1.58 | 1.92 |
| μ (eV) | −4.21 | −4.34 | −4.36 | −4.145 | −4.105 | −4.48 | −4.18 |
| χ (eV) | 4.21 | 4.34 | 4.36 | 4.145 | 4.105 | 4.48 | 4.18 |
| ω (eV) | 5.182 | 5.998 | 6.295 | 5.009 | 5.00 | 6.351 | 4.562 |
| ω+ (eV) | 3.291 | 4.025 | 4.303 | 3.151 | 3.158 | 4.309 | 2.700 |
| ω− (eV) | 7.501 | 8.365 | 8.663 | 7.296 | 7.263 | 8.789 | 6.880 |
| σ (eV−1) | 0.855 | 0.785 | 0.755 | 0.8575 | 0.8425 | 0.7900 | 0.5208 |
| ΔEn (eV) | 1.827 | 2.444 | 2.670 | 1.722 | 1.7378 | 2.661 | 1.334 |
| ΔEe (eV) | 10.247 | 11.124 | 11.410 | 10.011 | 9.948 | 11.621 | 9.715 |
Ma = Theoretical data of the compound from Ref.28.
The absorption peaks, oscillator strength and percentage of the molecular orbitals involved in transitions, calculated for M1–M6 at B3LYP/6-31G (p,d), are displayed in Table 5. The transition probability, as measured by oscillator strength (OS), corresponded to the fraction of negative charges (electrons) that accomplished a given transition. OS values <0.005 were considered to indicate transitions emanating from low absorption bands in the studied compounds; thus, only transitions with OS > 0.005 were considered in this study. M1 showed four strong absorptions, at 304.69, 333.69, 349.07 and 408.24 nm, arising from HOMO-2 → LUMO (94%), HOMO-1 → LUMO (96%), HOMO → LUMO + 1 (89%) and HOMO → LUMO (98%), respectively; the longest λmax was characterized as a π–π∗ transition arising from HOMO → LUMO. For M2, five strong absorption peaks were identified, at 305.98, 337.57, 341.62, 356.73 and 453.95 nm, with an OS of 0.0463, 0.0081, 0.2840, 0.0359 and 0.0285, respectively. The molecular orbitals’ percentage contributions to these transitions were as follows: HOMO-3 → LUMO (42%) and HOMO-4 → LUMO (35%) for 305.98 nm; HOMO-2 → LUMO (87%) for 337.57 nm; HOMO → LUMO+1 86% for 342 nm; HOMO-2 → LUMO 99% for 358 nm; and HOMO → LUMO 99% for 453.95 nm.
Table 5.
Calculated absorption peaks, oscillation strength and molecular orbitals involved in transitions for M1–M6.
| λmax (nm) | Oscillation strength | MO involved in transitions |
|---|---|---|
| M1 | ||
| 304.60 | 0.0349 | HOMO-2 → LUMO 94% |
| 333.69 | 0.0125 | HOMO-1 → LUMO 96% |
| 349.07 | 0.2918 | HOMO → LUMO+1 89% |
| 408.24 | 0.0105 | HOMO → LUMO 98% triplet |
| M2 | ||
| 305.98 | 0.0463 | HOMO-3 → LUMO 42% HOMO-4 → LUMO 35% |
| 337.57 | 0.0081 | HOMO-2 → LUMO 87% |
| 341.62 | 0.2840 | HOMO → LUMO+1 86% |
| 356.73 | 0.0359 | HOMO-1 → LUMO 99% |
| 453.97 | 0.0285 | HOMO → LUMO 99% triplet |
| M3 | ||
| 408.96 | 0.0131 | HOMO-2 → LUMO 81% |
| 420.81 | 0.0177 | HOMO-5 → LUMO 67% |
| 426.26 | 0.0243 | HOMO → LUMO+1 44% HOMO-1 → LUMO+1 33% |
| 439.54 | 0.0394 | HOMO-1 → LUMO+1 45% HOMO → LUMO+1 38% |
| 445.90 | 0.0082 | HOMO-1 → LUMO 88% |
| 494.49 | 0.0059 | HOMO → LUMO 96% triplet |
| M4 | ||
| 309.69 | 0.0983 | HOMO-1 → LUMO+1 94% |
| 332.48 | 0.0142 | HOMO-2 → LUMO 92% |
| 341.18 | 0.0057 | HOMO-1 → LUMO 95% |
| 347.16 | 0.3138 | HOMO → LUMO+1 86% |
| 409.31 | 0.0109 | HOMO → LUMO 98% triplet |
| M5 | ||
| 313.41 | 0.0901 | HOMO-1 → LUMO+1 96% |
| 337.82 | 0.0253 | HOMO-2 → LUMO 92% |
| 347.01 | 0.2753 | HOMO → LUMO+1 79% |
| 351.40 | 0.0416 | HOMO-1 → LUMO 93% |
| 417.91 | 0.0115 | HOMO → LUMO 98% triplet |
| M6 | ||
| 329.32 | 0.0199 | HOMO-1 → LUMO+1 97% |
| 342.96 | 0.2651 | HOMO → LUMO+2 89% |
| 349.68 | 0.0460 | HOMO-1 → LUMO 99% |
| 407.32 | 0.0125 | HOMO → LUMO+1 99% triplet |
| 451.04 | 0.0267 | HOMO → LUMO 99% triplet |
In addition, M3 showed five strong absorption peaks, with HOMO-2 → LUMO (81%) for 408.96 nm; HOMO-5 → LUMO (67%) for 420.81 nm; HOMO-2 → LUMO+1 (44%) and HOMO-1 → LUMO +1 (33%) for 426.26 nm; HOMO-1 → LUMO+1 (45%) and HOMO → LUMO+1 (38%) for 439.54 nm; HOMO-1 → LUMO (88%) for 445.90 nm; and HOMO → LUMO (96%) for 494.49 nm, arising from a low absorption band of 0.0059 OS. For M4, 309.69, 332.48, 347.16 and 409.31 nm were the four absorption peaks with OS higher than 0.005, arising from HOMO-1 → LUMO+1 (94%), HOMO-2 → LUMO (92%), HOMO → LUMO+1 (86%) and HOMO → LUMO (98%), respectively. Likewise, 313.41, 337.82, 347.01, 351.47 and 417.91 nm were five absorption peaks with OS higher than 0.005 arising from HOMO-1 → LUMO+1 (96%), HOMO-2 → LUMO (92%), HOMO → LUMO+1 (79%), HOMO-1 → LUMO (93%) and HOMO → LUMO (98%), respectively, for M5. The 417.91 nm absorption peak with the highest OS value was characterized as a π–π∗ transition. For M6, five strong absorption peaks at 329.32, 342.96, 349.68, 407.32 and 451.04 nm were identified with OS values of 0.0199, 0.2651, 0.0460, 0.0125 and 0.0267, respectively: HOMO-1 → LUMO+1 (97%) for 329 nm; HOMO → LUMO+2 (89%) for 324.9 nm; HOMO-1 → LUMO (99%) for 349.68 nm; HOMO → LUMO+1 (99%) for 407 nm; and HOMO → LUMO (99%) for 451 nm. The absorption peak with the highest OS was characterized as a π–π∗ transition, and the absorption peak next to the highest OS was characterized as an n–π∗ transition. All the compounds had one or two triplet transitions, thus potentially indicating that they possessed both orbital unpaired and spin unpaired electrons. Consequently, singlet transitions with sufficiently long lifetimes might have led to de-inversion of the spin of some of the electrons, thus generating a triplet.
Molecular electrostatic potential analysis
The static distribution of charge density is associated with the electrostatic potential map (MEP) distribution of charge on a molecule, and is a useful parameter for analyzing and predicting the responsiveness of a molecule toward an incoming electrophile or nucleophile during reaction initiation.47 This parameter has been successfully used to explain stacking and self-assembly of polymeric molecules and dyes, and the orientation of molecules in three-dimensional crystals.48 The MEP was simulated according to the optimized geometry obtained from DFT calculation to predict electrophilic and nucleophilic sites of attack. In Figure 2, blue indicates a positive region indicating electrophilic centers/electron-deficient areas; red represents the negative regions (areas with excess electrons) for nucleophilic reactivity/electron-rich centers of a molecule; and green represents regions with essentially zero potential.49, 50, 51 Generally, the MEP ranged from red to orange to yellow to green to blue (Figure 1). The negative (red) regions were located at the carbonyl oxygen and cyano group, thus indicating the most probable sites for electrophilic attack. The regions with excess electrons could result in intermolecular pulling of positive regions of nearby molecules, thus distorting the π–π stacking arrangement of these molecules.
Figure 2.

Molecular electrostatic potential (MEP) diagram of compounds M1–M6.
Vibration frequencies
Vibration frequency is a powerful and effective technique for identification of the organic functional groups of organic compounds; it can distinguish a molecular conformer form either its tautomer or isomer. Comparison of experimental and calculated vibrational modes with appropriate functional group assignment provides meaningful and useful information for understanding fairly complex systems.34 In addition, when experimental data are not available, the theoretical calculated vibration frequencies can be used with a reasonable level of accuracy to understand the effects of functional groups on molecules. Scaling of the calculated wave numbers increases the reliability and utility of the calculated frequency28,52; thus, a 0.9608 scaling factor was used in this work. The DFT-B3LYP/6-31G(d,p) level of calculation was used to determine the vibration frequencies of M1–M6 compounds, and the results were compared with those of 4-(3-(2-amino-3,5-dibromophenyl)-1-(benzoyl)-4,5-dihydro-1H-pyrazol-5-yl)benzonitriles.28 The scaled and unscaled vibration frequencies calculated with the DFT method are shown in Table 6. The calculated symmetric and asymmetric N–H stretching vibrations were 3394 and 3551 cm−1 for M1; 3399 and 3552 cm−1 for M2; 3385 and 3542 cm−1 for M3; 3389 and 3549 cm−1 for M4; 3391 and 3551 cm−1 for M5; and 3399 and 3551 cm−1 for M6. The value has been experimentally observed to be 3433 cm−1 and calculated to be 3452 cm−1 for Ma.28 Experimental observations have been conducted at 3390, 3378 and 3360 cm−1 in 1-((1,3-diphenyl-1H-pyrazol-4-yl)methylene)-4-phenylsemicarbazide, 4-phenyl-1-((1-phenyl-3-p-tolyl-1H-pyrazol-4-yl)methylene)semicarbazide and 1-((3-(4-hydroxyphenyl)-1-phenyl-1H-pyrazol-4-yl)methylene)-4-phenylsemicarbazide, respectively.8 The impure in plane bending N–H vibration (βN-H) was blended with vC = C stretching, as shown in Table 6; thus, the βN-H was calculated to be 1345, 1394, 1349, 1344, 1346 and 1345 cm−1 for M1–M6, respectively, but has been reported to be 1398 and 1402 cm−1 for Ma.28
Table 6.
Calculated IR frequencies (cm−1) at B3LYP/6-31G(d,p).
| Assignment |
M1 |
M2 |
M3 |
M4 |
M5 |
M6 |
Ma |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Unscaled | Scaled | Unscaled | Scaled | Unscaled | Scaled | Unscaled | Scaled | Unscaled | Scaled | Unscaled | Scaled | Expt | Scaled | |
| νN-Hsyn | 3532 | 3394 | 3538 | 3399 | 3523 | 3385 | 3527 | 3389 | 3529 | 3391 | 3538 | 3399 | 3433 | 3452 |
| νN-HAsyn | 3696 | 3551 | 3697 | 3552 | 3687 | 3542 | 3694 | 3549 | 3696 | 3551 | 3696 | 3551 | – | |
| νC-Hsyn | 3110 | 2988 | 3102 | 2980 | 3099 | 2978 | 3106 | 2984 | 3113 | 2991 | 3106 | 2984 | 2924 | 2980 |
| νC-HAsyn | 3239 | 3112 | 3250 | 3123 | 3195 | 3070 | 3234 | 3107 | 3233 | 3106 | 3231 | 3104 | 3071 | 3104 |
| νC = O | 1723 | 1655 | 1730 | 1662 | 1686 | 1620 | 1720 | 1653 | 1725 | 1657 | 1731 | 1663 | 1655 | 1646 |
| νC = N | 1642 | 1578 | 1637 | 1573 | 1632 | 1568 | 1634 | 1570 | 1634 | 1570 | 1639 | 1575 | 1619 | 1622 |
| νC = C, βN-H | 1345 | 1292 | 1347 | 1294 | 1349 | 1296 | 1344 | 1291 | 1346 | 1293 | 1345 | 1292 | 1398 | 1402 |
| vC = C | 1618 | 1555 | 1614 | 1551 | 1579 | 1517 | 1617 | 1554 | 1618 | 1555 | 1616 | 1553 | 1329 | 1353 |
| βC-H | 1192, 1177, 1172 | 1145, 1131, 1127 | 1213, 1188, 1176 | 1165, 1142, 1130 | 1197, 1189, 1178 | 1150, 1143, 1132 | 1203, 1188, 1176 | 1156, 1141, 1130 | 1201, 1183, 1171 | 1154, 1137, 1125 | 1203, 1183, 1177 | 1155, 1137, 1131 | 1165 | 1175 |
| τC-H | 998 920, 886 | 959, 884, 851 | 991, 948, 843 | 952, 910, 809 | 996, 952 856, | 957, 915, 822 | 966, 956, 886 | 928, 918, 790 | 979, 964, 895 | 941, 926, 860 | 998, 964, 890 | 959, 926, 855 | 987 | 980 |
| νC-F | 1287 | 1237 | 1287 | 1237 | – | – | – | – | – | – | – | – | 1223 | – |
| νC-NO2 | 1397 | 1342 | 1397 | 1342 | 1392 | 1337 | 1397 | 1342 | 1397 | 1342 | 1397 | 1342 | 1450a, 1394a | 1489a, 1397a |
| νNO2 | 1671 | 1605 | 1668 | 1603 | 1631 | 1567 | 1671 | 1605 | 1670 | 1605 | 1669, 1619, 1616 | 1604, 1556, 1553 | ||
| vNH2 | 1624 | 1560 | 1623 | 1559 | 1624 | 1560 | 1623 | 1559 | 1820 | 1556 | 1623 | 1559 | ||
| vC-NH2 | 1658 | 1593 | 1662 | 1597 | 1654 | 1589 | 1664 | 1599 | 1663 | 1598 | 1661 | 1596 | ||
| νOH | 3819 | 3669 | ||||||||||||
ν = stretching; β = in-plane bending: τ = out-of-plane bending, Sci = scissors; Ma and a = 4-(3-(2-amino-3,5-dibromophenyl)-1-(4-nitrobenzoyl)-4,5-dihydro-1H-pyrazol-5-yl)benzonitrile.28.
The aromatic C–H stretching vibrations for the compound were in the range of 3250–3102 cm−1, but were scaled to be in the region 2984–2978 cm−1. The C–H asymmetric stretching vibration for compounds M1–M6 was calculated to be 3112, 3195, 3070, 3107, 3106 and 3104 cm−1; the asymmetric C–H symmetric vibrations were 2988, 2980, 2978, 2984, 2991 and 2984 cm−1 for M1–M6, respectively. The C–H in-plane bending (βC-H) vibrations were 1145, 1131 and 1127 cm−1 for M1; 1165, 1142 and 1130 cm−1 for M2; 1150, 1143 and 1132 cm−1 for M3; 1156, 1141 and 1130 cm−1 for M4; 1154, 1137 and 1125 cm−1 for M5; and 1155, 1137 and 1131 cm−1 for M6. The βC-H vibration for 4-(3-(2-amino-3,5-dibromophenyl)-1-(benzoyl)-4,5-dihydro-1H-pyrazol-5-yl)benzonitriles has been experimentally observed at 1165 cm−1 and calculated to be 1175 cm−1.28 The C–H out-of-plane (τC-H) vibrations appeared in the region of 959–851 cm−1 for M1; 952–809 cm−1 for M2; 957–822 cm−1 for M3; 928–790 cm−1 for M4; 941–860 cm−1 for M5; and 959–855 cm−1 for M6.
The C = O stretching vibrations appeared in the region of 1655 cm−1 for M1; 1662 cm−1 for M2; 1620 cm−1 for M3; 1653 cm−1 for M4; 1657 cm−1 for M5; and 1663 cm−1 for M6. These vibrations have been observed at 1662 cm−1 for N-[(4-chlorophenyl)]-4-oxo-4-[oxy] butane amide,53 1637 cm−1 for diethyl 1H-pyrazole-3,5-dicarboxylate,25 1660 cm−1,28 1701 cm−1 for 4-chloro-3,5-dimethyl-1H-pyrazol-1-yl) (p-tolyl) methanone26 and 1683 cm−1 for 4-phenyl-1-((1-phenyl-3-p-tolyl-1H-pyrazol-4-yl)methylene)semicarbazide.8 The calculated vC-NO2 stretching was 1337 cm−1 for M3 but was 1342 cm−1 for other compounds. The νNO2 (acceptor group) stretching was calculated to be 1605, 1603, 1567, 1605 and 1605 cm−1 for M1–M5, respectively; meanwhile, M6 showed three vibrational modes for νNO2, at 1604, 1556 and 1553 cm−1. However, these bands were calculated for N-methyl-N-(2,4,6-trinitrophenyl) nitramide to be in the region of 1633–1591 cm−1 and 1388–1348 cm−1, and were assigned to vNO2 asymmetric and symmetric stretching vibrations, respectively.54 The vNH2 group (electron donor) stretching was 1560 cm−1 for M1 and M3; 1559 cm−1 for M2, M4 and M6; and 1556 cm−1 for M5. Generally, aromatic compounds containing fluorine show vC–F stretching in the region 1000–1400 cm−1.55 However, compounds M1 and M2 showed these vibrations at 1237 cm−1 and have been reported at 1223 cm−1.28
Polarizability and hyperpolarizability
The static polarizability (α), hyperpolarizability (β) and electric dipole moment (μ) based on the finite field method were calculated for the six compounds at DFT B3LYP/6-31G(d,p). The total static dipole moment (μ), mean polarizability (αo) and mean first hyperpolarizability (β0) were defined by using the x,y,z component, as shown in equations 8, 9, 10, 11, 12, 13, 14):
| 8 |
| 9 |
| 10 |
| 11 |
| 12 |
| 13 |
| 14 |
The presence of electron donating and electron withdrawing groups on π-conjugated molecules changes the ground state charge distribution and enhances asymmetric polarization of the molecules. Consequently, large nonlinear responses are correlated with a rapid response time; therefore, these molecules are desirable candidates for NLO applications.56,57 Any molecule with a minimum value of 4.187944 × 1−30 esu for the first hyperpolarizability is considered a good candidate for NLO applications.58 Therefore, these molecules’ NLO properties, polarizability and hyperpolarizability were assessed. The dipole moment, an essential parameter explaining the intermolecular interactions of molecules, is expected to have a higher value with stronger intermolecular interactions. The dipole moment values calculated for M1–M6 were 4.57, 4.06, 5.47, 6.45, 5.32 and 5.14 D (1D = 3.34 × 10−34 C m), respectively. The αo calculated for M1–M6 was 3.29, 3.28, 3.26, 3.26, 3.28 and 3.63 × 10−23 esu, respectively. The αo for Ma (4-[3-(2-amino-3,5-dibromophenyl)-1-(4-fluorobenzoyl)-4,5-dihydro-1H-pyrazol-5-yl]benzonitrile) has been reported to be 3.31 × 10−23 esu.28 However, the β0 for M1–M6 was 6.04, 5.21, 6.15, 6.74, 6.02 and 7.26 3.31 × 10−30 esu, respectively, and has been reported to be 8.47 × 10−30 esu for Ma.28 The β0 values for the studied compounds were lower than that of Ma, but approximately 16 times higher than that of urea (0.372 × 10−30 esu).57 The model compounds had higher β0 values than those of diphenylmethylidene-5-methyl-1H-pyrazole-3-carbohydrazide (β0 = 2.08 × 10−30 esu)59 and diethyl-1-H-pyrazole-3,5-dicarboxylate (β0 = 1.01 × 10−30 esu).24 Interchanging the position of NO2 and F, as shown in M1 and M2, led to a decrease in β0 by 0.83 × 10−30 esu and an increase in absorption wavelength (λmax) by 75 nm in M2. Thus, the position, nature and point of attachment of substituents on the model compound strongly affect the properties of the compound.57 In addition, M6 with two NO2 groups increased β0; therefore, the presence of an electron withdrawing group (nitro) on the phenyl ring contributed to higher hyperpolarizability values, possibly because of an inductive effect of the electron withdrawing group on the electronic density in the molecule.28 Our results showed that M1–M6 compounds may be suitable for NLO applications, and the magnitude of molecular hyperpolarizability was improved by functional group modification (Table 7).
Table 7.
Dipole moment, polarizability and hyperpolarizability of M1–M6.
| Dipole moment | ||||||
|---|---|---|---|---|---|---|
| Parameter | M1 | M2 | M3 | M4 | M5 | M6 |
| μx | 3.2717 | 2.4264 | −4.4201 | 4.22 | 1.2246 | −3.7596 |
| μy | −2.0652 | −2.9515 | −2.3989 | −4.2089 | 4.9235 | 1.4025 |
| μz | −2.4399 | 1.3697 | 2.1446 | −2.4754 | 1.5946 | 3.2168 |
| μtot | 4.57 | 4.06 | 5.47 | 6.45 | 5.32 | 5.14 |
| Polarizability/a.u. | ||||||
| αxx | −242.284 | −236.429 | −258.740 | −246.939 | −237.601 | −255.538 |
| αxy | 11.3659 | 14.9274 | −13.1037 | 6.41700 | −31.0350 | −6.7400 |
| αyy | −218.433 | −225.704 | −184.970 | −203.456 | −211.066 | −262.856 |
| αxz | 12.0644 | −4.6114 | 10.8756 | 12.2329 | −4.94560 | 13.6039 |
| αyz | −16.3197 | −2.0474 | 10.8167 | −16.7704 | −25.3816 | 6.80760 |
| αzz | −206.567 | −202.342 | −216.967 | −208.855 | −215.010 | −217.261 |
| αtot | −222.428 | −221.491 | −220.225 | −219.749 | −221.226 | −245.218 |
| α × 10−23 esu | 3.29 | 3.28 | 3.26 | 3.26 | 3.28 | 3.63 |
| Hyperpolarizability/a.u. | ||||||
| βxxx | 415.4398 | 307.9092 | −499.813 | 399.1294 | 296.4138 | −451.768 |
| βxxy | −143.263 | −61.1607 | −94.8057 | −99.1944 | 215.4269 | −151.772 |
| βxyy | 160.3521 | 223.159 | −36.6282 | 215.598 | 85.0875 | −278.063 |
| βyyy | −5.6029 | −50.0772 | −72.1537 | −181.966 | 262.2293 | 244.9397 |
| βxxz | −37.1159 | 8.9984 | 42.1252 | −29.5710 | −7.1656 | 55.6498 |
| βxyz | 84.942 | −9.2125 | 34.8642 | 81.0249 | 105.6754 | 53.0815 |
| βyyz | −31.9896 | 53.7171 | −11.3035 | −51.069 | 27.7063 | 100.8334 |
| βxzz | 103.2154 | 62.224 | −149.912 | 109.4585 | 108.3569 | −91.1906 |
| βyzz | −4.1993 | 13.766 | −18.9092 | 2.0788 | 18.0156 | −6.0768 |
| βzzz | 5.8459 | −20.1728 | −5.7925 | 4.1569 | −6.0768 | −0.3585 |
| βtot | 698.9145 | 602.749 | 711.516 | 779.86 | 697.037 | 840.259 |
| β × 10−30 esu | 6.04 | 5.21 | 6.15 | 6.74 | 6.02 | 7.26 |
For (α): 1 a.u. = 0.1482 × 10−24 esu and for (β): 1 a.u. = 8.6393 × 10−33 esu).
Molecular docking
The antihypertensive and antioxidant properties of the studied pyrazole derivatives (M1–M6) were investigated via molecular docking, and the obtained results were compared with the results for rolipram and taurine. The antihypertensive activity of the compounds was evaluated by considering their inhibitory activity against phosphodiesterases (PDEs). PDEs are enzymes participating in cAMP and cGMP homeostasis by acting on phosphodiester bonds.60,61 When PDE is inhibited, the cAMP and cGMP levels increase, thereby decreasing calcium levels in cells. Consequently, blood vessels are vasodilated with relaxing of the blood vessels, and the risk of hypertension markedly decreases.62 However, phosphodiesterase 4 (PDE4) has been reported to be the main enzyme in the hydrolysis of cAMP, another mediator that controls pro-inflammation and anti-inflammation.61,62 To investigate the PDE inhibitory activity of the compounds, we performed molecular docking of the compounds on PDE4 (PDB ID: 1RO6) downloaded from the Protein Data Bank. The downloaded 1RO6 was complexed with the rolipram drug, a selective PDE4 inhibitor that increases the quantity of cAMP in immune and nerve cells63,64 and compared the docking results.
Antioxidants are common food additives that inhibit cellular damage mainly through their free radical scavenging ability.64,65 Free radicals are reactive oxygen species produced in the body through various metabolic processes, in phagocytosis, in prostaglandin synthesis and in the cytochrome P-450 system, as a result of exposure to different physiochemical conditions or pathological states.66 Excessive free radicals in the body lead to a condition known as oxidative stress, which harmfully alters proteins, lipids and DNA, and can initiate the progression of pathologies including immune system deterioration, atherosclerosis and abnormal cell growth leading to nucleofugality cancer.66 Analysis of pyrazoline derivatives has indicated that they are promising antioxidants.67, 68, 69 Therefore, we examined the model compounds for their antioxidant ability by docking them against a dehydrogenase inhibitor downloaded from the Protein Data Bank (PDB ID: 5ADH). Taurine is an antioxidant involved in protection of hepatic tissue by deactivating reactive oxygen species, thereby removing formation of osmoregulation, calcium homeostasis, lipid peroxidation and protein carbonyl formation, detoxification, cytoprotection and neuromodulation.70, 71, 72 Reports have indicated that taurine concentrations are inversely associated with diabetes complications.73, 74, 75 The optimized structures of the model compounds were used for docking simulations. The docking was performed with AutoDock Tool 1.5.6 and AutoDock Vina 1.1.2; proteins were treated; and molecular interactions between receptors and ligands were visualized with Edupymol version 1.7.4.4 and BIOVIA Discovery studio 2019, as previously reported.76, 77, 78, 79, 80, 81, 82, 83, 84 The grid box using AutoDock tool before use of AutoDock Vina for docking calculations was as follows: center (X = 4.834, Y = 15.305, Z = 24.227) and size (X = 64, Y = 52, Z = 74) for 5ADH, and (X = 32.382, Y = 72.334, Z = 31.711) and size (X = 62, Y = 58, Z = 66) for 1RO6, with default exhaustiveness (exhaustiveness = 8) for steady docking calculation speed.
A recent study has indicated that molecular docking of pyrazole derivatives such as carboxy pyrazole derivatives with various cancer cells (breast, MCF-7; bone marrow, K-562; and cervix, HeLa),6 aryl pyrazoles with tyrosinase enzyme,85 pyrazole-phenyl semicarbazone derivatives with α-glucosidase,8 imidazole–pyrazole conjugates with α-glucosidase10 and 4-aryl-N-(5-methyl-1H-pyrazol-3-yl)benzamides with Acinetobacter baumannii protein11 was in agreement with experimental observations, and has also detailed the nature of the protein-ligand interactions. The use of molecular docking for in silico screening of bioactivity of heterorganic compounds, as well as drug design and discovery, has become frequent and relevant in pharmacology. Therefore, docking serves as a reliable and time saving method for simulation of binding poses of ligand conformations in the active sites of receptors, and calculation of the binding affinity and interactions of protein-ligand complexes.86
The binding affinity of the stable ligands docked with dehydrogenase inhibitor (PDB ID: 5ADH) ranged from −8.8 to 9.3 kcal/mol: M1 (−9.0 kcal/mol), M2 and M3 (−9.3 kcal/mol) and M4, M5 and M6 (−8.8 kcal/mol). Similar binding affinities (−8.3 to −9.5 kcal/mol) have been reported for 1-benzyl-2-phenyl-1H-benzimidazole derivatives docked with dehydrogenase (PDB ID: 5ADH).87 The binding affinity calculated for taurine was −3.7 kcal/mol, as shown in Table 8, thus indicating that these compounds may be excellent inhibitors for APO-liver dehydrogenase and thus possess good antioxidant properties. Ligand interactions with the binding pocket of dehydrogenase (PDB ID: 5ADH) revealed that ARG 369 and ARG 202 are involved in hydrogen bond interactions with the NO2 group of M1; ARG 295 and ARG 47 are involved in a π-alkyl interaction; Val 294 is involved in a π-cation interaction, and VAL 203 is involved in π-sigma interactions with M1. The amino acid residues GLY 202, VAL 203 and ILE 269 are involved in hydrogen bond interactions; PRO 295, ARG 47 and PRO 296 are involved in π-alkyl interactions; VAL 203 is involved in π-sigma interactions; and CYS 46 is involved in π-sulfur interactions with M2. In addition, GLY 202, VAL 203, ARG 47 and ILE 269 are involved in hydrogen bonding, and ILE 269, VAL 203, GLY 202, PRO 295 and ARG 47 form π-alkyl interactions with M3. PRO 295 and ARG 47 are involved in π-alkyl interactions; VAL 203 is involved in π-sigma interactions; ARG 369 and GLY 202 form hydrogen bond interactions, and HIS 51 is involved in π-cation interactions with M4. Amino acid residues ARG 47, PRO 295 and PRO 296 are involved in π-alkyl interactions; SER 48 and ARG 369 are involved in hydrogen bonding; VAL 203 forms a π-sigma interaction; and CYS 46 forms π-sulfur interactions with M5. Furthermore, SER 48, VAL 203 and ILE 269 are involved in hydrogen bonding; ARG 47, PRO 295 and PRO 296 form π-alkyl interactions; and VAL 203 is involved in a π-sigma interaction with M6 (Table 8 and Figure 3). Taurine is involved in hydrogen bonding with SER 367, ARG 369 and ARG 47, and also participates in Van der Waals interactions with VAL 203 and CYS 46. Similar binding modes for protein-ligand interactions have been observed for 1-benzyl-2-phenyl-1H-benzimidazole derivatives docked with dehydrogenase (PDB ID: 5ADH), and ILE 269, VAL 203, GLY 202, PRO 295 and ARG 47 amino acid residues have been found to be involved in the interactions.87
Table 8.
Binding affinity (ΔG) and hydrogen bonding interactions of the 5ADH and 1RO6 receptors with compounds M1–M6.
| Ligand | 5ADH receptor |
1RO6 receptor |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Binding affinity ΔG (kcal/mol) | Inhibition constant Ki (μM) | H-bond with ligands | H-bond distance (Å) | Binding affinity ΔG (kcal/mol) | Inhibition constant Ki (μM) | H-bond with ligands | H-bond distance (Å) | ||
| M1 | −9.0 | 0.25 | ILE'269 VAL'294 GLY'202 ARG'269 ILE'368 |
2.9 2.4 2.3 2.1 3.3 |
−9.7 | 0.07 | GLU'304 ASP'392 ASP'275 |
3.2 3.2 3.5 |
|
| M2 | −9.3 | 0.15 | ILE'269 VAL'203 |
2.7 2.5 |
−9.2 | 0.17 | SER'282 GLN'417 |
2.7 2.4 |
|
| M3 | −9.3 | 0.15 | ILE'269 ARG'47 SER'367 ILE'368 GLY'202 |
2.7 2.4, 2.6 3.2 3.2 2.7 |
−9.4 | 0.12 | HIS'234 ASN'395 GLN'443 |
2.2 2.0 3.3, 2.0 |
|
| M4 | −8.8 | 0.35 | ILE'269 VAL'294 ARG'369 ILE'368 GLY'202 |
3.0 2.8 2.1 2.3 2.3 |
−9.9 | 0.05 | GLU'304 GLN'417 GLY'280 ASP'392 ASP'275 |
3.5 2.8 2.8 3.1 3.5 |
|
| M5 | −8.8 | 0.35 | ARG'369 | 2.2 | −8.5 | 0.58 | GLU'304 ASP'275 HIS'234 |
2.4 3.5 2.5 |
|
| M6 | −8.8 | 0.35 | SER'48 ILE'269 VAL'203 |
2.6 2.6 2.5 |
−8.9 | 0.29 | THR'345 HIS'307 HIS'234 ASN'395 |
3.3 2.5 2.4 2.4 |
|
| Taurine | −3.7 | SER 367 ARG 369 ARG 47 |
2.6 2.4 2.3 |
Rolipram | −8.8 | 0.35 | ASP'392 HIS'234 GLN'443 |
3.6 2.1 3.3, 2.4, 3.3 |
|
Figure 3.




Docked complexes of the 5ADH receptor with compounds M1–M6, showing interactions in the binding pocket.
The ligand bound at the active site of the 1RO6 receptor (Figure 4) revealed that MET 347 and ILE 410 are involved in alkyl-π interactions; PHE 446, PHE 414 and VAL 281 form π-π stacking, amide-π stacking and π-π T-stacking interactions; and GLU 413, GLY 280 and GLN 417 form fluorine-π interactions with M1. Amino acid residues PHE 446 and PHE 414 are involved in π-π stacking and form π-π T-stacking interactions; MET 347 and ILE 310 are involved in alkyl-π interactions; CYS 432, SER 282 and GLN 417 participate in hydrogen bonding; ASP 392 and ASP 275 form fluorine-π interactions; and HIS 234 and ASP 392 are involved in π-cationic and anionic interactions with M2. In addition, MET 347, PHE 446, ILE 410, TYR 233, LEU 303, HIS 278 and HIS 234 all form alkyl-π interactions, and ASN 395, HIS 234 and GLN 443 are involved in hydrogen bond interactions with M3. Amino acids PHE 414, VAL 281 and PHE 446 are involved in amide-π and π-π T-stacking interactions; GLN 417 forms hydrogen bond interactions; MET 347 forms alkyl-π interactions; and VAL 281 is involved in carbon hydrogen bond interactions with M4. MET 347, PHE 414 and ILE 410 form alkyl- π interactions; PHE 446 and TYR 233 are involved in π-π T-stacking and π-π T-stacking interactions; and HIS 234 forms hydrogen bond and π-cationic interactions with M5. Moreover, MET 347 and ILE 410 form alkyl-π interactions; ASN 395, HIS 234 and HIS 307 are involved in hydrogen bond interactions; TYR 233 forms π-π stacking interactions; and HIS 234 is involved in π-cationic interactions with M6. Rolipram is involved in π-π and π-π T-stacking interactions with PHE 446 and TYR 233; hydrogen bonding with GLN 443 and HIS 234; π-sigma interactions with ILE 410; and alkyl-π interactions with PHE 446 and MET 431 (Table 8 and Figure 4). The free energy for binding of rolipram with PDE (PDB ID: 1RO6) in the active pocket was −9.7, −9.2, −9.4, −9.9, −8.5 and −8.9 kcal/mol for M1–M6, respectively, whereas −8.8 kcal/mol was calculated for rolipram. These findings suggest that all model compounds except M5 exhibit PDE inhibitory activity than rolipram drug.
Figure 4.




Docked complexes of the 1RO6 receptor with compounds M1–M6, showing interactions in the binding pocket.
Conclusion
DFT and molecular docking methods were performed on a set of (4-fluorophenyl)[5-(4-nitrophenyl)-3-phenyl-4,5-dihydro-1H-pyrazol-1-yl]methanone derivatives to calculate their molecular properties such as global electrophilicity, electron donating and accepting power, electrostatic charge distribution, and anti-hypertensive and antioxidant activity. The calculated ω+ revealed that the electrophilic nature of the compounds was increased by the presence of electron withdrawing groups, and the effect was particularly pronounced in the compound with two NO2 groups, M6. MEP analysis showed that amide and nitro groups on the compounds were centers of electrophilic attacks, and the magnitude of the molecular hyperpolarizability suggested that the compounds might have NLO properties.
The binding affinity for the protein-ligand complexes ranged from −8.8 to −9.3 kcal/mol—values higher than that of taurine, an antioxidant involved in inhibition of dehydrogenase (PDB ID: 5ADH) in hepatic tissue. For PDE (PDB: 1RO6), the binding affinity ranged from −8.5 to −9.9 kcal/mol, whereas that of rolipram, a selective inhibitor of PDE4, was −8.8 kcal/mol, thereby indicating that only M5 (−8.5 kcal/mol) had a lower binding affinity than rolipram. Thus, the docking results showed that these compounds may be excellent antioxidant and anti-inflammatory agents.
Source of funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Conflict of interest
The authors have no conflict of interest to declare.
Ethical approval
Not applicable.
Authors contributions
SB, OOA, OAK: Conceptualization; OAK, IAO, OTE, AMD: Methodology; IAO, SB, OTE, LDF, OAK, OAD, AIO, AMD and OOA: Writing- Original draft preparation; OAK and AMD: Software and Visualization; OAD and AIO: Data curation; IAO, SB, OTE, LDF, OAK, OAD, AIO, AMD and OOA: Reviewing and Editing; SB, OOA: Supervision: SB: Validation: OAK. All authors have critically reviewed and approved the final draft and are responsible for the content and similarity index of the manuscript.
Footnotes
Peer review under responsibility of Taibah University.
References
- 1.Holzer W., Gruber H. N1-Substituted 3,5-dimethoxy-4-halogeno-1H-pyrazoles: synthesis and NMR study. J Heterocycl Chem. 1995;32(4):1351–1354. [Google Scholar]
- 2.Haddad N., Baron J. Novel application of the palladium-catalyzed N-arylation of hydrazones to a versatile new synthesis of pyrazoles. J Baron Tetrahedron Lett. 2002;43:2171–2173. doi: 10.1016/S0040-4039(02)00245-9. [DOI] [Google Scholar]
- 3.Zheng Y., Zheng M., Ling X., Liu Y., Xue Y., An L., Gu N., Jin M. Design, synthesis, quantum chemical studies and biological activity evaluation of pyrazole–benzimidazole derivatives as potent Aurora A/B kinase inhibitors. Bioorg Med Chem Lett. 2013;23:3523–3530. doi: 10.1016/j.bmcl.2013.04.039. [DOI] [PubMed] [Google Scholar]
- 4.Fustero S., Sánchez-Roselló M., Barrio P., Simón-Fuentes A. From 2000 to Mid-2010: a fruitful decade for the synthesis of pyrazoles. Chem Rev. 2011;111:6984–7034. doi: 10.1021/cr2000459. [DOI] [PubMed] [Google Scholar]
- 5.Ansari A., Ali A., Asif M. Biologically active pyrazole derivatives. New J Chem. 2017;41:16–41. doi: 10.1039/C6NJ03181A. [DOI] [Google Scholar]
- 6.Channar P.A., Afzal S., Ejaz S.A., Saeed A., Larik F.A., Mahesar P.A., Lecka J., Sévigny J., Erben6 M.F., Iqbal J. Exploration of carboxy pyrazole derivatives: synthesis, alkaline phosphatase, nucleotide pyrophosphatase/phosphodiesterase and nucleoside triphosphate diphosphohydrolase inhibition studies with potential anticancer profile. Euro J Med Chem. 2018 doi: 10.1016/j.ejmech.2018.07.002. [DOI] [PubMed] [Google Scholar]
- 7.Hes R.V., Wellinga K., Grosscurt A.C. 1-Phenylcarbamoyl-2-pyrazolines: a new class of insecticides. 2. Synthesis and insecticidal properties of 3,5-diphenyl-1-phenylcarbamoyl-2-pyrazolines. J Agric Food Chem. 1978;26:915–918. [Google Scholar]
- 8.Azimi F., Ghasemi J.B., Azizian H., Najafi M., Faramarzi M.A., Saghaei L., Sadeghi-aliabadi H., Larijani B., Hassanzadeh F., Mahdavi M. Design and synthesis of novel pyrazole-phenyl semicarbazone derivatives as potential α-glucosidase inhibitor: kinetics and molecular dynamics simulation study. Int J Biol Macromol. 2018 doi: 10.1016/j.ijbiomac.2020.10.263. [DOI] [PubMed] [Google Scholar]
- 9.Rahman M.A., Siddiqui A.A. Pyrazoline derivatives: a worthy insight into the recent advances and potential pharmacological activities. Int J Pharmaceut Sci Drug Res. 2010;2:165–175. [Google Scholar]
- 10.Hampp C., Hartzema A.G., Kauf T.L. Cost-utility analysis of rimonabant in the treatment of obesity. Value Health. 2008;11:389–399. doi: 10.1111/j.1524-733.2007.00281.x. [DOI] [PubMed] [Google Scholar]
- 11.Chaudhry F., Shahid W., al-Rashida M., Ashraf M., Munawar M.A., Khan M.A. Synthesis of imidazole-pyrazole conjugates bearing aryl spacer and exploring their enzyme inhibition potentials. Bioorg Chem. 2021;108 doi: 10.1016/j.bioorg.2021.104686. [DOI] [PubMed] [Google Scholar]
- 12.Ahmad G., Rasool N., Qamar M.U., Alam M.M., Kosar N., Mahmood T., Imran M. Facile synthesis of 4-aryl-N-(5-methyl-1H-pyrazol-3-yl)benzamides via Suzuki Miyaura reaction: antibacterial activity against clinically isolated NDM-1-positive bacteria and their Docking Studies. Arab J Chem. 2021;14 [Google Scholar]
- 13.Szukalski A., Sznitko L., Cyprych K., Miniewicz A., Mysliwiec J. Light amplification in derivatives of pyrazoline-based system. J Phys Chem C. 2014;118:8102–181010. [Google Scholar]
- 14.Mysliwiec J., Sznitko L., Szukalski A., Parafniuk K., Bartkiewicz S., Miniewicz A., Sahraoui B., Kajzar I. Rau F. Amplified spontaneous emission of 3-(1,1-dicyanoethenyl)-1-phenyl-4,5-dihydro-1H-pyrazole molecule embedded in various polymer matrices. Opt Mater. 2012;34:1725–1728. [Google Scholar]
- 15.Sznitko L., Mysliwiec J., Parafiniuk K., Palewska K., Bartkiewicz S., Miniewicz A. Amplified spontaneous emission in polymethyl methacrylate doped with 3-(1,1- dicyanoethenyl)-1-phenyl-4,5-dihydro-1H-pyrazole (DCNP) Chem Phys Lett. 2011;512:247–250. [Google Scholar]
- 16.Moylan C.R., Miller R.D., Twieg R.J., Betterton K.M., Lee V.Y., Matray T.J., Nguyen C. Synthesis and nonlinear-optical properties of Donor−Acceptor-substituted triaryl azole derivatives. Chem Mater. 1993;5:1499–1508. [Google Scholar]
- 17.Szukalski A., Sznitko L., Cyprych K., Miniewicz A., Mysliwiec J. Light amplification in derivatives of pyrazoline-based systems. J Phys Chem C. 2014;118:8102–8110. [Google Scholar]
- 18.Jin M., Liang Y.J., Lu R., Chuai X.H., Yi Z.H., Zhao Y., Zhang H.J. Synthesis and properties of photoluminescence and electroluminescence of pyrazoline derivatives. Synth Met. 2004;140:37–41. [Google Scholar]
- 19.Miniewicz A., Palewska K., Karpinski P., Sznitko L., Zielinski M. Fluorescence and SHG in organic nanocrystals of DCNP. Proc SPIE. 2012;8464 [Google Scholar]
- 20.Fu H.-B., Yao J.-N. Size effects on the optical properties of organic nanoparticles. J Am Chem Soc. 2001;123:1434–1439. [Google Scholar]
- 21.Fuxing G., Lei Z., Xuefeng Y., Limin T. Polymer single-nanowire optical sensors. Nano Lett. 2008;8:2757–2761. doi: 10.1021/nl8012314. [DOI] [PubMed] [Google Scholar]
- 22.Chandrakantha B., Isloor A.M., Sridharan K., Philip R., Shetty P., Padaki M. Novel N-substituted-5-phenyl-1H-pyrazole-4-ethyl carboxylates as potential NLO materials. Arab J Chem. 2013;6:97–102. [Google Scholar]
- 23.Papagiannouli I., Szukalski A., Iliopoulos K., Mysliwiec J., Courisa S., Sahraoui B. Pyrazoline derivatives with a tailored third order nonlinear optical response. RSC Adv. 2015;5:48363–48367. [Google Scholar]
- 24.Zhao P.-S., Wang H.Y., Li R.Q., Guo H.M. Synthesis, crystal structure, electronic spectra and density functional studies on 1N-phenyl-3-(3,4-dichlorophenyl)-5-phenyl-2-pyrazoline. J Chin Chem Soc (Taipei, Taiwan) 2008;5:183–188. [Google Scholar]
- 25.Udaya Sri N., Chaitanya K., Prasad M.V.S., Veeraiah V., Veeraiah A. Experimental (FT-IR, FT-Raman and UV-Vis spectra) and density functional theory calculations of diethyl 1Hpyrazole-3,5-dicarboxylate. J Mol Struct. 2012;1019:68–79. [Google Scholar]
- 26.Channar P.A., Saeed A., Erben M.F., Larik F.A., Riaz S., Florke U., Arshad M. Synthesis, conformational studies and NBO analysis of (4-chloro-3,5-dimethyl-1H-pyrazol- 1-yl)(p-tolyl)methanone. J Mol Struct. 2019;1191:152–157. [Google Scholar]
- 27.Saeed A., Khurshid A., Floerke U., Echeverría G.A., Piro O.E., Gil D.M., Rocha M., Frontera A., Mumtaz A., El-Seedi H., Erben M.F.F. Intermolecular interactions in antipyrine-like derivatives 2-halo-N-(1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1-H-pyrazol-4-yl)benzamides: X-ray structure, Hirshfeld surface analysis and DFT calculations. New J Chem. 2020 doi: 10.1039/D0NJ03958F. [DOI] [Google Scholar]
- 28.Bharathi R., Santhi N. Combined experimental and theoretical studies on molecular structures, spectroscopy of 4-(3-(2-amino-3,5-dibromophenyl)-1-(benzoyl)-4,5-dihydro-1H-pyrazol-5-yl)benzonitriles through NBO, FT-IR, HOMO-LUMO and NLO analyzes. J Theor Comput Chem. 2017;16(7) doi: 10.1142/S0219633617500572. [DOI] [Google Scholar]
- 29.Spartan user's guide, Wave function, Inc, Irvine, CA 92612 USA.
- 30.Hehre W.J., Radom L., Schleyer P.V.R., Pope J.A. Wiley; New York: 1988. Ab initio molecular orbital theory. [Google Scholar]
- 31.Becke A.D. Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 32.Lee C., Yang W., Parr R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 33.Spartan’14 Wavefunction, Inc. Irvine, CA.
- 34.Andersson M.P., Uvdal P. New scale factors for harmonic vibrational frequencies using the B3LYP density functional method with the triple-œ basis set 6–311+G(d,p) J Phys Chem A. 2005;109:2937–2941. doi: 10.1021/jp045733a. [DOI] [PubMed] [Google Scholar]
- 35.Delgado-Montiel T., Baldenebro-Lopez J., Soto-Rojo R., Glossman-Mitnik D. Quantum chemical study of the effect of π-bridge on the optical and electronic properties of sensitizers for DSSCs incorporating dioxythiophene and thiophene units. Theor Chem Acc. 2016;135:235. [Google Scholar]
- 36.Delgado-Montiel T., Baldenebro-Lopez J., Soto-Rojo R., Glossman-Mitnik D. Theoretical study of the effect of π-bridge on optical and electronic properties of carbazole-based sensitizers for DSSCs. Molecules. 2020;25:3670. doi: 10.3390/molecules25163670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parr R.G., Szentpaly L., Liu S. Electrophilicity index. J Am Chem Soc. 1999;121(9):1922–1924. [Google Scholar]
- 38.Gazquez J.L., Cedillo A., Vela A. Electrodonating and electroaccepting powers. J Phys Chem. 2007;111:1966–1970. doi: 10.1021/jp065459f. [DOI] [PubMed] [Google Scholar]
- 39.Zhao D., Lu Q., Su R., Li Y., Zhao M. Light harvesting and optical-electronic properties of two quercitin and rutin natural dyes. Appl Sci. 2019;9(2019):2567. [Google Scholar]
- 40.Shimaa A.H. TD-DFT calculations, electronic structure, natural bond orbital analysis, nonlinear optical properties electronic absorption spectra and antimicrobial activity application of new bis-spiropipridinon/pyrazole derivatives. Eur J Chem. 2018;9(4):287–302. [Google Scholar]
- 41.El-shishtawy R.M., Abdullah M.A., Saadullah G.A., Shaaban A.K.E. Molecular design of donor-acceptor dyes for efficient dye-sensitized solar cells I: a DFT study. J Mol Model. 2014;20(6):2241–2245. doi: 10.1007/s00894-014-2241-5. [DOI] [PubMed] [Google Scholar]
- 42.Irfan A., Muhammad S., Alsehemi A.G., Al-Assiri M.S., Chaudhry A.R. The effect of anchoring groups on the electro-optical and charge injection in triphenylamine derivatives-Ti6O12. J Theor Comput Chem. 2015;14(4) doi: 10.1142/S0219633615500297. [DOI] [Google Scholar]
- 43.Kim B.-G., Zhen C.-G., Jeong E.J., Kieffer J., Kim J. Organic dye design tools for efficient photocurrent generation in dye-sensitized solar cells: exciton binding energy and electron acceptors. Adv Funct Mater. 2012;22:1606–1612. [Google Scholar]
- 44.Semire B., Oyebamiji A.K., Odunola A.O. Tailoring of energy levels in (2Z)-2-cyano-2-[2-[(E)-2-[2-[(E)-2-(p-tolyl)vinyl]thieno[3,2-b]thiophen-5-yl]vinyl]pyran-4-ylidene]acetic acid derivatives via conjugate bridge and fluorination of acceptor units for effective D-π-A dye-sensitized solar cells: DFT-TDDFT approach. Res Chem Intermed. 2017;43:1863–1879. doi: 10.1007/s11164-016-2735-0. [DOI] [Google Scholar]
- 45.Omri N., Yahyaoui M., Banani R., Messaoudi S., Moussa F., Abderrabba M. Ab-initio HF and density functional theory investigations on the synthesis mechanism, conformational stability, molecular structure and UV spectrum of N0-Formylkynurenine. J Theor Comput Chem. 2016;15 [Google Scholar]
- 46.Kosar B., Albayrak C. Spectroscopic investigations and quantum chemical computational study of (E)-4-methoxy-2-[(p-tolylimino)methyl]phenol. Spectrochim Acta. 2011;78:160–167. doi: 10.1016/j.saa.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 47.Bouzayen N., Mbarek M., Alimi K. Solvent effects on optical and electronic properties of carbazolebenzothiazole based bipolar compound: TD-DFT/PCM approach. Comput Theor Chem. 2015;3(1):28–39. [Google Scholar]
- 48.Kumar M.D., Rajesh P., Dharsini R.P., Inban M.E. Molecular geometry, NLO, MEP, HOMO-LUMO and mulliken charges of substituted piperidine phenyl hydrazines by using density functional theory. Asian J Chem. 2020;32(2):401–407. [Google Scholar]
- 49.Semire B., Bello I.A. Density functional theory (DFT) study on structural and electronic properties of disperse dyes derived from 2-amino-4-trifluoromethylbenzothiazole and N, N-alkylanilines. Int J Appl Chem. 2013;9:151–163. [Google Scholar]
- 50.Semire B., Odunola O.A. DFT study on low molecular weight α,α-Ditert-butyl-4h-cyclopenta[2,1-b,3;4-b']dithiophene and α,α-ditert-butyl-4h-Cyclopenta[2,1-B,3;4-B']Dithiophene S-oxide bridged derivatives. Quim Nova. 2014;37(5):833–8384. [Google Scholar]
- 51.Raftani M., Abram T., Bennani M.N., Bouachrine M. Theoretical study of new conjugated compounds with a low bandgap for bulk heterojunction solar cells: DFT and TD-DFT study. Res Chem. 2020;2 doi: 10.1016/j.rechem.2020.100040. [DOI] [Google Scholar]
- 52.Arockia doss M., Savithiri S., Rajarajan G., Thanikachalam V., Saleem H. Synthesis, spectroscopic (FT-IR, FT-Raman, UV and NMR) and computational studies on 3tpentyl-2r,6c diphenylpiperidin-4-one semicarbazone. Spectrochim Acta. 2015;148:189–202. doi: 10.1016/j.saa.2015.03.117. [DOI] [PubMed] [Google Scholar]
- 53.Chahkandi M., Bhatti M.H., Yunus U., Shaheen S., Nadeem M., NawazTahir M. Synthesis and comprehensive structural studies of a novel amide based carboxylic acid derivative: non covalent interactions. J Mol Struct. 2017;1133:499–509. [Google Scholar]
- 54.Anbu V., Vijayalakshmi K.A. Quantum chemical studies on the spectroscopic, electronic structural and nonlinear properties of an organic N-methyl-N-(2,4,6-Trinitrophenyl) nitramide energetic molecule. J Curr Phys Chem. 2019;9:5–21. [Google Scholar]
- 55.Silverstein R.M., Webster F.X. 7th ed. Wiley; New York: 2005. Spectroscopic identification of organic compounds. [Google Scholar]
- 56.Srinivasan P., David S.A. DFT and Bader's AIM analysis of 2, 5, diphenyl-1, 3, 4-oxadizole molecule: a organic light emitting diode (OLED) J Theor Comput Chem. 2015;14 doi: 10.1142/S0219633615500388. [DOI] [Google Scholar]
- 57.Jin Z.M., Zhao B., Zhou W., Jin Z. X-ray powder diffraction analysis of a nonlinear optical material o–chlorobenzol–benzoyl thiourea. Power Diffra. 1997;12:47–48. doi: 10.1017/S0885715600009428. [DOI] [Google Scholar]
- 58.Semire B., Larayetan A.R. Ab initio and DFT study on molecular structure, reactivity indices and nonlinear properties of 4-(4-aminophenylethynyl)picolinic acid and 4-(3-aminophenylethynyl)picolinic acid. Iraqi Nat J Chem. 2016;16:224–242. [Google Scholar]
- 59.Karrouchi K., Brandán S.A., Sert Y., El Karbane M., Radi S., Ferbinteanu M., Garcia Y., Ansar M. Synthesis, structural, molecular docking and spectroscopic studies of (E)-N’-(4-methoxybenzylidene)-5-methyl-1H-pyrazole-3-carbohydrazide. J Mol Struct. 2021;1225 [Google Scholar]
- 60.Zaccolo M., Movsesian M.A. cAMP and cGMP signaling cross-talk: role of phosphodiesterases and implications for cardiac pathophysiology. Circ Res. 2007;100(11):1569–1578. doi: 10.1161/CIRCRESAHA.106.144501. [DOI] [PubMed] [Google Scholar]
- 61.Bender A.T., Beavo J.A. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev. 2006;58:488–520. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- 62.Xu R.X., Rocque W.J., Lambert M.H., Vanderwall D.E., Luther M.A., Nolte R.T. Crystal structures of the catalytic domain of phosphodiesterase 4B complexed with AMP, 8-Br-AMP, and rolipram. J Mol Biol. 2004;337:355–365. doi: 10.1016/j.jmb.2004.01.040. [DOI] [PubMed] [Google Scholar]
- 63.Kim H.K., Kwon J.Y., Yoo C., Abdi S. The analgesic effect of rolipram, a phosphodiesterase 4 inhibitor, on chemotherapy-induced neuropathic pain in rats. Anesth Analg. 2015;121:822–828. doi: 10.1213/ANE.0000000000000853. [DOI] [PubMed] [Google Scholar]
- 64.Raker V.K., Becker C., Steinbrink K. The cAMP pathway as therapeutic target in autoimmune and inflammatory diseases. Front Immunol. 2016;7(2016):123. doi: 10.3389/fimmu.2016.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lobo V., Patil A., Phatak A., Chandra N. Free radicals, antioxidants and functional foods. Impact Human Health. 2010;4(8):118–126. doi: 10.4103/0973-7847.70902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Young I.S., Woodside J.V. Antioxidants in health and disease. J Clin Pathol. 2001;54(2001):176–186. doi: 10.1136/jcp.54.3.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ser H.-L., Palanisamy U.D., Yin W.-F., AbdMalek S.N., Chan K.-G., Goh B.-H., Lee L.-H. Presence of antioxidative agent, Pyrrolo[1,2-a]pyrazine-1,4-dione, hexahydro- in newly isolated Streptomyces mangrovisoli sp. nov. Front Microbiol. 2015;6:854. doi: 10.3389/fmicb.2015.00854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stepanić V., Matijašić M., Horvat T., Verbanac D., Kučerová-Chlupáčová M., Saso L., Žarković N. Antioxidant activities of alkyl substituted pyrazine derivatives of chalcones—in vitro and in silico study. Antioxidants. 2019;8:90. doi: 10.3390/antiox8040090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kitawata B.S., Singh M. Synthesis, characterization, antibacterial, antioxidant, DNA binding and SAR study of novel pyrazine moiety bearing 2-pyrazoline derivatives. New J Chem. 2014;38:4290–4299. [Google Scholar]
- 70.Bouckenooghe T., Remacle C., Reusens B. Is taurine a functional nutrient? Curr Opin Clin Nutr Metab Care. 2006;9:728–733. doi: 10.1097/01.mco.0000247469.26414.55. [DOI] [PubMed] [Google Scholar]
- 71.Sirdah M.M. Protective and therapeutic effectiveness of taurine in diabetes mellitus: a rationale for antioxidant supplementation. Diabetes Metabol Syndr. 2015;9:55–64. doi: 10.1016/j.dsx.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 72.Murakami S. Role of taurine in the pathogenesis of obesity. Mol Nutr Food Res. 2015;59:1353–1363. doi: 10.1002/mnfr.201500067. [DOI] [PubMed] [Google Scholar]
- 73.Franconi F., Bennardini F., Mattana A., Miceli M., Ciuti M., Mian M., Gironi A., Anichini R., Seghieri G. Plasma and platelet taurine are reduced in subjects with insulin-dependent diabetes mellitus: effects of taurine supplementation. Am J Clin Nutr. 1995;61(5):1115–1119. doi: 10.1093/ajcn/61.4.1115. [DOI] [PubMed] [Google Scholar]
- 74.Sak D., Erdenen F., Müderrisoglu C., Altunoglu E., Sozer V., Gungel H., Guler P.A., Sak T., Uzun H. The relationship between plasma taurine levels and diabetic complications in patients with type 2 diabetes mellitus. Biomolecules. 2019;9:96. doi: 10.3390/biom9030096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Maleki V., Mahdavi R., Hajizadeh-Sharafabad F., Alizadeh M. The effects of taurine supplementation on oxidative stress indices and inflammation biomarkers in patients with type 2 diabetes: a randomized, double-blind, placebo-controlled trial. Diabetol Metab Syndrome. 2020;12:9. doi: 10.1186/s13098-020-0518-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Trott O., Olson A.J. AutoDockVina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010;31:455–561. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Oyebamiji A.K., Fadare O.A., Akintelu S.A., Semire B. Biological studies on Anthra[1,9-cd]pyrazol-6(2D)-one analogues as anti-vascular endothelial growth factor via in silico mechanisms. Chem Afr. 2021;4:955–963. doi: 10.1007/s42250-021-00276-2. [DOI] [Google Scholar]
- 78.Adegbola P.I., Semire B., Fadahunsi O.S., Adegoke A.E. Molecular docking and ADMET studies of Allium cepa, Azadirachta indica and Xylopia aethiopica isolates as potential anti-viral drugs for Covid-19. Virus Dis. 2021;32:85–97. doi: 10.1007/s13337-021-00682-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Adegbola A.E., Fadahunsi O.S., Alausa A., Abijo A.Z., Balogun T.A., Aderibigbe T.S., Semire B., Adegbola P.I. Computational prediction of nimbanal as potential antagonist of respiratory syndrome coronavirus. Inform Med Unlocked. 2021;24 doi: 10.1016/j.imu.2021.100617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yonas B., Alfred M., Paballo L., Oyebamiji A.K., Adedapo S.A., Thierry Y.F., Lesetja R.M. Molecular hybrid of 1,2,3-triazole and schiff base as potential antibacterial agents: DFT, molecular docking and ADME studies. J. Mol. Struct. 2023;1286:135617. [Google Scholar]
- 81.Kramer B., Rarey M., Lengauer T. Evaluation of the FlexX incremental construction algorithm for protein ligand docking. Proteins Struct Funct Genet. 1999;37:228–241. doi: 10.1002/(sici)1097-0134(19991101)37:2<228::aid-prot8>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 82.El-Azab A.S., Mary Y.S., Panicker C.Y., Abdel-Aziz A.A.M., El-Sherbeny M.A., Van Alsenoy C. DFT and experimental (FT-IR and FT-Raman) investigation of vibrational spectroscopy and molecular docking studies of 2-(4-oxo-3-phenethyl-3,4-dihyroquinazolin-2-ylthio)-N-(3,4,5-trimethoxyphenyl) acetamide. J Mol Struct. 2016;1113:133–145. doi: 10.1016/j.molstruc.2016.02.038. [DOI] [Google Scholar]
- 83.Adegbola P.I., Fadahunsi O.S., Adegbola A.E., Semire B. In silico studies of potency and safety assessment of selected trial drugs for the treatment of COVID-19. Silico Pharmacol. 2021;9:45. doi: 10.1007/s40203-021-00105-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Adepoju A.J., Latona D.F., Olafare O.G., Oyebamiji A.K., Abdul-Hammed M., Semire B. Molecular docking and pharmacokinetics studies of (Curcumin) potency against Ebola virus. Ovidius Univ Ann Chem. 2022;33:22–35. [Google Scholar]
- 85.Mahmudov I., Demir Y., Sert Y., Abdullayev Y., Sujayev A., Alwasel S.H., Gulcin I. Synthesis and inhibition profiles of N-benzyl- and N-allyl aniline derivatives against carbonic anhydrase and acetylcholinesterase – a molecular docking study. Arab J Chem. 2022;15 [Google Scholar]
- 86.Channar P.A., Afzal S., Ejaz S.A., Saeed A., Larik F.A., Mahesar P.A., Lecka J., Sévigny J., Erben M.F., Iqbal J. Exploration of carboxy pyrazole derivatives: synthesis, alkaline phosphatase, nucleotide pyrophosphatase/phosphodiesterase and nucleoside triphosphate diphosphohydrolase inhibition studies with potential anticancer profile. Euro J Med Chem. 2018 doi: 10.1016/j.ejmech.2018.07.002. [DOI] [PubMed] [Google Scholar]
- 87.Ibrahim A.O., Semire B., Adepoju A.J., Latona D.F., Oyebamiji A.K., Owonikoko A.D., Oladuji T.E., Odunola O.A. In Silico investigations on structure, reactivity indices, NLO properties, and bio-evaluation of 1-benzyl-2-phenyl-1H-benzimidazole derivatives using DFT and molecular docking approaches. Biointer Res Appl Chem. 2023;13:233. doi: 10.33263/BRIAC133.233. [DOI] [Google Scholar]