

Abstract
Tumor cells surviving hypoxic stress acquire the ability to drive cancer progression. To explore the contribution of dehydrogenases to the low oxygen concentration response, we used siRNAs targeting 163 dehydrogenase‐coding genes and discovered that glutamate dehydrogenase 1 (GDH1) plays a critical role in regulating colorectal cancer (CRC) cell survival under hypoxia. We observed that GDH1 deficiency had an inhibitory effect on CRC occurrence and impaired hypoxia‐inducible factor 1‐alpha (HIF‐1α) stability even under hypoxia. Mechanistically, hypoxia triggered p300 recruitment to GDH1, promoting its acetylation at K503 and K527. GDH1 acetylation at K527 induced the formation of a GDH1 complex with EGLN1/HIF‐1α; in contrast, GDH1 acetylation at K503 reinforced its affinity for α‐ketoglutarate (αKG), and glutamate production. In line with this view, αKG is a product of GDH1 under normoxia, but hypoxia stimulation reversed GDH1 enzyme activity and αKG consumption by the EGLN1/HIF‐1α complex, increasing HIF‐1α stability and promoting CRC progression. Clinically, hypoxia‐modulated GDH1 AcK503/527 can be used as a biomarker of CRC progression and is a potential target for CRC treatment.
Keywords: acetylation, colorectal cancer, GDH1, HIF1α, α‐ketoglutarate
Subject Categories: Cancer, Metabolism
An siRNA screen identifies αKG metabolic enzyme GDH1 as acetylation‐dependent control factor for intestinal cancer progression.
Introduction
When the stress signal of extracellular oxygen exhaustion is transduced to intracellular EGLN1, a process of stress called hypoxia occurs. Tumor cells that survive hypoxia acquire the ability to metastasize, resist drugs, or tolerate nutrient limitation. However, how tumor cells survive in response to hypoxia is still unclear. Upon hypoxia, HIF1α is reported to be stabilized and accumulated in various cancer cells and is associated with the progression and adverse clinical outcome of many different tumor types, including colorectal cancer (CRC) (Brahimi‐Horn et al, 2007; Böğürcü et al, 2018). Hence, discovery of a potential strategy to modulate the tumor cell response to hypoxia would be of great significance.
The activity of EGLN1 is tightly associated with HIF1α stability and is affected by the EGLN1 substrate oxygen and cofactor αKG. αKG is not only an intermediate metabolite in the Krebs cycle but also a direct product of glutamate metabolism (Stillman et al, 1993). Multiple enzymes are involved in αKG metabolism. For example, glutamic‐oxaloacetate transaminase (GOT) and members of the glutamic‐pyruvic transaminase (GPT) family produce αKG via glutamate deamination or a transaminase reaction. Members of the GDH family produce αKG via glutamate dehydrogenation, while members of the isocitrate dehydrogenase (IDH) family produce αKG via isocitrate dehydrogenation. In glioma cells, the absence of GDH1 was found to reduce the cellular level of αKG by approximately 65%, indicating that GDH1 might play a major role in αKG production in glioma cells (Yang et al, 2009). However, there is no clear association between αKG and CRC progression under hypoxia.
Generally, αKG is maintained at a steady state within the cell (Wang et al, 2019b), but dramatic changes in intracellular αKG levels profoundly impact gene transcription and signal transduction. Given that αKG is a cofactor for dioxygenases, such as TETs, KDMs, and EGLNs, it is reasonable to question whether changes in intracellular αKG levels can alter the activities of these enzymes. For example, the absence of BCAT1 was shown to cause αKG to accumulate to twice its basal level, and this increase in αKG activated EGLN1, which in turn inhibited the stability of HIF1α (Raffel et al, 2017). However, the Km value of EGLN1 for αKG is 1.3 μM (Lorenzo et al, 2014), and the intracellular αKG concentration varies from approximately 10–200 μM depending on the cell context (Jin et al, 2015). Also, a prolyl hydroxylases in vitro assay showed 2‐Hydroxyglutarate (2HG) could activate EGLN1 (Tarhonskaya et al, 2014) probably because 2HG has a similar structure with αKG and meanwhile in vitro assay facilitated the enriched 2HG to bind EGLN1. Therefore, we suspect that the global change in αKG concentration induced by BCAT1 or other αKG‐producing or ‐consuming enzymes should be insufficient to affect the enzyme activity of EGLN1 by exchanging of αKG/2HG.
In this study, we found that once cells suffer hypoxia stress, GDH1 targets the EGLN1/HIF1α complex after GDH1 acetylation by p300. In detail, AcK527 induced the formation of the GDH1/EGLN1/HIF1α axis, while GDH1 acetylation at K503 alleviated the restriction of αKG binding to GDH1, AcK503‐GDH1 resulted in glutamate production using αKG as substrate. As a consequence of the combined GDH1 acetylation at K503 and K527, the reversed GDH1 activity likely consumed αKG around the GDH1/EGLN1/HIF1α complex, which led to the inhibition of EGLN1 activity and in turn stabilized HIF1α. Of note, the synchronic acetylation of GDH1‐K503/K527 can be used as a biomarker of CRC progression, and thus, blockade of AcK503/527‐GDH1 would benefit CRC treatment.
Results
GDH1 is required for the response to hypoxia and is associated with a poor prognosis in CRC
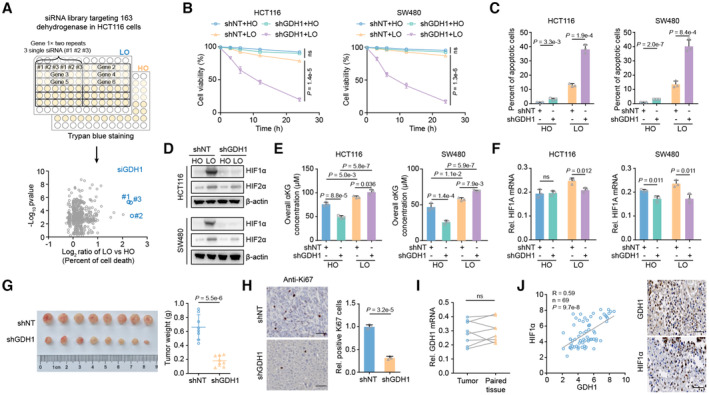
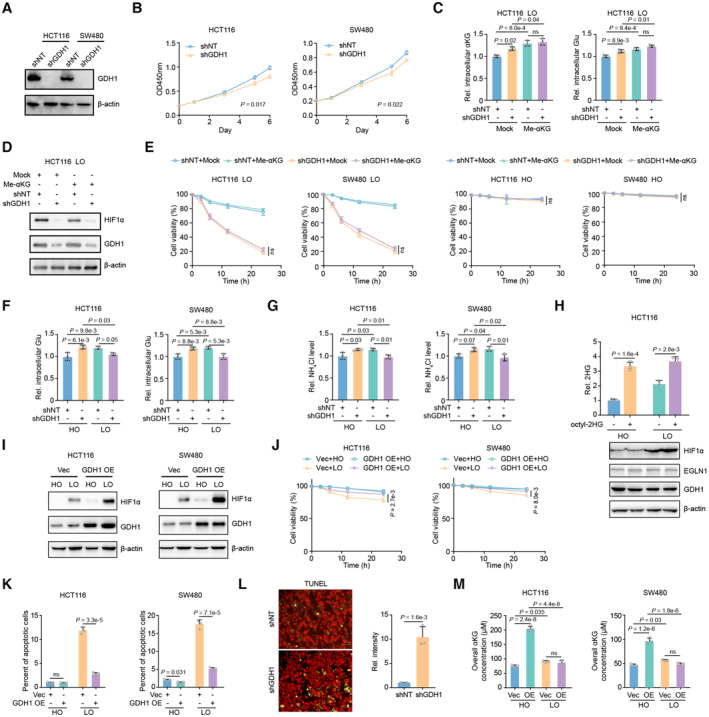
To discover the contribution of tumor metabolism, especially dehydrogenases, to the hypoxia response, we screened 163 dehydrogenase‐coding genes under hypoxia and observed that GDH1 depletion dramatically induced cell death under hypoxia (Fig 1A). We depleted GDH1 in HCT116 and SW480 cells (Fig EV1A) and found that CRC cells lacking GDH1 were more sensitive to hypoxia than those expressing GDH1, while loss of GDH1 slightly reduced cell proliferation under normoxic conditions (Fig EV1B). In addition, compared with the control cells, the percentage of apoptotic cells increased in the GDH1 depletion group, especially under hypoxia (Fig 1B and C). Supplementation with cell‐permeable αKG was successfully executed (Fig EV1C), but the restored αKG did not rescue the effect on HIF1α protein levels (Fig EV1D) and cell viability (Fig EV1E) in the cells lacking GDH1 under hypoxia, indicating that GDH1 might play an intricate role under hypoxia. Measurement of glutamate and ammonia levels reflects the possible conversion of GDH1 enzyme activity under normoxia and hypoxia because the increased glutamate and ammonia concentrations after GDH1 depletion under normoxia were consumed under hypoxia (Fig EV1F and G). Supplementation with octyl‐2‐hydroxyglutarate (2HG) also had no significant effect on HIF1α protein stability, suggesting that globally increased 2HG does not efficiently compete with αKG for EGLN1 binding (Fig EV1H). Under hypoxia, HIF1α protein levels in the control groups were higher than those in GDH1‐depleted cells, while HIF2α protein levels were slightly decreased after GDH1 depletion (Fig 1D). Compared with control cells, HIF1α protein levels were higher in CRC cells overexpressing GDH1, and overexpression of GDH1 conferred CRC cell resistance to hypoxia and resulted in a lower percentage of apoptotic cells in vitro and in vivo (Fig EV1I–L). The overall αKG content increased under hypoxia compared with normoxia. Loss of GDH1 impaired the increase in the overall αKG level under normoxia but not hypoxia. Consistent results were observed in cells overexpressing GDH1 under normoxia and hypoxia (Figs 1E and EV1M). These data suggest that GDH1 may not exhibit dehydrogenase activity under hypoxia.
Figure 1. GDH1 confers tumor cell resistance to hypoxia and promotes tumorigenesis.
-
AThe workflow for screening the dehydrogenase‐coding genes required for resisting hypoxia in HCT116 cells. In detail, HCT116 cells were collected and plated in 96‐well plates. Cells were transiently transfected with siRNAs separately targeting 163 dehydrogenase‐coding genes identified in the KEGG database using RNAiMAX for 36 h, treated with or without hypoxia for 24 h, and finally stained with Trypan blue. The origin of siRNA library was released from horizondiscovery website. The x‐axis represents the log2‐transformed fold changes in the percentage of dead cells among LO‐ vs. HO‐treated cells. Normoxia, 20% oxygen, is also termed HO. Hypoxia, 0.5% oxygen, is also termed LO.
-
BHCT116 and SW480 cells with GDH1 knockdown were incubated under normoxia and hypoxia, respectively. Then, cell viability was assessed by trypan blue staining (n = 3).
-
CThe cells in (B) were collected after incubation under normoxia or hypoxia, and PI/Annexin V double staining was then used to measure the percentage of apoptotic cells via FACS (n = 3).
-
DThe cells in (B) were collected after incubation under normoxia or hypoxia induction for 6 h, and the HIF1α protein level was measured via immunoblot analysis.
-
EHCT116 and SW480 cells with GDH1 knockdown were incubated under normoxia and hypoxia, respectively. Then, overall αKG was assessed using an αKG assay kit (Abcam) (n = 3).
-
FHIF1A gene transcription was slightly impaired after GDH1 depletion. The cells in (B) were collected after incubation under normoxia or hypoxia, and the HIF1A mRNA level was analyzed via quantitative real‐time PCR (qRT–PCR) (n = 3; ns, not significant).
-
G, HTumor formation study. HCT116‐shNT and HCT116‐shGDH1 cells were injected into the left groin of nude mice. When the tumor size reached 300 mm3, the mice were sacrificed, and the tumors were removed for further experiments (n = 8) (G). Solid tumors were stained with an antibody against Ki67 (Scale bars: 50 μm) (n = 3) (H).
-
IThe relative mRNA level of GDH1 was determined in 8 paired CRC and adjacent tissues (n = 8).
-
JPositive correlation between GDH1 and HIF1α protein expression in CRC tissue.
Data information: Data are mean ± SD from the biological replicates (B, C, E–H). Statistics: two‐way ANOVA with Tukey's HSD post hoc test (B); unpaired two‐tailed student's t‐test (C, F–H, J); one‐way ANOVA followed by with Tukey's HSD post hoc test (E); paired two‐tailed Student's t‐test (I).
Source data are available online for this figure.
Figure EV1. GDH1 depletion sensitizes CRC cells to hypoxia.
-
AConstruction of GDH1 depleted cell lines.
-
BCell proliferation assay was performed using cell lines constructed in Fig S1A (n = 3).
-
C, DHCT116 and SW480 cells with or without GDH1 were supplemented with cell permeable methyl‐αKG under hypoxia. Then, the intracellular level of αKG was tested by αKG determination kit (C) (n = 3). Cells were collected and indicated antibodies were applied to test HIF1α, EGLN1 and GDH1 proteins (D).
-
EHCT116 and SW480 cells with or without GDH1 were supplemented with cell permeable methyl‐αKG under normoxia and hypoxia. Then, cell viability was counted by trypan blue staining (n = 3).
-
F, GHCT116 and SW480 cells with or without GDH1 were cultured under normoxia or hypoxia. Cells were collected for determining glutamate (Glu, F) and ammonia (NH4Cl, G) (n = 3).
-
HHCT116 were incubated with or without cell permeable octyl‐2HG under normoxia or hypoxia. Indicated antibodies were applied to test HIF1α, EGLN1 and GDH1 proteins (n = 3).
-
IHIF1α and GDH1 were tested by western blot in HCT116 or SW480 cells with Vec or GDH1 overexpression under normoxia and hypoxia.
-
JHCT116 and SW480 cells with or without GDH1 overexpression were incubated under normoxia and hypoxia, respectively. Then, cell viability was counted by trypan blue staining (n = 3).
-
KThe apoptotic percent of cells in Fig S1D was counted by PI/Annexing V double staining (n = 3).
-
LTUNEL analysis of GDH1 knockdown HCT116 cells under hypoxia. The left was the representative microscopic data, the right was the calculated relative intensity data (n = 3).
-
MThe overall αKG level in HCT116 or SW480 cells with or without GDH1 overexpression under normoxia and hypoxia (n = 3).
Data information: Data are mean ± SD from the biological replicates (B, C, E–H, J–M). Statistics: two‐way ANOVA with Tukey's HSD post hoc test (B, E, J); one‐way ANOVA with Tukey's HSD post hoc test (C, F–H, M); unpaired two‐tailed Student's t‐test (K, L).
Source data are available online for this figure.
Furthermore, we ruled out the effect of GDH1 on HIF1A transcription levels (Fig 1F), suggesting that GDH1‐induced upregulation of the HIF1α protein level is mainly achieved by facilitating the stabilization of HIF1α under hypoxia rather than by affecting the level of gene transcription. The ability of CRC cells lacking GDH1 to form tumors also decreased drastically (Fig 1G and H). To examine GDH1 expression at the clinical level, we assessed the mRNA levels of GDH1 in 16 pairs of CRC and matched paracancerous samples and found that GDH1 did not exhibit high tumor tissue‐specific expression (Fig 1I). In tumor tissues, we observed a positive correlation between the GDH1 and HIF1α protein levels (Fig 1J).
Taken together, our primary data suggest that GDH1 participates in regulating HIF1α stability, potentially offering a novel way to stabilize HIF1α.
GDH1 depletion in the mouse intestine impairs CRC progression
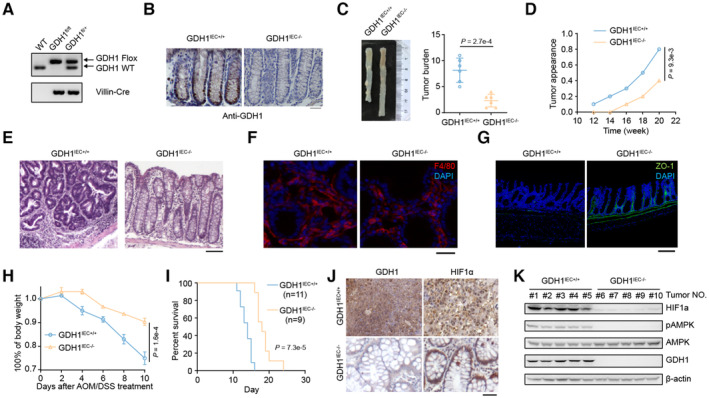
To investigate the role of GDH1‐mediated oncogenesis in vivo, we attempted to construct GDH1 knockout mice but found that mouse embryos showed serious defects and frequently died after GDH1 deletion. Therefore, tissue‐specific knockout of GDH1 in the intestine was performed using the Cre‐loxP recombination system and was verified by PCR, immunohistochemistry (IHC), and immunoblotting (Figs 2A and B, and EV2A).
Figure 2. Depletion of GDH1 in the mouse intestine impairs colorectal cancer formation.
-
AIdentification of GDH1 knockout (KO) in mouse intestines by PCR.
-
BIdentification of GDH1 KO in mouse intestines by immunohistochemistry (IHC).
-
C–EThe AOM/DSS‐induced inflammatory cancer switch was reduced by the loss of GDH1. (C) Tumor burden (n = 6). (D) Tumor appearance. (E) H&E staining of colon cancer tumors from GDH1IEC+/+ and GDH1IEC−/− mice (scale bar, 200 μm).
-
FF4/80 staining in a noninflamed portion of the colon after AMO/DSS treatment (scale bar, 50 μm).
-
GZO‐1 staining in colon sections of mice treated with sequential AMO/DSS treatment (scale bar, 200 μm).
-
HBody weight of GDH1IEC+/+ and GDH1IEC−/− mice after AOM/DSS treatment (n = 3).
-
ISurvival percentages of GDH1IEC+/+ mice (n = 11) and GDH1IEC−/− mice (n = 9) during progression of the inflammatory cancer switch.
-
JGDH1 and HIF1α protein expression in CRC tissue from GDH1IEC+/+ mice and GDH1IEC−/− mice.
-
KBoth HIF1α protein levels and AMPK activity were assessed in the intestine of GDH1IEC−/− mice after AOM/DSS treatment using the indicated antibodies.
Data information: Data are mean ± SD from the biological replicates (C, H). Statistics: unpaired two‐tailed Student's t‐test (C); two‐way ANOVA with Tukey's HSD post hoc test (D, H); log‐rank test (I).
Source data are available online for this figure.
Figure EV2. GDH1 depletion did not significantly affect cytokine and HIF1A expression.
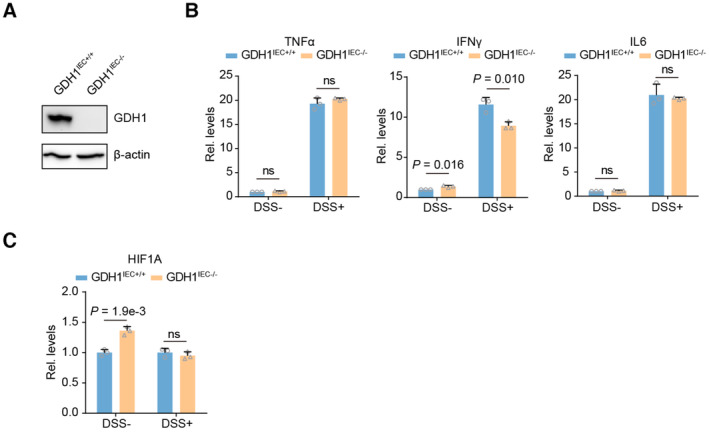
- The identification of GDH1 knock out in the intestine by western blot.
- qRT‐PCR analysis of whole colon homogenates to assess cytokines production after AOM/DSS treatment (n = 3).
- qRT‐PCR analysis of whole colon homogenates to assess HIF1A expression after AOM/DSS treatment (n = 3).
Data information: Data are mean ± SD from the biological replicates (B, C). Statistics: unpaired two‐tailed Student's t‐test (B, C).
Source data are available online for this figure.
The azoxymethane (AOM)/dextran sodium sulfate (DSS) model is a widely used inflammation‐associated colon cancer model in rodents (De Robertis et al, 2011; Thaker et al, 2012). Loss of GDH1 in the intestine significantly reduced the occurrence of CRC in adult mice (Fig 2C). We also observed delayed appearance of tumors after depletion of GDH1 in the mouse intestine (Fig 2D). Furthermore, H&E staining verified the presence of tumors in the intestine (Fig 2E). Of note, equal infiltration of macrophages or microglial cells in the control and GDH1IEC−/− mice was discovered by assessing approximate F4/80 positivity (Fig 2F), and no significant difference was found in the production of proinflammatory cytokines, such as TNFα, IL6, and IFNγ, after AOM/DSS induction in the control and GDH1IEC−/− mice (Fig EV2B). However, focal regions of continuous ZO‐1 staining were destroyed in control wild‐type mice but remained relatively intact in GDH1‐depleted mouse colons (Fig 2G). Mice without GDH1 in the intestine exhibited tolerance to AOM/DSS and a delayed decrease in body weight (Fig 2H). Consistently, GDH1 depletion significantly prolonged the survival time of mice after AOM/DSS treatment (Fig 2I). IHC of GDH1 and HIF1α and H&E staining showed that GDH1 depletion in the intestine limited CRC development and HIF1α levels (Fig 2J). Furthermore, HIF1α protein was almost undetectable in GDH1IEC−/− mice, whereas high levels of HIF1α were found in the intestinal cancer tissues of WT mice treated with AOM/DSS (Fig 2K); however, no significant difference in HIF1A mRNA expression was observed in GDH1IEC−/− and GDH1IEC+/+ mice without or with DSS treatment (Fig EV2C). Hence, GDH1 can promote intestinal cancer development, possibly by increasing HIF1α stability once hypoxia occurs during CRC progression.
Hypoxia‐induced acetylation of GDH1 is required for HIF1α stability
To explore the correlation between GDH1 and HIF1α stability, we first excluded the possibility that hypoxia could affect GDH1 protein levels in CRC cells (Fig 3A). Then, we inserted a FLAG tag into the GDH1 C‐terminus, which had no effect on GDH1 activity (Fig EV3A and B). Using this system, we enriched GDH1 and observed decreased GDH1 dehydrogenase activity under hypoxia (Fig 3B). Under hypoxia, the level of GDH1 panlysine (K) acetylation, but not K methylation and S/T/Y phosphorylation, was significantly increased (Fig 3C), suggesting that GDH1 lysine acetylation is involved in the hypoxia response. The enriched GDH1‐FLAG was identified by liquid chromatography–mass spectrometry (LC–MS). GDH1 acetylation sites at K503 and K527 were detected under hypoxia but not under normoxia (Fig EV3C and D). Both the mutations of K503 to R503 and K527 to R527 impaired GDH1 acetylation under hypoxia. When these two lysine sites were simultaneously mutated to arginine residues, GDH1 acetylation was heavily impaired (Fig 3D and E). Furthermore, after mutation of K503 and K527 to R503 and R527, respectively, the transcriptional activity of HIF1α decreased by approximately 75% (Fig 3F). Meanwhile, the protein levels of HIF1α under hypoxia decreased by more than half after the loss of GDH1 acetylation at K503 or K527 (Fig 3G).
Figure 3. Hypoxia‐induced GDH1 acetylation was required for HIF1α stability.
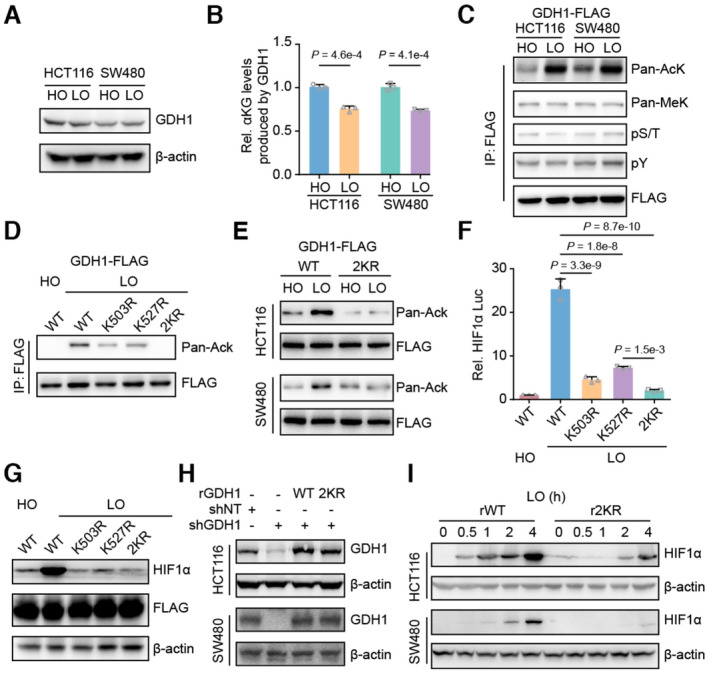
- HCT116 and SW480 cells were incubated under normoxia and hypoxia, respectively. Then, the cells were collected, and the GDH1 protein level was analyzed via immunoblotting.
- Hypoxia reduced the activity of GDH1 in producing αKG. A FLAG tag was inserted into the GDH1 C‐terminus in HCT116 and SW480 cells. The cells were collected after incubation under normoxia or hypoxia, and M2 flag beads were used to enrich FLAG‐tagged GDH1. GDH1‐FLAG was used to assess the enzymatic activity of GDH1 in producing αKG (n = 3).
- GDH1‐FLAG in (B) was examined using antibodies against pan‐lysine acetylation and methylation and pan‐serine/threonine and tyrosine phosphorylation.
- GDH1 acetylated at both K503 and K527 under hypoxia. K503R and K527R were mutated endogenously. GDH1‐FLAG was enriched, and an antibody against pan‐lysine acetylation was used to examine lysine acetylation of GDH1 when K503 or K527 was mutated to R503 or R527. 2KR represents K503/K527R.
- HCT116 or SW480 cells containing WT or GDH1 with K503/K527R mutations were incubated under normoxia and hypoxia, respectively. GDH1‐FLAG was enriched, and an antibody against pan‐lysine acetylation was used to test GDH1‐lysine acetylation.
- Loss of acetylation at GDH1‐K503 or GDH1‐K527 impaired the transcriptional activity of HIF1α (n = 3).
- HIF1α protein stability under hypoxia depends on GDH1‐K503 and GDH1‐K527 acetylation.
- GDH1‐depleted cell lines were constructed and then rescued with the expression of wild‐type GDH1 or K503/K527R mutant GDH1.
- HIF1α protein stability in rWT or r2KR mutant cells was induced over time under hypoxia.
Data information: Data are mean ± SD from the biological replicates (B, F). Statistics: unpaired two‐tailed Student's t‐test (B); one‐way ANOVA with Tukey's HSD post hoc test (F).
Source data are available online for this figure.
Figure EV3. The enrichment of GDH1 for acetylation identification.
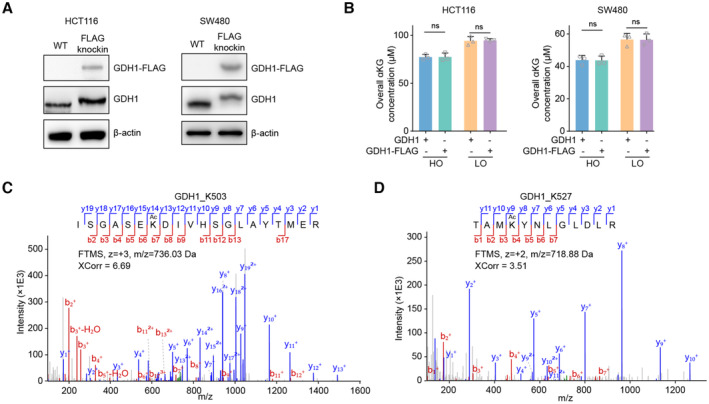
-
AThe successful knockin of FLAG into C terminal of GDH1 in HCT116 or SW480 cells was tested by western blot.
-
BThe overall αKG level in HCT116 or SW480 cells with or without FLAG tagging endogenous GDH1 C terminal under normoxia and hypoxia (n = 3).
-
C, DMS/MS spectra identified K503 and K527 acetylation of GDH1 purified from HCT116 cells.
Data information: Data are mean ± SD from the biological replicates (B). Statistics: unpaired two‐tailed Student's t‐test (B).
Source data are available online for this figure.
The data showed delayed and weak stabilization of HIF1α protein levels when K503/527 was mutated to R503/527, offering solid evidence that the protein stability of HIF1α is regulated by GDH1 lysine acetylation (Fig 3H and I). Based on these data, we preliminarily ascertained that hypoxia‐induced acetylation of GDH1 is required for HIF1α stability.
Hypoxia‐induced GDH1 acetylation at K503 reverses GDH1 dehydrogenase activity
How does hypoxia‐mediated GDH1 acetylation modulate the protein stability of HIF1α? As shown in Fig 4A and B, GDH1 with the K503R mutation increased the formation of GDH1 hexamers compared with WT GDH1. K527R‐GDH1 polymer formation was more or less consistent with that of WT‐GDH1 (Fig 4A and B). Hence, we speculated that GDH1 acetylation at K503 regulates GDH1 dehydrogenase activity.
Figure 4. GDH1 acetylation at K503 reverses GDH1 dehydrogenase activity through decreased formation of GDH1 hexamers.
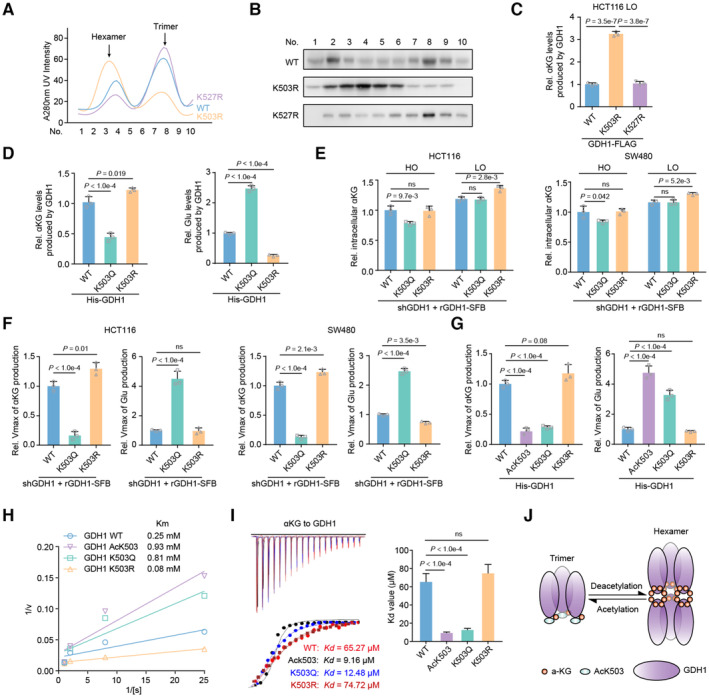
-
AGDH1‐FLAG (including WT and K503R and K527R mutants) was enriched and analyzed via fast protein liquid chromatography (FPLC).
-
BImmunoblot analysis of GDH1‐FLAG levels at different time points.
-
CThe K503R mutation enhances GDH1 activity in producing αKG. FLAG‐tagged GDH1 was purified from HCT116 cells (n = 3).
-
DThe K503Q mutation reduces GDH1 activity leading to αKG production while enhancing the enzymatic activity leading to Glu production. His‐tagged GDH1 was purified from E. coli (n = 3).
-
E, FHCT116 or SW480 cells were depleted of endogenous GDH1 and rescued with SFB‐tagged rGDH1 WT, K503Q or K503R. The relative intracellular αKG concentration was measured under normoxia and hypoxia (E). rGDH1‐SFB proteins were enriched using M2‐FLAG beads to determine the Vmax of αKG production (F) (n = 3).
-
G–IHis‐GDH1 proteins (including WT, AcK503, K503Q and K503R) were purified from E. coli to determine the Vmax of αKG and Glu production (G). A Lineweaver–Burk plot was used to determine the Michaelis–Menten constant (Km) value for Glu production by the GDH1 proteins (H). V, reaction rate; [S], substrate concentration. ITC assays were performed with precipitated His‐GDH1 proteins and αKG (I) (n = 3).
-
JCartoon illustrating the working mechanism of the GDH1 acetylation‐mediated decrease in hexamers and the enhanced binding affinity to αKG.
Data information: Data are mean ± SD from the biological replicates (C–G, I). Statistics: one‐way ANOVA with Tukey's HSD post hoc test (C–G, I).
Source data are available online for this figure.
Consistent with this speculation, upon hypoxia stimulation, the relative level of αKG produced by K503R‐GDH1 was significantly increased while The K527R mutation did not affect GDH1 enzyme activity (Fig 4C). However, compared with that of WT‐GDH1, the ability of K503R‐GDH1 to produce αKG was reduced by around 50%, while the ability of the K503Q‐GDH1 mutant to produce glutamate was increased by approximately 130% (Figs EV4A and 4D). In line with the above in vitro assay, the relative intracellular αKG content decreased in cells expressing the K503Q mutant, while that in cells expressing the K503R mutant increased under hypoxia compared with that in control cells (Figs EV4B and 4E).
Figure EV4. GDH1‐K503 acetylation modulates its enzyme activity.
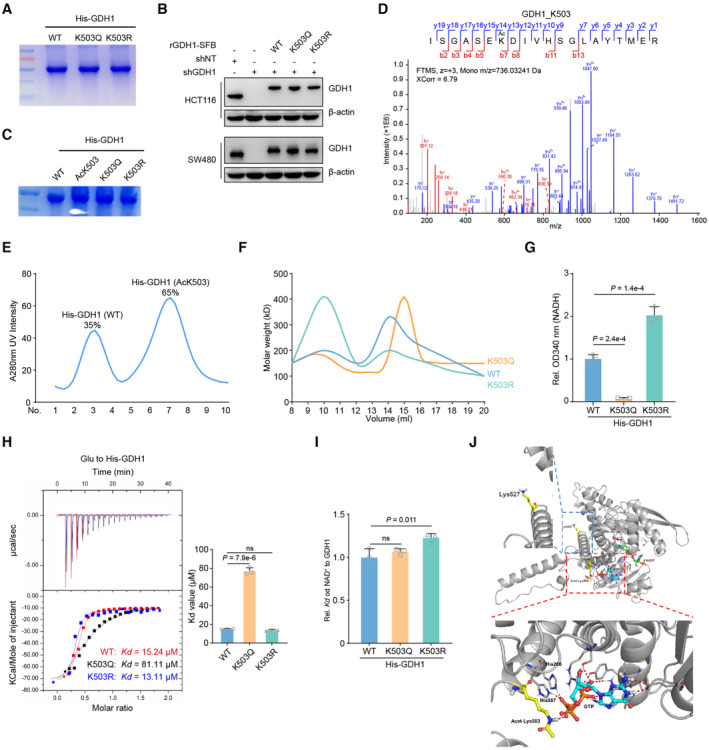
- His‐GDH1 (including WT, K503Q and K503R mutants) were purified from E. coli and separated by SDS‐PAGE. The coomassie blue staining was shown.
- Construction of GDH1‐depleted cell lines, which were rescued with expression of WT GDH1 or K503Q, or K503R mutant GDH1.
- His‐GDH1 (including WT, AcK503, K503Q and K503R mutants) were purified from E. coli and separated by SDS‐PAGE. The coomassie blue staining was shown.
- The acetyl modification of AcK503 His‐GDH1 from E. coli was confirmed by LC–MS/MS.
- His‐GDH1 (including WT and AcK503) purified from E. coli were separated and analyzed by FPLC.
- His‐GDH1 (including WT and AcK503) purified from E. coli were analyzed by dynamic light scattering.
- Measure the NADH level by the OD340 absorbance value, respectively (n = 3).
- His‐GDH1 proteins (including WT, K503Q or K503R) were purified from E. coli to determine Kd value. ITC assays were performed with precipitated His‐GDH1 proteins and αKG (n = 3)
- ITC assays were performed with precipitated His‐GDH1 proteins and NAD+ (n = 3)
- The complex of GDH1 (PDB code: 4ED5) with GTP is superimposed on the GDH1‐GTP complex. The protein and metabolites are shown as cartoon and are colored gray and orange or yellow, respectively. Acetyl‐K503, K527 and GTP are shown as sticks.
Data information: Data are mean ± SD from the biological replicates (G–I). Statistics: one‐way ANOVA with Tukey's HSD post hoc test (G–I).
Source data are available online for this figure.
The results of an enzyme kinetics experiment showed that K503Q‐GDH1 had a much lower maximum reaction rate (Vmax) of αKG production than GDH1‐WT, whereas K503R‐GDH1 had a moderately higher Vmax of αKG production than GDH1‐WT, suggesting that K503Q‐GDH1 has a heavily impaired ability to use glutamate (Glu) as a substrate (Fig 4F). To exclude the possibility that the K to Q mutation does not effectively mimic acetylation modification, we also purified AcK503‐GDH1, K503Q‐GDH1, and K503R expressed in E. coli (Fig EV4C). The qualities and activities of the above proteins were ascertained by a series of in vitro experiments, such as LC–MS for modification identification, FPLC and dynamic light scattering (DLC) to determine dimerization and NADH measurement to assess GDH1 dehydrogenase activity (Fig EV4D–G). AcK503‐GDH1 had a Vmax of αKG or Glu production similar to that of K503Q‐GDH1, demonstrating that the K503Q mutant mimics acetylation at K503 (Fig 4G). Moreover, we experimentally defined the Michaelis–Menten constant (Km) value of these GDH1 variants for Glu. The calculated Km value indicated that K503Q‐GDH1 had a much lower Glu affinity than GDH1‐WT, whereas K503R‐GDH1 had a higher Glu affinity than GDH1‐WT (Fig 4H). Isothermal titration calorimetry (ITC) further showed that AcK503‐ and K503Q‐GDH1 showed increased GDH1 to αKG binding affinity compared with WT or K503R‐GDH1, which was consistent with the results of previous enzyme assays in which the K503Q mutation reduced the ability of GDH1 to produce αKG (Fig 4I). We also summarized the effect of K503 acetylation on the exchange of the GDH1 trimer and hexamer, which indicates the conversion of GDH1 activity (Fig 4J).
Notably, the K503Q mutation reduced the binding affinity of Glu to GDH1 but had no effect on NAD+ binding to GDH1 (Fig EV4H and I). Molecular simulation revealed that GDH1‐His (H) 266, ‐H507 and GTP form a hydrogen bond network and that hypoxia‐induced acetylation at K503 of GDH1 locks GTP in the hydrogen bond network of H266‐H507, in turn reinforcing αKG in GDH1 as a substrate. Otherwise, the position of K527 is located in a large flat groove and facilitates GDH1 association with other partner proteins (Fig EV4J). Thus, we preliminarily conclude that under hypoxia, GDH1 enzyme activity is reversed by acetylation at K503.
Hypoxia‐induced GDH1 acetylation at K527 anchors GDH1 to the EGLN1/HIF1α complex
To find the correlation between HIF1α stability and the overall αKG content, we overexpressed certain enzymes that produce or consume αKG, and we observed that HIF1α protein was not regulated by global changes in αKG (Fig 5A and B). Furthermore, to elucidate the mechanism by which GDH1 mediates HIF1α protein stability, we applied liquid chromatography coupled with tandem mass spectrometry (LC–MS/MS) to verify both HIF1α and EGLN1 as GDH1‐interacting proteins under hypoxia (Fig 5C). Reverse Co‐IP was performed to test whether EGLN1 interacts with other αKG producers or consumers, such as BCAT1/2, GOT1/2, IDH1/2, and GPT (Fig EV5A). Using fraction assays and immunofluorescence (IF) experiments, we clarified that GDH1 interacts and colocalizes with EGLN1 in the cytosol upon hypoxia (Fig 5D and E). Under hypoxia, GDH1 accumulated in the cytosol but showed a slightly decreased presence in mitochondria. More importantly, cytosolic GDH1 presented lower dehydrogenase activity (Fig 5F). Furthermore, we performed a Co‐IP assay and found that loss of GDH1 acetylation at K527 abolished the interaction between GDH1 and EGLN1 (Fig 5G). Mimicked acetylation at K503 or K527 reduced EGLN1 hydroxylase activity, while the loss of acetylation at K503 or K527 dramatically enhanced EGLN1 hydroxylase activity even under hypoxia (Fig 5H). We purified GDH1 with lysine acetylation or with mimicked acetylation expressed in E. coli or HCT116 cells, respectively, and then tested whether antibodies specifically targeted the corresponding lysine acetylation (Fig EV5B). By incubating the above purified GDH1 proteins with EGLN1, we found that GDH1 acetylation at K503 but not at K527 constrained EGLN1 hydroxylase activity (Fig 5I). A potential explanation for this result is that GDH1 combined with EGLN1 in this in vitro reaction system and the effect of K527 acetylation was not reflected. As hypoxia drives GDH1 acetylation and targets EGLN1, the overexpression of WT‐GDH1 dramatically impaired EGLN1 activity upon hypoxia (Fig EV5C).
Figure 5. Tumor cells require both GDH1‐K503 and GDH1‐K527 acetylation to simultaneously reduce EGLN1 activity under hypoxia.
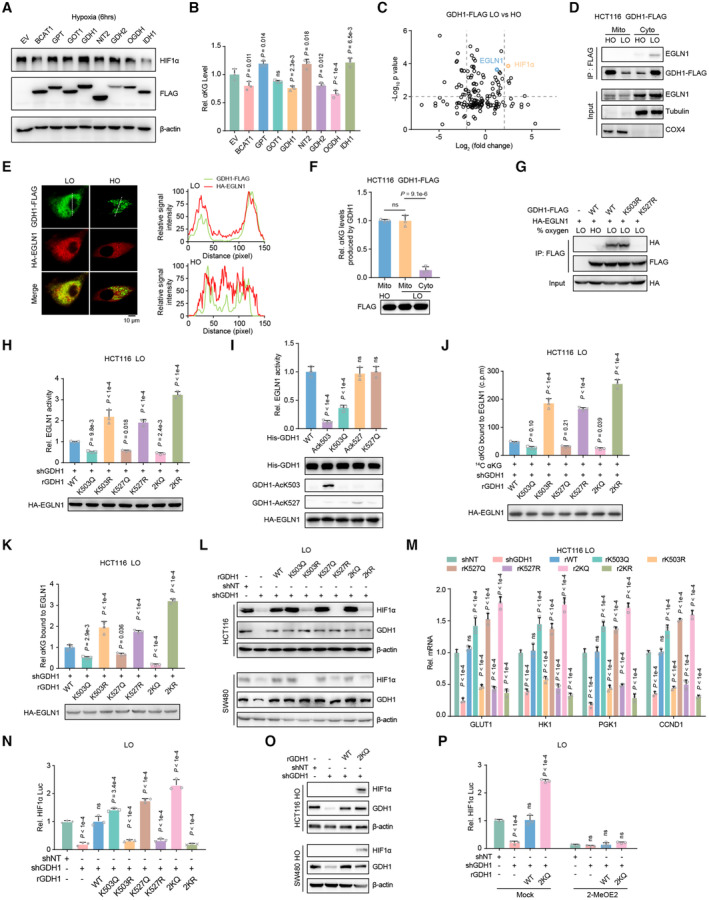
-
A, BImmunoblot of the HIF1α protein level under hypoxia for 6 h when enzymes related to the αKG level were overexpressed in cells, including GDH1 (A). Relative αKG levels in contrast to the control (B) (n = 3).
-
CHCT116‐GDH1‐FLAG cells were incubated under normoxia and hypoxia. Cells were then collected, and M2 flag beads were used to enrich FLAG‐tagged GDH1. GDH1‐FLAG‐associated proteins were identified by LC–MS/MS. The fold change was evaluated as the ratio of expression under hypoxia to that under normoxia. The x‐axis represents the log2‐transformed fold change, and the y‐axis represents the minus log10‐transformed P‐value.
-
DA Co‐IP assay was performed to test the interaction between GDH1 and EGLN1 in the cytosolic and mitochondrial fractions from HCT116‐GDH1‐FLAG cells.
-
EImmunofluorescence analysis of GDH1‐FLAG and HA‐EGLN1 was performed to assess their colocalization under normoxia and hypoxia.
-
FGDH1‐FLAG was enriched in the cytosolic and mitochondrial fractions from HCT116‐GDH1‐FLAG cells, and the enzymatic activity of GDH1 in producing αKG was evaluated (n = 3).
-
GHCT116 cells were cotransfected with plasmids encoding GDH1‐FLAG and HA‐EGLN1. A Co‐IP assay was performed to examine whether the K503R or K527R mutation affected the interaction between GDH1 and EGLN1.
-
H, IEGLN1 activity test. After hypoxia stimulation for 1 h, HA‐EGLN1 was enriched in HCT116‐shGDH1 cells restored with WT or mutant GDH1 (H). His‐GDH1 with K503 or K527 acetylation (AcK503 or AcK527) and mimic acetylation at K503 or K527 (K503Q or K527Q) were purified from E. coli. Then, His‐GDH1 was incubated for 1 h with HA‐EGLN1 (I). EGLN1 activity was tested by converting αKG to succinate (n = 3).
-
J, KRelative αKG content in EGLN1. HA‐EGLN1 was enriched in shNT or shGDH1 cells under hypoxia. A radiometric 14C‐αKG‐EGLN1 binding assay was performed using HA‐EGLN1 incubated with 14C‐αKG (J). Alternatively, the HA‐EGLN1 protein was digested by trypsin, and αKG was determined by LC–MS/MS analysis (K) (n = 3).
-
L–NDetection of HIF1α protein stability (L); the mRNA expression levels of the HIF1α target genes GLUT1, HK1, PGK1 and CCND1 (M); and HIF1α transactivation (N) in the indicated cell lines under hypoxia (n = 3).
-
ODetection of HIF1α protein stability in the indicated cell lines under normoxia.
-
PHIF1α transactivation was examined in the indicated cell lines under hypoxia with or without treatment with the HIF1α transactivation inhibitor 2‐MeOE2 (1 μM) for 12 h (n = 3).
Data information: Data are mean ± SD from the biological replicates (B, F, H–K, M, N, P). Statistics: one‐way ANOVA with Tukey's HSD post hoc test (B, F, H–K, M, N, P); unpaired two‐tailed Student's t‐test (C).
Source data are available online for this figure.
Figure EV5. Acetylation modification at both K503 and K527 regulates the survival of CRC cells under hypoxia.
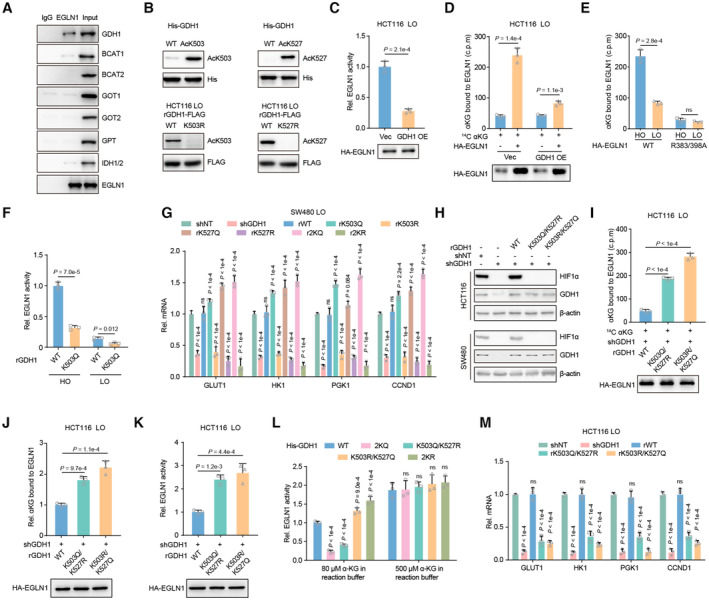
-
AEGLN1 was enriched from hypoxia pretreated HCT116 cells and indicated antibodies were applied to test the association of EGLN1 with αKG producing or consuming metabolic enzymes.
-
BThe specificity of antibodies against GDH1‐AcK503 or ‐AcK527 was analyzed by western blot. His‐ GDH1‐AcK503 or ‐AcK527 was prepared from E. coli. Restored expression of GDH1‐FLAG (including WT, K503R and K527R) was enriched from HCT116 cells.
-
CHA‐EGLN1 was enriched from HCT116 cells with or without GDH1 overexpression under hypoxia and then EGLN1 activity was tested by converting αKG to succinate (n = 3).
-
DRelative αKG in EGLN1. HA‐EGLN1 was enriched from Vec or GDH1 OE cells under hypoxia. A radiometric 14C‐αKG‐EGLN1 binding assay was performed using HA‐EGLN1 incubating with 14C‐αKG (n = 3).
-
ENegative control used for testing relative αKG in EGLN1. HA‐EGLN1 (including WT and R383/398A mutant) was enriched from HCT116 under normoxia and hypoxia. A radiometric 14C‐αKG‐EGLN1 binding assay was performed using HA‐EGLN1 incubating with 14C‐αKG (n = 3).
-
FMeasure the relative EGNL1 activity affected by restored WT or K503Q GDH1 under normoxia or hypoxia in vitro (n = 3).
-
GThe mRNA expression of HIF1α target genes, GLUT1, HK1, PGK1 and CCND1 was tested in indicated cell lines (n = 3).
-
HDetection of HIF1α protein stability in GDH1 depleted cell lines which were rescued with expression of WT GDH1 or K503Q/K527R or K503R/K527Q mutant GDH1.
-
I, JRelative αKG in EGLN1. HA‐EGLN1 was enriched from indicated cells under hypoxia. A radiometric 14C‐αKG‐EGLN1 binding assay was performed using HA‐EGLN1 incubating with 14C‐αKG (H). EGLN1 was digested by trypsin and LC–MS was applied to detect αKG eluted from EGLN1 (I) (n = 3).
-
KHA‐EGLN1 was enriched from indicated genetic manipulated HCT116 cells under hypoxia and then EGLN1 activity was tested by converting αKG to succinate (n = 3).
-
LHA‐EGLN1 was enriched from HCT116 cells under hypoxia and then incubated with indicated His‐GDH1 in the buffer containing 80 or 500 μM αKG. EGLN1 activity was tested by converting αKG to succinate (n = 3).
-
MThe mRNA expression of HIF1α target genes, GLUT1, HK1, PGK1 and CCND1, in indicated cell lines (n = 3).
Data information: Data are mean ± SD from the biological replicates (C–G, I–M). Statistics: unpaired two‐tailed Student's t‐test (C–F); one‐way ANOVA with Tukey's HSD post hoc test (G, I–M).
Source data are available online for this figure.
To test whether EGLN1‐bound αKG is regulated by GDH1 acetylation at K503 or K527, we developed a radiometric 14C‐αKG‐EGLN1 binding assay and LC–MS‐based protein‐bound metabolite assay. The consistent results of these two assays confirmed that acetylation at the K503 and K527 sites is indispensable for reducing the possibility of αKG binding to EGLN1 under hypoxia (Fig 5J and K). The specificity of the above assays to measure EGLN1‐bound αKG was confirmed using an EGLN1 R383/398A mutant, which lost the ability to capture αKG, and we observed a competitive effect of WT‐GDH1 overexpression on EGLN1‐bound αKG (Fig EV5D and E). Otherwise, EGLN1 activity was reduced in K503Q‐GDH1 cells (Fig EV5F). The acetylation of GDH1 at K503 or K527 is essential for maintenance of the stability of HIF1α under hypoxia. In detail, the individual K503R and K527R mutations resulted in HIF1α instability, and the 2KR double mutation reinforced this effect (Fig 5L). The expression of the HIF1α downstream genes GLUT1, HK1, PGK1, and CCND1 (Semenza, 2003; Luo et al, 2006) was also significantly reduced when GDH1 deacetylation occurred or was mimicked (Figs 5M and EV5G). In line with this, HIF1α transactivation was impaired by deacetylation at K503 or K527, and this impairment was reinforced by the 2KR mutant in GDH1 (Fig 5N). Simultaneously, mutating K503 and K527 to mimic constitutive acetylation significantly enhanced the stability of HIF1α under normoxia (Fig 5O). The increased HIF1α stability induced by GDH1 acetylation at K503 and K527 was abolished by the HIF1α transactivation inhibitor 2‐MeOE2 (Fig 5P).
We also assessed the effect of individual mimic acetylation mutations on HIF1α and found that mimic acetylation at K503 or K527 alone did not have a sufficient capacity to stabilize HIF1α under hypoxia (Fig EV5H) by exhausting αKG in EGLN1 (Fig EV5I and J) and impaired EGLN1 activity (Fig EV5K and L). Finally, the individual mimic acetylation mutations in GDH1 did not drive gene expression mediated by HIF1α (Fig EV5M).
Hence, upon hypoxia exposure, GDH1‐K527 acetylation targets EGLN1, while GDH1‐K503 acetylation produces glutamate using αKG as a substrate. The conversion of GDH1 enzyme activity prompts HIF1α stability by consuming αKG in the GDH1/EGLN1/HIF1α axis.
p300 acetylates GDH1 in cytosol
Using the K503R‐GDH1 or K527R‐GDH1 mutant to exclude nonspecific recognition, both antibodies were found to specifically identify GDH1 acetylation under hypoxia (Fig 6A) and indicated that the extent of AcK503 and AcK527 increased with prolonged hypoxia treatment time (Fig 6B). GDH1‐AcK503 and GDH1‐AcK527 were present in both the cytosol and nucleus (Fig 6C).
Figure 6. p300 acetylates GDH1 at K503 and K527 under hypoxia.
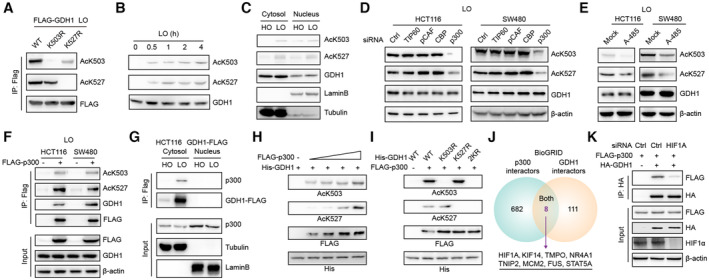
-
AImmunoblot analysis identified the specificity of the antibody against GDH1‐K503 or GDH1‐K527 acetylation.
-
BGDH1‐K503 or GDH1‐K527 acetylation increased with the time course of hypoxic stress.
-
CGDH1‐K503 or GDH1‐K527 acetylation occurred in both the cytosol and nucleus.
-
D, EThe levels of both AcK503 and AcK527 were analyzed when cells were transfected with small interfering RNA (siRNA) targeting TIP60, PCAF, CBP or p300 (D) or when HCT116 or SW480 cells were treated with the p300 inhibitor A‐485 (1 μM) for 12 h under hypoxia (E).
-
FHCT116 and SW480 cells were transfected with or without FLAG‐p300. The p300‐associated proteins were enriched with M2‐FLAG beads and subjected to immunoblot analysis using antibodies against GDH1, GDH1‐K503 or GDH1‐K527 acetylation.
-
GA Co‐IP assay was performed to examine the interaction between GDH1 and p300 in the cytosolic and nuclear fractions of HCT116 GDH1‐FLAG cells.
-
Hp300 acetylated GDH1 at K503 and K527 in a dose‐dependent manner.
-
Ip300 specifically acetylates GDH1 at K503 and K527.
-
JThe GDH1 and p300 interacting proteins were extracted from the BioGrid database, and the shared interacting proteins are shown in a Venn diagram.
-
KThe HIF1A gene was transiently knocked down, and HCT116 cells were cotransfected with FLAG‐p300 and HA‐GDH1 for a Co‐IP assay.
Source data are available online for this figure.
TIP60, PCAF, CBP, and p300 are four major acetyltransferases responsible for lysine acetylation (Wapenaar & Dekker, 2016). The knockdown efficiency of small interfering RNAs specifically targeting the genes encoding the above acetyltransferases is shown in Appendix Fig S1A. p300 depletion induced a decrease in AcK503 and AcK527, while the knockdown of the other acetyltransferases did not have this effect (Fig 6D). Furthermore, using an inhibitor specifically targeting p300, we observed a result consistent with that obtained by p300 depletion with specific siRNA (Fig 6E). Co‐IP experiments showed that the FLAG‐p300‐associated complex contained GDH1 acetylated at K503 and K527 (Fig 6F). Co‐IP analyses of cytosolic and nuclear fractions identified GDH1 and p300 interacting in the cytosol but not in nuclei, suggesting that nuclear AcK503‐GDH1 and AcK527‐GDH1 first occurred in the cytoplasm (Fig 6G). As shown in the BioGRID database, approximately 30% of p300 interactors are localized in the cytosol (Appendix Fig S1B), and IF staining using an antibody against p300 showed that p300 localized in both the cytosol and nucleus (Appendix Fig S1C). Moreover, the HIF1α protein level was blocked by p300 knockdown in WT‐GDH1‐overexpressing cells but not in 2KQ‐GDH1‐overexpressing cells (Appendix Fig S1D). These results demonstrate that p300 is required for GDH1 acetylation.
We also performed a semi‐in vitro biochemical experiment in which gradually increased FLAG‐p300 purified from HCT116 cells was incubated with His‐GDH1 purified from E. coli, and the levels of AcK503‐GDH1 and AcK527‐GDH1 increased in this in vitro acetylation reaction system (Fig 6H). When we mutated K503 to R503 or K527 to R527, no signals for GDH1 acetylated at K503 or K527 were detected, even when p300 was added to the acetylation reaction system (Fig 6I). To clarify how GDH1 recruits p300, we evaluated the common interacting proteins of GDH1 and p300 in the BioGrid database. As shown in Fig 6J, eight proteins, including HIF1α, were potential bridges linking p300 to GDH1. We first transiently knocked down the HIF1A gene and found that HIF1A depletion resulted in an obvious reduction in the interaction between GDH1 and p300 (Fig 6K). The above results further confirm that p300 forms an axis with GDH1/HIF1α and is responsible for acetylating GDH1 at K503 and K527.
GDH1 acetylation at both K503 and K527 is required to form solid tumors and is correlated with a poor CRC prognosis
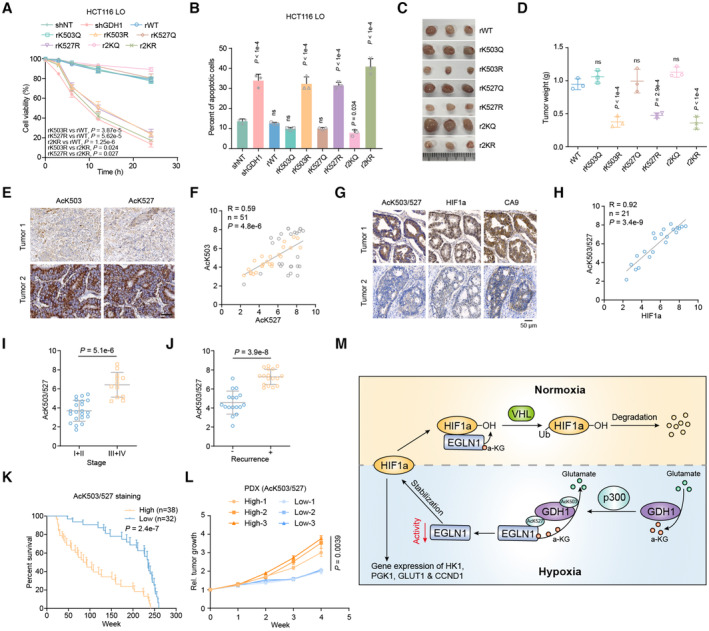
Under hypoxia, the expression of the individual GDH1 K503R or GDH1 K527R mutant sensitized cells to hypoxia, and the GDH1 2KR mutant reinforced this effect (Fig 7A and Appendix Fig S2A). Consistently, only approximately 7% of 2KQ‐GDH1 mutant cells were apoptotic. Under the same conditions, more than 30% of K503R‐, K527R‐ or 2KR‐GDH1 mutant cells were apoptotic (Fig 7B, Appendix Fig S2B and C). Moreover, HIF1α knockdown blocked the increased resistance to apoptosis of WT or 2KQ‐GDH1 cells under hypoxia but not of GDH1 knockdown cells, further supporting the existence of the GDH1/EGLN1/HIF1α axis (Appendix Fig S2D). Experiments in tumor‐bearing nude mice showed that GDH1 acetylation was required for the formation of solid tumors. A lack of GDH1 acetylation at K503 or K527 significantly decreased the ability of cells to form solid tumors (Fig 7C and D).
Figure 7. AcK503/527‐GDH1 indicates a poor prognosis, mainly dependent on HIF1α stabilization.
-
A, BCell viability and percentage of apoptosis in the indicated cell lines under hypoxia were analyzed by trypan blue (A) and PI/Annexin V double staining (B), respectively (n = 3).
-
C, DThe xenograft tumor formation efficiency of the indicated cell lines was assessed, and tumor weight measurements were performed (n = 3).
-
ERepresentative images of CRC samples are shown after antibody staining of AcK503‐GDH1 and AcK527‐GDH1.
-
FThe correlation between AcK503‐GDH1 and AcK527‐GDH1 expression in CRC samples. The tissue sections were quantitatively scored according to the percentage of positive cells and the staining intensity (H‐score) (n = 51).
-
GRepresentative images of the same CRC samples are shown after staining with antibodies against AcK503/527‐GDH1 and HIF1α.
-
HCorrelation between AcK503/527‐GDH1 and HIF1α expression in CRC samples (n = 21).
-
IDetection of AcK503/527‐GDH1 expression in CRC grades I + II and III + IV (grade I + II, n = 21; grade III + IV, n = 12).
-
JDetection of AcK503/527‐GDH1 expression in patients with (+) or without (−) recurrence (−, n = 17; +, n = 17).
-
KPatient survival corresponding to different levels of AcK503/527‐GDH1 expression (High, n = 38; Low, n = 32).
-
LPDX growth related to high or low levels of AcK503/527‐GDH1 expression.
-
MSchematic model described in the discussion section.
Data information: Data are mean ± SD from the biological replicates (A, B, D, I, J, L). Statistics: two‐way ANOVA with Tukey's HSD post hoc test (A, L); one‐way ANOVA with Tukey's HSD post hoc test (B, D); unpaired two‐tailed Student's t‐test (F, H); Mann–Whitney U‐test (I, J); log‐rank test (K).
Source data are available online for this figure.
Because the simultaneous acetylation of GDH1 at K503 and K527 is required to maintain HIF1α stability, we first examined the correlation between AcK503 and AcK527 and then selected samples in which AcK503 and AcK527 were highly positively correlated for subsequent experiments (Appendix Fig S2E, Fig 7E and F). To investigate the correlation between AcK503/527‐GDH1 and HIF1α at the tissue level, we prepared an antibody against AcK503/527‐GDH1 by mixing the two acetylated peptides (Appendix Fig S2F–H). The AcK503/527‐GDH1 and HIF1α signals in tissues were highly positively correlated, indicating that HIF1α protein stability is upregulated when GDH1 is diacetylated at K503/527 in the clinic (Fig 7G and H). The tumor tissue from AOM/DSS mice also presented a similar correlation between AcK503/527‐GDH1 and HIF1α in hypoxic regions (Appendix Fig S2I). The above experiments revealed that AcK503‐GDH1/K527 is positively correlated with the degree of CRC malignancy (Fig 7I and J) and is expected to be a biomarker to characterize CRC progression and prognosis. Furthermore, we calculated the survival duration of patients in the two groups depending on their level of acetylated K503/K527‐GDH1. Patients with high AcK503/527‐GDH1 expression clearly presented a shorter survival time than those with low expression (Fig 7K). We observed a higher tumor growth rate in patient‐derived xenografts (PDXs) from patients with a high AcK503/527‐GDH1 expression (Fig 7L). The above data suggest that GDH1 acetylation at K503/K527 can indicate CRC progression under hypoxia.
Discussion
In this study, through siRNA screening targeting 163 dehydrogenase‐coding genes, we found that GDH1 could confer tolerance to hypoxia. The effects of metabolic enzyme defects on organ development and pathology have been poorly studied. Using the Cre‐loxP recombination system to selectively deplete metabolic enzymes in specific organs, we obtained a mouse model lacking the GDH1 gene in the small intestine. Although GDH1 knockout had no significant effect on intestinal development, AOM/DSS treatment did not effectively induce CRC in mice with GDH1 deletion in the intestine. Hence, first, through a series of in vitro and in vivo experiments, we ascertained the oncogenic role of GDH1 in CRC development by adapting tumor cells to hypoxic stress.
The formation of blood vessels occurs later in the process of solid tumor formation, which subjects solid tumors to many stresses, such as hypoxia and nutrient limitation (Hanahan & Weinberg, 2011). Solid tumors have developed strategies to address these stresses and survive. Here, we examined the stress response signaling pathways associated with hypoxia and nutrient limitation. AMPK phosphorylation and HIF1α levels were significantly lower in CRC tissues of GDH1‐deficient mice than in those of WT mice. We previously reported that GDH1 regulates AMPK phosphorylation under glucose deficiency (Wang et al, 2019a). In the present study, we found that GDH1 also plays a role in modulating HIF1α stability. HIF1α regulates a wide range of target genes involved in tumor cell growth, such as GLUT1, HK1, PGK1, and CCND1 (Semenza, 2003). Consequently, the expression of these genes was significantly decreased in GDH1‐deficient CRC cells.
In both cell lines and animal models, we preliminarily verified that GDH1 is required to stabilize HIF1α under hypoxia. In different cellular contexts, GDH1 depletion results in a 25–60% decrease in intracellular αKG levels (Yang et al, 2009; Jin et al, 2015). Given that αKG is a cofactor for members of the TET, DNMT, and EGLN families, it is reasonable to hypothesize that changes in the level of αKG would alter the activity of these enzymes. EGLN family depends on multiple substrates and cofactors for activity, including αKG, 2HG, and iron availability, and on the presence of oxidative stress (Ke & Costa, 2006; Kaelin & Ratcliffe, 2008). It is reported that EGLN1 activity is not only regulated by oxygen but also regulated by the intracellular metabolite αKG, which competes with its analogue 2HG. For example, BCAT1 is reported to restrict αKG levels and thus promote glioma occurrence and survival by decreasing EGLN1 enzyme activity and increasing HIF1α protein stability (Raffel et al, 2017); therefore, a correlation between BCAT1 and EGLN1 can be proposed. However, biochemistry indicators were not examined in their study. In fact, the Km value of EGLN1 for αKG is 1.3 μM (Lorenzo et al, 2014), while in different cell contexts, the intracellular αKG concentration changed in the range of 10–200 μM, which was insufficient to affect EGLN1 enzyme activity. Meanwhile, although 2HG is a well‐established competitive inhibitor of αKG, the global increase in 2HG induced by supplementation with cell‐permeable octyl‐2HG had no effects on EGLN1 activity in this study. This result was supported by a previous report showing that the IDH1‐R132H mutant utilized αKG as a substrate and produced R‐2HG, which did not stabilize HIF1α protein (Koivunen et al, 2012). Conversely, forced expression of an IDH1‐R132H mutant reduced αKG but increases HIF1α protein (Zhao et al, 2009). The inconsistent results of the above studies suggest that global changes in intracellular αKG or 2HG do not uniformly affect EGLN1 activity. Thus, we should shift our perspective toward signal transduction‐mediated local changes in αKG.
Under hypoxia, GDH1 undergoes simultaneous acetylation at K503 and K527, which regulate GDH1 enzyme activity and target the EGLN1/HIF1α complex, respectively. Lysine acetylation modifies enzyme activity, protein stability, and protein–protein interactions, among other effects (Choudhary et al, 2014). Hypoxia‐induced GDH1 acetylation at K503 likely confers resistance to αKG feedback inhibition of GDH1, after which GDH1 can use αKG as a substrate to produce glutamate. The direction of the reaction will be determined when the concentrations of the substrate and product are no longer changed by the enzyme. A reversible reaction would occur if the accumulation of the product or the affinity of the enzyme to the product exhibits a significant increase. We previously reported that PGK1 phosphorylation caused a reversible reaction upon the stimulation of resident tissue macrophages secreting IL‐6 (Zhang et al, 2018).
To ascertain the effect of the hypoxia‐induced interaction between GDH1 and EGLN1 and of the decreased GDH1 αKG production activity on EGLN1 activity, we performed a series of experiments, as shown in Fig 5F–L. We first determined EGLN1 activity in cells with different genetic manipulations and used an in vitro assay in which different GDH1 mutants were incubated with EGLN1, and the results preliminarily linked GDH1 K503 and K527 acetylation to EGLN1 activity. Next, we applied both a radiometric 14C‐αKG‐EGLN1 binding assay and IP‐LC/MS determination to identify the relative quantity of EGLN1 binding to αKG. Taken together, the data showed that a reverse GDH1 enzyme reaction occurred upon hypoxia and exhausted αKG around EGLN1 or impaired the affinity of EGLN1 for αKG.
p300 has been reported to positively regulate HIF1α protein stability (Geng et al, 2012), and MUC1 has been shown to physically interact with HIF1α and p300 to stabilize HIF1α at the protein level (Chaika et al, 2012). Consistently, we confirmed that p300 acetylated GDH1 under hypoxia and linked p300 to EGLN1 activity and HIF1α stability. Moreover, HIF1α significantly promoted the interaction between GDH1 and p300, and upregulated GDH1 acetylation. The initiation of GDH1 acetylation primarily promoted HIF1α stabilization, which in turn promoted the interaction between GDH1 and p300, forming a positive cycle for fast accumulation of HIF1α and a rapid response to hypoxic stress. This should be an important positive feedback loop which allows tumor cells to survive when facing hypoxia.
Although metabolic reprogramming is a hallmark of tumorigenesis and progression, little is known about the metabolic enzymes and oncometabolites that regulate CRC resistance to hypoxia and facilitate survival, and very few metabolic enzymes or molecules have been identified as potential biomarkers in situ (Sullivan et al, 2016). In this study, GDH1 acetylation at K527 facilitated GDH1 anchoring of EGLN1/HIF1α, while GDH1 acetylation at K503 drove GDH1 to use αKG as a substrate to produce glutamate, the progression of which reduced the concentration of αKG around/in the EGLN1/HIF1α complex or impaired the capacity of EGLN1 to bind αKG, both of which in turn stabilized HIF1α. Therefore, the hypoxia‐modulated acetylation of GDH1 at K503 and K527 is a potential biomarker of CRC progression, and the GDH1/EGLN1 axis is a potential target for CRC treatment (Fig 7M).
Materials and Methods
Patient specimens
Tumors and adjacent colorectal tissues were obtained from patients with CRC who underwent surgery or biopsy at The First Affiliated Hospital and The Third Affiliated Hospital of Sun Yat‐sen University between 2012/08 and 2017/03. The patients had not received any chemotherapies or other treatments before surgery treatment or diagnosis at the hospital, harboring high or low AJCC stage level. The clinicopathological information was supplied in Tables [Link], [Link]. The study protocol was approved by the Ethics Committee of Sun Yat‐sen University (Guangzhou, China, ethics approval: #fahsysu‐038). Written informed consent was obtained from all participants in this study. All the research was carried out in accordance with the provisions of the Declaration of Helsinki of 1975 and the Department of Health and Human Services Belmont Report.
IHC analysis
Tissue sections from paraffin‐embedded human CRC tissues were stained with antibodies as indicated. We quantitatively scored the tissue sections according to the percentage of positive cells and the staining intensity. We rated the intensity of staining on a scale of 0–3: 0, negative; 1, weak; 2, moderate; and 3, strong. We assigned the following proportion scores: X indicates the percentage of the tumor cells that were stained (0 ≤ [X1 + X2 + X3] ≤ 10), where X3 indicates strong staining, X2 moderate staining and X1 weak staining. The score (H‐score) was obtained using the following formula: 3 × X1 + 2 × X2 + 1 × X3, yielding scores ranging from 0 to 10. Scores were compared with overall survival, defined as the time from the date of diagnosis to death or the last known date of follow‐up. The use of human CRC specimens was approved by the IRB at Sun Yat‐sen University Guangzhou Cancer Center and complied with all relevant ethical regulations. Informed consent was obtained from all patients.
AOM/DSS‐induced colitis‐associated colon cancer model
All mice were maintained under a 12‐h light/dark cycle with free access to water and standard mouse chow in a specific‐pathogen‐free (SPF) facility, receiving humane care. All experimental procedures were performed in compliance with the Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee (IACUC) in Guangzhou University (Guangzhou, China, ethics approval: #gzhu2018039).
In the present study, the effect of AOM injection and DSS dietary administration was evaluated in GDH1IEC−/− mice and control wild‐type mice in C57BL/6 background. On Day 0, mice were given a single intraperitoneal injection of AOM (10 mg/kg body weight) at the age of 7 weeks old. Seven days after the AOM injection, the animals received 2% DSS (w/v) in the drinking water for 7 days, and then, drinking water alone was supplied for 7 days. The DSS‐to‐water cycle was repeated a total of three times. All animals were sacrificed on Day 80. At the time of sacrifice, a complete necropsy was performed on all mice. Histopathological examination was performed on paraffin‐embedded sections after hematoxylin and eosin (H&E) staining.
Plasmids
pCDH‐GDH1 and pColdI‐His‐GDH1 were constructed in a previous study (Wang et al, 2019a), as was pCDH‐p300 (Wang et al, 2017). PCR‐amplified HIF1α was cloned into pCDH‐GDH1. K503R/Q and K527R/Q mutations were generated using a QuickChange site‐directed mutagenesis kit (Stratagene, La Jolla, CA, USA).
The pGIPZ control and pGIPZ GDH1 shRNA were generated as previously described (Wang et al, 2019a). The HIF1α reporter plasmid was generated using the sequences cloned into the pGL3‐TA promoter, and the sequence is listed in a previous report (Wang et al, 2019a).
Cell culture, transfection and designed antibody
Human CRC HCT116 and SW480 cell lines were purchased from American Type Culture Collection (ATCC). The cell lines were authenticated by STR‐profiling and tested for mycoplasma contamination to ensure that they were mycoplasma‐free. To induce hypoxic stress, the cells were cultured in a hypoxia incubator chamber (STEMCELL Technologies Inc., #27310).
Colorectal cells were transfected with small interfering RNAs (siRNAs, 50 nM) against human dehydrogenase and acetyltransferase‐coding genes using MAX siRNA transfection reagent (Thermo Scientific Dharmacon Inc., USA), while nonspecific siRNA (siNC) was used as a negative control. All siRNAs were purchased from GenePharma Technology (Shanghai, China). siRNA sequences were listed in Appendix Table S1.
Antibodies against GDH1 acetylation at K503, K527, or K503/527 were custom designed and constructed by ABclonal Technology (Wuhan, China). Briefly, the acetyl‐peptides AcK503‐GDH1 (ISGASEAcKDIVHSG), AcK527‐GDH1 (IMRTAMAcKYNLGL) and their respective control peptides without acetyl groups were first synthesized. Then, New Zealand White (NZW) rabbits were immunized by intradermal injection of 500 μg of indicated polypeptides which were mixed with Freund's adjuvant to 2 ml volume. This process was repeated three times, and the dose was reduced to half in the fourth time. Serum samples were taken to test the titers of indicated antibodies at 2 weeks after the fourth immunization. The antibodies were purified and obtained by affinity chromatography.
Quantitative RT–PCR (qRT–PCR)
Total RNA was extracted with an RNA high‐purity total RNA rapid extraction kit (QIAGEN). cDNA was prepared using a GoScript reverse transcription system (Promega). qRT–PCR was performed using SYBR PCR premixture (Promega) and an ABI 7500 fast system under the following conditions: 5 min at 95°C followed by 38 cycles at 95°C for 30 s, 60°C for 40 s, and 72°C for 1 min. Data were normalized to the expression of the control gene (β‐actin) for each experiment. Data are presented as the mean ± SD of three independent experiments. The sequences of the primer pairs used for qRT–PCR were listed in Appendix Table S2.
Western blotting and antibodies
Western blot analysis was performed using standard protocols. An anti‐β‐actin (Merck, CAS: ABT264) antibody was used as a control for whole‐cell lysates. Endogenous antibodies including HIF1α (Clone number: D1S7W, CAS: #36169), HIF2α (Clone number: E2N9W, CAS: #57921), EGLN1 (Clone number: D31E11, CAS: #4835), COX4 (Clone number: 3E11, CAS: #4850), p300 (Clone number: D8Z4E, CAS: #86377), AMPK (Clone number: D63G4, CAS: #5832), and Phospho‐AMPK (Clone number: 40H9, CAS: #2535), GDH1 (Clone number: D9F7P, CAS: #12793), LaminB (Clone number: D9V6H, CAS: #13435), BCAT1 (Clone number: D4V2T, CAS: #16431), BCAT2 (Clone number: D8K3O, CAS: #79764), GOT1 (Clone number: E4A4O, CAS: #34423), GOT2 (CAS: #71692), GPT (CAS: AV45710) were purchased from Cell Signaling Technology. Antibodies including His‐tag (Clone number: HIS.H8, CAS: ab18184), Pan‐Methyl‐Lysine (CAS: ab7315), Phospho‐Ser/Thr (CAS: ab117253), Phospho‐Tyrosine (Clone number: EPR16871, CAS: ab179530) were purchased from Abcam. Antibody IDH1/2 (CAS: sc‐373816) was purchased from Santa Cruze. The dilution of these primary antibodies was 1:1,000. Tag antibodies against FLAG (CAS: AE005) or HA (CAS: A0208) and β‐Tubulin (CAS: AC008) antibody were purchased from Abclonal (Wuhan, China) and used at a dilution of 1:5,000. IP and IB analyses were performed with the indicated antibodies.
Cell viability assay
HCT116 or SW480 cells with or without genetic modifications were plated in DMEM (10% FBS) under hypoxia or normoxia for the indicated times. Cells were collected at the indicated time points, and cell viability was determined by trypan blue staining.
PI/Annexin V
We measured the percentage of apoptotic cells under hypoxic stress as previously described (Shao et al, 2019).
Mass spectrometry analysis of GDH1‐interacting proteins and GDH1 acetylation
GDH1‐FLAG and its interacting proteins was enriched by immunoprecipitation with an antibody against the Flag tag. The proteins were eluted and separated using SDS–PAGE followed by Coomassie blue staining. The gel lane was dissected and subjected to in‐gel digestion. Briefly, the gel was chopped into 1 mm3 pieces and decoloured using 50 mM NH4HCO3 in 30% ACN under oscillation. Then, the gel was subjected to vacuum freeze lyophilization and alkylated using 10 mM TCEP+ 50 mM CAA in 50 mM NH4HCO3 at 95°C for 5 min. The supernatant was removed, and the gel was again subjected to vacuum freeze lyophilization. Then, the gel was treated with trypsin (20 ng/μl in 50 mM NH4HCO3) and incubated at 37°C for 16 h. The peptides were extracted three times in an ultrasonic water bath by 0.1% TFA in 90% ACN. The resulting peptides were subjected to vacuum freeze lyophilization, purified using C18 stage tips and then analyzed via liquid chromatography–tandem mass spectrometry (LC–MS/MS). Peptides were loaded and separated on a homemade microtip C18 column (ReproSil‐Pur C18‐AQ, 3 μm, 75 μm × 250 mm) on a nanoflow HPLC Easy‐nLC 1000 system (buffer A, 0.1% FA in H2O; buffer B, 0.1% FA in 100% ACN; Thermo Fisher Scientific). The LC gradient was set as follows: 2–4% B for 1 min; 4–30% B for 47 min; 30–45% B for 5 min; 45–90% B for 1 min; and 90% B for 6 min. MS data were collected via data‐dependent acquisition in the profile mode on a Q Exactive mass spectrometer set as follows: MS1 resolution, 70,000 @ m/z 200; mass range, 300–1,600 m/z; AGC Target, 3e6; maximum IT, 50 ms; topN, 10. MS2 resolution, 17,500 @ m/z 200; normalized collision energy (NCE), 27%; isolation width (m/z), 1.6 m/z; AGC Target, 1e5; maximum IT, 120 ms; charge states include 2–5; dynamic exclusion, 30 s. The mass spectrometric data were analyzed using Proteome Discoverer 2.3 against the human Swiss‐Prot database with carbamidomethyl cysteine set as a fixed modification and oxidized methionine, protein N‐term acetylation and lysine acetylation set as variable modifications.
Lysine‐acetylated GDH1 expression and purification
To obtain site‐directed GDH1‐AcK, we constructed pET22b‐GDH1 encoding GDH1 with a C‐terminal 6×His tag. We cotransformed E. coli BL21 (DE3) cells with pAcKRS and pET22b‐GDH1 with a C‐terminal 6×His tag. As described previously (Neumann et al, 2008), GDH1‐AcKs were overexpressed in E. coli BL21 (DE3) following the addition of Ara and IPTG in the presence of ampicillin, chloramphenicol, 10 mM NAM and AcK. GDH1‐AcKs were purified by Ni‐NTA affinity chromatography, followed by gel‐filtration chromatography with a Superdex 75. The purity of the preparations was assessed by SDS–PAGE, and the protein concentration was determined using a Bradford protein assay kit.
Measurement of reversible GDH1 activity and GDH1 enzyme kinetics
GDH1 activity associated with αKG production was measured at room temperature. GDH1 was immunoprecipitated from the lysates of CRC cells or purified from E. coli and subjected to GDH1 enzymatic activity assays in reaction buffer containing 100 mM Tris (pH 8.0), 50 mM L‐Glu, and 1 mM NAD+. The change in absorbance at 340 nm due to an increase in NADH was measured using a BioTek Synergy Neo Multi‐Mode Plate Reader (BioTek, USA).
GDH1 activity associated with Glu production was measured at room temperature in reaction buffer containing 100 mM Tris (pH 8.0), 10 mM αKG, 5 mM NADH, and 500 μM NH4Cl. The change in absorbance at 340 nm due to a decrease in NADH was measured using a BioTek Synergy Neo Multi‐Mode Plate Reader (BioTek, USA).
The continuous change in absorbance at 340 nm due to an increase or decrease in NADH was used to calculate the Vmax of αKG or Glu, and the Km was determined by nonlinear regression analysis, which was performed using Prism 7 (GraphPad).
Measurement of αKG
The indicated cells were seeded into six‐well plates, then collected and lysed, and the lysate was examined using an αKG Assay Kit (Abcam, αKG Assay kit (colorimetric), CA, USA) according to the manufacturer's instructions.
Luciferase reporter assay
Transcriptional activation of HIF1α in HCT116 cells was measured as previously described (Wang et al, 2017). Relative levels of luciferase activity were normalized to the levels in untreated HCT116 cells expressing control vector and then normalized to the levels of luciferase activity induced by the Renilla control plasmid.
HIF peptide hydroxylation assay for assessing EGLN1 activity
An N‐terminally biotinylated peptide corresponding to HIF1 residues 556–575 (DLDLEALAPYI PADDDFQLR) was synthesized by changing two methionines (M) to alanine (A). Equal amounts of HA‐EGLN1 protein (10 nM) in HA beads were purified from the indicated HCT116 cells under hypoxia and incubated with the above peptide (100 nM) for 2 h in PHA buffer containing FeCl2 (100 μM), ascorbate (2 mM), and αKG (50 μM). The HA agarose beads were washed three times with NETN (20 mM Tris–HCl [pH 8.0], 100 mM NaCl, 1 mM EDTA, 0.5% NP‐40), and peptide hydroxylation was determined by LC–MS.
To measure GDH1 activity using αKG as a substrate and the consequence of αKG exhaustion in EGLN1, we first incubated His‐GDH1 (including WT or mutant) with GST‐EGLN1 (in the GST beads) in the adjusted buffer, changing 10 mM αKG to 500 μM (called high αKG) or 80 μM (called low αKG, which is likely close to the cytoplasmic αKG level under normal oxygen conditions) αKG. Then, EGLN1 was precold washed twice and incubated with HIF1α to assess EGLN1 activity.
Measurement of EGLN1‐bound αKG
To assess EGLN1‐bound αKG, we first developed a radiometric 14C‐αKG‐EGLN1 binding assay. In detail, bead‐bound HA‐EGLN1 purified from the indicated HCT116 cells under hypoxia was washed with Tris‐buffered saline (TBS) buffer (50 mM Tris, 150 mM NaCl [pH 7.5]), followed by incubation with 0.2 mCi 14C‐αKG for 30 min in TBS buffer. The beads were then washed with TBS buffer containing 1% NP‐40. The bead‐bound EGLN1 protein was eluted with 10 mg of HA peptides, and radioactivity was detected by liquid scintillation counting. We also developed another method for testing EGLN1‐bound αKG using LC–MS. In detail, we enriched HA‐EGLN1 from the indicated HCT116 cells under hypoxia and incubated HA‐EGLN1 in TBS buffer containing 100 μM αKG. HA‐EGLN1 beads were quickly washed with TBS buffer containing 1% NP‐40 but not αKG. Then, HA‐EGLN1 was digested using protease to release αKG from the EGLN1 protein, and the αKG quantity was measured via liquid chromatography–mass spectrometry (LC–MS). The above results were normalized based on the total protein concentration in each sample.
Subcellular fractionation analyses
HCT116 cells were harvested and washed three times with cold PBS. Cytosolic or mitochondrial fractions were prepared using a Nuclear/Cytosol Fraction Kit (Active Motif).
Isothermal titration calorimetry
Thermodynamic parameters of the binding of GDH1 WT, K503R, or K503Q to Glu or αKG were measured by isothermal titration calorimetry (ITC) assays using an ITC200 Microcalorimeter (MicroCal) at 25°C. GDH1 was expressed in E. coli and purified via His tag affinity purification. In all experiments, the initial injection of 0.5 ml of Glu or αKG solution was discarded to eliminate the effect of titrant diffusion across the syringe tip during the equilibration process, and each dataset consisted of 20 injections of 2 ml each of 1 mM Glu or αKG into the sample cell containing 250 ml of 0.05 mM GDH1. The heat of dilution was negligible in all cases. The binding constant and other thermodynamic parameters were determined by fitting the integrated titration data using the single binding site model via the nonlinear least‐squares method, which was implemented in MicroCal Origin software (version 7.0).
In vitro acetyltransferase activity assay
FLAG‐tagged p300 was purified from HCT116 cells and eluted with the FLAG peptide. p300 was quantified as 50 ng/μl, and we added GDH1 to the p300 acetyltransferase activity reaction system according to the manufacturer's instructions. The acetyltransferase activity was measured using a kit (ab204536).
Subcutaneous xenograft model
All mice were maintained under a 12‐h light/dark cycle with free access to water and standard mouse chow in a specific‐pathogen‐free (SPF) facility, receiving humane care. All experimental procedures were performed in compliance with the Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee (IACUC) in Guangzhou University (Guangzhou, China, ethics approval: #gzhu2018039).
Briefly, 2 × 106 HCT116‐derived stable cells were subcutaneously injected into the left dorsal region of randomized 6‐week‐old male athymic nude mice. After inoculation for 28 days, the mice were euthanized, and the tumors were dissected. Differences in the weight of xenograft tumors were statistically analyzed.
Statistical analysis
All results are presented as the mean ± standard deviation (SD), unless stated otherwise. Statistical analyses were performed using two tailed Student's t‐test for two groups comparison, one‐way or two‐way analysis of variance (ANOVA) for multiple comparisons on GraphPad Prism version 8.3 (GraphPad Software, Inc.). About clinical patients, Mann–Whitney U‐test was used for two groups comparison, Pearson's correlation analysis was used to calculate the regression and correlation, log‐rank test was used to assess the patient survival curve. P < 0.05 was considered statistically significant. No statistical methods were applied to pre‐evaluate sample size. The sample size was determined based on our experiences and similar with reported literature for specific experiments. No data were excluded from the analyses. All experiments were performed with at least three biologically independent samples or three biologically independent experiments. n values indicated biologically independent samples and experiments. Except the IHC scoring, the investigators were not blinded to allocation during experiments and outcome assessment for consistent of experimental procedures. For in vivo and in vitro assays, groups were randomly allocated.
Author contributions
Xiongjun Wang: Resources; supervision; funding acquisition; writing – original draft; project administration; writing – review and editing. Kunhua Hu: Conceptualization; data curation; formal analysis; validation; investigation; methodology. Yufeng Ding: Resources; formal analysis; investigation; project administration; writing – review and editing. Hongwen Zhu: Data curation; software; formal analysis; validation; investigation; visualization; methodology. Xiaoqian Jing: Resources; formal analysis; validation; investigation; visualization. Weiling He: Resources; methodology. Hua Yu: Resources; formal analysis; supervision; validation; investigation; methodology; project administration.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Source Data for Expanded View
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
This work was granted by the National Natural Science Foundation of China (92053113) and mainly supported by the Guangdong Basic and Applied Basic Research Foundation (2021B1515020016). In addition, we thanked a lot for the substantial support offered by the molecular biology platform at Precise Genome Engineering Center of Guangzhou University.
The EMBO Journal (2023) 42: e112675
Contributor Information
Weiling He, Email: hewling@mail.sysu.edu.cn.
Hua Yu, Email: yuhua@sibcb.ac.cn.
Xiongjun Wang, Email: xiongjunwang@sibs.ac.cn, Email: wangxiongjun@gzhu.edu.cn.
Data availability
This study contains no data amenable to large‐scale repository deposition.
References
- Böğürcü N, Seidel S, Garvalov BK, Acker T (2018) Analysis of hypoxia and the hypoxic response in tumor xenografts. Methods Mol Biol 1742: 283–300 [DOI] [PubMed] [Google Scholar]
- Brahimi‐Horn MC, Chiche J, Pouysségur J (2007) Hypoxia and cancer. J Mol Med (Berl) 85: 1301–1307 [DOI] [PubMed] [Google Scholar]
- Chaika NV, Gebregiworgis T, Lewallen ME, Purohit V, Radhakrishnan P, Liu X, Zhang B, Mehla K, Brown RB, Caffrey T et al (2012) MUC1 mucin stabilizes and activates hypoxia‐inducible factor 1 alpha to regulate metabolism in pancreatic cancer. Proc Natl Acad Sci USA 109: 13787–13792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M (2014) The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol 15: 536–550 [DOI] [PubMed] [Google Scholar]
- De Robertis M, Massi E, Poeta ML, Carotti S, Morini S, Cecchetelli L, Signori E, Fazio VM (2011) The AOM/DSS murine model for the study of colon carcinogenesis: from pathways to diagnosis and therapy studies. J Carcinog 10: 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng H, Liu Q, Xue C, David LL, Beer TM, Thomas GV, Dai MS, Qian DZ (2012) HIF1α protein stability is increased by acetylation at lysine 709. J Biol Chem 287: 35496–35505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646–674 [DOI] [PubMed] [Google Scholar]
- Jin L, Li D, Alesi GN, Fan J, Kang HB, Lu Z, Boggon TJ, Jin P, Yi H, Wright ER et al (2015) Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer Cell 27: 257–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG, Ratcliffe PJ (2008) Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 30: 393–402 [DOI] [PubMed] [Google Scholar]
- Ke Q, Costa M (2006) Hypoxia‐inducible factor‐1 (HIF‐1). Mol Pharmacol 70: 1469–1480 [DOI] [PubMed] [Google Scholar]
- Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S et al (2012) Transformation by the (R)‐enantiomer of 2‐hydroxyglutarate linked to EGLN activation. Nature 483: 484–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo FR, Huff C, Myllymäki M, Olenchock B, Swierczek S, Tashi T, Gordeuk V, Wuren T, Ri‐Li G, McClain DA et al (2014) A genetic mechanism for Tibetan high‐altitude adaptation. Nat Genet 46: 951–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo F, Liu X, Yan N, Li S, Cao G, Cheng Q, Xia Q, Wang H (2006) Hypoxia‐inducible transcription factor‐1alpha promotes hypoxia‐induced A549 apoptosis via a mechanism that involves the glycolysis pathway. BMC Cancer 6: 26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H, Peak‐Chew SY, Chin JW (2008) Genetically encoding N(epsilon)‐acetyllysine in recombinant proteins. Nat Chem Biol 4: 232–234 [DOI] [PubMed] [Google Scholar]
- Raffel S, Falcone M, Kneisel N, Hansson J, Wang W, Lutz C, Bullinger L, Poschet G, Nonnenmacher Y, Barnert A et al (2017) BCAT1 restricts αKG levels in AML stem cells leading to IDHmut‐like DNA hypermethylation. Nature 551: 384–388 [DOI] [PubMed] [Google Scholar]
- Semenza GL (2003) Targeting HIF‐1 for cancer therapy. Nat Rev Cancer 3: 721–732 [DOI] [PubMed] [Google Scholar]
- Shao J, Lu J, Zhu W, Yu H, Jing X, Wang YL, Wang X, Wang XJ (2019) Derepression of LOXL4 inhibits liver cancer growth by reactivating compromised p53. Cell Death Differ 26: 2237–2252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stillman TJ, Baker PJ, Britton KL, Rice DW (1993) Conformational flexibility in glutamate dehydrogenase. Role of water in substrate recognition and catalysis. J Mol Biol 234: 1131–1139 [DOI] [PubMed] [Google Scholar]
- Sullivan LB, Gui DY, Vander Heiden MG (2016) Altered metabolite levels in cancer: implications for tumour biology and cancer therapy. Nat Rev Cancer 16: 680–693 [DOI] [PubMed] [Google Scholar]
- Tarhonskaya H, Rydzik AM, Leung IK, Loik ND, Chan MC, Kawamura A, McCullagh JS, Claridge TD, Flashman E, Schofield CJ (2014) Non‐enzymatic chemistry enables 2‐hydroxyglutarate‐mediated activation of 2‐oxoglutarate oxygenases. Nat Commun 5: 3423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaker AI, Shaker A, Rao MS, Ciorba MA (2012) Modeling colitis‐associated cancer with azoxymethane (AOM) and dextran sulfate sodium (DSS). J Vis Exp 10.3791/4100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Qiao Y, Xiao M, Wang L, Chen J, Lv W, Xu L, Li Y, Wang Y, Tan MD et al (2017) Opposing roles of acetylation and phosphorylation in LIFR‐dependent self‐renewal growth signaling in mouse embryonic stem cells. Cell Rep 18: 933–946 [DOI] [PubMed] [Google Scholar]
- Wang X, Liu R, Qu X, Yu H, Chu H, Zhang Y, Zhu W, Wu X, Gao H, Tao B et al (2019a) α‐ketoglutarate‐activated NF‐κB signaling promotes compensatory glucose uptake and brain tumor development. Mol Cell 76: 148–162 [DOI] [PubMed] [Google Scholar]
- Wang Y, Bai C, Ruan Y, Liu M, Chu Q, Qiu L, Yang C, Li B (2019b) Coordinative metabolism of glutamine carbon and nitrogen in proliferating cancer cells under hypoxia. Nat Commun 10: 201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapenaar H, Dekker FJ (2016) Histone acetyltransferases: challenges in targeting bi‐substrate enzymes. Clin Epigenetics 8: 59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Sudderth J, Dang T, Bachoo RM, McDonald JG, DeBerardinis RJ (2009) Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res 69: 7986–7993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yu G, Chu H, Wang X, Xiong L, Cai G, Liu R, Gao H, Tao B, Li W et al (2018) Macrophage‐associated PGK1 phosphorylation promotes aerobic glycolysis and tumorigenesis. Mol Cell 71: 201–215 [DOI] [PubMed] [Google Scholar]
- Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, Yu W, Li Z, Gong L, Peng Y et al (2009) Glioma‐derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF‐1alpha. Science 324: 261–265 [DOI] [PMC free article] [PubMed] [Google Scholar]