


Abstract
To investigate molecular controls of cardiomyocyte proliferation, we utilized cardiomyocytes induced to proliferate indefinitely by SV40 large T antigen (T-ag). In the T-ag-immortalized AT-1, AT-2 and HL-1 cardiomyocytes, normal cellular proteins associating with T-ag and p53 were identified, isolated and microsequenced. Peptide sequencing revealed that proteins of 90, 100 and 160 kDa were homologs of MRE11, NBS1 and RAD50, respectively. These three proteins play critical roles in the detection and repair of DNA double-strand breaks, activation of cell cycle checkpoints and telomere maintenance. In this report, we describe the cDNA cloning and double-strand sequencing of the rat homologs of MRE11, NBS1 and RAD50. We also determined the mRNA and protein levels of MRE11, NBS1 and RAD50 at different stages of heart development and in different tissues. MRE11 mRNA was only detected in the immortalized cardiomyocytes and in the testes. Although the 90 kDa MRE11 protein was seen in most samples examined, it was only detected at extremely low levels in proliferating cardiomyocytes (normal and immortalized). The 6.0 kb MRE11-related mRNA transcript (MRT) was seen in all samples examined. Levels of both NBS1 and RAD50 mRNA transcripts peaked in the heart at postnatal day 10. NBS1 mRNA levels were at very low levels in the T-ag-immortalized AT-1, AT-2 and HL-1 cells but NBS1 protein was observed at extremely high levels. We propose that SV40 large T antigen’s interaction with the MRE11-NBS1-RAD50 pathway and with p53 ablates critical cell cycle checkpoints and that this is one of the major factors involved in the ability of this oncoprotein to immortalize cardiomyocytes.
INTRODUCTION
The heart is the first organ to form during embryogenesis. During fetal development, growth of the heart results from proliferation of differentiating cardiomyocytes. In mammals, cardiomyocyte proliferation occurs during fetal and early postnatal development, ceasing irreversibly in mice and rats at approximately postnatal day three (1–3) and possibly by the third week in humans (4). After this period, the increasing functional demand placed on the heart by the growing organism is met by cardiac muscle cell enlargement (hypertrophy) rather than proliferation (3). Because adult mammalian cardiomyocytes cannot divide (2,3,5,6), cardiac injury results in loss of functional muscle tissue with attempted compensation occurring by hypertrophy of the remaining myocytes and proliferation of non-myocytes (6). However, mammalian cardiomyocyte proliferation has been induced by the expression of the SV40 large T antigen (T-ag) oncoprotein, as shown in transgenic mice (7–9), cultured neonatal rat cardiomyocytes (10,11) and human fetal cardiomyocytes (12). In transgenic mice, cardiomyocyte proliferation was also induced by the expression of c-MYC (13), calmodulin (14) and insulin-like growth factor-1 (15). These examples of induced cardiomyocyte proliferation support the possibility of stimulating cardiac myocyte proliferation in vivo subsequent to cardiac trauma as a means of replacing lost functional cardiac tissue. Thus, an understanding of the molecular mechanism(s) by which T-ag, or any factor, induces cardiomyocyte proliferation could be of great potential therapeutic value.
Extensive research indicates some of the transforming activity of T-ag resides in its ability to physically complex with and repress the activity of pRb and p53, two tumor suppressor proteins involved in the control of the cell cycle (16,17). In a similar manner, both the adenovirus E1a and E1b proteins and the oncogenic human papilloma virus E7 and E6 proteins induce cell proliferation by complexing with pRb and p53, respectively (18). By binding pRb, T-ag induces the release of the E2F transcription factor, which activates promoters of genes required for S phase transition (19). However, induced proliferation of terminally differentiated cells forces inappropriate DNA synthesis, resulting in apoptosis (20). By complexing with p53, T-ag and E1b block the apoptotic function of p53 and allow proliferation (18). T-ag-induced proliferation is also related to its ability to associate with p300 and CBP (21). Thus, T-ag can induce proliferation, in many cases, by physically associating with endogenous cellular proteins involved in cell cycle control and apoptosis. To further investigate possible molecular controls of proliferation of cardiomyocytes, we identified other endogenous cellular proteins associating with p53 and T-ag in cardiomyocytes.
For this study T-ag-immortalized AT-1 (22,23), AT-2 (24,25) and HL-1 (26) cardiomyocyte cell lines were used. These were derived from proliferating atrial cardiomyocytes of transgenic mice expressing T-ag in the atrium of the heart (8,9). We immunoprecipitated proteins from AT-1 and AT-2 cardiomyocytes using anti-T-ag and anti-p53 antibodies. We identified, isolated and microsequenced proteins of 90, 100 and 160 kDa. Partial peptide sequences revealed these proteins were the rat homologs of MRE11, NBS1 and RAD50, respectively. In this report, we describe the cloning, sequencing and developmental expression of these genes in the heart and discuss their possible involvement in the immortalization of cardiomyocytes by T-ag.
MATERIALS AND METHODS
Nucleotide sequences of the rat MRE11, NBS1 and RAD50 homologs were deposited in GenBank™/EBI Data Bank with accession nos AF218574, AF218575 and AF218576, respectively.
Reagents
Antibodies PAb 101 and PAb 419 were from American Type Culture Collection (Rockville, MD) (27,28) and PAb 421 was from Oncogene Research Products (Cambridge, MA) and was also generously provided by Dr Ed Harlow (Massachusetts General Hospital Cancer Center, Charlestown, MA) (28). Antibody UCHL-1 was from Boehringer-Mannheim (Indianapolis, IN).
Cell culture
AT-1 cells are derived from right atrial tumors of transgenic mice in which T-ag expression was targeted to atrial myocytes by the atrial natriuretic factor promoter (22,23). They exhibit myocyte-specific genetic and ultrastructural characteristics, proliferate in culture and form a synchronously beating monolayer (22,23,29). AT-1 cells were isolated by an overnight tryptic digestion and cultured in gelatin-coated flasks at a density of 4 × 105 cells/ml PC1 medium (Ventrex, Portland, ME) containing 10% fetal bovine serum (FBS) (ICN Biomedicals, Costa Mesa, CA) (23,29).
AT-2 cells are derived from atrial tumors arising in transgenic mice in which the expression of T-ag was targeted to the heart by the α-myosin heavy chain promoter (9). AT-2 cells lost most of their myocyte-specific markers after multiple passages but are easily grown in culture (9). AT-2 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM)/F12 medium containing 10% FBS, 10 µg/ml insulin and 0.4 µg/ml glutamine.
The HL-1 cardiomyocyte cell line, derived from AT-1 cells, can be serially passaged in culture and stored cryogenically (26). They contract spontaneously and retain genetic, morphological, biochemical and electrophysiological properties characteristic of differentiated cardiac myocytes (26). HL-1 cells were grown in Ex-Cell 320 medium (JRH Biosciences, Lenexa, KS) containing 10% FBS, 10 µg/ml insulin, 50 µg/ml endothelial cell growth supplement (Upstate Biotechnology, Lake Placid, NY), 1 µM all trans retinoic acid, 10 µM norepinephrine (Sigma) and an additional 1× non-essential amino acids (all Life Technologies, Rockville, MD).
Metabolic labeling and immunoprecipitation
AT-1 cells were cultured in methionine-free DMEM containing 2% dialyzed FBS, 4 mM glutamine, 5 µg/ml insulin and 500 µCi/ml Tran35S-label (>1000 Ci/mmol, ICN) and incubated for 4 h. Cell monolayers were washed twice with ice-cold phosphate-buffered saline. Lysis buffer [50 mM HEPES pH 7.3, 250 mM NaCl, 0.1% Nonidet P-40, 10 mM NaF, 300 µM Na3VO4, 5 µg/ml phenylmethylsulfonyl fluoride, 5 µg/ml aprotinin and 5 µg/ml leupeptin (all from Sigma); 1.2 ml/T25 flask] was added for 30 min (30). Lysate was cleared by centrifugation at 12 000 g at 4°C for 15 min. Each milliliter of lysate was additionally cleared by adding 30 µl of a 1:1 slurry of Protein A–Sepharose beads (Pharmacia, Piscataway, NJ): lysis buffer, mixing on a rocking platform at 4°C for 30 min and centrifuging for 2 min at 4°C at 2000 g. Incubation of 200 µl of cleared lysate, 500 µl of lysis buffer and 2 µg of antibody was done for 4–16 h at 4°C on a rocking platform. Twenty microliters of a 1:1 slurry of Protein A–Sepharose beads: lysis buffer was added and the incubation continued for 30–60 min. Immune complexes bound to Protein A–Sepharose beads were pelleted by centrifugation at 4°C for 2 min at 2000 g and washed three times with 1 ml of lysis buffer at 4°C. A control immunoprecipitation was done in an identical manner with antibody UCHL-1, which is of the same subclass as PAb 421.
SDS–PAGE and autoradiography
35S-radiolabeled proteins of the immune complexes were separated by SDS–PAGE and visualized by autoradiography using standard protocols (31). Labeled proteins were visualized by autoradiography with Kodak Biomax MR film or a PhosphorImager (Molecular Dynamics, Sunnyvale, CA).
Preparative protein isolation and peptide microsequencing
Sufficient quantities of proteins complexing with T-ag and p53 in cardiomyocytes were isolated for microsequencing by preparative immunoprecipitations from AT-2 cells using anti-p53 antibody PAb 421 (30). In preliminary assays, in addition to T-ag and p53, proteins of 90, 100 and 160 kDa were consistently seen in AT-1 and AT-2 cells. AT-2 cells were used because they are much easier to grow in culture.
AT-2 cells were grown in 100 T150 flasks in 30 ml of DMEM/F12 medium containing 10% FBS and 10 µg/ml insulin. Lysate was prepared from ~80% confluent cultures by rinsing the cells twice with ice-cold PBS and incubating in 2.4 ml lysis buffer for 30 min on ice. Lysate was pre-cleared as above. Immunoprecipitations were done with 80 µl of PAb 421–agarose beads per 15 ml of lysate (7 µg antibody per µl of beads). Immune complexes were washed three times with ice-cold lysis buffer and the proteins separated by 7% SDS–PAGE (31).
Proteins were electrotransferred onto Immobilon-Psq membranes. The membrane was stained 1 min in 0.1% Amido Black 10B (ICN) in 1% acetic acid/40% methanol and destained in H2O. Sections of membrane binding proteins of interest were excised and sent to John Leszyk at the Core Laboratory for Protein Sequencing and Mass Spectrometry at the University of Massachusetts Medical Center, Shrewsbury, MA for protein microsequencing.
Preparation of anti-p90, anti-p100 and anti-p160 polyclonal antibodies
The most antigenic regions of the p90, p100 and p160 peptides were determined using the PROTEAN software program (DNASTAR, Madison, WI). Antisera against p90 were produced by the Core Facility of the Biotechnology Unit at LSU Health Sciences Center based on the p90 sequence ILLGGDLFHENKPSR. This peptide, coupled to keyhole limpet hemocyanin, was used to prepare antisera in two New Zealand White rabbits by a program of multiple intradermal injections. Polyclonal anti-p90 antibodies were purified by Protein A chromatography.
Antisera against the p100 peptide KQPPDIESFYPPIDE and p160 peptide NFHELVKERQEREAK were produced by Genosys Biotechnologies (The Woodlands, TX) and purified in the same manner as described above. In addition, these antibodies were affinity-purified using either a p100- or a p160-immunoaffinity column, respectively (30).
Western blot analysis
Protein lysates were prepared from AT-1, AT-2 and HL-1 cells in an identical manner as described for immunoprecipitations. Tissue lysates were prepared using 10 volumes of lysis buffer per gram tissue. Proteins separated by SDS–PAGE were transferred to Hybond-C nitrocellulose membranes (Amersham Life Sciences, Arlington Heights, IL). Membranes were blocked overnight at 4°C in 5% non-fat dried milk in Tris-buffered saline (20 mM Tris pH 7.6, 137 mM NaCl) containing 0.02% Tween 20 (TBS-T), washed with TBS-T and incubated for 1 h at room temperature in a 1:1500 dilution of anti-MRE11, anti-NBS1 or anti-RAD50 polyclonal antibody in TBS-T with 5% dried milk. Blots were washed in TBS-T, incubated at room temperature for 1 h with a 1:1500 dilution of horseradish peroxidase-labeled anti-rabbit IgG (Amersham Life Sciences) in TBS-T with 5% dried milk and washed in TBS-T. Antibody-bound proteins were visualized with the ECL western blotting detection system (Amersham Life Sciences).
cDNA cloning and sequencing of the rat homologs of MRE11, NBS1 and RAD50
Database homology searches using the National Center for Biotechnology Information (NCBI) BLAST Network Service revealed all four p90 peptide sequences were 100% identical to murine MRE11 and 89% identical to human MRE11 (see Results) (32,33).
To clone the rat homolog of MRE11, two primers (5′-ACTATAAAAATGAGCCCCACAG-3′ and 5′-ACATTATCTTCGGTTTCTTCTTG-3′) spanning the coding region of murine MRE11 were used in the PCR. The DNA template was single-stranded cDNA reverse-transcribed from total RNA of fetal-day 17 Sprague–Dawley rat heart ventricles. Total RNA was isolated using TRIzol reagent (Life Technologies).
The PCR program consisted of 38 cycles of 94°C for 25 s, 47°C for 1 min and 72°C for 3 min in a GeneAmp PCR System 9600 machine (Perkin-Elmer, Norwalk, CT). The 2.1 kb PCR product was cloned into the pGEM-T vector (Promega, Madison, WI). Three independent clones were double-strand sequenced. The amino acid sequence of the rat MRE11 homolog consensus was compared to other MRE11 homologs with the CLUSTALV program using a PAM 250 scoring matrix (34).
Database homology searches revealed the p100 peptides were 85% identical to human NBS1 (see Results) (35–37). In addition, one p100 peptide was 100% identical to a protein coded for by a murine expressed sequence tag (EST) (accession no. AA239763). Because this murine EST overlapped the 5′ untranslated region and start of coding of NBS1, it could be used to design a PCR primer to clone full-length NBS1. Fortuitously, a rat EST (accession no. H33522) was 84% identical to the 5′ portion of this murine EST (but did not overlap the coding region of NBS1). Based on this EST the primer 5′-CGTCGCATGTCGCAGTG-3′ was used with the Adapter Primer 1 provided in the rat heart Marathon-Ready cDNA kit (Clontech, Palo Alto, CA). PCR amplifications were done using the Advantage cDNA Kit (Clontech). The PCR program consisted of 1 cycle at 94°C for 15 s, 5 cycles of 94°C for 15 s and 70°C for 5 min, 28 cycles of 94°C for 15 s and 68°C for 5 min and 1 cycle of 72°C for 10 min. The 2.6 kb product was cloned into the pGEM-T Easy vector. Three independent clones were double-strand sequenced. The amino acid sequences of the rat and human NBS1 homologs were compared as described for MRE11.
Database homology searches with the five p160 peptide sequences revealed all were 100% identical to murine RAD50 and 80% identical to human RAD50 (38,39). PCR amplifications were performed using primers based on the murine RAD50 sequence to obtain five overlapping fragments of RAD50. These five fragments were then used as templates in a PCR reaction using the upper primer of the most 5′ fragment and the lower primer of the most 3′ fragment to obtain the complete 4.4 kb rat homolog of RAD50.
To obtain the most 5′ fragment of 286 bp, the primers 5′-GCGGGGCCGGAAGTGCTCTC-3′ and 5′-GTCGTCTTCCCCGCCCCATTG-3′ were used in the PCR program of 1 cycle of 94°C for 1 min, 40 cycles of 94°C for 20 s, 67°C for 30 s and 72°C for 2 min and 1 cycle of 72°C for 10 min. For the next overlapping fragment of 1583 bp the PCR primers 5′-TCTGGGCGTGCGAAGTTT-3′ and 5′-GTGTGCGGGTTGTTGTATGAT-3′ were used with a program of 94°C for 1 min, 20 cycles of 94°C for 20 s, 58°C for 30 s and 72°C for 3 min and 1 cycle of 72°C for 10 min. The template for these two PCR amplifications was the adult rat heart Marathon-Ready cDNA (Clontech). The middle fragment of 878 bp was obtained using the primer pair 5′-CATTTCATCACGGCGCCGCTC-3′ and 5′-AGCGGACTGGGGAGGACGATTGA-3′ in a program of 94°C for 1 min, 38 cycles of 94°C for 20 s, 64°C for 30 s and 72°C for 3 min, and 1 cycle of 72°C for 10 min. The fourth fragment of 1487 bp was obtained using the primers 5′-GAGCGGCGCCGTGATGAAATG-3′ and 5′-GCCAGGGCCAGTCGGATGATG-3′ with a program of 38 cycles of 94°C for 30 s, 65°C for 1 min and 72°C for 2 min and 1 cycle of 72°C for 10 min. The fifth and most 3′ fragment of 823 bp was obtained with the primers 5′-ATCTTTGGCGGAGTACCTATCGTG-3′ and 5′-AGCCCCGTTCAATGACTGTGGTTTC-3′. Its program was 94°C for 1 min, 38 cycles of 94°C for 20 s and 68°C for 4 min and 1 cycle of 68°C for 3 min. The DNA template for these last three amplifications was single-strand cDNA synthesized from total RNA isolated from fetal-day 17 Sprague–Dawley rat heart ventricles.
To obtain full-length RAD50 the five PCR products were used as templates in a single PCR reaction with the primers 5′-GCGGGGCCGGAAGTGCTCTC-3′ and 5′-AGCCCCGTTCAATGACTGTGGTTTC-3′. The amplification program was 1 min at 94°C, 40 cycles of 94°C for 20 s, 64°C for 30 s and 72°C for 4.5 min and 1 cycle of 72°C for 10 min. The 4.4 kb product was cloned into the pGEM-T Easy vector and three independent clones were double-strand sequenced. The amino acid sequence of rat RAD50 was compared to RAD50 of human, Saccharomyces cerevisiae and Arabidopsis thaliana in the same manner as described for MRE11.
Northern blot analysis
Total RNA samples (12 µg) containing 1 µg ethidium bromide were fractionated on 1.2% agarose, 2.2 M formaldehyde gels and transferred to Hybond-N nylon membranes (Amersham Life Sciences). Template MRE11, NBS1 and RAD50 DNAs for random-prime labeling were the full-length PCR products described. DNA was 32P-labeled using the Prime-a-Gene random-prime labeling system (Promega). Hybridizations were overnight at 42°C in 50% formamide, 7% SDS, 1 mM EDTA, 100 mM Na2HPO4, pH 7.2, 250 mM NaCl and 0.1 mg/ml denatured herring testes DNA. Washes were at 62°C in 1% SDS, 1 mM EDTA, 40 mM NaH2PO4 pH 7.2. Autoradiography and analysis were done using a PhosphorImager and ImageQuant software. To normalize the 32P signals, RNA on the membranes was stained in 0.5 µg/ml ethidium bromide in water for 25 min with agitation, de-stained in four changes of water for 10 min, scanned on a Bio-Rad Gel Doc 1000 and quantified using ImageQuant software. The values obtained were used to normalize the 32P signals to account for differences in RNA loading. This was done for two northern blots, and the values were averaged and graphed.
RESULTS
As one means to gain an understanding of the mechanism by which T-ag immortalizes cardiac myocytes, proteins were identified in complexes with T-ag and p53 in AT-1 and AT-2 cells (Fig. 1). We report here on the 90, 100 and 160 kDa proteins whose cDNAs were isolated and sequenced, and whose patterns of RNA and protein expression were examined.
Figure 1.
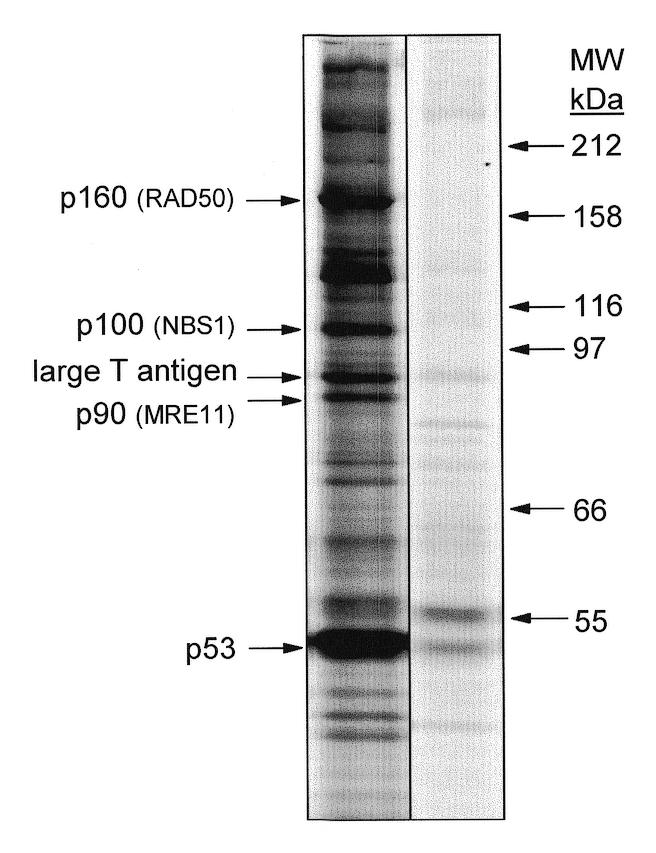
SV40 large T antigen- and p53-associated proteins in cultured SV40 large T antigen-transformed AT-1 murine cardiomyocytes. Protein complexes in NP-40 lysates from [35S]methionine metabolically-labeled cultured AT-1 cardiomyocytes were immunoprecipitated with anti-p53 monoclonal antibody PAb 421. Proteins separated on denaturing SDS–polyacrylamide gels were visualized by autoradiography. In addition to SV40 large T antigen and p53, proteins of 90, 100 and 160 kDa were consistently identified. Positions of molecular weight standards are shown in kDa on the right. The right panel is a control immunoprecipitation in which an irrelevant antibody of the same subclass as PAb 421 (IgG2a) was used in an identical manner as for PAb 421.
Identification of the 90, 100 and 160 kDa proteins as the rat homologs of MRE11, NBS1 and RAD50, respectively
From p90 four peptide sequences were obtained: LALENEVDFILLGGDLEFHENKPSR, IGPIKNEQQLFYVSQPGSSV, GKTGEEINFGMLITKPASEG and GMGEAVQEFVDKEEKDAIEELVK. Evaluations of these sequences using the NCBI BLAST Network Service (40) and FASTA Email Server on GenomeNet (41) revealed all four p90 sequences were 100% identical to murine MRE11 (accession nos U58987 and U60318) and 88% identical to human MRE11 (accession nos U37359, AF073362 and AF022778) (32,33,42).
In the same manner of analysis, the two peptide sequences obtained from p100, KQPPDIESFYPPIDEPAIGS and NHAVLTVNFPVTSLSQTDEI, were found to be 85% identical to human NBS1 (accession nos AF049895, AF058696 and AF069291) (35–37).
The five peptide sequences obtained from p160 were DEIFSATRYIK, NFHELVKERQEREAK, ASQLLSDLTDKEALK, ILELDQELTK and AKEQISPLETALEK. Homology searches revealed all five p160 peptide sequences were 100% identical to murine RAD50 (accession no. U66887) (38) and 80% identical to human RAD50 (accession nos U63139 and NM_005732) (39).
Cloning and amino acid sequence analysis of the rat homolog of MRE11
Having identified p90 as an MRE11 homolog, PCR primers were used to amplify the rat MRE11 homolog. The 2.1 kb product was cloned and three independent clones were double-strand sequenced (accession no. AF218574). Homology searches identified, in addition to two murine and three human MRE11 homologs mentioned above, a putative Xenopus laevis homolog (accession no. AF134569), five homologs from S.cerevisiae (accession nos D11463, P32829, NC_001145, U60829 and Z49939) (43,44) and RAD32, the MRE11 homolog of Schizosaccharomyces pombe (accession nos S58097, X82322 and Z50112) (45) (Fig. 2). Rat MRE11 also has significant similarity to a putative MRE11 homolog from Drosophila melanogaster (accession no. AF132144). A comparison of the amino acid sequence of the rat MRE11 homolog with the human, X.laevis, D.melanogaster, S.pombe and S.cerevisiae MRE11 amino acid sequences revealed the strong conservation of the N-terminal half of MRE11, as has been previously reported (Fig. 2) (32,42). This conservation is revealed in the observation that amino acids 12–251 of human and rat MRE11 are 96.7% identical. In this region are four highly conserved amino acid sequence motifs characteristic of phosphoesterases from a variety of proteins (46).
Figure 2.

Amino acid sequence comparison of MRE11 homologs of R.norvegicus, human, X.laevis, D.melanogaster, S.pombe and S.cerevisiae. The MRE11 amino acid sequences from R.norvegicus (accession no. AF218574), human (accession no. AF073362), X.laevis (accession no. AF134569), D.melanogaster (accession no. AF132144), S.pombe (RAD32, accession no. S58097) and S.cerevisiae (accession no. D11463) were compared using the CLUSTALV program with a PAM 250 scoring matrix. Amino acids at a single position scored as identical by the CLUSTALV program are highlighted in black. If at the same position there were additional amino acids that were similar to those scored as identical, the similar amino acids are highlighted in gray. If at a given position, there were no identical amino acids, but similar amino acids were present, those similar amino acids are also highlighted in gray. The following amino acids were considered similar: D,E,N,Q; F,W,Y; K,R; A,G; I,V; L,M; S,T. Murine MRE11 amino acid sequences (accession nos U58987 and U60318) were not included due to their close identity to the rat sequence (~95%). The epitopes recognized by our anti-MRE11 rabbit polyclonal antibody reside in amino acids 55–69 (ILLGGDLFHENKPSR) of human and rat MRE11.
Northern blot analysis of MRE11
In human tissues, MRE11 mRNA had previously been identified as a 2.5 kb transcript (32,33). In the present study the 2.5 kb MRE11 transcript was only seen in rat testes and in the T-ag-immortalized AT-1, AT-2 and HL-1 cardiomyocytes (Fig. 3A, B, F and G). It was not detected in any other tissue examined or in the rat heart at any age of development (Fig. 3A, B, F and G). The inability to see the 2.5 kb MRE11 transcript may be due to the use of 12 µg of total RNA in the present study in contrast to the 1.5–2.0 µg of poly(A) RNA in previous studies (32,36,42). A 6.0 kb transcript, the same size as a previously reported MRE11-related transcript (MRT), was seen in all samples examined (Fig. 3A, B, D and G) (32,36,42).
Figure 3.
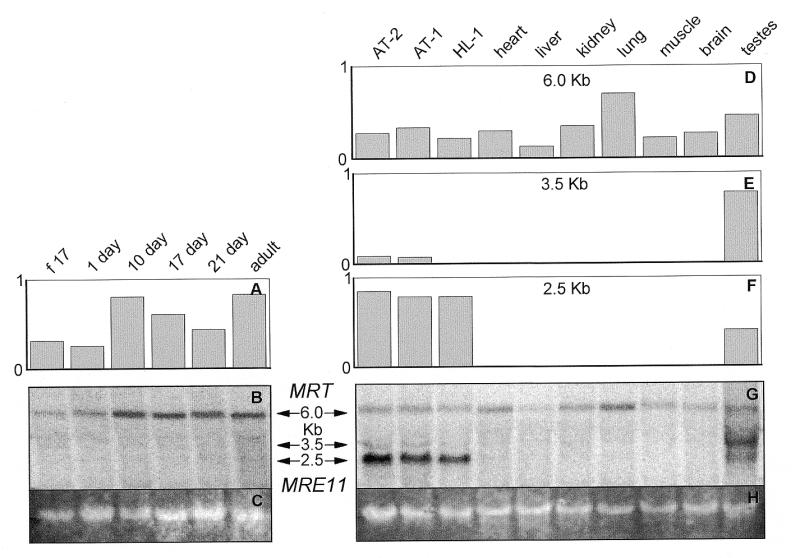
Northern blot analysis of MRE11 in (A and B) ventricles of the rat heart at different ages of development and (D–G) SV40 large T antigen-immortalized AT-2, AT-1 and HL-1 murine cardiomyocytes and adult rat tissues. Twelve micrograms per lane of total RNA from the indicated samples was probed with [32P]dCTP random prime-labeled full-length rat MRE11 cDNA. Hybridizations, washes and exposures were identical for both sets of samples. Signals were visualized on a Molecular Dynamics STORM PhosphorImager and quantified using ImageQuant software. RNA on the membranes was subsequently stained with ethidium bromide and visualized on a Bio-Rad Gel Doc 1000 (C and H) and quantified using the ImageQuant analysis program. The 32P values (B and G) were normalized against the ethidium bromide values (C and H). The graphs (A, D, E and F) are based on the average of two northern blots. The 2.5 kb transcript is MRE11. The 6.0 kb transcript is MRT, the MRE11-related transcript. (f 17: fetal-day 17 rat heart; 1 day, 10 day, 17 day, 21 day: rat heart, number of days postnatal; adult: 60 days postnatal rat heart; AT-2, AT-1, HL-1: SV40 large T antigen-immortalized murine cardiomyocytes.)
Western blot analysis of MRE11
The 90 kDa MRE11 protein was undetectable in fetal-day 17 rat heart but was seen in the heart at postnatal days 10, 17, 21 and 60 (adult) (Fig. 4A). MRE11 protein was also undetectable in T-ag-immortalized AT-1, AT-2 and HL-1 cardiomyocytes and in adult rat brain (Fig. 4B). MRE11 protein was seen at high levels in adult rat heart ventricles, liver, lung and testes (Fig. 4B). In addition, a second protein of ~94 kDa was detected in liver (Fig. 4B).
Figure 4.
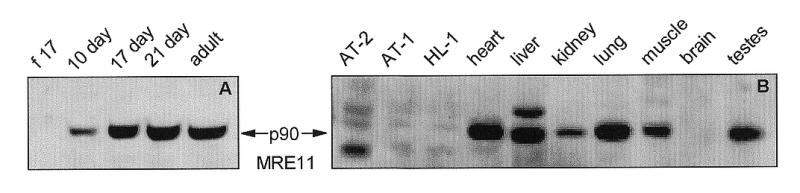
Western blot analysis of MRE11 in (A) ventricles of the rat heart at different ages of development and (B) SV40 large T antigen-immortalized AT-2, AT-1 and HL-1 murine cardiomyocytes and adult rat tissues. Proteins in NP-40 lysates of the indicated samples were separated by denaturing SDS–PAGE and detected by a chemiluminescent technique using our anti-MRE11 rabbit polyclonal antibody and a horseradish peroxidase-linked secondary anti-rabbit IgG antibody. (f 17: fetal-day 17 rat heart; 1 day, 10 day, 17 day, 21 day: rat heart, number of days postnatal; adult: 60 days postnatal rat heart; AT-2, AT-1, HL-1: SV40 large T antigen-immortalized murine cardiomyocytes.)
Cloning and amino acid sequence analysis of the rat homolog of NBS1
Having identified p100 as a homolog of NBS1, PCR primers were used to amplify the rat NBS1 homolog. The 2.6 kb product was cloned and three independent clones were double-strand sequenced (accession no. AF218575). Database homology searches identified, in addition to three human NBS1 homologs mentioned above, three murine NBS1 homologs (accession nos AB016988, AF076687 and AF092840) (47). The amino acid sequence alignment of the human and rat NBS1 proteins revealed regions of strong sequence conservation (Fig. 5).
Figure 5.

Amino acid sequence comparison of NBS1 homologs of R.norvegicus and human. The NBS1 amino acid sequences of rat (accession no. AF218575) and human (accession no. AF049895) were aligned as described for MRE11 in Figure 2. Three murine NBS1 amino acid sequences (accession nos AB016988, AF076687 and AF092840) were not included because of their close identity (86%) to the rat sequence. The epitopes recognized by our anti-NBS1 rabbit polyclonal antibody reside in amino acids 188–202 (KQPPDIESFYPPIDE) of the rat NBS1 sequence amino acid sequence.
Northern blot analysis of NBS1
In the ventricles of the heart, a 2.5 kb NBS1 transcript was present at low levels at fetal-day 17, slightly higher levels at postnatal day 1, highly abundant levels at postnatal day 10 and at decreased levels at day 21 and in the adult rat heart (Fig. 6A and B). The 2.5 kb NBS1 transcript was barely detectable in AT-1, AT-2 and HL-1 cardiomyocytes and in the brain (Fig. 6D and E). High levels were seen in testes and variable levels in the other rat tissues examined. In contrast to the single 2.5 kb mRNA transcript seen in murine and rat tissues, two NBS1 mRNA transcripts of 2.5 and 4.5 kb are observed in human tissues (35–37).
Figure 6.

Northern blot analysis of NBS1 in (A and B) ventricles of the rat heart at different ages of development and (D and E) SV40 large T antigen-immortalized AT-2, AT-1 and HL-1 murine cardiomyocytes and adult rat tissues. Twelve micrograms per lane of total RNA from the indicated samples was probed with [32P]dCTP random prime-labeled full-length rat NBS1 cDNA. Analysis was performed in an identical manner as for MRE11 as detailed in Figure 3. (f 17: fetal-day 17 rat heart; 1 day, 10 day, 17 day, 21 day: rat heart, number of days postnatal; adult: 60 days postnatal rat heart; AT-2, AT-1, HL-1: SV40 large T antigen-immortalized murine cardiomyocytes.)
Western blot analysis of NBS1
NBS1 protein was observed at approximately equal levels in the fetal-day 17 rat heart, at postnatal days 10, 17 and 21 and in the adult heart (Fig. 7A). NBS1 protein was seen at extremely high levels in the T-ag-immortalized AT-1, AT-2 and HL-1 cardiomyocytes (Fig. 7B). No NBS1 protein was detected in kidney, very low levels were observed in liver and skeletal muscle and moderate levels were observed in heart, lung and brain (Fig. 7B).
Figure 7.

Western blot analysis of NBS1 in (A) ventricles of the rat heart at different ages of development and (B) SV40 large T antigen-immortalized AT-2, AT-1 and HL-1 rat cardiomyocytes and adult rat tissues. Proteins in NP-40 lysates of the indicated samples were separated by denaturing SDS–PAGE and detected by a chemiluminescent technique using our anti-NBS1 rabbit polyclonal antibody and a horseradish peroxidase-linked secondary anti-rabbit IgG antibody. (f 17: fetal-day 17 rat heart; 1 day, 10 day, 17 day, 21 day: rat heart, number of days postnatal; adult: 60 days postnatal rat heart; AT-2, AT-1, HL-1: SV40 large T antigen-immortalized murine cardiomyocytes.)
Cloning and amino acid sequence analysis of RAD50
Having identified p160 as a RAD50 homolog, PCR primers were used to amplify the rat homolog. The 4.4 kb PCR product was cloned and three independent clones were double-strand sequenced (accession no. AF218576). Homology searches identified, in addition to two human and a single murine RAD50 homolog mentioned above, three additional human RAD50 homologs (accession nos AF057299, AF057300 and AC004041) (39,48), an additional murine homolog (accession no. AC005742) (38) and three RAD50 homologs from S.cerevisiae (accession nos X14814, Z71526, Y13139 and X96722) (49,50). Potential RAD50 homologs were seen in Caenorhabditis elegans (accession nos Z75312 and Z78200) (51) and A.thaliana (accession no. AC006223) (52).
The RAD50 amino acid sequences of Rattus norvegicus, human, S.cerevisiae and A.thaliana were aligned (Fig. 8). As previously observed, the RAD50 N- and C-termini are highly conserved (Fig. 8) (38,39,48). It was unexpected that a potential RAD50 homolog from A.thaliana would seemingly be more closely related to human RAD50 at the protein level than RAD50 from S.cerevisiae.
Figure 8.

Amino acid sequence comparison of RAD50 homologs of R.norvegicus, human, S.cerevisiae and A.thaliana. The RAD50 amino acid sequences of R.norvegicus (accession no. AF218576), human (accession no. U63139), S.cerevisiae (accession no. X14814) and A.thaliana (accession no. AC006223) were aligned as described for MRE11 in Figure 2. Two murine RAD50 amino acid sequences (accession nos U66887 and AC005742) were not included as they were nearly identical (~96%) to the rat sequence. The epitopes recognized by our anti-RAD50 rabbit polyclonal antibody reside in amino acids 395–409 (NFHELVKERQEREAK) of rat RAD50.
Even though the MRE11 amino acid sequences of rat and mouse are ~95% identical and the RAD50 amino acid sequences of rat and mouse are ~95% identical, the NBS1 amino acid sequences of rat and mouse are only 86% identical.
Northern blot analysis of RAD50
The levels of the 5.5 kb mRNA transcript of RAD50 in the fetal-day 17 rat heart, postnatal days 10, 17 and 21 and in the adult heart (Fig. 9A and B) were very similar to that seen for NBS1 (Fig. 6A and B). RAD50 was seen at low levels in AT-1, AT-2 and HL-1 cardiomyocytes and in all other tissues examined, with its greatest abundance in the testes (Fig. 9D and F). An additional 4.0 kb transcript was seen in the testes (Fig. 9E and F).
Figure 9.
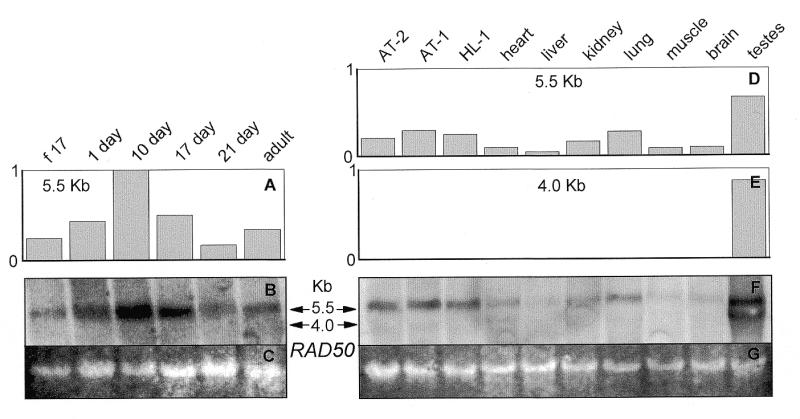
Northern blot analysis of RAD50 in (A and B) ventricles of the rat heart at different ages of development and (D–F) SV40 large T antigen-immortalized AT-2, AT-1 and HL-1 murine cardiomyocytes and adult rat tissues. Twelve micrograms per lane of total RNA from the indicated samples was probed with [32P]dCTP random prime-labeled full-length rat RAD50 cDNA. Analysis was performed in an identical manner as for MRE11 as detailed in Figure 3. (f 17: fetal-day 17 rat heart; 1 day, 10 day, 17 day, 21 day: rat heart, number of days postnatal; adult: 60 days postnatal rat heart; AT-2, AT-1, HL-1: SV40 large T antigen-immortalized murine cardiomyocytes.)
Western blot analysis of RAD50
The 160 kDa RAD50 protein was present at low levels in the heart at fetal-day 17, at relatively constant levels at postnatal days 10, 17 and 21 and at slightly lower levels in the adult rat heart (Fig. 10A and B). RAD50 was observed in the T-ag-immortalized AT-1, AT-2 and HL-1 cells and in liver, kidney and lung (Fig. 10B). RAD50 protein was barely detectable in skeletal muscle with slightly higher levels observed in brain and the ventricles of the heart (Fig. 10B).
Figure 10.

Western blot analysis of RAD50 in (A) ventricles of the rat heart at different ages of development and (B) SV40 large T antigen-immortalized AT-2, AT-1 and HL-1 murine cardiomyocytes and adult rat tissues. Proteins in NP-40 lysates of the indicated samples were separated by denaturing SDS–PAGE and detected by a chemiluminescent technique using our anti-RAD50 rabbit polyclonal antibody and a horseradish peroxidase-linked secondary anti-rabbit IgG antibody. (f 17: fetal-day 17 rat heart; 1 day, 10 day, 17 day, 21 day: rat heart, number of days postnatal; adult: 60 days postnatal rat heart; AT-2, AT-1, HL-1: SV40 large T antigen-immortalized murine cardiomyocytes.)
Thus, at the different ages of heart tissue examined, NBS1 and RAD50 mRNA levels were very similar, as were their protein levels. NBS1 and RAD50 mRNA levels peaked at postnatal day 10 (Figs 6B and 9B) but their protein levels were relatively constant (Figs 7A and 10A).
DISCUSSION
To examine possible molecular controls of cardiac myocyte proliferation we utilized the T-ag-immortalized AT-1, AT-2 and HL-I cardiomyocytes (26,29). By immunoprecipitations of protein complexes from AT-2 cells with the anti-p53 antibody PAb 421, we identified, besides p53 and T-ag, additional proteins of 90, 100 and 160 kDa (Fig. 1). Partial peptide sequencing revealed they were homologs of MRE11, NBS1 and RAD50, respectively.
These three proteins, acting in a complex, play critical roles in: (i) detection and repair of DNA double-strand breaks (DSBs) (53,54); (ii) activation of cell cycle checkpoints in response to DSBs (53,55,56); (iii) initiation of meiotic recombination (43,53,57); and (iv) maintenance of telomere length (53,58–61). In response to DNA damage, mammalian cells withdraw from the cell cycle to allow for DNA repair. Within 30 min of DNA DSBs, nuclear foci appear which contain BRCA1, NBS1, MRE11 and RAD50 proteins and the foci remain until the bulk of the DSBs are repaired (53,62–64). Mutations in NBS1 or MRE11 block the formation of foci (36,65). To examine their potential role in regulating cardiomyocyte proliferation, we cloned and characterized the rat homologs of MRE11, NBS1 and RAD50.
Because MRE11 protein was immunoprecipitated from AT-1 and AT-2 cells by antibody PAb 421, it was assumed that western blot analysis would detect MRE11 protein in AT-1 and AT-2 cells (Fig. 1). However, MRE11 protein was undetectable in AT-1 or AT-2 cells (Fig. 4B). This indicates MRE11 protein was present at very low levels in AT-1 and AT-2 cells. It also suggests that most of the MRE11 protein was in complexes that could be immunoprecipitated by antibody PAb 421. The very low levels of MRE11 protein in the AT-1 and AT-2 cells was also surprising as these cells contained the highest levels of MRE11 mRNA observed in any tissue examined (Fig. 3). The presence of at least some MRE11 protein would seem necessary, as the loss of MRE11 from embryonic stem cells is lethal (62,66). In the samples examined in the present study, in addition to the very low levels of MRE11 in the AT-1, AT-2 and HL-1 cells, MRE11 protein was either not present or present at undetectable levels in proliferating cardiomyocytes from fetal-day 17 heart tissue and the adult rat brain (Fig. 4). Thus in both examples of proliferating cardiomyocytes examined, i.e. fetal cardiac tissue and T-ag-immortalized cardiomyocytes, MRE11 protein was present at very low levels, if at all. However, MRE11 is compatible with proliferation because in embryonic stem cells, primary human fibroblasts and immortalized cells, MRE11 protein is readily observed by both immunoprecipitation and in situ immunocytochemistry (36,54,63,64,66–68). However, the very low level of MRE11 protein seen in the present study in both examples of proliferating cardiomyocytes suggests low levels of MRE11 may be necessary to allow proliferation of cardiomyocytes. A role for MRE11 in controlling proliferation is supported by the recent observation that in cells subjected to ionizing radiation, MRE11 is necessary for the suppression of DNA synthesis (67). This is also supported by the recent finding that ATM cannot suppress DNA synthesis without MRE11 (65) and NBS1 (69,70).
NBS1 also seems to be involved in the control of the cell cycle because cultured cells from Nijmegen Breakage Syndrome (NBS) patients, who have a mutation in NBS1, have a perturbed G1/S cell cycle checkpoint, and NBS cells fail to halt DNA synthesis after ionizing radiation-induced DSBs (71,72). In addition, the normal up-regulation of p53 protein levels observed in response to ionizing radiation is reduced in NBS cells (36,72). The involvement of NBS1 protein with cell cycle regulation is also supported by the presence of a forkhead-associated domain and a breast cancer C-terminal domain, both of which are commonly found in cell cycle regulatory and DNA repair proteins (35). Because the MRE11 and NBS1 proteins function together in DSB repair, it was expected that NBS1 protein would also be present at low levels in the T-ag-immortalized cardiomyocytes, similar to the low levels of MRE11 observed. However, NBS1 protein was extremely abundant (Fig. 7B). The increased levels of NBS1 protein may result from the stabilization of NBS1 by T-ag in a manner similar to the stabilization and increased levels of p53 commonly seen in other T-ag-immortalized cells (73). NBS1 protein stabilization is also suggested by the observation that although AT-1, AT-2 and HL-1 cells had the highest levels of NBS1 protein, they had the lowest levels of NBS1 mRNA (Fig. 6A and B). The data presented here suggest that although NBS1 protein was observed at very high levels in the AT-1, AT-2 and HL-1 cells, the usual function of NBS1 in halting cell cycle progression was blocked by the presence of T-ag.
A different possible role for the MRE11-NBS1-RAD50 complex in cell immortalization is suggested by the role of this complex in telomere maintenance. In S.cerevisiae, MRE11, XRS2 and RAD50 are essential for maintaining normal telomere ends (74,75). This is seemingly a necessary requirement for continued cell proliferation as most immortalized mammalian cells have lengthened or stabilized telomeres (76,77). Although T-ag clearly perturbs the MRE11-NBS1-RAD50 and p53 pathways in immortalized cardiomyocytes, it remains to be determined if this interaction affects the potential role of the MRE11-NBS1-RAD50 protein complex in maintaining telomeres in mammalian cells.
We propose that SV40 large T antigen’s interaction with the MRE11-NBS1-RAD50 pathway ablates critical cell cycle checkpoints and this is one of the major factors involved in the ability of this oncoprotein to immortalize cardiomyocytes.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Chad Donaldson for excellent technical assistance and Mark Nienaber for the DNA sequencing of MRE11, NBS1 and RAD50 (Procter & Gamble Pharmaceuticals, Health Care Research Center, Mason, OH). This work was supported by NIH grant HL59879.
DDBJ/EMBL/GenBank accession nos AF218574–AF218576
REFERENCES
- 1.Rumyantsev P.P. (1991) In Gindina,K.A. (translator) and Carlson,B.M. (ed.), Growth and Hyperplasia of Cardiac Muscle Cells. Soviet Medical Reviews Supplement Series, Cardiology Vol. 3. Harwood Academic Publishers, New York, NY.
- 2.Claycomb W.C. (1991) In Oberpriller,J.O., Oberpriller,J.C. and Mauro,A. (eds), Proliferative Potential of the Mammalian Ventricular Cardiac Muscle Cell. The Development and Regenerative Potential of Cardiac Muscle. Harwood Academic Publishers, New York, NY, pp. 351–363.
- 3.Claycomb W.C. (1992) Trends Cardiovasc. Med., 2, 231–236. [DOI] [PubMed] [Google Scholar]
- 4.Mayhew T.M., Pharaoh,A., Austin,A. and Fagan,D.G. (1997) J. Anat., 191, 107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anversa P. and Kajstura,J. (1998) Circ. Res., 83, 1–14. [DOI] [PubMed] [Google Scholar]
- 6.Soonpaa M.H. and Field,L.J. (1998) Circ. Res., 83, 15–26. [DOI] [PubMed] [Google Scholar]
- 7.Behringer R.R., Peschon,J.J., Messing,A., Gartside,C.L., Hauschka,S.D., Palmiter,R.D. and Brinster,R.L. (1988) Proc. Natl Acad. Sci. USA, 85, 2648–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Field L.J. (1988) Science, 239, 1029–1033. [DOI] [PubMed] [Google Scholar]
- 9.Katz E.B., Steinhelper,M.E., Delcarpio,J.B., Daud,A.I., Claycomb,W.C. and Field,L.J. (1992) Am. J. Physiol., 262, 1867–1876. [DOI] [PubMed] [Google Scholar]
- 10.Sen A., Dunnmon,P., Henderson,S.A., Gerard,R.D. and Chien,K.R. (1988) J. Biol. Chem., 263, 19132–19136. [PubMed] [Google Scholar]
- 11.Jahn L., Sadoshima,J., Greene,A., Parker,C., Morgan,K.G. and Izumo,S. (1996) J. Cell Sci., 109, 397–407. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y.C., Neckelmann,N., Mayne,A., Herskowitz,A., Srinivasan,A., Sell,K.W. and Ahmed-Ansari,A. (1991) In Vitro Cell. Dev. Biol., 27, 63–74. [DOI] [PubMed] [Google Scholar]
- 13.Jackson T., Allard,M.F., Sreenan,C.M., Doss,L.K., Bishop,S.P. and Swain,J.L. (1990) Mol. Cell. Biol., 10, 3709–3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gruver C.L., DeMayo,F., Goldstein,M.A. and Means,A.R. (1993) Endocrinology, 133, 376–388. [DOI] [PubMed] [Google Scholar]
- 15.Reiss K., Cheng,W., Ferber,A., Kajstura,J., Li,P., Li,B., Olivetti,G., Homcy,C.J., Baserga,R. and Anversa,P. (1996) Proc. Natl Acad. Sci. USA, 93, 8630–8635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ludlow J.W. (1993) FASEB J., 7, 866–871. [DOI] [PubMed] [Google Scholar]
- 17.Ludlow J.W. and Skuse,G.R. (1995) Virus Res., 35, 113–121. [DOI] [PubMed] [Google Scholar]
- 18.Levine A.J. (1990) Virology, 177, 419–426. [DOI] [PubMed] [Google Scholar]
- 19.Herwig S. and Strauss,M. (1997) Eur. J. Biochem., 246, 581–601. [DOI] [PubMed] [Google Scholar]
- 20.Kirshenbaum L.A. and Schneider,M.D. (1995) J. Biol. Chem., 270, 7791–7794. [DOI] [PubMed] [Google Scholar]
- 21.Eckner R., Ludlow,J.W., Lill,N.L., Oldread,E., Arany,Z., Modjtahedi,N., DeCaprio,J.A., Livingston,D.M. and Morgan,J.A. (1996) Mol. Cell. Biol., 16, 3454–3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steinhelper M.E., Lanson,N.A.,Jr, Dresdner,K.P., Delcarpio,J.B., Wit,A.L., Claycomb,W.C. and Field,L.J. (1990) Am. J. Physiol., 259, 1826–1834. [DOI] [PubMed] [Google Scholar]
- 23.Delcarpio J.B., Lanson,N.A.,Jr, Field,L.J. and Claycomb,W.C. (1991) Circ. Res., 69, 1591–1600. [DOI] [PubMed] [Google Scholar]
- 24.Daud A.I., Lanson,N.A.,Jr, Claycomb,W.C. and Field,L.J. (1993) Am. J. Physiol., 264, 1693–1700. [DOI] [PubMed] [Google Scholar]
- 25.Borisov A.B. and Claycomb,W.C. (1995) Ann. NY Acad. Sci., 752, 80–91. [DOI] [PubMed] [Google Scholar]
- 26.Claycomb W.C., Lanson,N.A.,Jr, Stallworth,B.S., Egeland,D.B., Delcarpio,J.B., Bahinski,A. and Izzo,N.J.,Jr (1998) Proc. Natl Acad. Sci. USA, 95, 2979–2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gurney E.G., Harrison,R.O. and Fenno,J. (1980) J. Virol., 34, 752–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harlow E., Crawford,L.V., Pim,D.C. and Williamson,N.M. (1981) J. Virol., 39, 861–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lanson N.A. Jr, Glembotski,C.C., Steinhelper,M.E., Field,L.J. and Claycomb,W.C. (1992) Circulation, 85, 1835–1841. [DOI] [PubMed] [Google Scholar]
- 30.Harlow E. and Lane,D. (1988) Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, NY.
- 31.Sambrook J., Maniatis,T. and Fritsch,E.F. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, NY.
- 32.Petrini J.H., Walsh,M.E., DiMare,C., Chen,X.N., Korenberg,J.R. and Weaver,D.T. (1995) Genomics, 29, 80–86. [DOI] [PubMed] [Google Scholar]
- 33.Paull T.T. and Gellert,M. (1998) Mol. Cell, 1, 969–979. [DOI] [PubMed] [Google Scholar]
- 34.Higgins D.G., Bleasby,A.J. and Fuchs,R. (1992) Comput. Appl. Biosci., 8, 189–191. [DOI] [PubMed] [Google Scholar]
- 35.Varon R., Vissinga,C., Platzer,M., Cerosaletti,K.M., Chrzanowska,K.H., Saar,K., Beckmann,G., Seemanova,E., Cooper,P.R., Nowak,N.J. et al. (1998) Cell, 93, 467–476. [DOI] [PubMed] [Google Scholar]
- 36.Carney J.P., Maser,R.S., Olivares,H., Davis,E.M., Le Beau,M., Yates,J.R.,III, Hays,L., Morgan,W.F. and Petrini,J.H. (1998) Cell, 93, 477–486. [DOI] [PubMed] [Google Scholar]
- 37.Matsuura S., Tauchi,H., Nakamura,A., Kondo,N., Sakamoto,S., Endo,S., Smeets,D., Solder,B., Belohradsky,B.H., Der Kaloustian,V.M. et al. (1998) Nature Genet., 19, 179–181. [DOI] [PubMed] [Google Scholar]
- 38.Kim K.K., Daud,A.I., Wong,S.C., Pajak,L., Tsai,S.C., Wang,H., Henzel,W.J. and Field,L.J. (1996) J. Biol. Chem., 271, 29255–29264. [DOI] [PubMed] [Google Scholar]
- 39.Dolganov G.M., Maser,R.S., Novikov,A., Tosto,L., Chong,S., Bressan,D.A. and Petrini,J.H. (1996) Mol. Cell. Biol., 16, 4832–4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pearson W.R. and Lipman,D.J. (1988) Proc. Natl Acad. Sci. USA, 85, 2444–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chamankhah M., Wei,Y.F. and Xiao,W. (1998) Gene, 225, 107–116. [DOI] [PubMed] [Google Scholar]
- 43.Johzuka K. and Ogawa,H. (1995) Genetics, 139, 1521–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bowman S., Churcher,C., Badcock,K., Brown,D., Chillingworth,T., Connor,R., Dedman,K., Devlin,K., Gentles,S., Hamlin,N. et al. (1997) Nature, 387, 90–93. [PubMed] [Google Scholar]
- 45.Tavassoli M., Shayeghi,M., Nasim,A. and Watts,F.Z. (1995) Nucleic Acids Res., 23, 383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharples G.J. and Leach,D.R. (1995) Mol. Microbiol., 17, 1215–1217. [DOI] [PubMed] [Google Scholar]
- 47.Vissinga C.S., Yeo,T.C., Woessner,J., Massa,H.F., Wilson,R.K., Trask,B.J. and Concannon,P. (1999) Cytogenet. Cell. Genet., 87, 80–84. [DOI] [PubMed] [Google Scholar]
- 48.Kim K.K., Shin,B.A., Seo,K.H., Kim,P.N., Koh,J.T., Kim,J.H. and Park,B.R. (1999) Gene, 235, 59–67. [DOI] [PubMed] [Google Scholar]
- 49.Alani E., Subbiah,S. and Kleckner,N. (1989) Genetics, 122, 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sen-Gupta M., Guldener,U., Beinhauer,J., Fiedler,T. and Hegemann,J.H. (1997) Yeast, 13, 849–860. [DOI] [PubMed] [Google Scholar]
- 51.Wilson R., Ainscough,R., Anderson,K., Baynes,C., Berks,M., Bonfield,J., Burton,J., Connell,M., Copsey,T., Cooper,J. et al. (1994) Nature, 368, 32–38. [DOI] [PubMed] [Google Scholar]
- 52.Lin X., Kaul,S., Rounsley,S., Shea,T.P., Benito,M.I., Town,C.D., Fujii,C.Y., Mason,T., Bowman,C.L., Barnstead,M. et al. (1999) Nature, 402, 761–768. [DOI] [PubMed] [Google Scholar]
- 53.Haber J.E. (1998) Cell, 95, 583–586. [DOI] [PubMed] [Google Scholar]
- 54.Maser R.S., Monsen,K.J., Nelms,B.E. and Petrini,J.H. (1997) Mol. Cell. Biol., 17, 6087–6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee S.E., Moore,J.K., Holmes,A., Umezu,K., Kolodner,R.D. and Haber,J.E. (1998) Cell, 94, 399–409. [DOI] [PubMed] [Google Scholar]
- 56.Weinert T. (1998) Cell, 94, 555–558. [DOI] [PubMed] [Google Scholar]
- 57.Haber J.E. (1997) Cell, 89, 163–166. [DOI] [PubMed] [Google Scholar]
- 58.Boulton S.J. and Jackson,S.P. (1998) EMBO J., 17, 1819–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shore D. (1998) Science, 281, 1818–1819. [DOI] [PubMed] [Google Scholar]
- 60.Chamankhah M. and Xiao,W. (1999) Nucleic Acids Res., 27, 2072–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Haber J.E. (1999) Cell, 97, 829–832. [DOI] [PubMed] [Google Scholar]
- 62.Petrini J.H. (1999) Am. J. Hum. Genet., 64, 1264–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhong Q., Chen,C.F., Li,S., Chen,Y., Wang,C.C., Xiao,J., Chen,P.L., Sharp,Z.D. and Lee,W.H. (1999) Science, 285, 747–750. [DOI] [PubMed] [Google Scholar]
- 64.Nelms B.E., Maser,R.S., MacKay,J.F., Lagally,M.G. and Petrini,J.H. (1998) Science, 280, 590–592. [DOI] [PubMed] [Google Scholar]
- 65.Stewart G.S., Maser,R.S., Stankovic,T., Bressan,D.A., Kaplan,M.I., Jaspers,N.G., Raams,A., Byrd,P.J., Petrini,J.H. and Taylor,A.M. (1999) Cell, 99, 577–587. [DOI] [PubMed] [Google Scholar]
- 66.Xiao Y. and Weaver,D.T. (1997) Nucleic Acids Res., 25, 2985–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Trujillo K.M., Yuan,S.S., Lee,E.Y. and Sung,P. (1998) J. Biol. Chem., 273, 21447–21450. [DOI] [PubMed] [Google Scholar]
- 68.Dong Z., Zhong,Q. and Chen,P.L. (1999) J. Biol. Chem., 274, 19513–19516. [DOI] [PubMed] [Google Scholar]
- 69.Lim D.S., Kim,S.T., Xu,B., Maser,R.S., Lin,J., Petrini,J.H. and Kastan,M.B. (2000) Nature, 404, 613–617. [DOI] [PubMed] [Google Scholar]
- 70.Zhao S., Weng,Y.C., Yuan,S.S., Lin,Y.T., Hsu,H.C., Lin,S.C., Gerbino,E., Song,M.H., Zdzienicka,M.Z., Gatti,R.A. et al. (2000) Nature, 405, 473–477. [DOI] [PubMed] [Google Scholar]
- 71.Shiloh Y. (1997) Annu. Rev. Genet., 31, 635–662. [DOI] [PubMed] [Google Scholar]
- 72.Jongmans W., Vuillaume,M., Chrzanowska,K., Smeets,D., Sperling,K. and Hall,J. (1997) Mol. Cell. Biol., 17, 5016–5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Levine A.J. (1997) Cell, 88, 323–331. [DOI] [PubMed] [Google Scholar]
- 74.Nugent C.I., Bosco,G., Ross,L.O., Evans,S.K., Salinger,A.P., Moore,J.K., Haber,J.E. and Lundblad,V. (1998) Curr. Biol., 8, 657–660. [DOI] [PubMed] [Google Scholar]
- 75.Le S., Moore,J.K., Haber,J.E. and Greider,C.W. (1999) Genetics, 152, 143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim N.W., Piatyszek,M.A., Prowse,K.R., Harley,C.B., West,M.D., Ho,P.L., Coviello,G.M., Wright,W.E., Weinrich,S.L. and Shay,J.W. (1994) Science, 266, 2011–2015. [DOI] [PubMed] [Google Scholar]
- 77.Holt S.E. and Shay,J.W. (1999) J. Cell Physiol., 180, 10–18. [DOI] [PubMed] [Google Scholar]