

Abstract
Background
Necroptosis and pyroptosis, two types of proinflammatory programmed cell death, were recently found to play important roles in spinal cord injury (SCI). Moreover, cyclic helix B peptide (CHBP) was designed to maintain erythropoietin (EPO) activity and protect tissue against the adverse effects of EPO. However, the protective mechanism of CHBP following SCI is still unknown. This research explored the necroptosis- and pyroptosis-related mechanism underlying the neuroprotective effect of CHBP after SCI.
Methods
Gene Expression Omnibus (GEO) datasets and RNA sequencing were used to identify the molecular mechanisms of CHBP for SCI. A mouse model of contusion SCI was constructed, and HE staining, Nissl staining, Masson staining, footprint analysis and the Basso Mouse Scale (BMS) were applied for histological and behavioural analyses. qPCR, Western blot analysis, immunoprecipitation and immunofluorescence were utilized to analyse the levels of necroptosis, pyroptosis, autophagy and molecules associated with the AMPK signalling pathway.
Results
The results revealed that CHBP significantly improved functional restoration, elevated autophagy, suppressed pyroptosis, and mitigated necroptosis after SCI. 3-Methyladenine (3-MA), an autophagy inhibitor, attenuated these beneficial effects of CHBP. Furthermore, CHBP-triggered elevation of autophagy was mediated by the dephosphorylation and nuclear translocation of TFEB, and this effect was due to stimulation of the AMPK-FOXO3a-SPK2-CARM1 and AMPK-mTOR signalling pathways.
Conclusion
CHBP acts as a powerful regulator of autophagy that improves functional recovery by alleviating proinflammatory cell death after SCI and thus might be a prospective therapeutic agent for clinical application.
Keywords: Cyclic helix B peptide, Spinal cord injury, Proinflammatory cell death, Autophagy, AMPK signalling Pathway
Graphical abstract
Highlights
-
•
Cyclic helix B peptide (CHBP) is designed and synthesized from erythropoietin.
-
•
CHBP fosters functional restoration after traumatic spinal cord injury.
-
•
CHBP promotes autophagy to inhibit pyroptosis and necroptosis after SCI.
-
•
The effect of CHBP on autophagy is medicated by TFEB activation following SCI.
-
•
CHBP increases TFEB activity via AMPK-mTOR and AMPK-FoxO3a-SPK2-CARM1 signalling pathways after SCI.
Abbreviations
- SCI
spinal cord injury
- EPO
erythropoietin
- CHBP
cyclic helix B peptide
- GEO
gene expression omnibus
- BMS
Basso mouse scale
- 3-MA
3-methylladenine
- HPLC
high-performance liquid chromatography
- ACN
acetonitrile
- DAB
diaminobenzidine
- HE
haematoxylin and eosin
- SYN
synaptophysin
- HRP
horseradish peroxidase
- FITC
fluorescein isothiocyanate
- DAPI
4′,6-diamidino-2-phenylindole
- AAV
adenoassociated virus
- SEM
standard error of the mean
- LSD
least significance difference
- RIPK
receptor-interacting protein kinases
- OD
optical density
- GSEA
gene set enrichment analysis
- CC
compound C
- CEPO
carbamoylated erythropoietin
- GSDMD
gasdermin D
- IL
interleukin
- HBSP
helix B-surface peptide
- ALI
acute lung injury
- DEGs
differentially expressed genes
- SPPS
solid-phase peptide synthesis
- TFEB
transcription factor EB
- NS
normal saline
- PBS
phosphate-buffered saline
- PFA
paraformaldehyde
- WB
western blot
- qPCR
quantitative real-time polymerase chain reaction
- PVDF
polyvinylidene fluoride
- BSA
bovine serum albumin
- ECM
extracellular matrix
- I/R
ischemia/reperfusion
- CNS
central nervous system
1. Introduction
Spinal cord injury (SCI) is devastating, can be disabling, is one reason for increasing public health costs, and is a public health concern worldwide [1,2]. SCI refers to the detrimental changes resulting from direct or indirect trauma to the spine leading to sudden contusion or anatomical discontinuity of the spinal cord [3]. Mechanical insult to the spinal cord causes primary injury, and spinal cord transection can disrupt impulse conduction, contributing to transitory or persistent impairment of the spinal cord's normal sensory, motor, and autonomic functions [4]. In addition, subsequent cell death in nerve tissues and the activation of inflammatory cells in damaged areas cause secondary injury, which can induce proinflammatory cytokine and chemokine accumulation, axonal degeneration, demyelination and cell damage [5]. Primary SCI is unavoidable and irreversible, and therapeutic modalities are therefore primarily focused on attenuating the subsequent pathological changes caused by secondary injury to improve clinical outcomes and prognosis in SCI patients [6]. Although the mechanisms of secondary SCI are not entirely clear, numerous pathophysiological changes, such as inflammation, oxidative stress, apoptosis, and autophagy, have been found to occur after SCI. An increasing number of studies suggest that anti-inflammatory therapies and strategies that regulate cell death during secondary injury are potentially promising treatments for SCI [[7], [8], [9]].
Previous research has determined that pyroptosis is an important event that aggravates the neuroinflammatory response and cell death following SCI [10]. Pyroptosis, a novel type of proinflammatory cell death, is primarily regulated by Gasdermin D (GSDMD) and Caspase-1 and is accompanied by the generation of different inflammatory mediators, such as interleukin (IL)-18 and IL-1β [11]. Overproduction of proinflammatory factors (such as IL-1β) and oxidative stress amplify inflammation and aggravate secondary injury [12]. Pathologically, the inflammasome can further activate and cleave cysteine protease-1 precursor (Pro-Caspase-1) to produce bioactive Caspase-1 and activate downstream cascade reactions, resulting in increased cell death and inflammatory responses [13]. Necroptosis, another form of proinflammatory cell death, is initiated by receptor-interacting protein kinase (RIPK)1, RIPK3 and mixed-lineage kinase domain-like protein (MLKL) [14]. Failed suppression of necroptosis regulators leads to disruption of the plasma membrane and cell lysis, which have emerged as key drivers of neuroinflammation and degeneration in the central nervous system (CNS) [15,16]. Previous studies have discovered that the stimulation of necroptosis contributes to cell loss and tissue damage in several neurodegenerative diseases [17,18] as well as in SCI [19]. However, the potential mechanisms involved in the regulation of pyroptosis and necroptosis after SCI remain unknown. Autophagy is a physiological process that sustains homeostasis and energy conservation during times of stress via recycling of unnecessary or dysfunctional cellular components. Autophagy regulates microtubule dynamics and axonal regeneration to alleviate nerve injury after SCI [20], and inhibition of autophagy can result in cellular death [21]. Previously, studies found that inhibition of autophagic activity leads to increased levels of pyroptosis and necroptosis after SCI [21,22], and autophagy agonists ameliorate the impairment of neural function [23].
Erythropoietin (EPO) is an endocrine hormone produced by renal tubular interstitial cells that can be released into the bloodstream to produce physiological effects. Many studies have reported the neuroprotective effect of EPO in neurological disorders, as well as in SCI [3,24]. However, there are possible pitfalls associated with the administration of EPO to patients of various ages and with underlying chronic comorbidities, as EPO can potentially cause adverse effects such as hypertension and thrombosis, which restricts its clinical application [25]. Helix B-surface peptide (HBSP), a small peptide derived from the helix-B domain of EPO, maintains EPO activity and protects tissues from the adverse effects of EPO [26]. Furthermore, HBSP has been reported to protect against hypoxic injury by increasing the levels of autophagy [27]. However, its short half-life and ability to be rapidly degraded are major disadvantages of HBSP. Since the introduction of HBSP, the novel peptide cyclic helix B peptide (CHBP), which has increased resistance to proteolytic decomposition and an improved tissue-protective effect and is effective when administered at a lower frequency and dosage, has been designed on the basis of the amino acid sequence of HBSP [28,29]. CHBP displays significant tissue-protective and regenerative effects, including on the kidneys [30], heart [31], and brain [32]. CHBP protects renal tissue from ischemia/reperfusion (I/R) damage by stimulating autophagic activity and decreasing programmed cell death and inflammation [33,34]. CHBP mitigates sepsis-triggered acute lung injury (ALI) by decreasing NLRP3 inflammasome stimulation and downstream IL-1β secretion [35]. CHBP increases ischaemic flap viability by enhancing autophagy and decreasing necroptosis [36]. However, whether CHBP is neuroprotective remains unclear, and no research has reported the effect of CHBP on the progression of SCI. Hence, our team hypothesized that CHBP might exert a neuroprotective effect by enhancing autophagy-mediated suppression of pyroptosis and necroptosis and promote functional recovery after SCI.
2. Materials and methods
2.1. Tissue microarray construction and pathway analysis
To investigate the underlying causal mechanisms of SCI, the original microarray dataset GSE171441 was acquired from the Gene Expression Omnibus (GEO) database, and SCI model animals were classified into two subgroups based on the observation time point after injury. Subgroup I included 3 SCI model mice and 3 uninjured mice from which spinal cord samples were collected at 3 days postinjury, and subgroup II included 3 injured mice and 4 uninjured mice from which spinal cord tissues were collected at 35 days postinjury. R software (version 4.0.3) and Bioconductor packages were employed for data harvesting and statistics [37]. Differentially expressed genes (DEGs) were identified via the limma R package of GEO2R, and the thresholds for statistical significance were a modified P value < 0.05 and a |log2(fold-change)| > 1.2. Gene Ontology (GO) functional enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were performed using the DAVID online tool (version 6.8), and a P value less than 0.05 was considered the threshold for enriched GO function terms and signalling pathways [38]. In addition, gene set enrichment analysis (GSEA) was employed to assess the enrichment of specific genetic signatures in gene sets between the control and SCI groups.
2.2. RNA sequencing and functional enrichment analyses
Tissue was collected 3 days after SCI, and total RNA was extracted with TRIzol reagent according to the manufacturer's specifications. RNA purity and concentrations were assessed using a NanoDrop 2000 spectrophotometer (Thermo Scientific, USA). RNA integrity was evaluated with an Agilent 2100 Biological Analyzer (Agilent Technology, USA). Then, libraries were constructed using a TruSeq Stranded mRNA LT Sample Prep Kit (Illumina, San Diego, CA, USA) according to the manufacturer's instructions. Transcriptome sequencing and analysis were conducted by OE Biotech Co., Ltd. (Shanghai, China). Afterwards, libraries were constructed by sequencing with an Illumina HiSeq X Ten system, and 125 bp/150 bp paired-end reads were produced. The raw data were processed using Trimmomatic. Data that contained poly-N reads and low-quality reads were discarded, and clean reads were preserved for subsequent assays. The clean reads were mapped to the mouse genome (GRCm38.p6) via HISAT2. Fragments per kilobase of transcript per million mapped reads (FPKM) values were determined for every gene using Cufflinks, and the read counts of every gene were acquired using HTSeqcount. Differential expression analyses were conducted via the DESeq (2012) package for R. P < 0.05 and a fold change >2 or <0.5 were used as thresholds. Layer clustering analyses of 13 DEGs were performed to identify the gene expression features of different groups and specimens. GO enrichment analysis of the DEGs was performed using R based on the hypergeometric distribution.
2.3. Animals
Due to the shorter urethras of female animals, artificial voiding to prevent urine retention after SCI is easier in females than in males; thus, female animals are often used in experimental SCI studies. The C57BL/6 mice (6 weeks, 20–30 g, female) used for the animal experiments were provided by Wenzhou Medical University Experimental Animal Center (licence no. SCXK [39] 2015-0001), Wenzhou, China. All mice were housed in normal experimental cages on a 12 h/12 h light/dark cycle and provided access to food and water ad libitum.
2.4. Ethics statement
All experiments involving animals were conducted according to the ethical policies of and procedures approved by the Ethics Committee of Wenzhou Medical University, China (Approval No. wydw 2017-0022).
2.5. Peptide biosynthesis
Peptides were produced via Fmoc solid-phase peptide synthesis (SPPS) and subjected to purification using Varian ProStar 218 high-performance liquid chromatography (HPLC) equipment on an Agilent Venusil MP C18 reversed-phase column. They were eluted with a linear gradient of H2O and acetonitrile (ACN) containing 0.05% trifluoroacetic acid (TFA) at a flow velocity of 1 mL min-1. Isolation was supervised at 220 nm using a UV detection device. Next, the peptides were subjected to mass spectrometry analyses using the Voyager-DE STR device. HPLC elution was performed with a gradient of solvent A (0.05% TFA and 2% ACN) and solvent B (0.05% TFA and 90% ACN). Each peptide was dispersed in deionized water to a final concentration of 20 mg ml−1 and preserved at −20 °C for future assays. The purity of the purified peptides was assessed as 99.78% using HPLC.
2.6. Antibodies and reagents
The primary antibodies and reagents listed below were utilized in this study and obtained from the indicated source: diaminobenzidine (DAB), haematoxylin and eosin (HE) dyes, Masson staining reagent, pentobarbital sodium, and Nissl staining reagent were purchased from Solarbio Science & Technology (PRC). Cytoplasmic Extraction Reagent and NE-PER™ Nuclear and bicinchoninic acid (BCA) reagents were purchased from Thermo Fisher Scientific (USA). 3-Methyladenine (3-MA) was purchased from Sigma–Aldrich (USA; Cat# M9281). Goat anti-mouse IgG H&L (Alexa Fluor® 647), goat anti-rabbit IgG H&L (Alexa Fluor® 488), and anti-microtubule-associated protein-2 (MAP2), anti-p62/SQSTM1, anti-synaptophysin (SYN), anti-IL-18, anti-adaptor apoptotic speck-like protein with a CARD (ASC), anti-RIPK1, anti-RIPK3, and anti-NeuN antibodies were purchased from Abcam (England; Cat# ab106393, Cat# ab150115, Cat# ab104224, Cat# ab150077, Cat# ab183830, Cat# ab52636, Cat# ab207323, Cat# ab62344, Cat# ab180799, and Cat# ab56416, respectively). An anti-transcription factor EB (TFEB) antibody was purchased from Bethyl Laboratories (Montgomery, TX, USA; Cat# 134369.100). A horseradish peroxidase (HRP)-conjugated IgG secondary antibody was obtained from Santa Cruz Biotechnology (USA). Compound C (CC, C24H25N5O; purity ≥98.14%) was obtained from MedChemExpress (USA; Cat# HY-13418A). An anti-IL-1β antibody was obtained from ABclonal (USA; Cat# A1112). Primary antibodies against CARM1, AMPK, mTOR, Beclin-1, NLRP3, p-FOXO3a, FOXO3a, p-AMPK, C-Caspase-8, and p-mTOR were purchased from Cell Signaling Technology (USA; Cat# 3379, Cat# 5832, Cat# 5536, Cat# 3738, Cat# 15101, Cat# 9466, Cat# 12829, Cat# 2535, Cat# 97756, and Cat# 2983, respectively). An anti-LC3B antibody was purchased from Novus Biologicals (USA; Cat# NB600-1384). Antibodies against GAPDH, VPS34, SKP2, CTSD, Histone-H3, and Caspase-1 were purchased from Proteintech Group (USA; Cat# 10494-1, Cat# 12452-1, Cat# 15010-1-AP, Cat# 21327-1, Cat# 17168-1, and Cat# 22915-1, respectively). Antibodies against NLRP1, MLKL, GSDMD and p-TFEB (Ser221) were obtained from Affinity Biosciences (USA; Cat# DF13187, Cat# DF7412, Cat# AF4013, and Cat# AF3708, respectively). 4′,6-Diamidino-2-phenylindole (DAPI) was obtained from Beyotime Biotechnology (PRC).
2.7. Animal model of SCI
Before the assays, mice were anaesthetized via intraperitoneal injection of 1% (w/v) pentobarbital sodium (50 mg/kg). Subsequently, laminectomy of the T9-T10 spinal cord was performed to expose a circular area of dura. Next, weight drop was performed to induce spinal cord contusion as previously described [39]. In short, a bar (1.2 mm and 10 g) was dropped from 15 mm onto the spinal cord to cause moderate spinal cord contusion while maintaining the integrity of the dura. After damage, the skin, fascia, and muscle layers were sutured using 4–0 nonabsorbable silk threads. The animals in the sham group underwent identical treatment as described above except weight drop. After surgery, artificially voided was performed 3 times every day.
2.8. Adeno-associated virus (AAV) vector production
The AAV-TFEB shRNA utilized herein was produced and packaged by Shanghai Genetic Chemical Corporation (PRC). The TFEB shRNA was inserted into the pAV-U6-shRNA-CMV-EGFP plasmid by cloning to yield pAV-U6-shRNA (TFEB)-CMV-EGFP. AAV9-U6-shRNA (TFEB)-CMV-EGFP was obtained by transfecting pAV-U6-shRNA (TFEB)-CMV-EGFP, an auxiliary adenovirus (AD) plasmid, and an AAV Rep/Cap-expressing plasmid into AAV-293 cells. AAV9-U6-shRNA (scramble)-CMV-EGFP was produced via a similar procedure. The virus particles were subjected to purification via the iodixanol gradient approach. The titres of AAV9-U6-shRNA (TFEB)-CMV-EGFP and AAV9-U6-shRNA (scrambled)-CMV-EGFP were 1:243 × 1012 and 1:22 × 1012 genomic copies per ml, respectively, as determined by quantitative real-time polymerase chain reaction (qPCR).
2.9. Drug and AAV vector administration
Our team randomly divided 436 mice into 16 groups: the sham (n = 54), SCI (n = 59), SCI + CHBP (n = 59), sham+3-MA (n = 24), SCI+3-MA (n = 24), SCI + CHBP+3-MA (n = 48), SCI + scrambled shRNA control (n = 6), SCI + CHBP + scrambled shRNA control (n = 54), SCI + TFEB shRNA (n = 6), SCI + CHBP + TFEB shRNA (n = 54), SCI + CHBP + CC (n = 12), SCI + CHBP 1 d (n = 12), SCI + CHBP 5 d (n = 12), and SCI + CHBP 7 d (n = 12) groups. The CHBP-treated animals were administered CHBP (8 nmol/kg/day) by intraperitoneal injection every day for 1, 3, 5, or 7 days after SCI [36]. An equivalent volume of normal saline (NS) was administered to the sham group and SCI group. Intraperitoneal injection of 3-MA (15 mg/kg) or CC (1.5 mg/kg) was administered every day 0.5 h before CHBP treatment. The SCI + scrambled shRNA control group, SCI + TFEB shRNA group, SCI + CHBP + scrambled shRNA control group and SCI + CHBP + TFEB shRNA group received intravenous injection of 100 μl viral vector in phosphate-buffered saline (PBS) (1 × 1010 packaged viral genomes) 14 days prior to SCI. After 14 days, the SCI + CHBP + scrambled shRNA control group and SCI + CHBP + TFEB shRNA group received the same treatment as the CHBP group. The mice were euthanized via an overdose of pentobarbital sodium, and histological specimens were obtained for relevant assays on days 1, 3, 14 and 28.
2.10. Behavioural evaluation
The Basso Mouse Scale (BMS) was applied to assess mobility on days 0, 1, 3, 7, 14, 21, and 28 after SCI and to assess functional restoration [40]. BMS scores range from 0 to 9, with 0 reflecting overall paralysis and 9 reflecting normal motor function. Footprint analysis was performed 28 days after surgery. The hindlimbs (red) and forelimbs (blue) were stained with different coloured dyes. Two independent researchers blinded to the groups assessed the outcomes [41].
2.11. Preparation of tissues and HE, Nissl and Masson staining
On days 14 and 28 after SCI, the mice were anaesthetized with 2% (w/v) pentobarbital sodium and perfused with NS followed by 4% (w/v) PFA in PBS. A piece of rostral spinal cord (1 mm long, 4 mm away from the injury epicentre) and the whole injured segment (10 mm long, centred on the injury site) were isolated and postfixed in 4% (w/v) PFA for 24 h. The tissues were successively dehydrated in graded ethanol solutions. The specimens were cleaned and embedded in paraffin. Paraffin slices (4 μm) were placed on slides coated with gelatine. The rostral spinal cord pieces and whole segments were cut into longitudinal and transverse slices, respectively. The longitudinal and transverse slices were stained with haematoxylin and eosin as previously described [[42], [43], [44]]. The transverse slices were stained with 1% cresol purple for Nissl staining per the manufacturer's specification. Cells positive for Nissl in the anterior horns identified as motor neurons according to previous methods [45]. For Masson staining, longitudinal sections were permeabilized with 10% K2Cr2O7 and 10% trichloroacetic acid (TCAA), and the nuclei were stained using haematoxylin. Subsequently, the slides were differentiated with HCl and ethyl alcohol, and blued using weak NH3 and stained with Masson dye. The staining process was performed as previously described [46]. Eventually, bright-field images of HE, Masson and Nissl staining were acquired by a light microscope with a 4 × objective (Olympus, Japan). The thresholding method in ImageJ was used to quantify the Masson-stained lesion area (blue). The number of Nissl-positive motor neurons in the anterior horn was determined manually in a double-blind manner.
2.12. Immunofluorescence (IF) staining
On days 1, 3 and 28 after SCI, samples were harvested for IF staining. Our team performed IF staining of rostral spinal cord pieces (1 mm long, 4 mm from the injury epicentre) as described previously [47]. The samples were subjected to deparaffinization, rehydration, and washing and then incubated with 10.2 mM sodium citrate buffering solution for 20 min at 95 °C. Next, the slices were permeabilized using 0.1% (v/v) PBS-Triton X-100 (10 min). Afterwards, the slices were blocked in 10% (v/v) bovine serum albumin (BSA) in PBS (1 h) before overnight incubation at 4 °C with primary antibodies against TFEB (1:200), RIPK1 (1:200)/NeuN (1:400), Caspase-1 (1:200)/NeuN (1:400), SYN (1:200)/NeuN (1:400), LC3 (1:200)/NeuN (1:400), RIPK3 (1:200)/NeuN (1:400), GSDMD (1:150)/NeuN (1:400), p62 (1:200)/NeuN (1:400) and MAP2 (1:200). Finally, the samples were washed three times for 10 min at ambient temperature, incubated with secondary antibody at 37 °C for 1 h and counterstained with DAPI solution.
2.13. Quantification of IF staining
Specimens stained for Caspase-1, GSDMD, RIPK1, RIPK3, LC3 and p62 were photographed using a Zeiss LSM 800 confocal microscope. Image acquisition and processing were performed with Zen Blue software (Zeiss), and the images were prepared for presentation using ImageJ software (Version 1.52a). The target areas were photographed with a 20 × objective lens without optical zoom using confocal laser scanning microscopy at a resolution of 1024 × 1024 pixels. MAP2-, TFEB- and SYN-stained specimens were viewed and imaged using a fluorescence microscope with a 10 × objective lens (Olympus, Japan). The exposure time was kept constant for each marker. For transverse sections, images were captured 0.5–1 mm rostral to the lesion site; images of 6 random areas in the anterior horn in 3 random sections from each animal were obtained. The integrated density of MAP2, TFEB, Caspase-1, GSDMD, RIPK1, RIPK3 and p62 in each neuron labelled by NeuN was calculated using ImageJ software. The numbers of SYN-positive synapses and LC3 puncta in neurons were determined manually in a double-blind manner.
2.14. Western blot (WB) analyses
On days 1 and 3 after SCI, we euthanized the mice. Then, we obtained spinal cord samples (1.5 cm; including the injury epicentre) and stored them at −80 °C before performing WB analyses. Our team extracted protein from some of the samples in lysis buffer solution. Nuclear and cytoplasmic proteins were extracted from the remaining specimens using Cytoplasm Abstraction Reagent and NE-PER™ Nucleus. Our team purified the total protein obtained from the spinal cord samples using protein extraction reagents. Protein concentrations were quantified using the BCA assay. Equal amounts of protein (60 μg) were separated using 12% (w/v) gel electrophoresis. The proteins were subsequently transferred to PVDF membranes (Roche Applied Science, USA), which were blocked with 5% (w/v) ski, milk and incubated with antibodies against the following proteins overnight at 4 °C: AMPK (1:1000), p-AMPK (1:1000), mTOR (1:1000), p-mTOR (1:1000), p-TFEB (Ser221) (1:1000), FOXO3a (1:1000), p-FOXO3a (1:1000), CARM1 (1:1000), SKP2 (1:1000), histone-H3 (1:1000), Beclin-1 (1:1000), CTSD (1:1000), SQSTM1/p62 (1:1000), LC3B (1:1000), VPS34 (1:1000), RIPK1 (1:1000), RIPK3 (1:1000), ASC (1:1000), C-Caspase-8 (1:1000), MLKL (1:1000), Caspase-1 (1:1000), GAPDH (1:1000), NLRP3 (1:1000), GSDMD (1:1000), IL-18 (1:1000), NLRP1 (1:1000), and IL-1β (1:1000). We subsequently incubated the membranes with HRP-conjugated IgG secondary antibodies at ambient temperature for 120 min. Finally, we visualized the bands using a ChemiDoc™ XRS + Gel Imaging System (Bio-Rad) following the instructions of an enhanced chemiluminescence (ECL) immunodetection kit.
2.15. Immunoprecipitation
Immunoprecipitation was performed as previously described [48]. Spinal cord tissues were homogenized in IGEPAL CA-630 buffer (50 mM Tris-HCl, 1% IGEPAL CA-630, 10 mM EDTA, 150 mM NaCl, 50 mM NaF, 1 μM leupeptin and 0.1 μM aprotinin). After centrifugation, the homogenate supernatant was collected for immunoprecipitation. The primary antibody was covalently immobilized on protein A/G agarose with a Pierce Crosslink immunoprecipitation kit according to the manufacturer's instructions. The samples were incubated with immobilized antibody beads at 4 °C for 2 h. The samples were also subjected to immunoprecipitation with an IgG isotype control. After immunoprecipitation and washing with Tris-buffered saline (TBS), the samples were eluted with glycine-HCl (0.1 M, pH 3.5), and the immunoprecipitants were subjected to Western blotting using related primary antibodies.
2.16. qPCR
Total RNA was extracted from spinal cord tissues with TRIzol reagent. cDNA was prepared using a reverse transcription enzyme with PrimeScript II 1st Strand cDNA Synthesis Reagent (6210B, Takara) according to the manufacturer's specification. Next, using information from GenBank, the following primer sequences were produced by Nanjing Zoonbio Biological Technology Company: Beclin-1 5′-GGACCAGGAGGAAGCTCAGTACC-3’ (forward) and 5′-CGCTGTGCCAGATGTGGAAGG-3’ (reverse); Vps34 5′-GTCGGTTCCTGTCGCAGAAGTTC-3’ (forward) and 5′-TATCCAGGTGCCGGTCTCCAAC-3’ (reverse); Sqstm1 5′-ACAACCCGTGTTTCCTTT-3’ (forward) and 5′- TGCCACCTTTCACTCACTA-3’ (reverse); Ctsd 5′-GGGCATCCAGGTAGTTTT-3’ (forward) and 5′-CGTCTTGCTGCTCATTCT-3’ (reverse); and Lc3 5′-CTACGCCTCCCAAGAAACC-3’ (forward) and 5′-AGAGCAACCCGAACATGACT-3’ (reverse). Real-time PCR was performed with SYBR Premix Ex Taq (RR420A, TAKARA). The reaction conditions were as follows: 30 cycles of denaturation at 95 °C for 0.5 min, annealing at 65 °C for 0.5 min, and extension at 72 °C for 45 s and signal identification at 72 °C. Finally, the target mRNA expression levels were normalized to the mRNA expression levels of β-actin.
2.17. Statistics
Our team performed all statistical analyses using SPSS 19 (USA), and all analyses were performed in a double-blinded manner. The data are shown as the mean ± SEM. To control undesired variation, data standardization was conducted. Comparisons between two independent groups were performed using a two-tailed unpaired t-test. Two-way analysis of variance (ANOVA) with Tukey's multiple comparisons test was used to analyse differences among three or more groups when the data were normally distributed, and nonparametric Mann‒Whitney U tests were used to compare data among the groups if the data were not normally distributed. P < 0.05 indicated statistical significance.
3. Results
3.1. CHBP promotes functional restoration after SCI
Motor function after SCI was assessed by HE, Nissl, Masson, and IF staining; footprint analysis; and the BMS. To determine the optimal administration time of CHBP for the treatment of SCI, mice were intraperitoneally injected with CHBP for 0, 1, 3, 5, or 7 days after SCI, and we used the BMS to assess limb function on days 1, 3, 7, 14, 21, and 28 after SCI. We found that 3 days of CHBP treatment had a significant therapeutic effect, and there were no significant differences among the SCI + CHBP 3 d, SCI + CHBP 5 d and SCI + CHBP 7 d groups (Fig. S1). HE and Masson staining revealed a larger glial scar (p < 0.01) in the lesion area in the SCI model animals than in the sham animals. HE and Nissl staining showed that there were fewer motor neurons (p < 0.01) within the lesion area in the SCI model animals than in the sham animals. Additionally, the damaged region in the spinal cord was evaluated via IF staining, and the results revealed decreased expression levels of MAP2 (p < 0.01) and fewer SYN-positive synapses on neurons (p < 0.01) in the SCI group than in the sham group. The glial scar area was decreased, the number of motor neurons was increased, the expression levels of neuronal MAP2 were elevated, and the number of SYN-positive synapses was increased in the SCI + CHBP group compared to the SCI group that received no treatment (p < 0.01 for all; Fig. 1A–H). Next, functional recovery was assessed by footprint analysis, and the SCI + CHBP group performed better than the SCI group on the 28th day after injury (Fig. 1I). The BMS score of the sham group was markedly higher than that of the SCI group on days 1, 3, 7, 14, 21, and 28 after injury (p < 0.01 for all). There were no marked difference in BMS score between the SCI and SCI + CHBP groups on day 1 or 3. However, CHBP-treated mice exhibited higher BMS scores than untreated SCI animals on days 7, 14, 21, and 28 after injury (p = 0.031, <0.01, <0.01, and <0.01, respectively; Fig. 1J). Overall, these results indicated that CHBP facilitated functional restoration after SCI.
Fig. 1.

CHBP promotes functional restoration after SCI. (A) Longitudinal spinal cord sections from the indicated groups were subjected to Masson staining and HE staining on day 28 after SCI (scale bar = 1000 μm). (B) Quantitation of Masson staining in the spinal cord in the different groups. (C) Transverse spinal cord sections from the indicated groups were analysed on day 14 via HE staining and Nissl staining (scale bar = 500 μm and 125 μm). (D) Quantitation analysis of Nissl-positive motor neurons in the anterior horn of the spinal cord in the indicated groups. (E) Images (10 × ) of spinal cord slices from the indicated groups following incubation with antibodies against MAP2 (scale bar = 25 μm). (F) MAP2 OD on day 28 after SCI. (G) Images (10 × ) of SCI slices (T11-T12) stained with antibodies against SYN/NeuN collected on day 28 (scale bar = 5 μm). (H) Quantitation of nerve cell-contacting synapses. (I) Photographs of mouse footprints on day 28 after SCI. (J) BMS scores of the indicated groups at the different time points (n = 12). (K) Heatmap of upregulated and downregulated genes in the spinal cords of mice treated with CHBP for 3 days (n = 5 mice per group). (L) Volcano plot of DEGs in the SCI and SCI + CHBP groups. The downregulated DEGs are shown in blue, and the upregulated DEGs are shown in red. (M) GO enrichment analysis of the targeted genes revealing the biological processes affected by CHBP. The data are presented as the mean ± SEM; n = 6 (except for BMS). **p < 0.01 versus the sham group. #p < 0.05 and ##p < 0.01 versus the SCI group. For the data in Fig. 1B, D, F, H and J, statistical analysis was performed using two-way ANOVA followed by Tukey's multiple comparison test. For the data in Fig. 1K-M, statistical analysis was performed using an unpaired t-test. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.2. Potential molecular mechanisms of CHBP for SCI
To explore the potential therapeutic mechanism of SCI, we analysed the original microarray dataset GSE171441, which was acquired from the GEO database. According to predetermined thresholds (modified P value < 0.05 and |log2(fold-change)| > 1.2), we identified 1275 and 1051 DEGs in subgroups I and II, respectively. Volcano plots were used to visualize the differences between control and injured spinal cord tissues (Fig. S2A and F). Heatmaps of the DEGs are shown in Fig. S2B and G. To identify the potential biological processes and pathways that were altered after SCI, GO functional enrichment analyses of the DEGs identified in subgroups I and II were performed. As shown in Fig. S2C and H, biological process analyses revealed that the DEGs in subgroups I and II were most markedly enriched in positive modulation of cytokine production, autophagosome maturation and assembly, necroptotic progress, and pyroptosis. Cellular component analyses revealed that the DEGs in subgroups I and II were enriched primarily in extracellular matrix (ECM). Molecular function analyses revealed that the DEGs in subgroups I and II were most markedly enriched in the necroptotic signalling pathway and cysteine-type endopeptidase activity involved in apoptotic process. The GSEA results also revealed significant differences between control and injured spinal cord tissues in subgroups I and II that were related to the regulation of autophagy, positive regulation of protein autophosphorylation, necroptotic process, the regulation of necroptotic process, and pyroptosis (Fig. S2D and I). KEGG pathway analyses revealed that the DEGs were notably enriched in the NOD-like receptor signalling pathway, NF-kappa B signalling pathway, and TNF signalling pathway (Fig. S2E and J).
To further verify these mechanisms underlying the effect of CHBP for SCI, we performed transcriptome sequencing of samples on day 3 from the SCI group and SCI + CHBP group. A total of 408 genes were differentially expressed (360 were downregulated and 48 were upregulated) between the SCI + CHBP group and the SCI group (p < 0.05 for all, Fig. 1K-L). GO analysis showed that the DEGs were predominantly enriched in pyroptosis, synapse, regulation of autophagy, inflammatory response, necroptotic signalling pathway and AMP-activated protein kinase activity (Fig. 1M). The transcriptome sequencing results showed that CHBP may promote autophagy and suppress pyroptosis and necroptosis to promote functional recovery after SCI.
3.3. CHBP decreases pyroptosis after SCI
Transcriptome sequencing suggested that pyroptosis was one of the underlying mechanisms of CHBP. It has reported that pyroptosis activity after SCI was mainly increased on days 1 and 3 [49]. Moreover, some pyroptosis proteins following SCI are the same as pro-inflammatory mediators which are known to peak at 3dpi. Therefore, we evaluated pyroptotic activity by assessing ASC, IL-18, GSDMD, IL-1β, Caspase-1, NLRP1, and NLRP3 expression within the spinal cord on days 1 and 3 following SCI. IF staining revealed that the density of Caspase-1 and GSDMD in neurons was markedly elevated in the SCI model animals (p < 0.01 for all), while the density of Caspase-1 and GSDMD was reduced in CHBP-treated SCI model animals compared to untreated SCI model animals (p < 0.01 for all) at 1 and 3 days post-injury (dpi), as shown in Fig. S3A-C and Fig. 2A-D. The expression levels of ASC, IL-18, GSDMD, IL-1β, Caspase-1, NLRP1, and NLRP3 were analysed by Western blotting (Fig. 2E). Compared to the sham animals, the SCI model animals exhibited greater optical densities (ODs) of NLRP1, IL-18, NLRP3, IL-1β, Caspase-1, ASC, and GSDMD at 3 dpi (p < 0.01 for all). The ODs of pyroptosis-associated biomarkers were lower in the SCI + CHBP group than in the SCI group at 3 dpi (p = 0.018, = 0.016, <0.01, = 0.013, <0.01, <0.01, and <0.01, respectively; Fig. 2F). Furthermore, the ODs of NLRP1, IL-18, NLRP3, IL-1β, Caspase-1, ASC, and GSDMD on day 1 were lower in the group treated with CHBP (p = 0.016, <0.01, = 0.047, <0.01, <0.01, <0.01, and <0.01, respectively; Fig. S3D–E). These findings confirmed that CHBP decreases the levels of pyroptosis-related biomarkers and thus inhibits pyroptosis after SCI.
Fig. 2.

CHBP attenuates pyroptosis after SCI. (A) Images (20 × ) of spinal cord slices from the SCI + CHBP, SCI and sham groups on day 3 after SCI following incubation with antibodies against Caspase-1/NeuN (scale bar = 25 μm). (B) Quantitation of the average OD of Caspase-1 in neurons after SCI. (C) Images (20 × ) of spinal cord slices from the SCI + CHBP, SCI and sham groups on day 3 after SCI following incubation with antibodies against GSDMD/NeuN (scale bar = 25 μm). (D) Quantitation of the average OD of GSDMD in neurons after SCI. (E) ODs of the NLRP1, IL-1β, ASC, Caspase-1, IL-18, NLRP3, and GSDMD bands in the indicated groups on day 3 after SCI. The blots were subjected to the same experimental conditions, and cropped images of the blots are shown. (F) The expression levels of NLRP1, IL-1β, GSDMD, NLRP3, ASC, Caspase-1, and IL-18 in each group were quantified. The data are presented as the mean ± SEM; n = 6 per group. *p < 0.05 and **p < 0.01 versus the sham group. #p < 0.05 and ##p < 0.01 versus the SCI group. Statistical analysis was performed using two-way ANOVA followed by Tukey's multiple comparison test.
3.4. CHBP suppresses necroptosis after SCI
The results of transcriptome sequencing suggested that CHBP may decrease cell death and attenuate neuroinflammation by regulating necroptosis. In addition, previous studies have shown that the increase of necroptosis after SCI is mainly concentrated on days 1 and 3 [50,51]. Therefore, our team evaluated the levels of necroptosis via IF and Western blotting on days 1 and 3 following SCI. IF staining revealed that the density of RIPK1 and RIPK3 in neurons was markedly elevated in the SCI model animals (p < 0.01 for all), but the density of RIPK1- and RIPK3-positive neurons was reduced in the CHBP-treated SCI model animals compared to the untreated SCI model animals (p < 0.01 for all) at 1 and 3 dpi, as shown in Fig. 3A–D and S3F–H. WB analysis of RIPK1, MLKL, C-Caspase-8 and RIPK3 expression was performed (Fig. 3E). The SCI group displayed higher ODs of MLKL, RIPK3, and RIPK1 but a lower OD of C-Caspase-8 than the sham group at 1 dpi (p < 0.01 for all). The SCI + CHBP group exhibited lower ODs of MLKL, RIPK3, and RIPK1 but a higher OD of C-Caspase-8 than the SCI group at 1 dpi (p = 0.047, <0.01, <0.01, and <0.01, respectively; Fig. 3F). Moreover, the SCI + CHBP group also exhibited lower ODs of MLKL, RIPK3, and RIPK1 but a higher OD of C-Caspase-8 than the SCI group at 3 dpi (p = 0.048, <0.01, <0.01, and <0.01, respectively; Fig. S3I–J). In general, these findings revealed that the ability of CHBP to restore function after SCI was partially due to the inhibition of necroptosis.
Fig. 3.

CHBP inhibits necroptosis after SCI. (A) Images (20 × ) of spinal cord slices from the SCI + CHBP, SCI and sham groups on day 1 after SCI following incubation with antibodies against RIPK1/NeuN (scale bar = 25 μm). (B) Quantitation of the OD of RIPK1 in neurons after SCI. (C) Images (20 × ) of spinal cord slices from the SCI + CHBP, SCI and sham groups on day 1 after SCI following incubation with antibodies against RIPK3/NeuN (scale bar = 25 μm). (D) Quantitation of the OD of RIPK3 in neurons after SCI. (E) ODs of the of RIPK1, MLKL, RIPK3, and C-Caspase-8 bands in the SCI + CHBP, SCI, and sham groups on day 1 after SCI. The blots were subjected to the same experimental conditions, and cropped images of the blots are shown. (F) The expression levels of RIPK1, MLKL, RIPK3, and C-Caspase-8 in the abovementioned groups were quantified. The data are presented as the average ± SEM; n = 6 each group. *p < 0.05 and **p < 0.01 versus the sham group. #p < 0.05 and ##p < 0.01 versus the SCI group. Statistical analysis was performed using two-way ANOVA followed by Tukey's multiple comparison test.
3.5. CHBP promotes autophagy after SCI
According to previous studies, autophagy is an important target for the treatment of CNS diseases and exerts a neuroprotective effect against programmed cell death caused by neuroinflammation [52,53]. To evaluate autophagy activity after SCI, our team assessed the expression of an autophagy substrate protein (p62), an autolysosomal biomarker (CTSD), and autophagosome biomarkers (Vps34, Beclin-1 and LC3II). As revealed by IF staining, the density of p62 in neurons was notably elevated following SCI (p < 0.01); nevertheless, the SCI + CHBP group exhibited a comparatively lower density of p62 in neurons than the SCI group (p < 0.01; Fig. 4A–B). The number of LC3II puncta in each neuron was increased in the SCI model animals than in the sham animals (p < 0.01). Furthermore, the number of LC3II puncta in each neuron was increased in the CHBP-treated SCI model animals compared to the untreated SCI model animals (p < 0.01), as shown in Fig. 4C-D. The transcription of autophagy-related genes was increased in the SCI model animals compared to the sham animals, as determined by qPCR, and the transcription of autophagy-associated genes was notably increased in CHBP-treated SCI model animals compared to untreated SCI model animals (p < 0.01 for all; Fig. 4E). The expression levels of VPS34, Beclin-1, LC3II, and p62 were significantly higher in the SCI group than in the sham group, whereas the expression level of CTSD was lower in the SCI group (p < 0.01 for all). The expression levels of VPS34, Beclin-1, LC3II, and CTSD were significantly higher in the CHBP-treated SCI model animals than in the untreated SCI model animals, whereas the expression level of p62 was lower (p = 0.023, <0.01, <0.01, <0.01, and <0.01, respectively; Fig. 4F–G). These findings indicated that matrix proteins accumulated after SCI despite the upregulation of autophagosome- and autophagolysosome-associated biomarkers. Moreover, these results revealed that CHBP upregulated the expression of autophagic lysosome- and autophagosome-related biomarkers and decreased the levels of substrate proteins by effectively ameliorating autophagic flux after SCI.
Fig. 4.

CHBP promotes autophagy after SCI. (A) Images (20 × ) of spinal cord slices from the SCI + CHBP, SCI and sham groups on day 3 after SCI following incubation with antibodies against p62/NeuN (scale bar = 25 μm). (B) Quantitation of the average OD of p62 in neurons after SCI in the abovementioned groups. (C) Images (20 × ) of spinal cord slices from the SCI + CHBP, SCI and sham groups on day 3 after SCI following incubation with antibodies against LC3/NeuN (scale bar = 25 μm). (D) The number of LC3II puncta in each neuron in the abovementioned groups. (E) mRNA levels of Beclin-1, Vps34, Ctsd, Lc3 and Sqstm1 normalized to β-actin levels in the injured area in the abovementioned groups on day 3 after SCI. (F) ODs of the p62, LC3II, Beclin-1, VPS34, and CTSD bands in the sham group, SCI group, and CHBP group on day 3 after SCI. The blots were subjected to identical experimental conditions, and cropped images of the blots are shown. (G) The expression levels of p62, LC3II, Beclin-1, VPS34, and CTSD in the abovementioned groups were quantified. The data are presented as the average ± SEM; n = 6 each group. *p < 0.05 and **p < 0.01 versus the sham group. #p < 0.05 and ##p < 0.01 versus the SCI group. Statistical analysis was performed using two-way ANOVA followed by Tukey's multiple comparison test.
3.6. CHBP promotes autophagy to inhibit pyroptosis and necroptosis after SCI
3-MA belongs to a group of phosphatidylinositol 3-kinase (PI3K) inhibitors that inhibit type III PI3 kinase activity and the production of PI3P and is commonly used for autophagy research in vitro [54]. 3-MA was coadministered with CHBP to evaluate whether CHBP improves outcomes after SCI by stimulating autophagy. IF and neuron colocalization analyses revealed that the SCI + CHBP+3-MA group exhibited fewer LC3II puncta and a greater p62 density than the SCI + CHBP group (p < 0.01 for both; Fig. 5A–C). WB analysis was performed to determine the levels of p62, CTSD, LC3II, VPS34, and Beclin-1 (Fig. 5D). The results revealed that the ODs of VPS34, Beclin-1, LC3II, and CTSD were lower in the SCI + CHBP+3-MA group than in the SCI + CHBP group (p < 0.01, <0.01, = 0.012, and <0.01, respectively), while the OD of p62 was higher in the SCI + CHBP+3-MA group than in the SCI + CHBP group (p < 0.01; Fig. 5E), indicating that 3-MA markedly inhibited autophagy activity when coadministered with CHBP. For further validation of autophagy as the primary process through which CHBP promotes recovery of neuronal function after SCI, our team explored the impact of 3-MA on pyroptosis and necroptosis. IF analysis revealed that the density of Caspase-1 and GSDMD in neurons was higher in the SCI + CHBP+3-MA group than in the SCI + CHBP group at 3 dpi (p < 0.01 for both; Fig. 5F–H). Compared to the SCI + CHBP group, the SCI + CHBP+3-MA group exhibited higher ODs of pyroptotic proteins (NLRP1, IL-18, NLRP3, ASC, Caspase-1, GSDMD and IL-1β) at 3 dpi (p < 0.01, = 0.031, = 0.037, <0.01, <0.01, and <0.01, = 0.040, respectively; Fig. 5I–J). Moreover, IF staining and WB analysis revealed that the expression of pyroptosis-related biomarkers was higher in the SCI + CHBP+3-MA group than in the SCI + CHBP group at 1 dpi, as presented in Fig. S4A–E. IF analysis also revealed that the densities of RIPK3 and RIPK1 in neurons were higher in the SCI + CHBP+3-MA group than in the SCI + CHBP group at 1 dpi (p < 0.01 for both; Fig. 5K-M). Compared to those in the SCI + CHBP group, the expression levels of necroptotic proteins (RIPK1, RIPK3, and MLKL) in the SCI + CHBP+3-MA group were higher, while the expression of C-Caspase-8 was lower in the SCI + CHBP+3-MA group at 1 dpi (p < 0.01, = 0.043, = 0.031, and <0.01, respectively; Fig. 5N-O). Furthermore, IF staining and WB analysis revealed that the expression of necroptosis biomarkers was higher in the SCI + CHBP+3-MA group than in the SCI + CHBP group at 3 dpi, as presented in Fig. S4F–J. These findings revealed that coadministration of 3-MA with CHBP attenuated the effects of CHBP on necroptosis and pyroptosis, indicating that CHBP may promote autophagy by suppressing necroptosis and pyroptosis after SCI.
Fig. 5.

CHBP promotes autophagy to inhibit pyroptosis and necroptosis after SCI. (A) Images (20 × ) of spinal cord slices from the mentioned groups on day 3 after SCI following incubation with antibodies against p62/NeuN and LC3/NeuN (scale bar = 25 μm). (B–C) Quantitation of the average OD of p62 and the number of LC3II puncta in each neuron in the abovementioned groups. (D) ODs of the p62, LC3II, Beclin-1, VPS34, and CTSD bands in the indicated groups on day 3 after SCI. (E) The expression levels of p62, LC3II, Beclin-1, VPS34, and CTSD in the abovementioned groups were quantified. (F) Images (20 × ) of spinal cord slices from the aforementioned groups on day 3 after SCI following incubation with antibodies against Caspase-1/NeuN and GSDMD/NeuN (scale bar = 25 μm). (G–H) Quantitation of the average ODs of Caspase-1 and GSDMD in neurons after SCI. (I) ODs of the NLRP1, IL-1β, NLRP3, Caspase-1, ASC, GSDMD, and IL-18 bands in the indicated groups on day 3 after SCI. (J) The expression levels of NLRP1, IL-1β, NLRP3, Caspase-1, ASC, GSDMD, and IL-18 in the abovementioned groups were quantified. (K) Images (20 × ) of spinal cord slices from the abovementioned groups on day 1 after SCI following incubation with antibodies against RIPK1/NeuN and RIPK3/NeuN (scale bar = 25 μm). (L–M) Quantitation of the ODs of RIPK1 and RIPK3 in neurons after SCI. (N) ODs of the RIPK1, MLKL, RIPK3, and C-Caspase-8 bands in the indicated groups on day 1 after SCI. (O) The expression levels of RIPK1, MLKL, RIPK3, and C-Caspase-8 in the abovementioned groups were quantified. The data are presented as the average ± SEM; n = 6 each group. @p < 0.05 and @@p < 0.01 versus the SCI + CHBP group. Statistical analysis was performed using an unpaired t-test.
3.7. Autophagy suppression reverses the neuroprotective effect of CHBP in SCI
We performed histomorphological and functional experiments comparing the sham group, sham+3-MA group, SCI group, SCI+3-MA group, SCI + CHBP group and SCI + CHBP+3-MA group to further confirm that 3-MA is not toxic and assess the efficacy of CHBP in an SCI model. As revealed by HE and Masson staining, the glial scar area in the lesion site was larger in the SCI + CHBP+3-MA group than in the SCI + CHBP group (p < 0.01, Fig. S5A–B), and there were fewer motor neurons in the lesion area in the SCI + CHBP+3-MA group than in the SCI + CHBP group (p < 0.01, Fig. S5C–D). Additionally, the injured region was evaluated via IF staining, and the results showed lower MAP2 levels (p < 0.01, Fig. S5E–F) and fewer SYN-positive synapses (p < 0.01, Fig. S5G–H) in the SCI + CHBP+3-MA group than in the SCI + CHBP group. The glial scar area was decreased, the number of motor neurons was increased, the expression levels of neuronal MAP2 were elevated, and the number of synapses positive for SYN was increased in the SCI + CHBP+3-MA group compared to the SCI+3-MA group (p < 0.01 for all; Fig. S5A–H). On the 28th day after SCI, the SCI + CHBP group exhibited remarkable recovery of hindleg motion, with relatively good crawling, whereas the SCI+3-MA and SCI + CHBP+3-MA group animals continuously dragged their hindlegs (Fig. S5I). No marked differences in BMS scores were observed between the SCI + CHBP group and SCI + CHBP+3-MA group on day 1 or 3. The BMS score was significantly lower in the SCI + CHBP+3-MA group than in the SCI + CHBP group on days 7, 14, 21, and 28 after SCI (p = 0.024, <0.01, <0.01, and <0.01, respectively; Fig. S5J). The BMS score was also significantly lower in the SCI+3-MA group than in the SCI + CHBP+3-MA group on days 21 and 28 after SCI (p = 0.038, and <0.01, respectively; Fig. S5J). Therefore, the autophagy-ameliorating effect of CHBP might have improved outcomes after SCI.
3.8. CHBP promotes autophagy by increasing TFEB activity and subsequently suppresses necroptosis and pyroptosis after SCI
Previous studies have shown that TFEB rescues autophagosome-lysosome pathway dysfunction in neurological diseases, reducing neuronal death and alleviating neuroinflammation [55,56]. Therefore, our team subsequently investigated TFEB expression in the nucleus and cytoplasm to determine whether CHBP modulates TFEB expression. CHBP treatment significantly elevated TFEB expression in neurons, and CHBP effectively enhanced TFEB nuclear translocation, as shown in Fig. 6A–B (p < 0.01 for both). Next, in agreement with the WB findings, cytoplasmic TFEB expression, cytoplasmic p-TFEB (Ser211) expression and intranuclear TFEB expression were markedly elevated in the SCI + CHBP group (p < 0.01 for all; Fig. 6C–D). To further confirm that CHBP stimulates TFEB stimulation to enhance autophagy activity and attenuate necroptosis and pyroptosis, our team applied AAV-TFEB shRNA to silence TFEB and developed an assay to compare the SCI + CHBP, SCI + CHBP + scrambled shRNA, and SCI + CHBP + TFEB shRNA groups. The results revealed that both cytoplasmic TFEB expression and nuclear TFEB expression in the SCI + CHBP + TFEB shRNA group were remarkably lower than those in the SCI + CHBP + scrambled shRNA group (p < 0.01 for both), but there was no significant difference in nuclear TFEB expression between the SCI + CHBP and SCI + CHBP + scrambled shRNA groups (Fig. 6E–F). These findings revealed that transfection of TFEB shRNA inhibited TFEB expression and nuclear translocation following SCI.
Fig. 6.

CHBP promotes autophagy by increasing TFEB activity. (A) IF staining of TFEB in neurons in the SCI + CHBP, SCI and sham groups on day 3 after SCI (scale bar = 25 μm). (B) Quantitation of the integrated density of TFEB in spinal cord neurons and the integrated density of TFEB in the nucleus of each neuron. (C) WB analyses of cytoplasmic p-TFEB(Ser221), cytoplasmic TFEB, nuclear p-TFEB(Ser221) and nuclear TFEB expression in the sham group, SCI group and SCI + CHBP group on day 3 after SCI. (D) Quantitation of the ODs of cytoplasmic p-TFEB (Ser221), cytoplasmic TFEB, nuclear p-TFEB (Ser221) and nuclear TFEB. (E) WB analysis of cytoplasmic p-TFEB (Ser221), cytoplasmic TFEB, nuclear p-TFEB (Ser221) and nuclear TFEB expression in the SCI + CHBP group, SCI + CHBP + scrambled shRNA and SCI + CHBP + TFEB shRNA group on day 3 after SCI. (F) Quantitation of the expression levels of cytoplasmic p-TFEB (Ser221), cytoplasmic TFEB, nuclear p-TFEB (Ser221) and nuclear TFEB. (G) Images (20 × ) of spinal cord slices in the abovementioned groups on day 3 after SCI following incubation with antibodies against LC3/NeuN (scale bar = 25 μm). (H) The number of LC3II puncta in the abovementioned groups on day 3 after SCI. (I) mRNA levels of Beclin-1, Vps34, Ctsd, Lc3 and Sqstm1 normalized to that of β-actin in the injury site in the abovementioned groups on day 3 after SCI. (J) ODs of the p62, VPS34, LC3II, Beclin-1, and CTSD bands in the SCI + CHBP, SCI + CHBP + scrambled shRNA and SCI + CHBP + TFEB shRNA groups on day 3 after SCI. (K) The expression levels of p62, VPS34, LC3II, Beclin-1, and CTSD in the abovementioned groups were quantified. The data are presented as the average ± SEM; n = 6 each group. *p < 0.05 and **p < 0.01 versus the sham group. #p < 0.05 and ##p < 0.01 versus the SCI group. @p < 0.05 and @@p < 0.01 versus the SCI + CHBP group. &p < 0.05 and &&p < 0.01 versus the SCI + CHBP + scrambled shRNA group. Statistical analysis was performed using two-way ANOVA followed by Tukey's multiple comparison test.
Next, our team examined whether TFEB nuclear translocation triggered by CHBP regulates autophagy activity, necroptosis, and pyroptosis. As shown by IF staining, the number of LC3II puncta in the SCI + CHBP + TFEB shRNA group was significantly decreased (p < 0.01), while there was no marked difference in the number of LC3II puncta in each neuron between the SCI + CHBP group and the SCI + CHBP + scrambled shRNA group (Fig. 6G–H). The transcription of autophagy-associated genes was decreased after transfection with TFEB shRNA, as determined by qPCR (p < 0.01 for all; Fig. 6I). In addition, as shown by Western blotting, the ODs of VPS34, p62, CTSD, LC3II, and Beclin-1 were not markedly different between the SCI + CHBP group and SCI + CHBP + scrambled shRNA group, and the ODs of LC3II, CTSD, Beclin-1, and VPS34 were markedly lower in the SCI + CHBP + TFEB shRNA group than in the SCI + CHBP + scrambled shRNA group; however, the opposite results were observed for p62 (p = 0.019, <0.01, <0.01, <0.01, and <0.01, respectively; Fig. 6J–K). Additionally, IF staining revealed that the level of Caspase-1 was markedly elevated in the SCI + CHBP + TFEB shRNA group (p < 0.01), while the integrated density of Caspase-1 in neurons was not markedly different between the SCI + CHBP group and SCI + CHBP + scrambled shRNA at 3 dpi (Fig. 7A–B). As shown by WB analysis, the ODs of NLRP1, IL-18, NLRP3, IL-1β, Caspase-1, ASC, and GSDMD were not markedly different between the SCI + CHBP group and the SCI + CHBP + scrambled shRNA group; however, the ODs of NLRP1, IL-18, NLRP3, IL-1β, Caspase-1, ASC, and GSDMD were markedly higher in the SCI + CHBP + TFEB shRNA group than in the SCI + CHBP + scrambled shRNA group at 3 dpi (p < 0.01 for all; Fig. 7C–D). Moreover, IF staining and WB analysis revealed that the expression of pyroptosis-related biomarkers was higher in the SCI + CHBP + TFEB shRNA group than in the SCI + CHBP + scrambled shRNA group at 1 dpi, as shown in Fig. S6A–D. Subsequently, IF staining revealed that the level of RIPK1 was markedly elevated in the SCI + CHBP + TFEB shRNA group (p < 0.01), but the integrated density of RIPK1 in neurons was not markedly different between the SCI + CHBP group and the SCI + CHBP + scrambled shRNA group at 1 dpi (Fig. 7E–F). As shown by WB analysis, the ODs of RIPK1, RIPK3, MLKL, and C-Caspase-8 were not markedly different between the SCI + CHBP group and SCI + CHBP + scrambled shRNA group. The ODs of RIPK1, RIPK3, and MLKL were markedly higher in the SCI + CHBP + TFEB shRNA group than in the SCI + CHBP + scrambled shRNA group, whereas the opposite results were observed for C-Caspase-8 at 1 dpi (p < 0.01 for all; Fig. 7G–H). Furthermore, IF staining and WB analysis revealed that the expression of necroptosis biomarkers was higher in the SCI + CHBP + TFEB shRNA group than in the SCI + CHBP + scrambled shRNA group at 3 dpi, as shown in Fig. S6E-H. Additionally, we administered AAV-TFEB shRNA to mice not treated with CHBP to clarify the regulatory role of TFEB and conducted an assay comparing the SCI-only, SCI + scrambled shRNA, and SCI + TFEB shRNA groups. The SCI + TFEB shRNA group showed reduced expression of autophagy-related biomarkers and inhibited TFEB expression and nuclear translocation with enhanced expression of pyroptosis-related biomarkers [57] and necroptosis biomarkers [14], as presented in Fig. S7A–H.
Fig. 7.

TFEB inhibition reverses the suppressive effects of CHBP on pyroptosis and necroptosis following SCI. (A) Analysis GSDMD and NeuN colocalization in spinal cord lesions on day 3 after SCI by IF staining (scale bar = 25 μm). (B) Average OD of GSDMD in neurons in the spinal cord lesion. (C) WB analysis of IL-18, IL-1β, GSDMD, Caspase-1, ASC, NLRP3, and NLRP1 expression levels in each group on day 3 after SCI. (D) The ODs of IL-18, IL-1β, GSDMD, Caspase-1, ASC, NLRP3, and NLRP1 in the different groups were quantified. (E) Analysis of RIPK1 and NeuN colocalization in the spinal cord lesion on day 1 after SCI by IF staining (scale bar = 25 μm). (F) Quantification of the OD of RIPK1 in neurons in the spinal cord lesion. (G) WB analysis of C-Caspase-8, MLKL, RIPK3, and RIPK1 expression in the three groups on day 1 after SCI. (H) The ODs of C-Caspase-8, MLKL, RIPK3, and RIPK1 in the different groups were quantified. The data are expressed as the mean ± SEM; n = 6 per group. @p < 0.05 and @@p < 0.01 versus the SCI + CHBP group. &p < 0.05 and &&p < 0.01 versus the SCI + CHBP + scrambled shRNA group. Statistical analysis was performed using two-way ANOVA followed by Tukey's multiple comparison test.
Finally, we evaluated the therapeutic effect of CHBP after transfection with TFEB shRNA. Compared to the SCI + CHBP group, the SCI + CHBP + TFEB shRNA group exhibited a larger glial scar area (p < 0.01, Fig. S8A–B), fewer motor neurons (p < 0.01, Fig. S8C–D), lower expression of MAP2 (p < 0.01, Fig. S8E–F), and fewer SYN-positive synapses (p < 0.01, Fig. S8G–H) following SCI. The animals in the SCI + CHBP + TFEB shRNA group dragged their hindlegs continuously on day 28 following injury (Fig. S8I). There were no marked differences in BMS scores among the SCI + CHBP, SCI + CHBP + scrambled shRNA, and SCI + CHBP + TFEB shRNA groups on day 1 or 3. Compared to those of the SCI + CHBP and SCI + CHBP + scrambled shRNA groups, the BMS scores of the SCI + CHBP + TFEB shRNA group were notably reduced on days 7, 14, 21, and 28 after SCI (p = 0.027, <0.01, <0.01, and <0.01, respectively; Fig. S8J). In summary, these findings revealed that TFEB stimulation and nuclear translocation are the main mechanisms by which CHBP elevates autophagy activity and suppresses necroptosis and pyroptosis after SCI.
3.9. CHBP stimulates TFEB activity via the AMPK-mTOR and AMPK-FOXO3a-SPK2-CARM1 signalling pathways after SCI
As revealed in previous studies, there are 2 vital signalling pathways that regulate TFEB: the AMPK-mTOR and AMPK-FOXO3a-SPK2-CARM1 signalling pathways. Transcriptome sequencing demonstrated that CHBP upregulated DEGs related to AMP-activated protein kinase activity (Fig. 1M). Therefore, our team explored whether these 2 signalling pathways are stimulated in spinal cord tissues following treatment with CHBP. The results revealed that CHBP elevated the nuclear translocation of p-AMPK and TFEB and reduced the level of p-mTOR in the cytoplasm (p < 0.01 for all) but did not alter the expression of mTOR (Fig. 8A–B), revealing that CHBP stimulated the AMPK-mTOR signalling pathway. In the nucleus, CHBP elevated the expression of AMPK, p-AMPK, FOXO3a, p-FOXO3a, and CARM1 while suppressing the expression of SPK2 (p < 0.01 for all; Fig. 8C–D), revealing that CHBP also stimulated the AMPK-FOXO3a-SPK2-CARM1 signalling pathway. Furthermore, Immunoprecipitation following by Western blotting showed that the binding of CARM1 to TFEB was increased in the spinal cord after injury (p < 0.01; Fig. 8E–F). To further investigate whether CHBP-induced TFEB stimulation is mediated by the AMPK-FOXO3a-SPK2-CARM1 and AMPK-mTOR signalling pathways, our team investigated the effect of CC, an AMPK blocker, in the SCI + CHBP group. p-AMPK levels and TFEB nuclear translocation were lower in the SCI + CHBP + CC group than in the SCI + CHBP group (p = 0.012 and p < 0.01), whereas p-mTOR levels were higher in the SCI + CHBP + CC group (p = 0.012, Fig. 8A–B). In addition, the ODs of AMPK, p-AMPK, FOXO3a, p-FOXO3a, and CARM1 were markedly decreased, whereas the opposite results were observed for SKP2 (p < 0.01, <0.01, <0.01, <0.01, <0.01, and = 0.019, respectively; Fig. 8C–D). Moreover, the binding of CARM1 to TFEB was decreased in the SCI + CHBP + CC group (p < 0.01; Fig. 8E–F). We further evaluated whether the AMPK-FOXO3a-SPK2-CARM1 and AMPK-mTOR signalling pathways participate in CHBP-mediated regulation of pyroptosis-, necroptosis-, and autophagy-associated proteins. As revealed by WB analysis, the ODs of Caspase-1, GSDMD, RIPK1, RIPK3, and p62 were considerably higher in the SCI + CHBP + CC group than in the SCI + CHBP group, whereas the expression levels of LC3II exhibited the opposite trend (p < 0.01 for all; Fig. 8G–H). Collectively, these findings verified that CHBP stimulated TFEB activity via the AMPK-mTOR and AMPK-FOXO3a-SPK2-CARM1 signalling pathways after SCI.
Fig. 8.

CHBP stimulates TFEB activity via the AMPK-mTOR pathway and AMPK-FOXO3a-SPK2-CARM1 signalling pathway after SCI. (A) WB analysis of the cytoplasmic expression of p-TFEB (Ser221), AMPK, p-AMPK, mTOR, and p-mTOR normalized to the expression of GAPDH as an internal control on day 3 after SCI and the nuclear expression of TFEB normalized to the expression of histone H3 as an internal control on day 3 after SCI. (B) Densitometric quantification of the p-mTOR, mTOR, p-AMPK, AMPK, p-TFEB (Ser221), and nuclear TFEB bands. (C) WB analysis of the nuclear expression of CARM1, SKP2, FOXO3a, p-FOXO3a, p-AMPK, and AMPK normalized to the expression of Histone H3 as an internal control on day 3 after SCI. (D) Densitometric quantification of the CARM1, SKP2, FOXO3a, p-FOXO3a, p-AMPK, and AMPK bands. (E) The nuclear CARM1-TFEB complex in the indicated groups was detected by immunoprecipitation on day 3 after SCI. (F) The densities of the TFEB and CARM1 bands in E normalized to the density of the band for H3 as a loading control. (G) WB analysis of RIPK1, RIPK3, GSDMD, and Caspase-1 expression levels at 1 dpi and 3 dpi and the expression levels of LC3II and p62 on day 3 after SCI in the three groups. (H) The densities of the LC3II, p62, RIPK1, RIPK3, GSDMD, and Caspase-1 bands in G normalized to the density of the GAPDH band. The data are presented as the mean ± SEM; n = 6 per group. *p < 0.05 and **p < 0.01 versus the SCI group. @p < 0.05 and @@p < 0.01 versus the SCI + CHBP group. ns, not significant. Statistical analysis was performed using two-way ANOVA followed by Tukey's multiple comparison test.
4. Discussion
SCI is a severe and disabling disease that requires extensive medical care and imposes a socioeconomic burden. Regrettably, effective strategies to improve motor, sensory, and autonomic functions in patients after SCI have not yet been developed. Aberrant neuroinflammation and cell death play important roles in the pathology of secondary injury after SCI, and drugs that attenuate proinflammatory cell death, primarily pyroptosis and necroptosis, and improve nerve regeneration have displayed significant therapeutic effects [58,59]. The bioinformatics analyses performed in this study based on the GSE171441 dataset identified several signalling pathways that were altered in mice with acute and chronic SCI, including the NOD−like receptor signalling pathway, the NF−kappa B signalling pathway and the TNF signalling pathway, which are associated with pyroptosis and necroptosis. Although CHBP has been reported to alleviate the inflammatory response in several diseases, whether CHBP attenuates pyroptosis and necroptosis after SCI has not been studied. Our study is the first to reveal that the EPO-derived peptide CHBP enhances autophagy activity, improves functional recovery and alleviates pyroptosis and necroptosis in SCI model mice. In addition, we further demonstrated that the neuroprotective effect of CHBP is due to the stimulation of TFEB through AMPK-FOXO3a-SPK2-CARM1 and AMPK-mTOR signalling channels.
EPO is an endocrine hormone that plays a protective role in many organs [3]. Many studies have described the neurogenetic effects of EPO in several neuropathological disorders [60,61]. For example, a study involving transcriptome analysis reported that a chemically modified form of EPO, carbamoylated erythropoietin (CEPO), increased neurogenesis and memory formation in rodent models and humans [62]. In addition, after SCI, neural precursors spontaneously release EPO, which subsequently accumulates in the injured area to counteract secondary degeneration, attenuating neuroinflammation and myelin loss [63]. However, because a very high concentration of EPO, which can greatly increase the risk of high blood pressure, stroke and thrombogenesis [64], is required for tissue protection, the clinical application of EPO remains limited [35]. Data acquired from crystallography studies of EPO bound to the EPO receptor have revealed that the aqueous face of helix B comprises 11 amino acids (QEQLERALNSS) [65]. For that reason, peptides designed on the basis of surface emulation analyses of EPO should have the bioactivity of helix B. To verify this assumption, HBSP was produced, and it was demonstrated to exert tissue-protective effects similar to that of EPO itself but with fewer adverse side effects [66]. Nevertheless, the 2-min plasma half-life of HBSP limits its usage in vivo [28]. Notably, cyclization serves as a valid method to stabilize the secondary architecture of a linear peptide and increase its stability and effect [[67], [68], [69]]. CHBP, which has a set of conformationally constrained helix B peptides that elevates its tolerance to proteolytic decomposition, improves its renoprotective effect, and reduces the required administration duration and dosage, was developed. Yang et al. reported that administration of CHBP inhibits cardiomyocyte apoptosis and inflammation in acute myocardial infarction animal models [70]. Zhang et al. found that CHBP inhibits NLRP3 inflammasome stimulation via the NF-κB pathway in alveolar macrophages to ameliorate sepsis-induced ALI [35]. In addition, Qi et al. discovered that CHBP protects the kidneys from renal tubulointerstitial fibrosis in unilateral ureter obstruction model mice by disrupting the NLRP3/Caspase-1/IL-1β pathway [71]. Herein, our team discovered that CHBP administration markedly improved functional recovery in mice after SCI.
First, we investigated the effects of CHBP in regulating pyroptosis and necroptosis in model mice. Pyroptosis, a proinflammatory form of programmed cell death, is modulated by the stimulation of Caspase-1, Caspase-4/5/11, several inflammasomes and GSDMD via the release of inflammation-mediating factors, such as IL-1β and IL-18 [57,72]. Pyroptosis is a process that is linked to inflammation and participates in the modulation of cell death after SCI [57,72,73]. Our data revealed that the protein levels of Caspase-1, GSDMD, IL-18 and IL-1β were significantly elevated in the SCI model animals compared to the sham animals but that the increases in inflammatory cytokine levels were attenuated by CHBP administration. In addition, the NLRP1 and NLRP3 inflammasomes, two important sources of the proinflammatory cytokines IL-1β and IL-18, are composed of the effectors pro-Caspase-1 and ASC and are essential for pyroptosis-regulated cell death [74,75]. A recent study reported that the NLRP1 and NLRP3 inflammasomes are overactivated in microglia and neurons in injured areas after SCI [76], and inhibiting the activation of NLRP1, NLRP3 and other inflammasome components exerts potential neuroprotective effects after SCI [77]. Our WB results revealed that CHBP inhibited the expression of NLRP1, NLRP3 and ASC after SCI, indicating that CHBP may be effective in inhibiting pyroptosis after SCI. In addition, necroptosis, a proinflammatory form of cell death, leads to the oligomerization and rupture of the plasma membrane via RIPK3 phosphorylation, which activates pseudokinase MLKL, resulting in cell death. Studies have revealed that RIPK1-mediated activation of necroptosis induces proinflammatory gene expression [78,79]. The expression levels of proinflammatory cytokines and TNF are increased in the spinal cords of mice with amyotrophic lateral sclerosis but are reduced by RIPK1 inhibition [80]. Furthermore, active oligomerized MLKL disrupts plasma membrane integrity by forming lytic pores; MLKL also controls the production of proinflammatory cytokines during the process of necroptosis [81]. According to our IF staining and WB results, CHBP reduced the expression of RIPK1, RIPK3, MLKL and Caspase-8, which are necroptosis-dependent proteins, in adult mice. Thus, our results indicate that CHBP alleviates pyroptosis and necroptosis in SCI mice.
We further explored the potential mechanism by which CHBP ameliorates SCI in mice. Our previous studies revealed that autophagy is vital for human health and disease, and inhibition of autophagy activity results in poor functional restoration in SCI mice [22]. Herein, our team discovered that CHBP inhibited pyroptosis and necroptosis to ameliorate cell death and improve functional recovery in SCI mice primarily by activating autophagy. We confirmed this result by applying 3-MA, a specific autophagy inhibitor. Our data revealed that autophagy was inhibited in mice after SCI and that CHBP significantly increased the expression of autophagic markers, such as Beclin-1, VPS34, CTSD and LC3, revealing that CHBP might be an activator of autophagy after SCI. However, the increase in autophagic activity was inhibited after 3-MA administration, and CHBP-induced cell survival and functional recovery were reversed after 3-MA treatment. Therefore, autophagy inhibition plays a significant role in SCI, and CHBP treatment reverses cell death and functional recovery inhibition induced by pyroptosis and necroptosis by promoting autophagy.
Next, we found that CHBP promotes TFEB nuclear translocation to induce autophagy via the AMPK signalling pathway. TFEB plays a vital role in lysosome biogenetic processes, participates in autophagy and promotes autophagosome formation and autophagosome-lysosome fusion [82,83]. Mechanistically, inactive TFEB in the cytoplasm is transported to the nucleus, where it is phosphorylated and exerts its regulatory function. Our WB results revealed that the nuclear level of TFEB protein was markedly elevated after CHBP administration in injured spinal cord tissue. Therefore, we speculated that CHBP may exert its physiological effects in a TFEB-dependent manner. To assess this hypothesis, TFEB shRNA was designed for subsequent studies. The data revealed that TFEB shRNA reduced the expression of TFEB in the cytoplasm and nucleus in SCI mice after CHBP treatment. In addition, protective autophagy was inhibited after TFEB shRNA administration, indicating that TFEB plays a role in autophagy under physiological conditions. Furthermore, we found that TFEB shRNA blocked functional recovery and cell survival in SCI mice after CHBP treatment, suggesting that CHBP promotes autophagy by increasing the expression of TFEB in the nucleus to inhibit pyroptosis and necroptosis.
Finally, our team explored how CHBP regulates TFEB levels. In the cytoplasm, stimulation of the AMPK-mTOR signalling pathway triggers the dephosphorylation of Ser211 of TFEB, causing TFEB-14-3-3 complex dissociation and enabling the entry of free TFEB into the nucleus. In the nucleus, stimulation of the AMPK-FOXO3a-SPK2-CARM1 signal transduction cascade elevates the level of CARM1, a coactivator of TFEB, which binds to TFEB and promotes the methylation of promotor sequences, triggering effective transcription of endogenous autophagy-related genes. In this study, our team showed that the AMPK-mTOR and AMPK-FOXO3a-SPK2-CARM1 signalling pathways were stimulated after the treatment of spinal cord injury with CHBP. When CC, an AMPK blocker, was used in conjunction with CHBP, the nuclear translocation and transcriptional activity of TFEB were blocked, and autophagy activity and the anti-inflammatory effects of CHBP in SCI were diminished. Our results indicated that CHBP promotes TFEB intracellular translocation to activate autophagy via the AMPK-FOXO3a-SPK2-CARM1 and AMPK-mTOR signalling pathways.
There are several limitations of this study. First, the new thioether-cyclized peptide CHBP was demonstrated to be more stable than HBSP and to have more persistent and potent effects at a lower dose; however, the stability of CHBP and its clinical potential require additional research. Next, we used intraperitoneal injection, which has poor translation potential in humans, and effective delivery systems, such as hydrogels, are lacking. Moreover, we sought to determine whether CHBP affects other cell death types following SCI, such as parthanatos and ferroptosis. The present bioinformatic analysis revealed that the TNF signalling pathway plays a pivotal role after SCI, and this research identifies TNF as a primary hub of reactive inflammatory events after SCI [84]. Therefore, the TNF signalling pathway may also be pivotal for autophagy. In addition, 3-MA inhibits type III PI3 kinase activity and production of PI3P rather than directly inhibiting recruitment of autophagy proteins. It is not specific for type III PI3 kinase (also can inhibit type I PI3 kinase) and is not a definitive proof that autophagy is required. In future studies, we will apply KO/KD of autophagy mediators such as ATG5 or ATG7 in vivo or in vitro to assess the effect on autophagy.
In conclusion, the protective effect of CHBP in kidney and skin flap I/R injury models has been demonstrated [36,85]. In our study, CHBP alleviated proinflammatory cell death and improved functional recovery after traumatic SCI. Compared with EPO, CHBP has better proteolytic decomposition resistance and stronger tissue-protective effects, and the required administration frequency and dose are lower; however, there is a lack of clinical experimental studies on CHBP. Nevertheless, CHBP might be an effective therapeutic agent for clinical application.
5. Conclusions
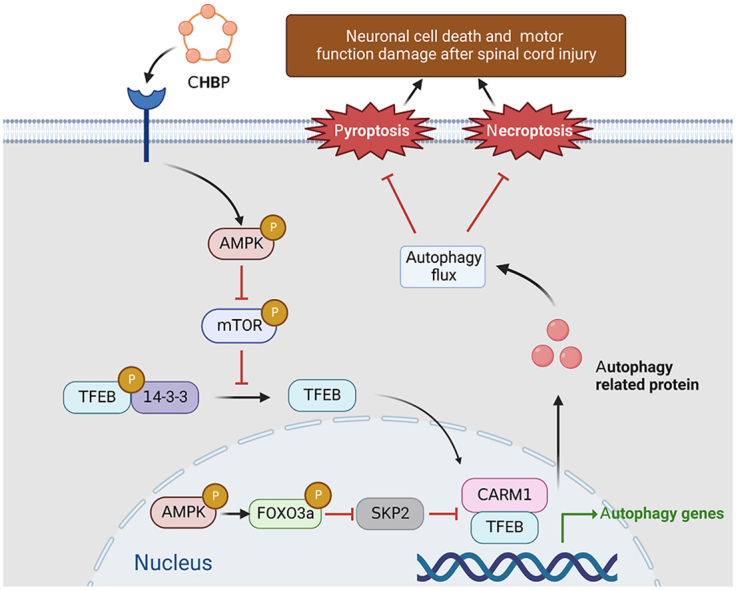
Our team found that two forms of proinflammatory cell death, pyroptosis and necroptosis, are dramatically increased in SCI, leading to cell death and inhibiting functional recovery. CHBP reverses these detrimental changes through TFEB-induced enhancement of autophagy by stimulating the AMPK-mTOR signalling pathway (Fig. 9). Hence, CHBP might be utilized as a novel therapeutic for SCI patients.
Fig. 9.

Schematic of the effects of TFEB, pyroptosis, necroptosis, and autophagy on CHBP induced neuronal cell death inhibition and motor function restoration at the molecular level.
Compliance with ethics requirements
All experiments involving animals were conducted according to ethical policies, and the procedures were approved by the Chinese Institutional Animal Care Committee of Wenzhou Medical University, China (approval No. wydw 2017-0022).
Credit author statement
Yu Xu, Yibo Geng, Hui Wang and Haojie Zhang performed experiments and analysed the data. Jianjun Qi, Feida Li, Xinli Hu, Yituo Chen, Haipeng Si and Yao Li provided critical appraisal. Yu Xu, Yibo Geng, Hui Wang and Haojie Zhang wrote the manuscript. Xiangyang Wang, Huazi Xu, Jianzhong Kong, Yuepiao Cai, Aimin Wu and Jian Xiao revised the manuscript. Wenfei Ni, Jian Xiao and Kailiang Zhou conceived the original idea and designed the experiments for this study. All authors read and approved the final manuscript.
Availability of data and materials
All data supporting the conclusions of this manuscript are provided in the text and figures. Please contact the author for data requests.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by grants from National Natural Science Foundation of China (No. 8207219 to Kailiang Zhou); Public Welfare Technology Research Project of Zhejiang Province (LGF20H150003 to Kailiang Zhou); Zhejiang Provincial Natural Science Foundation (No. LY17H060009 and No. Y21H060050 to Wenfei Ni); and Wenzhou Science and Technology Bureau Foundation (No. Y20210438 to Kailiang Zhou).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2023.102767.
Contributor Information
Wenfei Ni, Email: niwenfei@126.com.
Jian Xiao, Email: xfxj2000@126.com.
Kailiang Zhou, Email: zhoukailiang@wmu.edu.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
Data availability
Data will be made available on request.
References
- 1.Liu S., Sarkar C., Dinizo M., Faden A., Koh E., Lipinski M., et al. Disrupted autophagy after spinal cord injury is associated with ER stress and neuronal cell death. Cell Death Dis. 2015;6 doi: 10.1038/cddis.2014.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qin C., Feng H., Zhang C., Zhang X., Liu Y., Yang D., et al. Differential expression profiles and functional prediction of tRNA-derived small RNAs in rats after traumatic spinal cord injury. Front. Mol. Neurosci. 2019;12:326. doi: 10.3389/fnmol.2019.00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang H., Zhou W., Huang J., Zheng X., Tian H., Wang B., et al. Endocrine therapy for the functional recovery of spinal cord injury. Front. Neurosci. 2020;14 doi: 10.3389/fnins.2020.590570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fakhoury M. Spinal cord injury: overview of experimental approaches used to restore locomotor activity. Rev. Neurosci. 2015;26(4):397–405. doi: 10.1515/revneuro-2015-0001. [DOI] [PubMed] [Google Scholar]
- 5.Samantaray S., Das A., Matzelle D., Yu S., Wei L., Varma A., et al. Administration of low dose estrogen attenuates persistent inflammation, promotes angiogenesis, and improves locomotor function following chronic spinal cord injury in rats. J. Neurochem. 2016;137(4):604–617. doi: 10.1111/jnc.13610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carelli S., Giallongo T., Rey F., Colli M., Tosi D., Bulfamante G., et al. Neuroprotection, recovery of function and endogenous neurogenesis in traumatic spinal cord injury following transplantation of activated adipose tissue. Cells. 2019;8(4) doi: 10.3390/cells8040329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang C., Wang Q., Lou Y., Xu J., Feng Z., Chen Y., et al. Salidroside attenuates neuroinflammation and improves functional recovery after spinal cord injury through microglia polarization regulation. J. Cell Mol. Med. 2018;22(2):1148–1166. doi: 10.1111/jcmm.13368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leibinger M., Zeitler C., Gobrecht P., Andreadaki A., Gisselmann G., Fischer D. Transneuronal delivery of hyper-interleukin-6 enables functional recovery after severe spinal cord injury in mice. Nat. Commun. 2021;12(1):391. doi: 10.1038/s41467-020-20112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zbogar D., Eng J., Miller W., Krassioukov A., Verrier M. Physical activity outside of structured therapy during inpatient spinal cord injury rehabilitation. J. NeuroEng. Rehabil. 2016;13(1):99. doi: 10.1186/s12984-016-0208-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dai W., Wang X., Teng H., Li C., Wang B., Wang J. Celastrol inhibits microglial pyroptosis and attenuates inflammatory reaction in acute spinal cord injury rats. Int. Immunopharm. 2019;66:215–223. doi: 10.1016/j.intimp.2018.11.029. [DOI] [PubMed] [Google Scholar]
- 11.Shi J., Zhao Y., Wang K., Shi X., Wang Y., Huang H., et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 12.Swaroop S., Mahadevan A., Shankar S., Adlakha Y., Basu A. HSP60 critically regulates endogenous IL-1β production in activated microglia by stimulating NLRP3 inflammasome pathway. J. Neuroinflammation. 2018;15(1):177. doi: 10.1186/s12974-018-1214-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu W., Liu J., Wang W., Wang Y., Ouyang X. NLRP6 induces pyroptosis by activation of caspase-1 in gingival fibroblasts. J. Dent. Res. 2018;97(12):1391–1398. doi: 10.1177/0022034518775036. [DOI] [PubMed] [Google Scholar]
- 14.Liu S., Li Y., Choi H., Sarkar C., Koh E., Wu J., et al. Lysosomal damage after spinal cord injury causes accumulation of RIPK1 and RIPK3 proteins and potentiation of necroptosis. Cell Death Dis. 2018;9(5):476. doi: 10.1038/s41419-018-0469-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen H., Kankel M.W., Su S.C., Han S.W.S., Ofengeim D. Exploring the genetics and non-cell autonomous mechanisms underlying ALS/FTLD. Cell Death Differ. 2018;25(4):648–662. doi: 10.1038/s41418-018-0060-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ransohoff R.M. How neuroinflammation contributes to neurodegeneration. Science. 2016;353(6301):777–783. doi: 10.1126/science.aag2590. [DOI] [PubMed] [Google Scholar]
- 17.Ito Y., Ofengeim D., Najafov A., Das S., Saberi S., Li Y., et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science. 2016;353(6299):603–608. doi: 10.1126/science.aaf6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ofengeim D., Ito Y., Najafov A., Zhang Y., Shan B., Dewitt J., et al. Activation of necroptosis in multiple sclerosis. Cell Rep. 2015;10(11):1836–1849. doi: 10.1016/j.celrep.2015.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fan H., Tang H., Kang J., Shan L., Song H., Zhu K., et al. Involvement of endoplasmic reticulum stress in the necroptosis of microglia/macrophages after spinal cord injury. Neuroscience. 2015;311:362–373. doi: 10.1016/j.neuroscience.2015.10.049. [DOI] [PubMed] [Google Scholar]
- 20.He M., Ding Y., Chu C., Tang J., Xiao Q., Luo Z. Autophagy induction stabilizes microtubules and promotes axon regeneration after spinal cord injury. Proc. Nation. Academy Sci. U. S. A. 2016;113(40):11324–11329. doi: 10.1073/pnas.1611282113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Altman A., Miller M., Mahmud J., Smith N., Chan G. Human cytomegalovirus-induced autophagy prevents necroptosis of infected monocytes. J. Virol. 2020;94(22) doi: 10.1128/JVI.01022-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu C., Chen H., Zhuang R., Zhang H., Wang Y., Hu X., et al. Betulinic acid inhibits pyroptosis in spinal cord injury by augmenting autophagy via the AMPK-mTOR-TFEB signaling pathway. Int. J. Biol. Sci. 2021;17(4):1138–1152. doi: 10.7150/ijbs.57825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu Y., Hu X., Li F., Zhang H., Lou J., Wang X., et al. GDF-11 protects the traumatically injured spinal cord by suppressing pyroptosis and necroptosis via TFE3-mediated autophagy augmentation. Oxid. Med. Cell. Longev. 2021;2021 doi: 10.1155/2021/8186877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li J., Guo W., Xiong M., Zhang S., Han H., Chen J., et al. Erythropoietin facilitates the recruitment of bone marrow mesenchymal stem cells to sites of spinal cord injury. Exp. Ther. Med. 2017;13(5):1806–1812. doi: 10.3892/etm.2017.4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mccook O., Georgieff M., Scheuerle A., Möller P., Thiemermann C., Radermacher P. Erythropoietin in the critically ill: do we ask the right questions? Crit. Care. 2012;16(5):319. doi: 10.1186/cc11430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qu Y., Sun Q., Song X., Jiang Y., Dong H., Zhao W., et al. Helix B surface peptide reduces sepsis-induced kidney injury via PI3K/Akt pathway. Nephrology. 2020;25(7):527–534. doi: 10.1111/nep.13683. [DOI] [PubMed] [Google Scholar]
- 27.Tan R., Tian H., Yang B., Zhang B., Dai C., Han Z., et al. Autophagy and Akt in the protective effect of erythropoietin helix B surface peptide against hepatic ischaemia/reperfusion injury in mice. Sci. Rep. 2018;8(1) doi: 10.1038/s41598-018-33028-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang C., Xu Z., Zhao Z., Li L., Zhao T., Peng D., et al. A novel proteolysis-resistant cyclic helix B peptide ameliorates kidney ischemia reperfusion injury. Biochim. Biophys. Acta. 2014;1842(11):2306–2317. doi: 10.1016/j.bbadis.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 29.Yang C., Hosgood S., Meeta P., Long Y., Zhu T., Nicholson M., et al. Cyclic helix B peptide in preservation solution and autologous blood perfusate ameliorates ischemia-reperfusion injury in isolated porcine kidneys. Transplantation direct. 2015;1(2):e6. doi: 10.1097/TXD.0000000000000515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang C., Zhao T., Lin M., Zhao Z., Hu L., Jia Y., et al. Helix B surface peptide administered after insult of ischemia reperfusion improved renal function, structure and apoptosis through beta common receptor/erythropoietin receptor and PI3K/Akt pathway in a murine model. Exp. Biol. Med. 2013;238(1):111–119. doi: 10.1258/ebm.2012.012185. [DOI] [PubMed] [Google Scholar]
- 31.Ahmet I., Tae H., Juhaszova M., Riordon D., Boheler K., Sollott S., et al. A small nonerythropoietic helix B surface peptide based upon erythropoietin structure is cardioprotective against ischemic myocardial damage. Mol. Med. 2011;17:194–200. doi: 10.2119/molmed.2010.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patel N., Nandra K., Brines M., Collino M., Wong W., Kapoor A., et al. A nonerythropoietic peptide that mimics the 3D structure of erythropoietin reduces organ injury/dysfunction and inflammation in experimental hemorrhagic shock. Mol. Med. 2011;17:883–892. doi: 10.2119/molmed.2011.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zeng Y., Zheng L., Yang Z., Yang C., Zhang Y., Li J., et al. Protective effects of cyclic helix B peptide on aristolochic acid induced acute kidney injury. Biomed. Pharmacotherap. Biomed. Pharmacotherapie. 2017;94:1167–1175. doi: 10.1016/j.biopha.2017.07.131. [DOI] [PubMed] [Google Scholar]
- 34.Li L., Lin M., Zhang L., Huang S., Hu C., Zheng L., et al. Cyclic helix B peptide protects HK-2 cells from oxidative stress by inhibiting ER stress and activating Nrf2 signalling and autophagy. Mol. Med. Rep. 2017;16(6):8055–8061. doi: 10.3892/mmr.2017.7588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang X., Zhang W., Qiu Y., Ju M., Yang C., Tu G., et al. Cyclic helix B peptide alleviates sepsis-induced acute lung injury by downregulating NLRP3 inflammasome activation in alveolar macrophages. Int. Immunopharm. 2020;88 doi: 10.1016/j.intimp.2020.106849. [DOI] [PubMed] [Google Scholar]
- 36.Lou J., Zhang H., Qi J., Xu Y., Wang X., Jiang J., et al. Cyclic helix B peptide promotes random-pattern skin flap survival via TFE3-mediated enhancement of autophagy and reduction of ROS levels. Br. J. Pharmacol. 2021 doi: 10.1111/bph.15702. [DOI] [PubMed] [Google Scholar]
- 37.Ritchie M., Phipson B., Wu D., Hu Y., Law C., Shi W., et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang T., Wu B., Zhang X., Zhang M., Zhang S., Huang W., et al. Identification of gene coexpression modules, hub genes, and pathways related to spinal cord injury using integrated bioinformatics methods. J. Cell. Biochem. 2019 doi: 10.1002/jcb.27908. [DOI] [PubMed] [Google Scholar]
- 39.Byrnes K., Stoica B., Fricke S., Di Giovanni S., Faden A. Cell cycle activation contributes to post-mitotic cell death and secondary damage after spinal cord injury. Brain : J. Neurol. 2007;130:2977–2992. doi: 10.1093/brain/awm179. [DOI] [PubMed] [Google Scholar]
- 40.He Z., Zou S., Yin J., Gao Z., Liu Y., Wu Y., et al. Inhibition of endoplasmic reticulum stress preserves the integrity of blood-spinal cord barrier in diabetic rats subjected to spinal cord injury. Sci. Rep. 2017;7(1):7661. doi: 10.1038/s41598-017-08052-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang D., Gong F., Ge X., Lv C., Huang C., Feng S., et al. Neuron-derived exosomes-transmitted miR-124-3p protect traumatically injured spinal cord by suppressing the activation of neurotoxic microglia and astrocytes. J. Nanobiotechnol. 2020;18(1):105. doi: 10.1186/s12951-020-00665-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou K., Zhou Y., Wu K., Tian N., Wu Y., Wang Y., et al. Stimulation of autophagy promotes functional recovery in diabetic rats with spinal cord injury. Sci. Rep. 2015;5 doi: 10.1038/srep17130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu M., Lin A., Lee H., Ko H., Lee T., Kou Y. Paeonol attenuates cigarette smoke-induced lung inflammation by inhibiting ROS-sensitive inflammatory signaling. Mediat. Inflamm. 2014;2014 doi: 10.1155/2014/651890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu K., Zhou K., Wang Y., Zhou Y., Tian N., Wu Y., et al. Stabilization of HIF-1α by FG-4592 promotes functional recovery and neural protection in experimental spinal cord injury. Brain Res. 2016;1632:19–26. doi: 10.1016/j.brainres.2015.12.017. [DOI] [PubMed] [Google Scholar]
- 45.Wang Q., He Y., Zhao Y., Xie H., Lin Q., He Z., et al. A thermosensitive heparin-poloxamer hydrogel bridges aFGF to treat spinal cord injury. ACS Appl. Mater. Interfaces. 2017;9(8):6725–6745. doi: 10.1021/acsami.6b13155. [DOI] [PubMed] [Google Scholar]
- 46.Li H., Wang C., He T., Zhao T., Chen Y., Shen Y., et al. Mitochondrial transfer from bone marrow mesenchymal stem cells to motor neurons in spinal cord injury rats via gap junction. Theranostics. 2019;9(7):2017–2035. doi: 10.7150/thno.29400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li J., Wang Q., Cai H., He Z., Wang H., Chen J., et al. FGF1 improves functional recovery through inducing PRDX1 to regulate autophagy and anti-ROS after spinal cord injury. J. Cell Mol. Med. 2018;22(5):2727–2738. doi: 10.1111/jcmm.13566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Niu C., Chen Z., Kim K.T., Sun J., Xue M., Chen G., et al. Metformin alleviates hyperglycemia-induced endothelial impairment by downregulating autophagy via the Hedgehog pathway. Autophagy. 2019;15(5):843–870. doi: 10.1080/15548627.2019.1569913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li X., Yu Z., Zong W., Chen P., Li J., Wang M., et al. Deficiency of the microglial Hv1 proton channel attenuates neuronal pyroptosis and inhibits inflammatory reaction after spinal cord injury. J. Neuroinflammation. 2020;17(1):263. doi: 10.1186/s12974-020-01942-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu S., Li Y., Choi H.M.C., Sarkar C., Koh E.Y., Wu J., et al. Lysosomal damage after spinal cord injury causes accumulation of RIPK1 and RIPK3 proteins and potentiation of necroptosis. Cell Death Dis. 2018;9(5):476. doi: 10.1038/s41419-018-0469-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu C., Chen J., Liu Y., Zhang J., Ding W., Wang S., et al. Upregulation of PSMB4 is associated with the necroptosis after spinal cord injury. Neurochem. Res. 2016;41(11):3103–3112. doi: 10.1007/s11064-016-2033-7. [DOI] [PubMed] [Google Scholar]
- 52.Xue Z., Zhang Z., Liu H., Li W., Guo X., Zhang Z., et al. lincRNA-Cox2 regulates NLRP3 inflammasome and autophagy mediated neuroinflammation. Cell Death Differ. 2019;26(1):130–145. doi: 10.1038/s41418-018-0105-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rong Y., Liu W., Wang J., Fan J., Luo Y., Li L., et al. Neural stem cell-derived small extracellular vesicles attenuate apoptosis and neuroinflammation after traumatic spinal cord injury by activating autophagy. Cell Death Dis. 2019;10(5):340. doi: 10.1038/s41419-019-1571-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pasquier B. Autophagy inhibitors. Cell. Mol. Life Sci. : CM. 2016;73(5):985–1001. doi: 10.1007/s00018-015-2104-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu Y., Xue X., Zhang H., Che X., Luo J., Wang P., et al. Neuronal-targeted TFEB rescues dysfunction of the autophagy-lysosomal pathway and alleviates ischemic injury in permanent cerebral ischemia. Autophagy. 2019;15(3):493–509. doi: 10.1080/15548627.2018.1531196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhuang X.X., Wang S.F., Tan Y., Song J.X., Zhu Z., Wang Z.Y., et al. Pharmacological enhancement of TFEB-mediated autophagy alleviated neuronal death in oxidative stress-induced Parkinson's disease models. Cell Death Dis. 2020;11(2):128. doi: 10.1038/s41419-020-2322-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Al Mamun A., Wu Y., Monalisa I., Jia C., Zhou K., Munir F., et al. Role of pyroptosis in spinal cord injury and its therapeutic implications. J. Adv. Res. 2021;28:97–109. doi: 10.1016/j.jare.2020.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu S., Wang J., Jiang J., Song J., Zhu W., Zhang F., et al. TLR4 promotes microglial pyroptosis via lncRNA-F630028O10Rik by activating PI3K/AKT pathway after spinal cord injury. Cell Death Dis. 2020;11(8):693. doi: 10.1038/s41419-020-02824-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu S., Wang J., Zhong J., Shao M., Jiang J., Song J., et al. CD73 alleviates GSDMD-mediated microglia pyroptosis in spinal cord injury through PI3K/AKT/Foxo1 signaling. Clin. Transl. Med. 2021;11(1):e269. doi: 10.1002/ctm2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wakhloo D., Scharkowski F., Curto Y., Javed Butt U., Bansal V., Steixner-Kumar A., et al. Functional hypoxia drives neuroplasticity and neurogenesis via brain erythropoietin. Nat. Commun. 2020;11(1):1313. doi: 10.1038/s41467-020-15041-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ureña-Guerrero M., Castañeda-Cabral J., Rivera-Cervantes M., Macias-Velez R., Jarero-Basulto J., Gudiño-Cabrera G., et al. Neuroprotective and neurorestorative effects of epo and VEGF: perspectives for new therapeutic approaches to neurological diseases. Curr. Pharmaceut. Des. 2020;26(12):1263–1276. doi: 10.2174/1381612826666200114104342. [DOI] [PubMed] [Google Scholar]
- 62.Tiwari N., Sathyanesan M., Schweinle W., Newton S. Carbamoylated erythropoietin induces a neurotrophic gene profile in neuronal cells. Progress in Neuro-Psychopharmacol. Biolog. Psychiat. 2019;88:132–141. doi: 10.1016/j.pnpbp.2018.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Carelli S., Giallongo T., Gombalova Z., Merli D., Di Giulio A., Gorio A. EPO-releasing neural precursor cells promote axonal regeneration and recovery of function in spinal cord traumatic injury. Restor. Neurol. Neurosci. 2017;35(6):583–599. doi: 10.3233/RNN-170750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lin C., Zhang M., Zhang Y., Yang K., Hu J., Si R., et al. Helix B surface peptide attenuates diabetic cardiomyopathy via AMPK-dependent autophagy. Biochem. Biophys. Res. Commun. 2017;482(4):665–671. doi: 10.1016/j.bbrc.2016.11.091. [DOI] [PubMed] [Google Scholar]
- 65.Ueba H., Brines M., Yamin M., Umemoto T., Ako J., Momomura S., et al. Cardioprotection by a nonerythropoietic, tissue-protective peptide mimicking the 3D structure of erythropoietin. Proc. Nation. Academy Sci. U. S. A. 2010;107(32):14357–14362. doi: 10.1073/pnas.1003019107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brines M., Patel N., Villa P., Brines C., Mennini T., De Paola M., et al. Nonerythropoietic, tissue-protective peptides derived from the tertiary structure of erythropoietin. Proc. Nation. Academy Sci. U. S. A. 2008;105(31):10925–10930. doi: 10.1073/pnas.0805594105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Long Y., Guo R., Luo J., Yang D., Roller P. Potentiating effect of distant sites in non-phosphorylated cyclic peptide antagonists of the Grb2-SH2 domain. Biochem. Biophys. Res. Commun. 2003;310(2):334–340. doi: 10.1016/j.bbrc.2003.09.022. [DOI] [PubMed] [Google Scholar]
- 68.Song Y., Peach M., Roller P., Qiu S., Wang S., Long Y. Discovery of a novel nonphosphorylated pentapeptide motif displaying high affinity for Grb2-SH2 domain by the utilization of 3'-substituted tyrosine derivatives. J. Med. Chem. 2006;49(5):1585–1596. doi: 10.1021/jm050910x. [DOI] [PubMed] [Google Scholar]
- 69.Long Y., Xue T., Song Y., Liu Z., Huang S., Yu Q. Synthesis and utilization of chiral alpha-methylated alpha-amino acids with a carboxyalkyl side chain in the design of novel Grb2-SH2 peptide inhibitors free of phosphotyrosine. J. Med. Chem. 2008;51(20):6371–6380. doi: 10.1021/jm800487r. [DOI] [PubMed] [Google Scholar]
- 70.Yang C., Zhang C., Jia J., Wang L., Zhang W., Li J., et al. Cyclic helix B peptide ameliorates acute myocardial infarction in mice by inhibiting apoptosis and inflammatory responses. Cell death discovery. 2019;5:78. doi: 10.1038/s41420-019-0161-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Qi R., Zhang W., Zheng L., Xu M., Rong R., Zhu T., et al. Cyclic helix B peptide ameliorates renal tubulointerstitial fibrosis induced by unilateral ureter obstruction via inhibiting NLRP3 pathway. Ann. Transl. Med. 2020;8(5):167. doi: 10.21037/atm.2020.02.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu Z., Yao X., Jiang W., Li W., Zhu S., Liao C., et al. Advanced oxidation protein products induce microglia-mediated neuroinflammation via MAPKs-NF-κB signaling pathway and pyroptosis after secondary spinal cord injury. J. Neuroinflammation. 2020;17(1):90. doi: 10.1186/s12974-020-01751-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Swanson K., Deng M., Ting J. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019;19(8):477–489. doi: 10.1038/s41577-019-0165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.He Y., Hara H., Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 2016;41(12):1012–1021. doi: 10.1016/j.tibs.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen F., Jiang G., Liu H., Li Z., Pei Y., Wang H., et al. Melatonin alleviates intervertebral disc degeneration by disrupting the IL-1β/NF-κB-NLRP3 inflammasome positive feedback loop. Bone research. 2020;8:10. doi: 10.1038/s41413-020-0087-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Grace P., Strand K., Galer E., Urban D., Wang X., Baratta M., et al. Vol. 113. 2016. Morphine paradoxically prolongs neuropathic pain in rats by amplifying spinal NLRP3 inflammasome activation; pp. E3441–E3450. 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chevriaux A., Pilot T., Derangère V., Simonin H., Martine P., Chalmin F., et al. Cathepsin B is required for NLRP3 inflammasome activation in macrophages, through NLRP3 interaction. Front. Cell Dev. Biol. 2020;8:167. doi: 10.3389/fcell.2020.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Christofferson D.E., Li Y., Hitomi J., Zhou W., Upperman C., Zhu H., et al. A novel role for RIP1 kinase in mediating TNFα production. Cell Death Dis. 2012;3(6) doi: 10.1038/cddis.2012.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhu K., Liang W., Ma Z., Xu D., Cao S., Lu X., et al. Necroptosis promotes cell-autonomous activation of proinflammatory cytokine gene expression. Cell Death Dis. 2018;9(5):500. doi: 10.1038/s41419-018-0524-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ito Y., Ofengeim D., Najafov A., Das S., Saberi S., Li Y., et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science. 2016;353(6299):603–608. doi: 10.1126/science.aaf6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Douanne T., André-Grégoire G., Trillet K., Thys A., Papin A., Feyeux M., et al. Pannexin-1 limits the production of proinflammatory cytokines during necroptosis. EMBO Rep. 2019;20(10) doi: 10.15252/embr.201947840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mansueto G., Armani A., Viscomi C., D'orsi L., De Cegli R., Polishchuk E., et al. Transcription factor EB controls metabolic flexibility during exercise. Cell Metabol. 2017;25(1):182–196. doi: 10.1016/j.cmet.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mao K., Chen J., Yu H., Li H., Ren Y., Wu X., et al. Poly (ADP-ribose) polymerase 1 inhibition prevents neurodegeneration and promotes α-synuclein degradation via transcription factor EB-dependent autophagy in mutant α-synucleinA53T model of Parkinson's disease. Aging Cell. 2020;19(6) doi: 10.1111/acel.13163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhu W., Chen X., Ning L., Jin K. Network analysis reveals TNF as a major hub of reactive inflammation following spinal cord injury. Sci. Rep. 2019;9(1):928. doi: 10.1038/s41598-018-37357-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu A., Wu J., Yang C., Wu Y., Zhang Y., Zhao F., et al. TRPM7 in CHBP-induced renoprotection upon ischemia reperfusion-related injury. Sci. Rep. 2018;8(1):5510. doi: 10.1038/s41598-018-22852-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the conclusions of this manuscript are provided in the text and figures. Please contact the author for data requests.
Data will be made available on request.