


Abstract
Cancer cells differ from normal cells in many characteristics including loss of differentiation and uninhibited cell proliferation. Recent studies have focused on the identification of factors contributing to cell growth and differentiation. Gut-enriched Krüppel-like factor (GKLF or KLF4) is a newly identified eukaryotic transcription factor and has been shown to play a role in regulating growth arrest. We have previously shown that GKLF mRNA levels were significantly decreased in colon cancer tissues, and that overexpression of GKLF in colonic adenocarcinoma cells (HT-29) resulted in reduction of cyclin D1 (CD1) mRNA and protein levels. The current study was undertaken to determine the mechanisms by which GKLF inhibited CD1 expression. In a transient transfection system, GKLF suppressed CD1 promoter activity by 55%. Sequential deletion and site-directed mutation analysis of the CD1 promoter have identified the sequence between –141 and –66, a region containing an Sp1 response element, to be essential for GKLF function. By electrophoretic mobility gel shift assay, recombinant GKLF and nuclear extracts from HT-29 cells were found to bind to the Sp1 motif on the CD1 promoter. The inhibitory effect of GKLF on the CD1 promoter activity was completely abolished by excessive amount of Sp1 DNA and GKLF significantly reduced the stimulatory function of Sp1 suggesting that GKLF and Sp1 may compete for the same binding site on the CD1 promoter. These results indicate that GKLF is a transcriptional repressor of the CD1 gene and that the inhibitory effect of GKLF is, in part, mediated by interaction with the Sp1 binding domain on its promoter.
INTRODUCTION
Gut-enriched Krüppel-like factor (GKLF; also named Krüppel-like factor 4, KLF4) is a newly identified eukaryotic transcription factor (1) that has three C-terminal zinc fingers with a high degree of sequence homology to lung Krüppel-like factor (LKLF) and erythroid Krüppel-like factor (EKLF) (2,3). This class of protein was named for its homology to the Drosophilia Krüppel protein (1). Using northern blot analysis and in situ hybridization, GKLF was found to express extensively in the gastrointestinal tract (4,5). Although early studies have suggested a role of GKLF in mediating growth arrest (4,5), the physiological property of GKLF in the gastrointestinal system is unknown. Recent studies from this laboratory have shown that GKLF mRNA levels in the colonic polyps and cancer were significantly decreased, suggesting that down-regulation of GKLF expression in the colon might contribute to cellular hyperproliferation and malignant transformation (6). In a human colonic adenocarcinoma cell line (HT-29), overexpression of GKLF significantly inhibited cyclin D1 (CD1) mRNA level as well as CD1 promoter activity. These data suggest that GKLF may function as a transcriptional repressor of the cyclin D1 gene to control cell growth in the colon.
While previous studies have shown that EKLF and LKLF transactivate the β-globulin promoter through interaction with CACCC or CACCC-like motifs, the target gene(s) for GKLF is/are largely unknown. Jenkins et al. have recently demonstrated that GKLF specifically bound the CACCC-like motif in both the human keratin 4 and Epstein–Barr virus ED-L2 promoters and suggested a role of GKLF in promoting differentiation of the squamous epithelium in the esophagus (7). Using target detection assay, Shields and Yang had proposed a consensus binding sequence of 5′-(G/A)(G/A)GG(C/T)G(C/T)-3′ for GKLF (8). This sequence is similar but not identical to the CACCC element described above. The current study was undertaken to determine the mechanism(s) by which GKLF inhibited CD1 gene expression and to identify the regulatory element on the CD1 promoter with which GKLF interacted.
MATERIALS AND METHODS
Cell culture and transfection
The human embryonic kidney (L293) cells and human colonic adenocarcinoma (HT-29) cells (obtained from ATCC, Rockville, MD) were cultured in minimal essential and F-12 media (Sigma, St Louis, MO) at 37°C in a 95% air + 5% CO2 atmosphere. Media were supplemented with 10% fetal bovine serum, 100 µg/ml streptomycin and 100 U/ml penicillin. Cells were transfected with DNA (1 µg/105 cells) using the Lipofectamine method according to the manufacturer’s protocol (GIBCO, Gaithersburg, MD) and as described previously (6). After transfection, cells were incubated for an additional 48 h before analysis.
Construction of the epitope-tagged GKLF expression vector and cyclin D1-Luc plasmids
The sense human GKLF (cDNA; nucleotides –292 to +1565, consisting of the full-length coding sequence) and antisense GKLF (cDNA; nucleotides +1565 to –292) cDNAs were subcloned into a pcDNA3.1/His expression vector (Invitrogen, San Diego, CA) in frame and 3′ to the His6 and anti-Xpress epitope tag coding sequence (9).
The full-length human CD1 promoter reporter construct (pCD1-1745 Luc, consisting of 1745 bp upstream of the transcription initiation site) and various truncated constructs have been described previously (10,11). The CD1 promoters were ligated to a pA3-Luc plasmid containing firefly luciferase reporter gene (10). In the context of the –163 bp fragment of the CD1 promoter, an Sp1 site was mutated by PCR-directed mutagenesis (GGGCGGG to GtaCGGG) to create –163Sp1mutLuc, and an E2F site was mutated from TTTGGCGCC to TTTcttGaC to generate –163E2FmutLuc. In addition, the CD1 E2F sequences from –163 to –133 and the Sp1-like sequences from –130 to –99 of the CD1 promoter were synthesized as complementary strands and cloned into a TK81pA3Luc plasmid to create heterologous reporter plasmids CD1(E2F)Luc and CD1(Sp1)Luc respectively (11). The control CMVLuc, SV40Luc and TKLuc plasmids were purchased from Promega (Promega Life Science Inc., Madison, WI).
Luciferase and β-galactosidase measurements
To examine transcriptional regulation of the CD1 promoters by GKLF, L293 or HT-29 cells were transiently transfected with pCMV gal, CD1Luc DNAs in the presence of GKLF or control plasmid (pCDNA-3). For the luciferase assay, transfected cells were washed twice with phosphate buffered saline (PBS, pH 7.4) and then lysed in 500 µl of lysis buffer following the manufacturer’s instructions (Analytical Luminescence, San Diego, CA). β-Galactosidase activity in 40 µl of the cell lysate was determined after a 5–30 min incubation at 37°C with 2 mM chlorophenol red β-galactopyranoside (Boehringer Mannheim, Indianapolis, IN) in 2 nM MgCl2, 0.1 mM MnCl2, 45 mM 2-mercaptoethanol, and 100 mM NaHPO4, pH 8.0. The reactions were stopped by adding 500 µl of 0.5 M EDTA, pH 8.0, and the absorbency at 570 nm was measured using a spectrophotometer. With each experiment, luciferase activity was determined in duplicate and normalized to β-galactosidase activity for each dish.
Purification of recombinant GKLF fusion protein and nuclear extracts
GKLF fusion protein was prepared by transfecting non-GKLF expressing L293 cells with sense GKLF or pCDNA3.1/His (control) cDNAs using the Lipofectamine method as described above. Lysates from transfected 293 cells were prepared in a RIPA lysis buffer supplemented with protease inhibitors (10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.2 mM phenylmethanesulfonyl fluoride, 0.5 mM DTT). Cells were lysed with two cycles of freezing–thawing, followed by passage through an 18-gauge needle.
Purification of the histidine-tagged protein was performed using B-PERTM 6×His Fusion Protein Purification Kit according to the manufacturer’s protocol (Pierce, Rockford, IL). The eluted fusion protein was examined for the presence of GKLF by western blot analysis and the fraction(s) containing the highest concentration of GKLF was used for electrophoretic mobility shift assays (EMSAs). In addition, nuclear extracts were prepared from GKLH-expressing HT-29 cells according to previously described modifications (12) of a standard protocol (13).
For western blot analysis, the protein samples were separated on a 10% SDS–polyacrylamide gel and transferred to a nitrocellulose membrane (Hybond ECL, Amersham Life Science, Arlington Heights, IL). The membrane was incubated overnight in the blocking buffer (10 mM Tris, pH 7.5, 100 mM NaCl, 0.1% Tween-20) containing 5% non-fat powdered milk. The membrane was immunoblotted with anti-GKLF antiserum (6). Following incubation with the secondary antibody, the membranes were visualized with enhanced chemiluminescence (Amersham).
Electrophoretic mobility shift assays (EMSAs)
EMSAs were performed to identify the protein binding to the regulatory elements on the CD1 promoter. Purified recombinant GKLF protein and nuclear extracts were prepared as described above. Double-stranded oligonucleotides, corresponding to the sequence of regulatory elements, were synthesized by the GIBCO (BRL Lifetech Co., Gaithersburg, MD) and purified by gel electrophoresis. Double-stranded oligonucleotide (5 pmol) was radiolabeled by the Klenow fill-in reaction in a buffer consisting of 10 mM Tris–HCl, pH 7.5, 5 mM MgCl2, 7.5 mM dithiothreitol, 33 µM dATP, 33 µM dGTP, 33 µM dTTP, 33 µM [α-32P]dCTP (NEN Life Science Products, Boston, MA), and 1 U DNA polymerase I Klenow fragment (Amersham Pharmacia Biotech, Piscataway, NJ). EMSA was carried out by incubating 10 µg of recombinant GKLF fusion protein or nuclear extracts with 5 fmol of the α-32P-labeled oligonucleotide DNA probe in a 20 µl binding reaction containing 10 mM Tris–HCl, 50 mM NaCl, 1 mM DTT, 1 mM EDTA, 10% glycerol, and 1.0 µg of poly(dA-dT) (Amersham Pharmacia Biotech). After incubation at room temperature for 15 min, the samples were loaded onto a 4% polyacrylamide, 0.25× Tris borate gel and electrophoresed at 10 V/cm for 2 h. The gel was dried and exposed to X-ray film (Kodak X-AR) at –70°C for 12 h. For competition experiments, the recombinant protein or nuclear extract was preincubated with excess unlabeled wild-type or mutated double-stranded oligonucleotides before the addition of the α-32P-labeled oligonucleotide DNA probe. Furthermore, supershift assay was performed by the incubation of GKLF protein/nuclear extracts and oligonucleotide mixture with GKLF antiserum at room temperature for 30 min before electrophoresis.
Competition of GKLF and Sp1 binding on the CD1 promoter
To further examine a potential interaction between GKLF and Sp1 on the CD1 promoter, L293 cells were either transfected with pCD1-963 Luc, GKLF DNA and increasing amounts of Sp1 DNA, or with increasing amounts of GKLF in the presence of 1 µg of pCMV-Sp1 cDNA (Sp1 cDNA was kindly provided by Dr Grace Gill, University of California at Berkeley). The relative luciferase activity was analyzed 48 h after transfection.
Identification of DNA-binding site on GKLF
Most of the DNA-binding domains in zinc finger proteins are located within their zinc finger regions. To identify the DNA-binding region in GKLF, three truncated GKLF constructs were generated by PCR using specific primers to delete consecutive zinc finger regions. The GKLF-1780 mutant was created by deleting the third zinc finger and the GKLF-1680 mutant consisted of GKLF cDNA from which the second and third zinc fingers regions were deleted. The GKLF-1580 mutant contained GKLF cDNA without three zinc finger sequences. These constructs were individually cotransfected with pCD1-963 and the luciferase activity was measured.
Northern blot hybridization analysis
Total RNA from HT-29 cells was extracted using the acid/phenol method of Chomczynski and Sacchi (14), and was electrophoresed on a 1.5% agarose/6% formaldehyde gel. Hybridization was analyzed under stringent conditions with human GKLF cDNAs radiolabelled with [32P]dCTP, using the Klenow fragment of DNA polymerase I and random oligonucleotides as primers (Promega). The blots were washed and autoradiograms were developed after exposure to X-ray film at –70°C, using a Cronex intensifying screen (DuPont).
Statistics
Results were expressed as mean ± S.E.. Statistical analysis was performed using ANOVA and Student’s t-test. A P-value of <0.05 was considered to be statistically significant.
RESULTS
Repression of CD1 promoter activity by GKLF
To determine the mechanisms by which GKLF inhibited CD1 mRNA levels, the effect of GKLF on CD1 promoter activity was examined. Cotransfection of sense GKLF in L293 cells inhibited pCD1-1745 Luc reporter activity in a dose-dependent manner and antisense GKLF transfection did not induce any significant change in CD1 promoter activity (Fig. 1A). In addition, GKLF had no effect on the basal transcriptional activity of pA3Luc (data not shown) or three other luciferase reporter plasmids: CMVLuc, SV40Luc and TKLuc (Fig. 1B).
Figure 1.

Effect of GKLF on CD1 promoter or CMV-Luc, SV40-Luc and TK-Luc reporters activity. (A) The sense (black bar), antisense (gray bar) GKLF or pCDNA-3 (open bar) expression vector was transfected with pCD1-1745 Luc reporter plasmid into L293 cells. The ratio of expression vector to reporter plasmid is shown on the abscissa. (B) The sense GKLF (black bar) or pCDNA-3 (open bar) DNA was transfected with CMV-Luc, SV40-Luc, or TK-Luc reporter plasmids (ratio of 1:100) into L293 cells. Luciferase activity was determined 48 h later. pCMV-βgal (0.05 µg) was also cotransfected with each construct to correct for differences in transfection efficiency. Data are expressed as means ± S.E. of three separate experiments. *P < 0.05, compared to pCDNA-3-transfected cells.
The region of the CD1 promoter required for repression of GKLF
The region of the CD1 promoter required for regulation by GKLF was determined in L293 cells by using a series of CD1 promoter constructs (Fig. 2A). Overexpression of GKLF resulted in a 55% inhibition of the pCD1-1745 Luc promoter activity (Fig. 2B). Deletion of the CD1 promoter sequence from –1745 to –141 did not affect the repressive effect of GKLF. In contrast, deletion of the region between –141 and –66, which comprises a consensus Sp1 site, abolished the GKLF-mediated effect (Fig. 2B). When the Sp1 sequence was mutated within the context of the –163 bp fragment (–163Sp1mutLuc), GKLF-mediated repression was abolished (Fig. 3). Cotransfection of GKLF with the heterologous reporter plasmid encoding the CD1 Sp1 sequence [CD1(Sp1)Luc] resulted in a 60% decrease of its transcriptional activity (Fig. 3). These results suggest that the Sp1 binding domain of the CD1 promoter is required for the repressive effect of GKLF.
Figure 2.

Repression of CD1 promoter activity by GKLF. (A) Schematic representation of the CD1 promoter showing the location of the DNA sequences resembling Sp1 and E2F binding sites as well as multiple CACCC motifs. (B) Sense GKLF (black bar) or pCDNA-3 (open bar) expression vector was cotransfected with the pCD1-1745 Luc reporter or an equal amount of each of the other 5′ promoter constructs into L293 cells. pCMV-βgal (0.05 µg) was also cotransfected with each construct to correct for differences in transfection efficiency. Data are expressed as means ± S.E. of four separate experiments. *P < 0.05, compared to pCDNA-3-transfected cells in each individual construct.
Figure 3.

Effect of GKLF on the transcriptional activity of mutated CD1 and heterologous reporters plasmids. –163Sp1mutLuc, mutation of the Sp1 sequence within the context of the –163 bp fragment of the CD1 promoter; –163E2FmutLuc, mutation of the E2F sequence in the context of the –163 bp of the CD1 promoter; CD1(Sp1)Luc, heterologous reporter construct consisting of the CD1 Sp1 sequence; and CD1(E2F)Luc, heterologous reporter construct consisting of the CD1 E2F. Plasmids were cotransfected with sense GKLF (black bar) or pCDNA-3 (open bar) into L293 cells and luciferase activity was determined 48 h later. Data represent the mean ± S.E. of four separate experiments. *P < 0.05, compared to pCDNA-3-transfected cells in each individual construct.
In the original description of the human CD1 promoter, a sequence homologous to an E2F binding site had been identified at –142 (10) which was subsequently shown to bind pRB/E2F-1 (11). Recently, the family of E2F transcription factors has been shown to play an important role in cell cycle progression (15). To determine whether interaction between E2F-like sequence and GKLF occurred on the CD1 promoter, point mutation of the E2F sequence was performed in the context of the –163 bp fragment (–163E2FmutLuc). Mutation of the E2F-binding site did not affect the repression of the CD1 promoter activity by GKLF, and GKLF exhibited no inhibitory effect on the heterologous E2F reporter plasmid [CD1(E2F)Luc] (Fig. 3), indicating that the E2F binding domain on the CD1 promoter is not required for GKLF function.
The effect of GKLF expression on CD1 promoter activity was further examined under physiological conditions. As reported previously (5), cellular GKLF mRNA levels were significantly induced during growth arrest by serum deprivation. In these studies, GKLF-expressing HT-29 cells were transfected with pCD1-1745 Luc construct and then cultured in the serum-free medium for 48 h to induce GKLF expression. As shown in Figure 4, in the absence of serum, GKLF mRNA levels were significantly increased (lower panel). Overexpression of GKLF in the fasting cells significantly decreased CD1 promoter activity (Fig. 4, upper panel), consistent with the repressive effect of GKLF showing in the above transfection studies.
Figure 4.
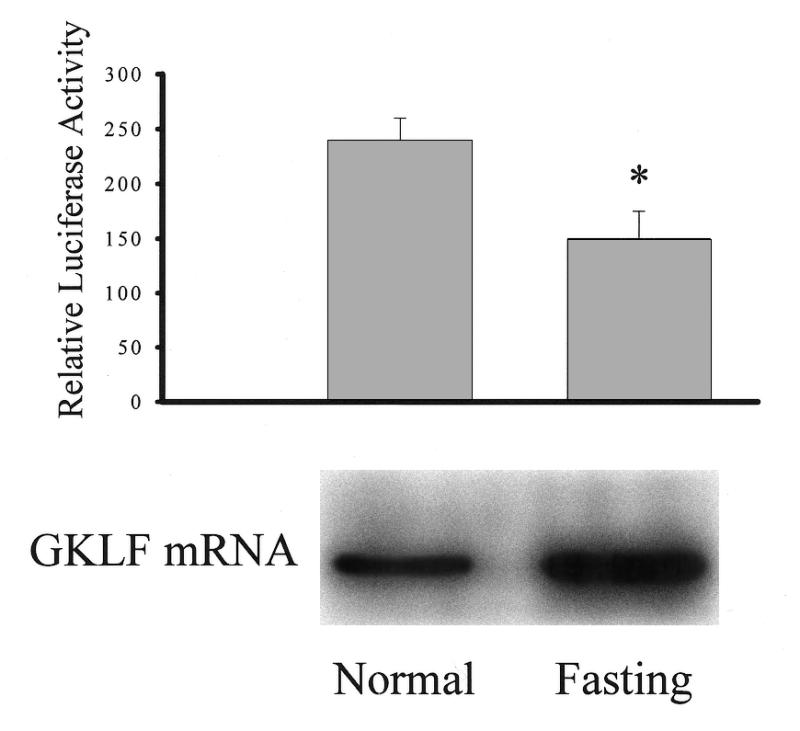
Effect of fasting on CD1 promoter activity and GKLF mRNA levels. HT-29 cells were transfected with pCD1-1745 Luc and pCMV-βgal DNAs. Cells were then cultured in medium without serum for 48 h and luciferase activity (upper panel) and GKLF mRNA (lower panel) were examined. Luciferase data represent the mean ± S.E. of four separate experiments. *P < 0.05 compared to normal. Total RNA (20 µg) was extracted from HT-29 cells and analyzed by formaldehyde gel electrophoresis. RNA blots were hybridized with 32P-labeled GKLF probe and a representative blot is shown on the lower panel.
Sp1 motif on the cyclin D1 promoter is specific for GKLF binding
Fusion protein lysates were harvested following transfection with GKLF or pCDNA3.1/His plasmids. As shown in Figure 5, cells transfected with GKLF cDNA expressed a prominent 66 kDa protein and a minor 34 kDa protein, corresponding to the expected molecular mass of GKLF (5,9). In contrast, no GKLF protein was identified in pCDNA3.1/His transfected cells. These findings are consistent with the results from Jenkins et al. (7). Moreover, as demonstrated by Jenkins et al. (7), the 34 kDa protein did not appear to play any significant role in GKLF-mediated function, no attempt was made to separate these two proteins for the following study.
Figure 5.
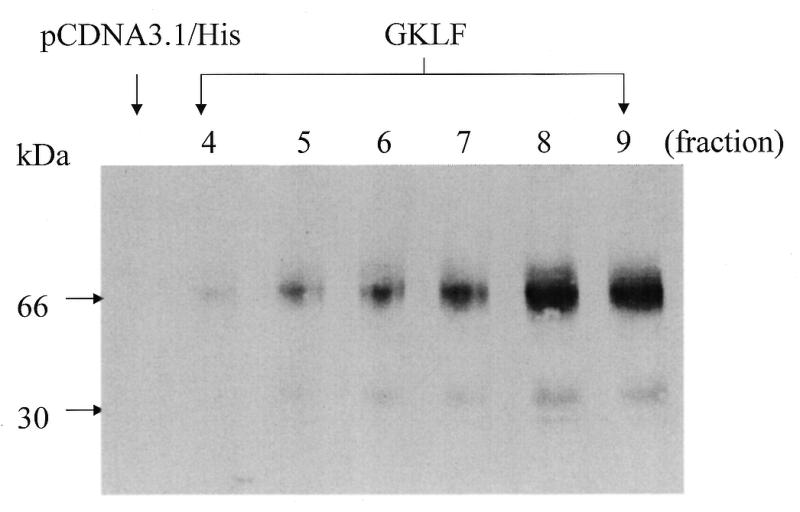
Histidine-tagged GKLF cDNA expressed in L293 cells yields a major 66-kDa and a minor 30-kDa fusion protein. L293 cells were transiently transfected with the pCDNA3.1/His or GKLF cDNAs. Fusion protein from L293 cells was harvested 48 h after transfection and was eluted through Ni-Chelated® columns. Protein (10 µg) from different eluted fractions was used for western blot analysis using GKLF-specific antibody. No GKLF immunoreactive protein was found in the pCDNA3.1/His-transfected cells.
To identify the regulatory element on the CD1 promoter, EMSA was performed using purified recombinant GKLF protein from fractions 8 and 9 (Fig. 5). The wild-type CD1 Sp1 oligonucleotide, containing the GGGGCGGGG motif, was used to generate radiolabeled probe. In addition, mutated double-stranded oligonucleotide, in which GG was substituted with tt, was used to determine the specificity of GKLF-Sp1 binding.
As shown below, GKLF was found to bind to the Sp1 motif on the CD1 promoter by EMSA (Fig. 6). Incubation of cell extracts with the wild-type Sp1 probe resulted in a DNA–protein complex (C). This complex was specific because it was repressed by excessive unlabeled wild-type Sp1 oligonucleotide (Fig. 6). Moreover, the DNA–protein complex was not repressed by mutant Sp1 oligonucleotide and no DNA–protein complex was identified when mutated Sp1 oligonucleotide was used as a probe (data not shown). Supershifts were conducted with antiserum to GKLF. The addition of the GKLF-specific antiserum supershifted the complex binding to the Sp1 oligonucleotide, indicating that the DNA–protein complex (band C) contained GKLF protein (Fig. 6).
Figure 6.
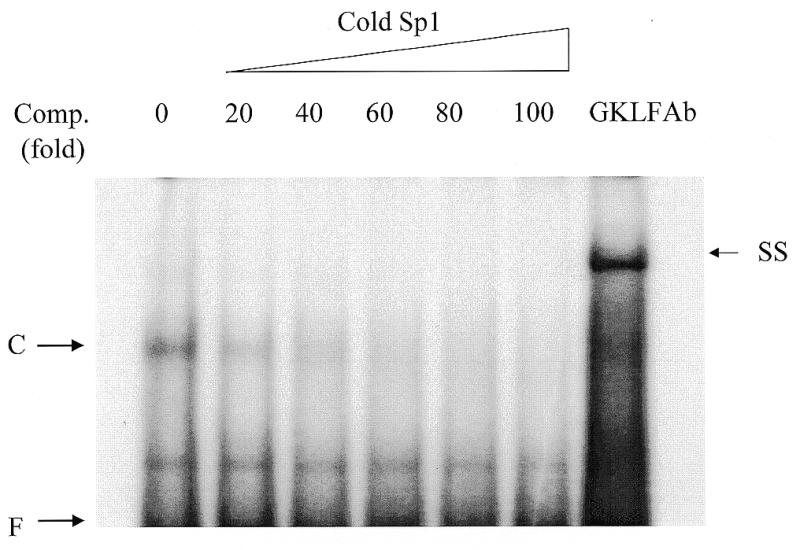
Affinity-purified GKLF protein binds specifically to the Sp1 motif on the CD1 promoter. Gel mobility shift assays were performed with purified GKLF protein (fractions 8 and 9) and a 25-bp doubled-stranded Sp1 oligonucleotide probe. The DNA–protein complex is indicated by C. The binding reactions were performed in the presence of excessive amount of unlabeled Sp1 oligonucleotide (comp. fold indicated fold increases in the amount of cold Sp1). The complex C was supershifted by the addition of GKLF antiserum in the reaction (SS). F, free probe.
Similar results were observed with nuclear extracts from HT-29 cells. As shown in Figure 7, incubation of cell extracts with wild-type Sp1 probe resulted in a DNA–protein complex (C, lane 1). This complex was specific because it was repressed by excess unlabeled wild-type (lane 2) but not by mutant Sp1 (lane 3) oligonucleotides. The addition of the GKLF-specific antiserum supershifted the complex binding to the Sp1 oligonucleotide (lane 4), indicating that complex C contained GKLF protein.
Figure 7.
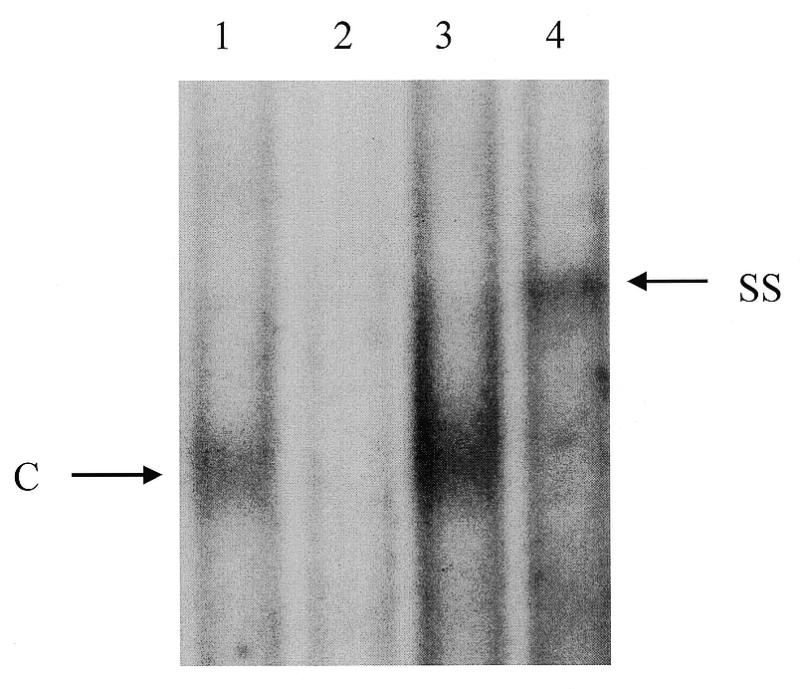
Gel mobility shift assays were performed with nuclear extracts from HT-29 cells and a 25-bp doubled-stranded Sp1 oligonucleotide probe in the absence (lane 1), or the presence of unlabeled wild-type (lane 2), or mutated Sp1 oligonucleotide (lane 3). The DNA–protein complex (C) was supershifted by the addition of GKLF antiserum in the reaction (SS, lane 4). Unlabeled competitors were added at 100-fold molar excess.
DNA-binding domain on GKLF
To identify the DNA-binding domain in GKLF, CD1 promoter plasmid was cotransfected with truncated GKLF DNA. As shown in Figure 8, cotransfection with wild-type GKLF resulted in a 45% repression of the CD1 promoter activity and this inhibitory effect was preserved when wild-type GKLF was replaced by either GKLF-1780 or GKLF-1680 mutant. However, deletion of all three zinc finger regions (GKLF-1580) completely abolished GKLF-repressed CD1 activity, indicating that the first zinc finger domain of the GKLF is essential for its function.
Figure 8.

Effect of wild-type GKLF and truncated GKLF-1580, -1680 and -1780 on pCD1-963 Luc promoter activity. L293 cells were transfected with 1 µg/well of pCD1-963 Luc, 0.05 µg/well pCMV β-Gal and 0.01 µg/well wild-type or truncated GKLF DNA. Data are expressed as % of control (cells transfected with pCD1-963 Luc only) and presented as means ± S.E. of five separate experiments.
Interaction between GKLF and Sp1 proteins
Results from the above studies suggest that GKLF exhibits its function by binding to the Sp1 consensus sequence on the CD1 promoter. The following studies were performed to determine if GKLF and Sp1 compete for the same binding site. In L293 cells, GKLF transfection induced a significant decrease in the CD1 promoter activity and this effect was attenuated by cotransfection with increasing amounts of Sp1 DNA (Fig. 9). Furthermore, Sp1 induced a dose-dependent increase on the CD1 promoter activity (data not shown), and this effect was abolished in the presence of increasing amount of wild-type GKLF but not truncated GKLF-1580 (Fig. 10). These data suggest that GKLF and Sp1 may compete for the same binding domain on the CD1 promoter.
Figure 9.

Effect of GKLF and Sp1 cotransfection on the CD1 promoter activity. L293 cells were transfected with pCD1-963 Luc construct and expression plasmids containing GKLF and Sp1 as indicated. Luciferase and β-galactosidase activities were determined 48 h later. Data were expressed as % of control (cells transfected with pCD1-963 Luc only) and presented as means ± S.E. of three separate experiments.
Figure 10.

Induction of CD1 promoter activity by Sp1 was abolished by GKLF but not by truncated GKLF-1580. L293 cells were transfected with pCD1-963 Luc construct and expression plasmids containing GKLF and Sp1 as indicated. Luciferase and β-galactosidase activities were determined 48 h later. Data were expressed as fold increases over control (cells transfected with pCD1-963 Luc only) and presented as means ± S.E. of three separate experiments.
DISCUSSION
Colorectal malignancy is one of the most common cancers encountered in the United States. Carcinogenesis of the colon is believed to begin with damage to normal cellular genes, the activation of oncogenes or the loss of tumor suppressor genes (16). A series of genetic events has been identified in the progression from normal to hyperproliferative epithelium, adenoma, carcinoma and invasive carcinoma of the colon, but the molecular events that regulate cellular hyperproliferation have not been fully examined. Recently, c-MYC has been found to be a downstream target of the adenomatous polyposis coli tumor suppressor gene (APC) (17). Mutations of APC cause loss of proliferate control of the colonic epithelium and result in cancer formation. Although APC mutations were identified in the majority of non-hereditary sporadic colon carcinoma, approximately one-third of colon cancer patients did not possess these mutations suggesting that other factors may be involved in the induction of hyperproliferation of colonic mucosa (18). Our laboratory has recently examined the expression of GKLF in the human colon, and demonstrated that GKLF mRNA levels were significantly decreased in the dysplastic epithelium, including adenomatous polyps and carcinoma (6). In the transfected HT-29 cells, overexpression of GKLF resulted in decreased DNA synthesis (6). These data suggest that GKLF may play an important role in governing cell growth, and that down-regulation of GKLF may cause colonic epithelium to become hyperproliferative. In our other report, we have shown that overexpression of the GKLF in the HT-29 cells resulted in a decrease in CD1 mRNA levels. In the present study, we report that the decrease in CD1 mRNA level is, in part, generated through the suppression of the CD1 promoter activity by GKLF. These observations were unlikely to be due to an experimental artifact of transfection, as we have demonstrated a similar inhibitory effect on CD1 promoter activity through physiological stimulation of GKLF expression (to fasting) in HT-29 cells. Furthermore, although our results suggest that GKLF may function as a transcriptional repressor of the CD1 gene to control cell growth in the colon, these studies do not exclude the possibility that other transcription factors, such as Sp3 or LKLF, may operative independently or synergistically with GKLF to control CD1 gene expression. Likewise, in addition to the GKLF–CD1 interaction, other mechanisms, such as the APC-β-catenin pathway, may also play significant roles in controlling cell growth in the gastrointestinal tract.
Although the mechanisms responsible for the inhibitory function of GKLF on CD1 promoter are unknown, our data suggests that this suppression is based on a competitive interaction between GKLF and Sp1. This is supported by our data showing that the effect of GKLF on the CD1 promoter is abolished by Sp1 cotransfection and that the stimulatory effect of Sp1 on the CD1 promoter activity is attenuated by GKLF transfection. Furthermore, cotransfection with the GKLF-1580 mutant, in which the DNA-binding domain of GKLF was deleted, failed to suppress the stimulatory effect of Sp1 on the CD1 promoter. These results are consistent with findings from Zhang et al. who demonstrated a similar interaction between GKLF and Sp1 on the CYPA1 promoter (19).
The eukaryotic cell cycle is a carefully regulated series of events. Progression of the cell cycle requires the involvement of a cyclin-dependent kinase complex. The interactions among cyclins and kinases are believed to drive the cell from one stage to another. Recent studies in cancer biology have described two checkpoints, one at the G1/S transition and the other at the G2/M transition, which control and ensure the order of events in the cell cycle and integrate DNA repair with cell cycle progression. When injury occurs, cells have the capacity to arrest cell cycle progression and to prevent accumulation of the damaged DNA (20,21). Failure to arrest cells would result in the release of cells with unstable genomes, which eventually evolve into malignant cells. In a concurrent report, we have shown that overexpression of GKLF induced cell arrest at the G1 phase indicating that GKLF may function as a G1/S checkpoint regulator to control normal cell proliferation. Recently, the family of Sp1 transcription factors has been shown to become active during the late G1 phase of the cell cycle (22). These data are consistent with findings from the current report and suggest a potential role of GKLF in controlling cell cycle progression through the interaction with Sp1 on the CD1 promoter. Although not examined in this study, abnormal expression of cyclins D, E and A have been reported in association with human cancers including pancreas, liver, esophagus and breast; and the cyclin D1 gene was recently shown to be activated by β-catenin in many colon cancer cell lines (21). Whether GKLF plays a role in the APC/β-catenin pathway warrants further investigation.
Members of the Krüppel-like transcription factors including EKLF and LKLF have been shown to transactivate reporter plasmids via the CACCC motif or CACCC-like variants such as CACACCC or CCACACCCT (3,23). GKLF was recently found to bind the CACCC-like motif on the human keratin 4 and Epstein–Barr virus ED-L2 promoters (7). As shown in Figure 2, there are at least three CACCC motifs on the cyclin D1 promoter. Deletion of each individual CACCC motif on the CD1 promoter has no effect on the inhibitory effect of GKLF indicating that CACCC is not the main GKLF binding domain in this promoter. However, the effect of deletion or mutation of multiple CACCC motifs on the CD1 promoter has not been examined in the current study. Whether such modifications may alter GKLF function requires further investigation. In our study, mutation of the Sp1 site on the proximal portion of the CD1 promoter completely abolished GKLF function and the demonstration of interaction between GKLF and Sp1 by the gel-shift assay indicates that the Sp1 motif represents a GKLF binding domain. In addition, similar results obtained using the nuclear extracts from GKLF-expressing HT-29 cells further support the conclusion that this interaction occurs in vivo. Although differing from the CACCC motif, the Sp1 sequence 5′-GGGGCGG-3′ on the CD1 promoter displays a high degree of sequence homology to the consensus binding domain of GKLF, 5′-(G/A)(G/A)GG(C/T)G(C/T)-3′, proposed by Shields and Yang (8).
In summary, the results of this study demonstrate that GKLF is a negative regulator of the CD1 promoter and that its effect is mediated through competition with Sp1 on the promoter.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported in part by United States Public Health Services grants DK-52186 (to C.-C.T.) and R29CA70897 and RO1CA75503 (to R.G.P.). R.G.P. is a recipient of the Irma T. Hirschi award and an award from the Susan G. Komen Breast Cancer Foundation. Work conducted at the Albert Einstein College of Medicine was supported by Cancer Center Core National Institutes of Health grant 5-p30-CA13330-26.
REFERENCES
- 1.Anderson K.P., Kern,C.B., Crable,S.C. and Lingrel,J.B. (1995) Mol. Cell. Biol., 15, 5957–5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Black A.R., Jensen,D., Lin,S.Y. and Azizkhan,J.C. (1999) J. Biol. Chem., 274, 1207–1215. [DOI] [PubMed] [Google Scholar]
- 3.Feng W.C., Southwood,C.M. and Bieker,J.J. (1994) J. Biol. Chem., 269, 1493–1500. [PubMed] [Google Scholar]
- 4.Garrett-Sinha L.A., Eberspaecher,H., Seldin,M.F. and Crombrugghe,B.D. (1996) J. Biol. Chem., 271, 31384–31390. [DOI] [PubMed] [Google Scholar]
- 5.Shields J.M., Christy,R.J. and Yang,V.W. (1996) J. Biol. Chem., 71, 20009–20017. [Google Scholar]
- 6.Shie J.L., Chen,Z.Y., O’Brien,M.J., Pestell,R.G., Lee,M.E. and Tseng,C.-C. (2000) Am. J. Physiol., in press. [Google Scholar]
- 7.Jenkins T.D., Optiz,O.G., Okano,J. and Rustgi,A.K. (1998) J. Biol. Chem., 273, 10747–10754. [DOI] [PubMed] [Google Scholar]
- 8.Shields J.M. and Yang,V.W. (1998) Nucleic Acids Res., 26, 796–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yet S.F., McA’Nulty,M.M., Folta,S.C., Yen,H.W., Yoshizumi,M., Hsieh,C.M., Layne,M.D., Chin,M.T., Wang,H., Perrella,M.A., Jain,M.K. and Lee,M.E. (1998) J. Biol. Chem., 273, 1026–1031. [DOI] [PubMed] [Google Scholar]
- 10.Motokura T. and Arnold,A. (1993) Gene Chromosomes Cancer, 7, 89–95. [DOI] [PubMed] [Google Scholar]
- 11.Watanabe G., Albanese,C., Lee,R.J., Reutens,A., Vairo,G., Henglein,B. and Pestell,R.G. (1998) Mol. Cell. Biol., 18, 3212–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jackson S.P. and Tjian,R. (1989) Proc. Natl Acad. Sci. USA, 86, 1781–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dignam J.D., Lebovitz,R.M. and Roeder,R.G. (1983) Nucleic Acids Res., 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chomczynski P. and Sacchi,N. (1987) Anal. Biochem., 162, 156–159. [DOI] [PubMed] [Google Scholar]
- 15.Hirofumi H., Iavarone,A. and Reeves,S.A. (1998) Oncogene, 16, 1513–1523. [DOI] [PubMed] [Google Scholar]
- 16.Vogelstein B. and Kinzler,K.W. (1993) Trends Genet., 93, 138–141. [DOI] [PubMed] [Google Scholar]
- 17.He T.C., Sparks,A.B., Rago,C., Hermeking,H., Zawel,L., da Costa,L.T., Morin,P.J., Volgelstein,B. and Kinzler,K.W. (1998) Science, 281, 1509–1512. [DOI] [PubMed] [Google Scholar]
- 18.Vogelstein B., Fearon,E.R., Hamilton,S.R., Kern,S.E., Preisinger,A.C., Leppert,M., Nakamura,Y., White,R., Smits,A.M. and Bos,J.L. (1988) N. Engl. J. Med., 319, 525–532. [DOI] [PubMed] [Google Scholar]
- 19.Zhang W., Shield,J.M., Sogawa,K., Fujii-Kuriyama,Y. and Yang,V.M. (1998) J. Biol. Chem., 273, 17917–17925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hall M. and Peters,G. (1996) Adv. Cancer Res., 68, 67–108. [DOI] [PubMed] [Google Scholar]
- 21.Hartwell L.H. and Kastan,M.B. (1994) Science, 266, 1821–1828. [DOI] [PubMed] [Google Scholar]
- 22.Nasmyth K. (1993) Curr. Opin. Cell Biol., 5, 166–179. [DOI] [PubMed] [Google Scholar]
- 23.Miller L.J. and Bieker,J.J. (1993) Mol. Cell. Biol., 13, 2776–2786. [DOI] [PMC free article] [PubMed] [Google Scholar]