

Abstract

WEE1 is a checkpoint kinase critical for mitotic events, especially in cell maturation and DNA repair. Most cancer cells’ progression and survival are linked with elevated levels of WEE1 kinase. Thus, WEE1 kinase has become a new promising druggable target. A few classes of WEE1 inhibitors are designed by rationale or structure-based techniques and optimization approaches to identify selective acting anticancer agents. The discovery of the WEE1 inhibitor AZD1775 further emphasized WEE1 as a promising anticancer target. Therefore, the current review provides a comprehensive data on medicinal chemistry, synthetic approaches, optimization methods, and the interaction profile of WEE1 kinase inhibitors. In addition, WEE1 PROTAC degraders and their synthetic procedures, including a list of noncoding RNAs necessary for regulation of WEE1, are also highlighted. From the standpoint of medicinal chemistry, the contents of this compilation serve as an exemplar for the further design, synthesis, and optimization of promising WEE1-targeted anticancer agents.
1. Introduction
Genotoxic stress causes DNA damage, that triggers complex signaling pathways called DNA damage responses (DDRs) to detect and repair DNA. The compromised pathways lead to tumor and cancer formation.1 The breakage of DNA strands triggers the ATR-ATM (Ataxia-Telangiectasia mutated and Rad3-related-Ataxia-Telangiectasia mutated) cascade, which ultimately leads to apoptosis, senescence, or DNA patching (based on the degree of DNA damage). The ATR (single-strand DNA breakage) and the ATM (double-strand DNA breakage) activates distress signal messengers called the checkpoint kinases (CHK1 and CHK2, respectively).2 Kinases are catalytic agents that transfer the γ-phosphate group from ATP to its substrate protein to either activate or deactivate the substrate, and the action is reversed by phosphatases (Figure 1).3 The kinase, CHK1 activates the guardian of the G2-M cell checkpoint, the WEE1 kinase (via phosphorylation), simultaneously deactivating the CDC25 (cell division cycle kinase 25, responsible for promoting the cell cycle).4−6 Conversely, CHK2 activates the tumor suppressor P53.7,8 The activated guardians disarm CDK1/2 (cell division kinases 1/2) and induce mitotic arrest, which ensures enough time to mend the DNA. The three members of the WEE protein kinase family, WEE1, WEE2, and MYT1 are negative regulators of cell cycle (by inhibiting CDKs). (1) WEE1 or WEE1A gene expression causes mitotic arrest and contributes to daughter cell maturation. Russell and Nurse et al. identified the WEE1 gene product of 112 kilobases (kb) as a protein kinase in fission yeast.9 Later, Igarashi et al. discovered the 2.8 kb long WEE1 gene and its gene product (432 amino acids long WEE1 protein) in humans.10 The WEE1 gene in humans is located on the 11th chromosome at the band 11p15, more precisely at 11p15.4, which contains 19,788 bases.11,12 It is a somatic nuclear serine/threonine (S/T) kinase responsible for cell cycle arrest by inhibiting CDKs via phosphorylating Y15 (in humans) and Y15 and T14 residues of CDC2/28 (homologue of CDK1 of fission and budding yeast).4,13−15 Though it phosphorylates Y and T residues, it is still classified as a S/T kinase and not a tyrosine kinase due to its structural and sequential similarities with S/T kinases.16 (2) WEE1B or WEE2 kinase is an oocyte-specific tyrosine kinase conserved from yeast to humans, responsible for premature oocyte arrest in the GV (germinal vesicle) state during prophase-1 and mature oocyte arrest during metaphase-2 before fertilization.17−19 Both meiotic arrests result from WEE2-catalyzed CDK1 inhibition (pY15).20 (3) MYT1 (recently renamed as PKMYT1) is an odd membrane-associated kinase (endoplasmic reticulum and Golgi apparatus) that phosphorylates CDK1 at T14 and Y15 (preferentially T14 in humans) and causes cell cycle arrest at the G2-M checkpoint.21−24 Due to their role in cell cycle regulation, WEE1 and MYT1 have become potential anticancer targets.25 In contrast, WEE2 has a role in fertilization but none in tumor induction.
Figure 1.

Mechanism of phosphorylation and dephosphorylation by kinase and phosphatases, respectively (adopted from Ha et al.3).
1.1. Structure of WEE1 Kinase
Squire et al. gave a detailed account of the structure of the WEE1 kinase using PDB ID: 1X8B (the cocrystal structure of the WEE1 complexed with PD0407824).16 Initially, Watanabe et al. in 1995 constructed a full-length human WEE1 kinase containing 646 amino acids with an approximate molecular mass of 71,597 Da. They depicted the presence of the N-terminal regulatory domain (NRD), the central kinase domain (KD), and the C-terminal regulatory domain (CRD).26 The NRD is equipped with many phosphorylation sites (P-sites), including P-sites for PLK1 (polo-like kinase 1) (S53) and CDK1 (S123), as well as a nuclear localization signal (NLS) (252RRRKR256) and three PEST regions.26 This domain furnishes a P-site at S121 for casein kinase 2 (CK2).27 Squire et al. reported that the kinase domain of WEE1 contains a typical kinase structure, i.e., an N-terminal domain (NTD) and a C-terminal domain (CTD) linked by the active site cleft that contains a short catalytic segment (422–433) and a long activation segment (462–486). The catalytic segment retains an altered HRD motif at 424HMD426, while the activation segment conserves DFG and APE motifs with displaced residues, 463DLG465 and 484ANE486, respectively. These residue displacements are specific to WEE1.2 The peptide connecting β5-αD is the hinge region (376–379). Additionally, the CRD has an inhibitory P-site at S642 (via BRSK1/2, AKT, and CHK1) and the 14-3-3 chaperon-binding consensus (639RSVpSLT644), which regulate WEE1 stability and subcellular localization.16 The WEE1 catalytic domain possesses a pair of Mg2+ ions, one is bound to N431 and the other to D463, along with three water molecules. ATP or a ligand is bound to the C379 hinge backbone and N376 gatekeeper residues. The adenine moiety or ligand scaffolds are encased in the hydrophobic pocket created by I305, V313, A326, and F433, forming π–π interactions with F433. The phosphate groups of ATP are bound to K328 and K428 residues.16 The active state conformations of WEE1 show striking similarities with CHK1 kinase.16 The full-length WEE1 contains a WEE box (233VNINPFT239P240) that encloses an inhibitory cyclin A-CDK complex P-site, T239. The role of the WEE box is to assist the catalytic activity of WEE1. Additionally, WEE1 conserves four RxL motifs, and the most conserved RxL1 binds to cyclin A-CDK to facilitate T239 phosphorylation.28 Moreover, RxL1 is confined within the binding consensus of chromosomal maintenance 1 (CRM1) or exportin-1 or the nuclear export signal (NES) (175PHKTFRKLRL184).28 The kinase domain also includes the binding motif of the SH2 domain of adaptor protein, CRK (492YTHL495). The NRD is highly rich in S, T, and Y residues ready to phosphorylate.29,30 An acetylation motif (AcM)177KTFRK/R181 is incorporated within the NES, where acetylation and deacetylation of K177 activate and deactivate WEE1 kinase, respectively.31 Moreover, in humans, WEE1 is enriched with autophosphorylation sites like Y295 and Y362, whose roles are unknown. A summary of the full-length WEE1 structure is illustrated in Figure 2A.
Figure 2.

(A) Structure of WEE1 kinase (adopted from Watanabe et al.;26 Matheson et al.;2 Squire et al.;16 Li et al.;28 Zhu et al.31). (B) Checkpoint arrest via WEE1 (adopted from Matheson et al.;2 Elbaek et al.38) and (C) functions of WEE1 kinase.
1.2. Action of WEE1 Kinase
Human WEE1 freezes the cell cycle at the G2-M and G1-S checkpoints by inhibiting the CDKs via phosphorylation at Y15. As a result, the formed homodimer cyclin B/A-pCDK1 complex, also called MPF (mitosis promoting factor), is inactive at the G2-M checkpoint, and the cell cycle is ceased. At the G1-S checkpoint, the inactive cyclin A/E-pCDK2 complex that forms stalls the cell cycle. WEE1 is found to act only if CDKs are bound to their respective cyclins in subsequent studies (Figure 2B,C).32 In normal cells, WEE1-mediated G2-M arrest ensures the maturation of daughter cells. Moreover, in cells with DNA damage, it ensures DNA repair. Therefore, loss of WEE1 action results in abnormally small-sized daughter cells and loss of genetic integrity, ultimately leading to mitotic catastrophe.9 WEE1 also phosphorylates HSP90 (Heat Shock Protein 90) on Y38, disrupting its chaperon and ATPase activity.33,34 Additionally, WEE1 is an “Epigenic modifier” that inhibits histone H2B (pY37), blocks histone transcription, and ensures appropriate stoichiometry between histones and DNA (Figure 2C).35,36 Moreover, it also interacts, phosphorylates, and inhibits SKP2/FBXL1 (S-Phase Kinase Associated Protein 2/F-box and Leucine-Rich Repeat Protein 1) (pS99), an E3 ubiquitin ligase responsible for degrading P21 and P27 (CDK inhibiting proteins), resulting in inhibited cell cycle progression.37 Furthermore, it protects the replication forks by direct or indirect regulation of endonucleases, MUS81/EME1 (essential meiotic structure-specific endonuclease 1) via CDK1 inactivation.38−40
1.3. Regulation of WEE1 Kinase
In the cell cycle, during the S and G2-phases, where the mitotic delay is a prerequisite, WEE1 is highly expressed and tightly shielded. During interphase, S642 is phosphorylated and bound by 14-3-3, stabilizing and protecting WEE1 from degradation.41−43 Additionally, HSP90 also contributes to WEE1 stability, and its substrate, survivin, inhibits caspase-3 and, in turn, protects WEE1 from degradation.33,44,45 MIG6 (mitogen-inducible gene 6) binds at the kinase domain of WEE1 and physically obstructs and prevents WEE1 degradation.46 WEE1 is transactivated by c-Fos/AP1 (c-Fos/activator protein 1) during the G1-S phase.47 However, after cell maturation or patching the damaged DNA, the cell cycle should proceed into mitosis (M-phase). CDC25 controls the entry into mitosis by removing the inhibitory phosphate group on CDK1/2, thus setting it free to act.48,49 The cells proceed with mitosis only if WEE1 is nonfunctional. Hence, WEE1 sheds its armor and is ready for negative regulation by different mechanisms (inactivation and degradation).26 Upon G2-M propagation, inactivation of WEE1 is a result of phosphorylation of WEE1 by NIM1/CDR1 (nonexpresser of PR genes1/cerebellar degeneration related protein 1, on the N-terminal catalytic domain),50−52 BRSK1/2 (BR serine/threonine kinase 1/2, at S642, that regulates differentiation of neurons),53,54 and CDK1/2-cyclin A (at T239).28 During M-phase, ubiquitin-dependent protein degradation is responsible for the downfall of WEE1. Initially, during the G2-M transition, PLK1, CDK1, and CK2 phosphorylation at S53, S123, and S121 residues generate phosphodegrons (PDs), and CDC34 ubiquitination of WEE1 allows SCF (skp1/cul1/F-box) β-TrCP (β-transduction repeat-containing protein), Tome-1 (trigger-of-mitotic-entry 1), Smurf1 (Smad ubiquitination regulatory factor1), and Pof1/3 (Promoter of Filamentation1/3) (in fission yeast) mediated WEE1 degradation.55−61 However, in a later study, WEE1 was found to interact with the β-subunit of CK2, but this interaction showed no remarkable influence on WEE1 kinase activity.62 Another mechanism reported is the nuclear export of WEE1. (1) AKT (Protein Kinase B) phosphorylates WEE1 at S642, which promotes nuclear export via 14-3-3Θ binding.63 (2) A CRM1-dependent nuclear export may occur via phosphorylation of S/TX residues (CDK2-cyclin A) near the RxL1 motif embedded in NES.28 During the onset of a new cycle, WEE1 is positively regulated via increased synthesis and dephosphorylation (FCP1 on T239).64 In Xenopus WEE1, phosphorylation of T186 (human T329 equiv) of WEE box creates a pS/TP binding motif for PIN1 (peptidyl-prolyl cis–trans isomerase NIMA-interacting 1) that inhibits WEE1 activity during M-phase.65 Zhu et al. recently identified WEE1 as an inactive endogenous homodimer forming intermolecular H-bonds between NRD (K177, H176, and T178) and KD (D405) of WEE1.31 These interactions are disrupted by the acetylation of K177 by GCN5 (General Control Non-Depressible 5) after pS642 by CHK1 due to DNA damage, which induces depolymerization and WEE1 activation. This action is reversed after DNA repair when SIRT1 (sirtuin 1) binds and flings the acetyl group off K177.31 WEE1 expression also tightly follows the circadian rhythm.66 A summary of the phosphorylation pathways of WEE1 is illustrated in Figure 3.
Figure 3.
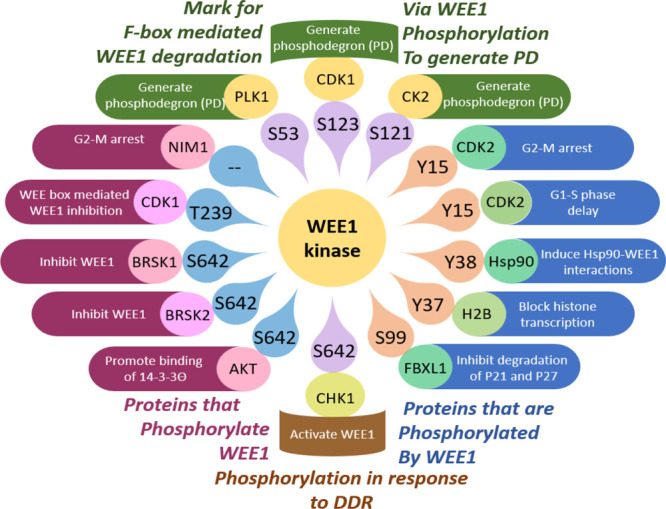
Proteins phosphorylation “of WEE1 and by WEE1”.
1.4. WEE1 and Cancer
In most cancer cells, the tumor-suppressor TP53 is nonfunctional, and the G1/S checkpoint is inoperative. Hence, the cells lose the ability to mend DNA before replication in S-phase and are thus allowed to carry mutations. Therefore, the G2/M checkpoint shoulders the responsibility for patching the DNA by employing cell cycle arrest and mending the DNA just enough to dodge apoptosis.2,67 Thus, getting rid of the restriction on the G2/M checkpoint will have a massive impact on the cancer cells. Apparently, WEE1 expression is highly elevated in many TP53 mutant cancer types like ovarian cancer, carcinomas, gliomas, leukemia, melanoma, breast cancer, medulloblastoma, etc., which further emphasizes its role in TP53 mutant onco-cell survival and growth. Moreover, WEE1 also regulates the G1/S checkpoint, so its inhibition not only disposes the cell cycle arrest at the G2/M gate but also at the G1/S gate, which further elevates γ-H2AX (histone family protein X) levels that imply DNA damage (double-strand breaks) and eventually cause mitotic catastrophe.2 Furthermore, a functional P21, the downstream regulator of P53, apparently shields the cancer cells from WEE1 inhibition-induced DNA damage and cell death during the S-phase, thus further supporting the notion that TP53 mutants are more susceptible to WEE1 inhibition.68 Later studies, however, unveiled that WEE1 inhibition is relatively less but productive on wild TP53 onco-cells. Further, AZD1775 radio-sensitizes oral tongue squamous cell carcinoma, irrespective of TP53 status.69
In contrast, the WEE1 gene was less expressed in colon cancer, nonsmall cell lung cancer (NSCLC), prostate cancer, and stromal breast cancer, which are associated with lamentable prognosis.67,70,71 Cancers lacking WEE1 expression are prone to genetic abnormalities and may be sensitive to DNA-damaging agents (DDAs), and treating with these agents induces WEE1 expression.2 Cells that either overexpress WEE1 or are induced to express WEE1, either way, depend on the G2/M checkpoint for escaping the mitotic catastrophe; thereby, inhibition of WEE1 provides a chance to treat both types of cancers.2,70 Additionally, monotherapy with WEE1 inhibitors (WEE1i) is effective against cells with active chromosomal instability (CIN), but a combination with DDAs is preferable for CIN-inactive cells.67 Targeting WEE1 also shows promising results in cancer types with BRCA (breast cancer), RAS (Rat Sarcoma virus), ATRX (ATP-dependent helicase ATRX, X-linked helicase II), EGFR (Epidermal Growth Factor Receptor) mutants, and CCNE1 (Cyclin E1)-amplified cancer types.72−78
PARP [poly(ADP-ribose) polymerase] is an essential target in tackling ovarian cancer, breast, pancreatic, and prostate cancers, including BRCA mutants. However, these cancers developed resistance to PARP inhibitors (PARPi) that undermined the therapeutic effects. The combination of PARPi with checkpoint regulating kinase inhibitors like WEE1i is a rather commendable strategy.79,80 Additionally, combination therapy of WEE1i with PARPi is in clinical trials for refractory, relapsed, and recurrent tumors (NCT02511795, NCT02576444, and NCT03579316, etc.). Moreover, exceptional synergistic cytotoxicity is observed when WEE1 and CHK1 are simultaneously inhibited. The cytotoxicity is due to the increased loading of CDC45 (the kinase responsible for replication initiation) caused by CHK1 inhibition, which causes unscheduled DNA replication and thus increased DNA damage, along with an increase in CDK activity due to WEE1 inhibition that further impairs DNA. Additionally, in small cell lung cancer (SCLC) with acquired resistance to CHK1i, WEE1 is overexpressed. Hence, simultaneous inhibition of WEE1 and CHK1 is a promising strategy.81,82 Inhibition of ATR, the upstream regulator of CHK1, with WEE1, also elevates cytotoxicity due to elevated replication stress in acute myeloid leukemia (AML) and triple-negative breast cancer (TNBC), in addition to the antimetastatic effect. Thus, clinical trials (NCT03330847) using combination therapies provide a therapeutic advantage due to the lower toxicity profile of ATR inhibitors (ATRi) compared to CHK1 inhibitors (CHK1i).83−85 However, this combination has dissimilar effects on different cancer cells; hence, further development may be restricted.86 Inhibiting WEE1 and RAD51 (responsible for repairing double-strand breaks of DNA) together also offers increased genotoxicity in a similar mechanism.87
HDAC (histone deacetylase) inhibition restrains the action of CHK1 and expression of c-MYC, and simultaneous inhibition of WEE1 results in elevated DNA damage and ultimately causes cell death, irrespective of TP53 status. This combination is in clinical trials (NCT02381548) and is effective against FLT3 mutants.88,89 Furthermore, c-MYC is an upregulated gene in many cancers, and such cancers get addicted to its presence, so suppressing MYC is a sound strategy. However, MYC is mostly considered nondruggable, but targeting its allies is feasible enough. Therefore, the above strategy to inhibit HDAC and WEE1 is productive.90,91 HSP90 is an essential chaperone and a potential target in cancer cells because it ensures proper folding of oncoproteins and protects them from degradation. However, HSP90 inhibitors (HSP90i) are not cytotoxic but cytostatic, thereby limiting their application, but WEE1 inhibition sensitizes oncocells to HSP90i. This combined inhibition lowers the expression of both WEE1 and survivin, clients of HSP90, along with the inactivation of AKT in both in vivo and in vitro studies, which contributes to the exceptional cytotoxicity.44,92 WEE1 inhibition has also elevated the expression of STAT1-mediated IFN-γ and PD-L1 (programmed cell death ligand 1) in SCLCs, thus combined WEE1 and PD-L1 may open a new path.93Figure 4 illustrates all the possible proteins that proffer a sound strategy when inhibited alongside WEE1 in combination therapy.
Figure 4.

All the possible proteins that proffer sound strategy when inhibited alongside WEE1 in combination therapy.
1.5. Kinase Inhibitors in the Market
Kinase-mediated signaling pathways in many cancers have emerged as hallmark targets for anticancer agents, and the FDA has approved more than 70 drugs to date.94 Most kinases are essential for vital cellular functions such as cell cycle progression, angiogenesis, cellular transport, cell adhesion, and signaling.3 Several kinase inhibitors on the market and clinical candidates have been targeting the phosphorylation site of kinase enzymes. The medicinal scaffolds such as aminopyrimidine, aminopyridine, fused pyrimidine, aminopyrazine, pyrazolopyrimidine, anthranilamide, thienopyridine, benzopyrimidine, quinoline, bipyridine, pyralopyrimidine, benzopyrazole, imidazopyrimdine, aminothiazole, pyralopyridine, pyridopyrimidine, pyrazotriazine, benzimidazole, indalopyrimidine, pyrazopyridine, pyrazolopyridine, quinazoline, and other miscellaneous heterocycles exist in the marketed drugs.
2. Small Molecule Inhibitors of WEE1 Kinase
WEE1 inhibitors are ATP competitive binders that bind in the ATP binding pocket of the kinase and prevent the ATP from binding, thus prohibiting WEE1 activation. Like all other kinase inhibitors, most WEE1i retain the pyrimidine core (I-XVI) and include scaffolds like pyridopyrimidines (VI–VIII), pyrazolopyrimidines (IV), pyrrolopyrimidines (III), pyrimidopyrimidines (X), and pyrimidine-based tricyclics (XI) and other micromolar scaffolds. Apart from these, WEE1i also include nonclassic kinase inhibitors like pyrrolocarbazoles (XVII), vanillates (4), pyrazoles (3), thiazoles (XIX), etc. Being a kinase, WEE1 also accommodates other kinase inhibitors in its ATP binding pocket, including bosutinib (7) and its isomer (8), PF03814735 (6), etc. Figure 5 summarizes all the scaffolds displaying WEE1 inhibition, including classic pyrimidine-based WEE1 kinase inhibitors, and other kinase inhibitors showing WEE1 inhibition. This review describes the above WEE1i, their binding modes, and synthesis procedures to provide an exemplar for the design and development of novel, more ideal WEE1i.
Figure 5.
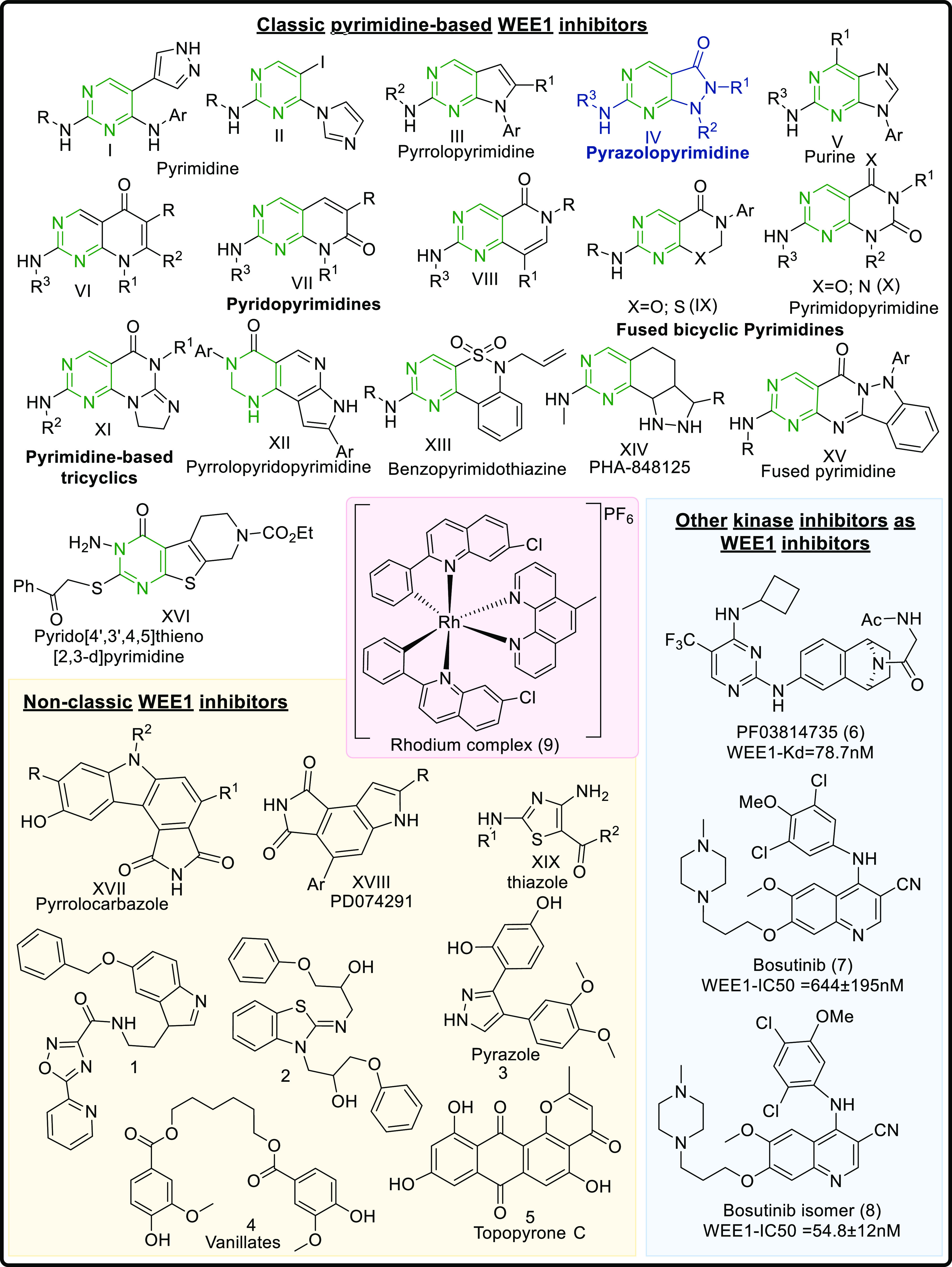
WEE1 kinase inhibitors from different heterocyclic scaffolds.
2.1. Pyridopyrimidine
Wang et al. identified PD0166285 (pyridopyrimidine) (10,Figure 6) as the first WEE1i through rapid screening of a chemical library. It is an ATP-competitive inhibitor of WEE1 and MYT1 with an IC50 value of 24 nM and 72 nM, respectively. It prevents phosphorylation of deactivation P-sites on CDK1 and abrogates the G2 checkpoint.95 This ligand has also shown activity against other kinases or off-targets like c-Src (IC50 = 0.009 μM), PDGFRB (IC50 = 0.1 μM), FGFR (0.43 μM), CDK1 (0.1 μM), EGFR (0.35 μM), and PF3D7 (0.35 μM). In 2004, Mizenina and Moasser et al. identified two c-Src kinase inhibitors, PD179483 (11) and PD166326 (12), as in vitro WEE1i that blocked S-phase.96 Some analogues and their activity values are mentioned in Table 1, and Figure 6A illustrates the structures of the very first WEE1 inhibitors.
Figure 6.

(A) Pyridopyrimidine analogues, (B) PD0166285 (10) in ATP binding pocket, and (C,D) PD0166285 (10) interaction profile in 3D and 2D, respectively (PDB ID: 5VC5).
Table 1. Some Pyridopyrimidine Analoguesa.
| structure | comp | R | R1 | R2 | R3 | IC50(μM) |
|---|---|---|---|---|---|---|
| VI | 35 | 2′,6′-diCl, 3′-OH Bn | Me | H | 3-phenoxypropanoic acid | 0.04 |
| VI | 36 | 2′-Cl Bn | Me | Me | N,N-diethyl-2-phenoxyethan-1-amine | 0.55 |
| structure | comp | R | R1 | R2 | R3 | Ki (nM) |
|---|---|---|---|---|---|---|
| VII | 37 | 2′,6′-diCl Bn | Me | – | 1-phenylpiperazine | 0.3 |
| VII | 38 | 2′,6′-diCl Bn | Me | – | N-methyl-1,2,3,4-tetrahydroisoquinoline | 0.2 |
| VIII | 39 | 2′,6′-diCl Bn | CH2OH | – | 2,4,4-trimethyl-1,2,3,4-tetrahydroisoquinoline | 0.3 |
| VIII | 40 | 2′,6′-diCl Bn | Me | – | 1-methyl-4-phenylpiperazine | 0.5 |
Bn stands for benzene.
2.1.1. Structure–Activity Relationship and Optimization Approaches of Pyridopyrimidines
The 2-aminopyrimidine ring of this scaffold acts like the adenine ring of ATP by forming 2 H-bond interactions (one pyrimidine “N” and 2-amino group) with backbone C376 of the hinge region and π–π interactions with F433 (Figures 6B–D).97 The aromatic substitution at position-6 remains perpendicular to the core moiety and forms catalytic π-interactions with K328 and H350 (Figure 6D).97 2′,6′-Disubstituted aromatic rings increase activity; however, the 2′,6′-dichloro group is optimal.98 3′-OH or 3′-NH2 groups are tolerated but not bulkier groups since they clash with E346.97,98 Similarly, small electronegative groups on 4′-position like −OH and −NH2 associate with D463 and contribute to increased activity.97N-Phenyl and its substitutions (R group from Table 1) protrude from the binding pocket and are solvent-exposed regions. Bulky hydrophilic substitutions with moderate length are highly sought after.97 Alkyl substitution on position-5 increases selectivity against c-Src (the alkyl group sterically clashes with T338 residue); however, this selectivity comes at the expense of potency.98 In the case of 8N-pyridopyrimidines, pyridine’s “N” (position-8) substituted with small alkyl groups like methyl or ethyl are more active, and these substituents are sandwiched in the cavity between F433 and V331; however, basic groups like amines are not tolerated.99 On the other hand, 6N-pyridopyrimidines show no significant difference in activity profile. In either case, position-5 carbonyl, if present, interacts with N376, the gatekeeper residue.99 Despite tremendous efforts, the optimization of this scaffold has yet to yield a WEE1i with high selectivity, restricting its clinical applications.
2.1.2. Synthesis of 8N-Pyridopyrimidine Core (VI, VII)
The substituted 8N-pyridopyrimidine (VII) scaffold can be made by a method in which 4-chloro-2-(methylthio)pyrimidine-5-carbonyl chloride (13) is reacted with Grignard reagent or aryl zinc halide. In the case of the Grignard reagent, low temperature is maintained in THF. In contrast, aryl zinc halide is first treated to a mixture of CuCN:2LiCl and then reacted with 13 in THF at low temperature to give keto aryl intermediate 14. In the first case, 14 reacts with an alkyl amine at high temperature in isopropanol and generates 15. Bicyclic intermediate 18 is cyclized, when intermediate 15 reacts with another alkyl amine along with Ν,Ν-dimethylformidedimethylacetal (DMF-DMA) at high temperature in a microwave reactor. In the second case, intermediate 14 is converted into bicyclic core 18 upon reaction with N-alkyl-substituted amide 19 in the presence of the (Pd2(dba)3) catalyst, xantphos ligand, and Ce2CO3 at high temperature in dioxane. Another route to generate 18 is when an amide 16 is reacted with 14 and affords 17, which in turn generates 18 when reacted with alkyl boronic acid at ambient temperature in a base Et3N and solvent CH2Cl2. The resultant product 18 is first oxidized with m-CPBA in acetonitrile and trifluoroacetic acid, followed by a reaction with aryl amines at elevated temperatures to give VI core (Table 1, Scheme 1).100
Scheme 1. Synthesis of 8N-Pyridopyrimidine (VI) Analogues.

Adapted from Mastracchio et al.100
Scaffold VII can be synthesized by reacting 20 with an alkyl amine in a basic medium, followed by reduction with lithium aluminum hydride (LAH) to produce an alcohol intermediate oxidized with MnO2 to give the corresponding aldehyde 21. Condensation of aldehyde 21 with aryl acetonitrile in a basic medium at 105 °C gives a cyclized imine 22, which, when undergoes acetylation, hydrolysis, followed by oxidation and amination, gives the 8N-pyridopyrimidine (VII) core (Table 1, Scheme 2).101
Scheme 2. Synthesis of 8N-Pyridopyrimidine (VII) Analogues.

Adapted from Klutchko et al.101
2.1.3. Synthesis of 6N-Pyridopyrimidine Core
When treated with 1,1-dimethoxy-N,N-dimethylmethanamine (24), ester 23 transforms into dimethyl analogue 25 at elevated temperatures, initially reacting with ammonia in an acidic condition with EtOH as a solvent gives a cyclized intermediate 26, which upon alkylation with an alkyl halide produces 6N-substituted intermediate 27. Dimethyl analogue 25 can also cyclize into 27 directly by reacting with a substituted amine in acidic media. Upon bromination of 27, 28 is produced, which undergoes Suzuki coupling and gives the arylated core 29 (Scheme 3).102
Scheme 3. Synthesis of 6N-Pyridopyrimidine Core and Crucial Intermediate 29.

Adapted from Penning et al.102
Similarly, 29 can also be generated when aqueous LiOH in THF and MeOH mixture hydrolyses the ester 30 to acid 31, which is converted into its respective amide 32 when reacted with thionyl chloride in DMF and dioxane to get an acid chloride that is then allowed to react with a substituted amine. The amide 32 is then reacted with (chloromethylene)dimethyliminium chloride (33) in DMF, which gives 29. Additionally, in an alternate method, key ester 30 is reacted with 24, and gives dimethylamine intermediate 34, which, upon reaction with an aryl amine, produces 29 (Scheme 4).102
Scheme 4. Synthesis of Key Intermediate 29.

Adapted from Penning et al.102
The resultant arylated 29 upon oxidation by m-CPBA and reaction with another aromatic amine yields 6N-pyridopyrimidine core (VIII) (Table 1, Scheme 5).102
Scheme 5. Synthesis of 6N-Pyridopyrimidine Analogues from 29.

Adapted from Penning et al.102
2.2. Pyrrolocarbazole
Pyrrolocarbazoles (XVII) were first synthesized and tested for WEE1 inhibition by Booth et al. in 2003, which exhibited IC50 < 1 μM, including activity against CHK1 and PKC (Protein kinase C).103 Squire et al. in 2005, described the human WEE1 structure with respect to the CHK1 structure using the PDB ID: 1X8B (a cocrystal structure of WEE1 complexed with Booth et al.’s pyrrolocarbazole, PD0407824 (41)).16 Later, Palmer et al. in 2006 confirmed PD0407824 (41) showing potent WEE1 inhibition (IC50 = 97 nM) in a high-throughput screening (HTS) program and proposed its structure–activity relationship (Figure 7).104 All analogues of 41 bind in the ATP binding pocket of WEE1 as shown in Figure 8A,D in the ATP competitive manner. Additionally, 41 displayed potent CHK1 inhibition (IC50 = 47 nM) due to its similar structure to the indolocarbazole scaffold of CHK1i. Despite systematic optimization to achieve selectivity, this scaffold displayed a broad-spectrum of kinase inhibition, which may have been the reason not to consider it for clinical trials. A few pyrrolocarbazole analogues are tabulated in Table 2, along with their IC50 values.
Figure 7.

Optimization of PD0407824 (41) to enhance selectivity, potency, and solubility (adopted from Palmer et al.;104 Smaill, Baker et al.;105 and Smaill, Lee et al.106).
Figure 8.

Pyrrolocarbazoles (XVII)-Pyrrolocarbazoles-WEE1 interaction profile in 3D. (A) Pyrrolocarbazoles and their binding modes in the WEE kinase pocket, (B) PDB ID: 1X8B, (C) PDB ID: 2NI6, (D) insight of overlapped poses of pyrrolocarbazoles, (E) PDB ID: 2IO6, (F) PDB ID: 2Z2W, (G) PDB ID: 3BI6, and (H) PDB ID: 3BIZ.
Table 2. Pyrrolocarbazole Analogues (XVII)a.
| comp | R | R1 | R2 | IC50 (nM) |
|---|---|---|---|---|
| 44 | H | Ph | H | 130 |
| 45 | H | 2-Cl Bn | H | 11 |
| 46 | H | 2,6-diCl Bn | H | 28 |
| 47 | H | 2-Cl Bn | (CH2)3OH | 9 |
| 48 | H | 2-Cl Bn | (CH2)2CONH2 | 6 |
| 49 | (CH2)4NMe2 | 2-Cl Bn | H | 49 |
| 50 | O(CH2)3Nmorph | 2-Cl Bn | (CH2)2OH | 15 |
Bn stands for benzene, Ph for phenyl, and morph for morpholine.
2.2.1. Structure–Activity Relationship and Optimization Approaches of Pyrrolocarbazoles
Palmer et al. proposed that the pyrrolo group is essential as 1-CO forms an H-bond with the −NH of C379 while the other 3-CO interacts with the gatekeeper residue N376 and a water molecule through H-bonding (Figure 8A–H).104 Additionally, pyrrolo 2-NH acts as a HBD (hydrogen bond donor) to E377. The following two rings form π–π stacking interactions with F433 (Figures 8 and 9).104 The presence of 9-OH facilitates the formation of H-bond with C379, G382, and occasionally with Y378; this results in a stronger binding affinity to the protein; hence, other substituents are not tolerated (Figure 8).104 The 2′-position of the aryl prefers “Cl” but can tolerate other substitutions with zero to little decline in activity while elevating selectivity (Figure 8E–H). However, 2′,6′-dichloro or 2′,6′-dimethyl increases potency and selectivity (Figure 7).104 5-Me substitution increases selectivity against CHK1, but alkanes larger than methyl are not tolerated. The carbazole “6N” interacts with S383 via three ordered water molecules. However, “N” can be replaced with either “S” or “O”. This replacement shows no significant change in WEE1 activity but increases selectivity if replaced with “O”.104 Smaill and Baker et al. introduced 6N-substitutions that can influence WEE1 and CHK1 selectivity, and slightly bulkier acidic groups increased selectivity and WEE1 potency; however, neutral polar groups like alcohols and amides are dual-specific inhibitors of WEE1 and CHK1 with elevated potencies (Figure 8C,E).105 Smaill and Lee et al. added bulky side chains containing amines like pyrrolidine at position-8 of 6N-methyl or 6N-alcohols to increase solubility with a slight increase in WEE1 potency (Figure 8G,H).106 The 4-aryl remains perpendicular to the pyrrolocarbazole moiety and fits into a small lipophilic cavity beside the gatekeeper (N376) (Figure 9A–F). A hydrophobic substitution at the 2′- position of the aryl enables an appropriate orientation to fit in the small pocket while forming a halogen bond with A326. Thus, this substitution, if absent, significantly reduces the potency of the inhibitor.
Figure 9.

Pyrrolocarbazoles (XVII)-WEE1 interaction profile. (A) PDB ID: 1X8B, (B) PDB ID: 2NI6, (C) PDB ID: 2IO6, (D) PDB ID: 2Z2W, (E) PDB ID: 3BI6, and (F) PDB ID: 3BIZ.
2.2.2. Synthesis of Pyrrolocarbazoles
Pyrrolocarbazoles (XVII) are synthesized by performing Wittig reactions of substituted 6-methoxy-1H-indole-2-carbaldehyde (51) with aryltriphenylphosphonium salt to give the corresponding 2-substituted dienes 52. Moreover, obtained diene 52 undergoes N-alkylation by an alkyl halide in the presence of NaH to give N-substituted diene 53 that is then reacted with 1H-pyrrole-2,5-dione or maleimide (54) at 175 °C directly to perform a Diels–Alder reaction or SnCl2 in toluene can also be used to form key cyclic compound 55. Aromatization of 55 using DDQ or MnO2 in dioxane followed by demethylation BBr3 in CH2Cl2 produce the pyrrolocarbazole moiety (XVII). Skipping N-alkylation produces XVII, where R2 = H (Table 2, Scheme 6).104
Scheme 6. Synthesis of Maleimide-Based Pyrrolocarbazole Analogues.

Adapted from Palmer et al.104
XVII can also be prepared by reacting the aldehyde 51 with Ph3PCHCOOBn to give a diene ester 56, which undergoes a Diels–Alder reaction with maleimide (54) to give the cyclic intermediate 57. Oxidation and reduction followed by amination of 57 give a free amine 58, which upon treatment with NaNO2 in an acidic medium followed by iodination with KI and CuI give iodo-analogue 59. Reduction by PyHCl followed by Suzuki coupling of 59 gives a R1-substituted intermediate, which upon alkylation with R2X in basic medium gives XVII (Table 2, Scheme 7).104
Scheme 7. Another route to synthesise maleimide-based pyrrolocarbazole analogues.

Adapted from Palmer et al.104
Another method to prepare the pyrrolocarbazole moiety is using a Grignard reagent to add alkyl or aryl to the aldehyde 51 to generate substituted alcohol 60. The corresponding alcohol 60 is oxidized with MnO2 to form a ketone, which upon reacting with alkyl or aryl phosphonium, the Wittig reagent gives the corresponding diene 61. XX can be generated when the diene 61 reacts with maleimide (54) at 180 °C for 40 min, followed by oxidation with MnO2, then demethylation by BBr3 (Scheme 8).104
Scheme 8. Synthesis of Maleimide-Based Multi-Substituted Pyrrolocarbazole Analogues.

Adapted from Palmer et al.104
Palmer et al. also provided an alternate method of synthesis where diene 52 reacted with maleic anhydride 62 gives the carbazole intermediate 63, which upon reaction with 2, 4-dimethoxybenzylamine (64) in the presence of acetic acid forms 65. Oxidation of benzylated analogue 65 with MnO2 followed by reaction with R2X in basic media gives the aromatic compound 66, which reacts with TFA in anisole then demethylation by BBr3 in DCM produces XVII (Table 2, Scheme 9).104
Scheme 9. Synthesis of Maleic Anhydride-Based Multi-Substituted Pyrrolocarbazole Analogues.

Adapted from Palmer et al.104
2.3. Pyrazolopyrimidine
Sagara et al. patented pyrazolopyrimidine (IV) analogues in 2007 as WEE1i.107 In 2009, high-throughput screening of a small chemical library discovered an initial hit, which was used to develop MK-1775 (67, Figure 13), a pyrazolopyrimidine, the first most potent and selective WEE1 kinase inhibitor with an IC50 value of 5.2 nM and an EC50 value of 49 nM (WiDr cells).108 MK-1775, later renamed AZD1775 or Adavosertib, was quickly promoted into phase-I, II clinical trials for monotherapy and combination therapies. It sensitizes the onco-cells to genotoxic agents like radiation, including antimetabolites (Gemcitabine, 5-Fluoro-uracil, Pemetrexed, and Capecitabine), alkylating agents (like Cis-platin, Temozolomide, and Carboplatin), antimicrotubular agent (like Paclitaxel and Docetaxel), and topoisomerase inhibitors (like Doxorubicin, Irinotecan, and Topotecan). And it has indeed reinforced carboplatin’s efficiency in TP53 mutants, including those with refractory or resistant ovarian cancer.109 Additionally, nonchemotherapeutic agents like PARPi (Olaparib) and ATRi (Ceralasertib) provide a better alternative for combinational therapy.110 HDAC inhibitor (Belinostat) and monoclonal antibodies (Durvalumab) are also studied with AZD1775. However, AZD1775 in combination therapy displayed fatigue, nausea, vomiting, diarrhea, thrombocytopenia, and neutropenia.110,111 As a single agent, AZD1775 initiates premature mitotic entry by WEE1 inhibition, but Guertin et al. stated that its cytotoxicity is a major consequence of AZD1775-induced DNA damage. Additionally, Guertin et al. reported AZD1775’s adverse events, including gastrointestinal disorders like diarrhea, vomiting, nausea, and hematologic toxicities (myelosuppression).112 Do et al. in 2015 also found myelosuppression and tachyarrhythmia as dose-limiting toxicities, limiting its clinical development.113 Later, Wright et al. discovered AZD1775’s equipotent PLK1 inhibitory action (WEE1, Kd = 3.2 nM; PLK1, Kd = 3 nM), and the binding mode is depicted in Figure 10. Though PLK1 is a negative regulator of WEE1, it regulates many pathways like FOXO, ATR signaling, FOXM1 pathways, APC/C-mediated degradations, etc.114 Therefore, off-target PLK1 inhibition by AZD1775 disrupts these pathways and might cause AZD1775’s cytotoxicity, including its adverse effects and toxicity profile.114 Moreover, AZD1775 also targets JAK2/3 (Janus kinase 2/3), ABL1, YES1, MAP3K4 (mitogen-activated protein kinase kinase kinase 4), etc. Hence, there is a need for new, more selective inhibitors.
Figure 13.

Optimization of AZD1775-based analogues (adopted from Matheson et al.;123,124 Qian et al.;121 Huang et al.;125 and Guler et al.126).
Figure 10.

Overlapping of AZD1775 and PLK1 inhibitor-I in the active site of PLK1 (PDB ID: 3DBD).
Some studies have observed the waning cytotoxicity of AZD1775. This phenomenon is not due to mutations in WEE1 but other mechanisms. Sen et al. identified a new pathway in SCLC that induces acquired resistance to WEE1i where AXL (AXL receptor tyrosine kinase) is overexpressed and so is its downstream protein mTOR (mechanistic target of rapamycin kinase), in addition to CHK1 activation that paves an alternate pathway to deactivate CDK1. Moreover, pS6 (phosphorylated ribosomal protein S6) and MET (mesenchymal–epithelial transition kinase) upregulation also desensitize SCLC cells to AZD1775.115,116 Lewis et al. observe that prolonged inhibition of WEE1 by AZD1775 promotes acquired resistance in HeLa cells due to overexpression of MYT1 (an alternate CDK1 regulator), and eliminating MYT1 has resensitized cells to AZD1775.117 Furthermore, in leukemia cells, HDAC activity is increased, which further increases c-MYC expression and confers acquired resistance to WEE1 inhibition.118 Additionally, Yes-associated protein (YAP) triggers the Fanconi anemia (FA) pathway and the corresponding transcriptional regulator, E2F1, which repair DNA damage and contribute to resistance. Further, the coinhibition of WEE1 and YAP with Dasatinib has sensitized the OVCAR-8 ovarian cancer cell line to AZD1775.119 Therefore, a parallel inhibition of WEE1 and CHK1, mTOR, AXL, HDAC, and YAP offers a good enough strategy to overcome the resistance (Figure 3).
Apart from AZD1775, ZN-c3, or Azenosertib (71), another pyrazolopyrimidine developed by Zentalis, is in phase-I, II trials in monotherapy for treating triple-negative breast cancer, ovarian cancer (NCT05368506), solid tumors (NCT04158336, NCT04972422), malignant tumors (NCT05128825), and uterine serous carcinoma (NCT04814108). In combination therapy, ZN-c3 is used to treat osteosarcoma with Gemcitabine (NCT04833582), Pt-resistant ovarian cancer along with PARPi (Niraparib; NCT05198804), and Carboplatin, Pegylated liposomal Doxorubicin, Paclitaxel, and Gemcitabine (NCT04516447); acute myeloid leukemia (AML) with BcL2 inhibitor (ZN-d5; NCT05682170); and metastatic colorectal cancer with BRAF (Encorafenib) and EGFR (Cetuximab) kinase inhibitors.
A monotherapy trial observed that ovarian cancer with amplified CCNE1 gene is hypersensitive to ZN-c3 in both in vitro and in vivo studies. Hypersensitivity toward ZN-c3 is also observed in combination therapies with chemotherapeutics like Paclitaxel.120 Nevertheless, another phase-I study (NCT05431582) is withdrawn due to lack of participants. Another analogue, SC0191 (70), demonstrated WEE1 inhibition and antiproliferative activity in TP53 mutant cells.121 The progress of detailed clinical trials of AZD1775 as monotherapy or in combination with other potential anticancer agents is provided in Table 3.
Table 3. Clinical Trials of AZD1775 and Their Status, Purpose, and Conditionsa.
| S. no. | NCT number | status | therapy | drugs | phase | conditions | purpose |
|---|---|---|---|---|---|---|---|
| 1 | NCT02508246 | C | CT | AZD1775, CDDP, Docetaxel | 1 | HNSCC | side effects and the best dose of AZD1775 in combination with CDDP, Docetaxel |
| 2 | NCT02666950 | C | MT, CT | AZD1775, AraC | 2 | advanced acute myeloid leukemia and myelodysplastic syndrome | competency of AZD1775 with and without Cytarabine |
| 3 | NCT02196168 | T | CT | CDDP, AZD1775 | 2 | recurrent or metastatic squamous cell cancer of the head and neck | competency of AZD1775 with and without CDDP |
| 4 | NCT02194829 | A | CT | AZD1775, Gemcitabine Hydrochloride, Nab-paclitaxel | 1 | metastatic adenocarcinoma of the pancreas | side effects and best dose of AZD1775 in combination with Nab-paclitaxel, Gemcitabine Hydrochloride |
| 5 | NCT02381548 | T | CT | AZD1775, Belinostat | 1 | relapsed and refractory myeloid malignancies and acute myeloid leukemia | side effects and best dose of AZD1775 and Belinostat |
| 6 | NCT01164995 | A | CT | AZD1775, Carboplatin | 2 | epithelial ovarian cancer | proof of concept (POC) study to prove WEE1 inhibition as an effective treatment in TP53 mutants |
| 7 | NCT02906059 | C | CT | AZD1775, Irinotecan | 1 | RAS or BRAF mutated metastatic colorectal cancer | safety and effectiveness in RAS or BRAF mutants |
| 8 | NCT03028766 | C | CT | AZD1775, CDDP, radiotherapy | 1 | head and neck cancer | best dose of AZD1775 in combination with CDDP |
| 9 | NCT02101775 | A | CT | AZD1775, Gemcitabine Hydrochloride | 2 | recurrent, platinum resistant epithelial ovarian, primary peritoneal, or fallopian tube cancers | comparing gemcitabine monotherapy to gemcitabine in combination with AZD 1775 |
| 10 | NCT02037230 | C | CT | AZD1775, Gemcitabine, radiotherapy | 1, 2 | adenocarcinoma of the pancreas | maximum tolerated dose (MTD) of AZD1775 |
| 11 | NCT04460937 | S | CT | AZD1775, radiotherapy | 1 | esophageal and gastresophageal junction cancer | maximally tolerated dose (MTD) of AZD1775 |
| 12 | NCT01748825 | C | MT | AZD1775 | 1 | advanced refractory solid tumors | safety, tolerability, and PK |
| 13 | NCT03253679 | S | MT | AZD1775 | 2 | advanced malignant solid neoplasm and refractory malignant solid neoplasm | objective response rate (ORR) to AZD1775 |
| 14 | NCT03668340 | R | MT | AZD1775 | 2 | uterine cancer | safety and effectiveness of AZD1775 |
| 15 | NCT03315091 | C | MT | AZD1775 | 1 | solid tumors | effect of food on the pharmacokinetics (PK) |
| 16 | NCT02207010 | C | MT | AZD1775 | 0 | glioblastoma multiforme (GBM) | CNS penetration |
| 17 | NCT02688907 | T | MT | AZD1775 | 2 | small cell lung cancer | competency of AZD1775 in MYC family amplification or CDKN2A mutation in TP53 mutants |
| 18 | NCT02593019 | C | MT | AZD1775 | 2 | small cell lung cancer | competency of AZD1775 in TP53 mutants |
| 19 | NCT02585973 | C | CT | AZD1775, CDDP, intensity modulated radiotherapy | 1b | carcinoma, squamous cell of head and neck | dose-escalation trial |
| 20 | NCT02791919 | W | CT | AZD1775, AraC, Fludarabine Phosphate | 1 | relapsed or refractory acute myeloid leukemia | dose-escalation trial, toxicity, PK |
| 21 | NCT03385655 | R | CT | AZD1775, Savolitinib, Darolutamide, CFI-400945, Ipatasertib, Durvalumab, and Tremelimumab, Carboplatin | 2 | prostate cancer | DNA abnormalities or biomarkers |
| 22 | NCT03313557 | C | MT | AZD1775 | 1 | solid tumors | safety and tolerability |
| 23 | NCT03333824 | C | CT | AZD1775, caffeine (CYP1A2), omeprazole (CYP2C19), midazolam (CYP3A), granisetron | 1 | advanced solid tumors | effect of AZD1775 on PK of other drugs |
| 24 | NCT03345784 | A | CT | AZD1775, CDDP, external beam radiation therapy | 1 | cervical, upper vaginal, and uterine cancers | recommended phase-II dose (RP2D) and safety profile of AZD1775 |
| 25 | NCT02610075 | C | MT | AZD1775 | 1 | solid tumors | determine maximum tolerant dose (MTD) |
| 26 | NCT02482311 | C | MT | AZD1775 | 1 | solid tumors | safety, tolerability, and PK |
| 27 | NCT02511795 | C | CT | AZD1775, Olaparib | 1 | refractory solid tumors and SCLC | dose-escalation trial |
| 28 | NCT02087176 | T | CT | AZD1775, antimitotic agent, Pegfilgrastim | 2 | previously treated SCLC | comparative study of AZD1775+antimitotic agent with placebo |
| 29 | NCT02341456 | C | MT, CT | AZD1775, Paclitaxel, Carboplatin | 1 | advanced solid tumors | multicenter study in adult asian patients |
| 30 | NCT02513563 | W | CT | AZD1775, Paclitaxel, Carboplatin | 1 | lung cancer | effects of AZD1775 |
| 31 | NCT02087241 | T | CT | AZD1775, Pemetrexed, Carboplatin | 2 | advanced NSCLC | comparative study of chemotherapy with and without AZD1775 |
| 32 | NCT03718143 | T | MT, CT | AZD1775,AraC | 2 | myelofibroses, acute myeloid leukemia, myelodysplastic syndromes | effects of AZD1775 as a single agent and in combination with AraC |
| 33 | NCT03012477 | C | CT | AZD1775, CDDP | 2 | breast cancer | effects of AZD1775 |
| 34 | NCT01047007 | T | MT, CT | AZD1775, CDDP, 5-FU | 1 | solid tumors | safety and MTD of AZD1775 |
| 35 | NCT02448329 | C | CT | AZD1775, Paclitaxel | 2 | advanced gastric adenocarcinoma | effects of AZD1775 |
| 36 | NCT04462952 | C | MT | AZD1775 | 1 | advanced solid tumors | safety, tolerability, pharmacokinetics (PK), and antitumor activity |
| 37 | NCT04439227 | A | MT | AZD1775 | 2 | lymphoma, neoplasm, myeloma | effect of AZD1775 on BRCA mutants |
| 38 | NCT01076400 | T | CT | AZD1775, Topotecan, CDDP | 1, 2 | cervical cancer | safety and MTD of AZD1775 |
| 39 | NCT01357161 | C | CT | AZD1775, Paclitaxel, CDDP | 2 | ovarian cancer | safety and efficacy of AZD1775 |
| 40 | NCT00648648 | C | CT | AZD1775, CDDP, Carboplatin, Gemcitabine | 1 | solid tumors | dose-escalation trial |
| 41 | NCT03284385 | A | MT | AZD1775 | 2 | SETD2-deficient solid tumors | effect of AZD1775 on SETD2 mutants |
| 42 | NCT04197713 | S | CT | AZD1775, Olaparib | 1 | solid tumors | side effects and best dose of AZD1775 |
| 43 | NCT02095132 | A | CT | AZD1775,Irinotecan | 1, 2 | solid tumors | side effects and best dose of AZD1775 |
| 44 | NCT01922076 | C | CT | AZD1775, radiotherapy | 1 | diffuse intrinsic pontine gliomas | side effects and best dose of AZD1775 |
| 45 | NCT01849146 | A | CT | AZD1775, radiotherapy, Temozolomide | 1 | glioblastoma | side effects and best dose of AZD1775 |
| 46 | NCT03579316 | R | MT, CT | AZD1775,Olaparib, Ceralasertib | 2 | recurrent fallopian tube, ovarian, primary peritoneal carcinomas | comparative study of AZD1775 with and without PARP inhibitors |
| 47 | NCT05008913 | T | MT | AZD1775 | 1 | advanced solid tumors | ADME study |
| 48 | NCT04949425 | T | MT | AZD1775 | 1 | advanced solid tumors | safety and tolerability study |
| 49 | NCT04590248 | A | MT | AZD1775 | 2 | uterine serous carcinoma | safety and efficacy of AZD1775 |
| 50 | NCT05212025 | W | CT | AZD1775, Gemcitabine | 2 | pancreatic cancer | safety and efficacy of the combination |
| 51 | NCT02546661 | A | MT,CT | AZD1775,AZD4547,MEDI4736, Olaparib, Vistusertib, AZD9150, Selumetinib | 1 | muscle invasive bladder cancer | safety, tolerability, pharmacokinetics (PK), and antitumor activity |
| 52 | NCT02937818 | A | CT | AZD1775, CBDP | 2 | platinum refractory extensive-stage SCLC | efficacy, safety, and tolerability |
| 53 | NCT02659241 | A | MT | AZD1775 | 1 | advanced fallopian tube, ovarian, primary peritoneal carcinomas | effects of AZD1775 |
| 54 | NCT02272790 | A | CT | AZD1775,CBDP, Paclitaxel, PLD, Gemcitabine | 2 | ovarian, fallopian tube, peritoneal cancer | efficacy and safety |
| 55 | NCT03330847 | A | CT | AZD1775,Olaparib, Ceralasertib | 2 | breast cancer | safety and efficacy of Olaparib in combination with AZD1775 and Ceralasertib |
| 56 | NCT01827384 | C | CT | AZD1775, Carboplatin, Everolimus, Temozolomide, Trametinib Veliparib | 2 | advanced malignant solid neoplasm | molecular profiling-based assignment of cancer therapy |
| 57 | NCT02465060 | A | CT | AZD1775, target-oriented inhibitors | 2 | solid tumors, lymphomas | genetic testing for targeted therapy |
C, completed; R, recruiting; A, active; S, suspended; T, terminated; W, withdrawn; CT, combination therapy; and MT, monotherapy.
2.3.1. Structure–Activity Relationship and Optimisation Approaches of Pyrazolopyrimidine
Pyrazolopyrimidines (IV) are Type-I kinase inhibitors that contain a 2-aminopyrimidine ring where 2-amino and 1N form an H-bond with C379 of WEE1 kinase. 6-carbonyl “O” also interacts with the N376 residue.122 The allyl attached to 5N is an important auxiliary group that enhances the interactions of carbonyl “O” with the gatekeeper. In addition, it may associate with K328 as it is within Vander Waals’ radii (Figures 11A-I and 12A–E). Allyl, when replaced with −Me and methoxybenzyl showed a significant reduction and complete loss of activity, respectively. Further, eliminating this group has resulted in an inactive inhibitor.123 The 4N is substituted with a 3′-isopropanol-substituted aromatic ring where the −OH group is necessary as it bonds with the D463 residue. The methyl groups, on the other hand, play a supporting role to lock the −OH group in an appropriate conformation.124 At last, the pyrazolopyrimidine ring forms π–π interactions with the F433 residue.124 Matheson et al. investigated the role of 2-amine substitutions that are solvent exposed, then proposed CM061 (68), and stated that substitution on the piperazine ring reduces toxicity while retaining its activity.124 Additionally, the presence of a flexible alkyl amine substitution on the para-position of the aniline (69) contributes to increased potency as a result of the induced H-bond with D386/E303.123 Despite enhanced potency and selectivity, 68 and 69 showed diminished cell viability and cytotoxicity, verifying that AZD1775’s cytotoxicity is not a product of WEE1 inhibition alone. Figure 13 illustrates the optimization of AZD1775.
Figure 11.

Pyrazolopyrimidine (IV)-WEE1 interaction profile in 3D; (A) Pyrazolopyrimidine and their binding modes in WEE kinase pocket; (B) insights of overlapped poses of Pyrazolopyrimidines, (C) PDB ID: 5V5Y, (D) PDB ID: 5VD4, (E) PDB ID: 7N3U and (F) PDB ID: 5VD5, (G) PDB ID: 5VD7, (H) PDB ID: 5VD8, (I) PDB ID: 5VD9.
Figure 12.

Pyrazolopyrimidine-WEE1 interaction profile in 2D. (A) PDB ID: 5VD4, (B) PDB ID: 5VD5, (C) PDB ID: 5VD7, (D) PDB ID: 5VD9, and (E) PDB ID: 5VDA.
Huang et al. investigated the effect of altering the 2-amino substitutions and pyridine group of AZD1775 on its selectivity, potency, and acceptable ADMET and PK properties. Introducing bicyclic substitutions on 2-amine has indelibly increased its potency but at the expense of intrinsic microsomal clearance with no significant increase in its selectivity.125 On the other hand, introducing a cyclopentapyridyl with a 3°-OH has indeed increased potency and selectivity while maintaining excellent druglike properties, including low intrinsic clearance.125 Additionally, the (R)-enantiomer of 3°-OH is more potent relative to its counterpart as the (R)-alcohol interacts with D463 (Figure 12D,E). Moreover, an ethyl group on cyclopentapyridyl (71) is more beneficial than the methyl group, while other bulkier alkyl groups showed a drastic decrease in potency (Figures 13 and 14A,B). Furthermore, modifications to the allyl group had a detrimental effect on activity, further validating the importance of this group. Eventually, ZN-c3 (71) was proposed, which is highly selective (IC50: WEE1 = 3.8 nM, PLK1 = 227 nM) and also shows sound cytotoxicity in H23 cell lines (IC50: ZN-c3 = 103 nM, AZD1775 = 122 nM).125 Moreover, cyclized AZD1775 has retained its allyl group as well as its tert-alcohol; hence, its binding mode ought to be similar to that of AZD1775. However, cyclization-associated intramolecular adjustments might restrain its binding efficiency in the WEE1 pocket. The cyclization approach may also disrupt the π–π stackings of SC0191 (70) with key F433. The cyclization of AZD1775 may improve the lipophilicity of the analogue, which might play a critical role in the determination of binding free energy (Figure 14C).
Figure 14.

(A) 2D Interaction profile of AZD1775 (67) (PDB ID: 5V5Y), (B) ZN-c3 (71) (PDB ID: 7N3U), respectively, and (C) interaction profile of SC0191 (70) with WEE1’s activie site residues.
A recent study by Guler et al. investigated the selectivity associated with the allyl group of AZD1775. In PLK1, the cavity accommodating the allyl group is narrower as the gatekeeper is L130. Therefore, the bulkier groups are not tolerated. Cyclic alkenes (67a) retained WEE1 activity while improving selectivity against PLK1. Other unsaturated and saturated groups displayed decreased WEE1 activity.126 Furthermore, this study determined myelosuppression as an on-target consequence (independent of PLK1 inhibition) of WEE1 inhibition using the Colony forming unit-megakaryocyte assay (CFU-Mk).126 Thus, this study offers a new scope for further optimization. A few analogues of pyrazolopyrimidine (80–83) and pyrrolopyrimide (84, 85), along with their inhibitory constant values, are incorporated in Figure 15.
Figure 15.

Pyrazolopyrimidine (80–83) and pyrrolopyrimidine (84 and 85) analogues.
2.3.2. Synthesis of Pyrazolopyrimidine Analogues
Alkyl hydrazine, when reacted with the ester 20 in the presence of a base like DIPEA (N,N-diisopropylethylamine) in THF results in the formation of an intermediate, which is reacted with EtOH to undergo cyclization in the basic medium to give the pyrazolopyrimidine moiety 72. Intermediate 72 is oxidized by PPMS (potassium peroxymonosulfate complex salt) in acetonitrile to give sulfoxide 73. In turn, 73 is reacted with an aryl-substituted amine in a basic medium, followed by a reaction with an aryl or alkyl bromide via Cu-catalyzed Ullman coupling or Chanlam coupling to give MK1775 analogues (IV) (Scheme 10).127
Scheme 10. Synthesis of Pyrazolopyrimidines (IV).

Adapted from Minh Pham et al.127
Another method to synthesize IV is by coupling the ester 20 with an alkyl bromide, which gives disubstituted intermediate 74, and copper acetate and K2CO3 in dioxane catalyze the reaction. Intermediate 74 undergoes oxidization by PPMS in acetonitrile to produce NA = not available, which in the presence of a base like DIPEA in toluene reacts with an aryl amine to form IV (Scheme 11).127
Scheme 11. Synthesis of Pyrazolopyrimidines.

Adapted from Minh Pham et al.127
Yu et al. patented a new synthesis method for the preparation of AZD1775. Here, the ester 20 is reacted with hydrazine; then an aryl is substituted at “N” to give mono aryl substituted intermediate 76, which is reacted with an allyl halide followed by oxidation and N-alkylation to produce AZD1775 (67) (Scheme 12).128
Scheme 12. Synthesis of AZD1775.

Adapted from Yu et al.128
Allyl pyrazolopyrimidine (77) is substituted with pyridinylhexenol (78) in the presence of 1,2-dimethylethylenediamine, methyl acetate, and potassium carbonate in dioxane to give 79. Alkene metathesis using Grubbs second generation catalyst in toluene, oxidation of the methylthio group, followed by reaction with methylphenylpiperazine in DIPEA generated the macrocyclic analogue, SC0191 (70) of AZD1775 (Scheme 13).129
Scheme 13. Synthesis of Macrocyclic Analogue of AZD1775 (70).
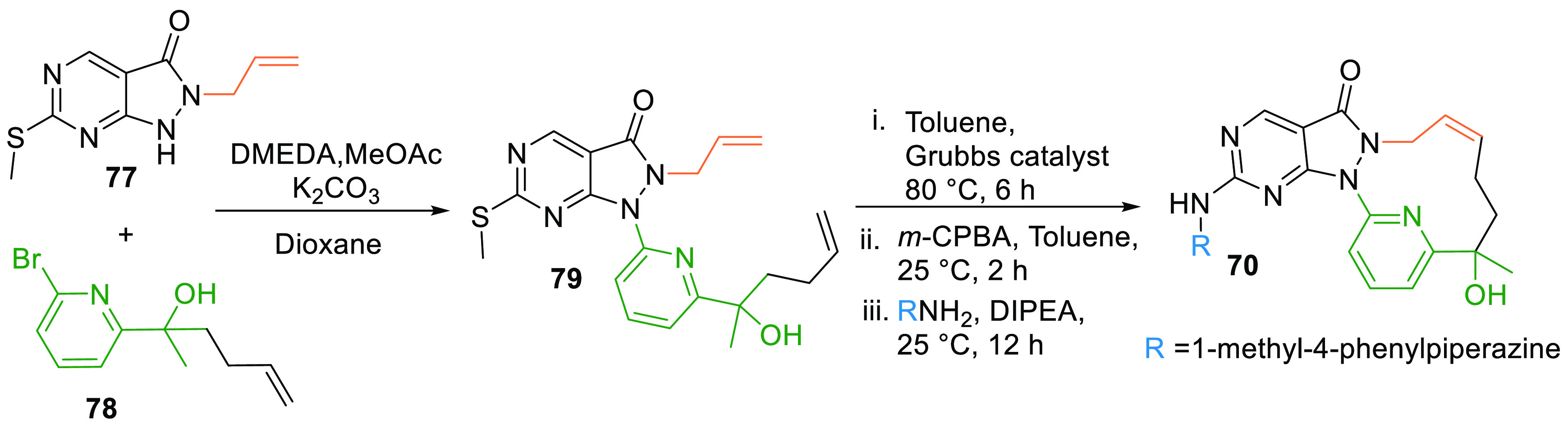
Adapted from Qian et al.129
2.4. Pyrrolopyrimidine Analogues
The significant binding of AZD1775, a pyrazolopyrimidine class of analogue, with ATP binding residues via H-bond formation with C379 and N379, as well as a hydrophobic network with pocket residues like I305, F443, and V313, led to the design of another series of WEE1 kinase inhibitors, pyrrolo[2,3-d]pyrimidine scaffold (XXI and XXII) (Figure 16). The pyrrolo[2,3-d]pyrimidine core proved to be a preferential scaffold with micromolar potency against WEE1 inhibition in biological enzyme assays and cellular anticancer screening methods.130 A few analogues (84 and 85) are shown in Figure 15.
Figure 16.

Optimization of the pyrrolo[2,3-d]pyrimidine scaffold as WEE1 kinase inhibitors (adopted from Chen et al.130).
2.4.1. Structure–Activity Relationship and Optimization Approaches of Pyrrolopyrimidines
SAR studies with various chemical modifications are being investigated following the optimization of the pyrrolo[2,3-d]pyridmidine scaffold as a potential WEE1i.130 WEE1 inhibition and anticancer activity of isopropyl and the effect of the sulfoximine group was investigated as fundamental in retaining the efficacy.
Also, isosterically similar alkyl, cyclopropyl, and alkoxy chains on pyrrolo[2,3-d]pyrimidines and piperazine incorporation, along with their potencies, were monitored (Figures 15, 16).130 Later, it was discovered that the fluoro-group adjacent to the alkyl chain on the core scaffold showed weak WEE1i (XXI) and loss of anticancer activity in the NCI-HI299 cell line. The design strategy, optimization of the pyrrolo[2,3-d]pyrimidine core as an emerging scaffold, and SAR studies against WEE1 kinase inhibition showed that the propyl alcohol and sulfoximine moieties on the pyridine (XXII) were crucial in imparting the anticancer activity, whereas the fluoro-group on the optimized core reduced the anticancer effect. Additionally, the pirazinyl and piperazinyl substituted amines occupy the hydrophobic cavity formed by I305, F443, and V313 residues of WEE1 kinase and furnish the H-bonds as well as charged interactions through the quaternary system, which is not seen in the case of morpholines/spirosystem.130 Pyrrolopyrimidines retain the aminopyrimidine ring of pyrazolopyrimidines, and thus the interactions with C379 are preserved. However, removing carboxyl group has resulted in a loss of interaction with the N376 residue, contributing to its decreased potency. On the other hand, the fluoro-substitution further reduced the potency due to harmful interactions with N376 and its surrounding residues. Replacing the isopropanol substitution on pyridine with iminodimethylsulfanone has decreased the potency because the bad interactions eclipse the good interactions with D463, K328, and other neighboring residues (Figure 17).130
Figure 17.

Relative interaction profile of pyrrolopyrimidine scaffolds (XXI and XXII) and AZD1775. (A) Pyrrolopyrimidine scaffolds with 3°-alcohol substituted pyridine and (B) pyrrolopyrimidine scaffolds with iminodimethylsulfanone-substituted pyridine.
2.4.2. Synthesis of Pyrrolopyrimidine analogues
The chemical synthesis of the pyrrolo[2,3-d]pyrimidine scaffold (XXI) involves the reaction of dichloro-substituted pyrimidine 86 and 2-amino-6-subsitituted (propyl alcohol or sulfoximine) pyridine 87 under reflux conditions to yield intermediate 88. The pyrimidine intermediate 88 upon CuI and Pd catalyzed reaction with an alkyne undergoes cyclization to afford desired scaffold 89, which on subsequent reaction with N-methylpiperpazinyl amine afforded the pyrrolo[2,3-d]pyridmidine class (XXI) of WEE1 kinase inhibitor with either propyl alcohol and sulfoximine substitution (Scheme 14).130
Scheme 14. Synthesis of Pyrrolopyrimidines (XXI).

Adapted from Chen et al.130
Fluorination of key pyrrolopyrimidine intermediate 89 using Selectfluor afforded the fluorinated derivative 90 and which, upon subsequent coupling with 4-(4-methylpiperazin-1-yl)aniline, afforded the weak WEE1 kinase inhibitor XXII (Scheme 15).130
Scheme 15. Synthesis of Fluoro-Pyrrolopyrmidines (XXII).

Adapted from Chen et al.130
2.5. Pyrimidopyrimidine
Pyrimidopyrimidine scaffold (Xb) was first mentioned in a patent as a WEE1i by Bamba et al. (2008).131 Later, other analogues of this scaffold were synthesized and tested for WEE1 inhibitory action (Figure 18). Additionally, Debio-0123 (91) (WEE1: IC50 = 41 nM, PLK1: IC50 = >10,000 nM) of this series is a potent and selective WEE1i from Debiopharm. It is in phase-I clinical trials for monotherapy (NCT05109975) and combination therapy with Carboplatin (NCT03968653) against advanced solid tumors.
Figure 18.

Pyrimidopyrimidine analogue and pyrimidine-based tricyclic analogue.
Debio-0123 (91) showed excellent target engagement with dose and time-dependent increase in γH2AX and histone H3 concentrations (indicating DNA damage). Though all tested doses were well tolerated, studies on various cell lines and animal species recommended a dosage of 30 mg/kg.132 Another study on debio-0123 combined with carboplatin and etoposide observed an increase in apoptosis and a decline in tumor growth in SCLC in vitro and in vivo. Moreover, the study concluded that debio-0123 sensitize SCLC tumors to the corresponding DDAs.133 Lately, a new phase-I, II trial is launched to identify its recommended phase-II dose (RP2D), safety, tolerability, and efficacy in combination with Temozolomide and radiation against Glioblastoma (IDH wild type) and grade-III Astrocytoma (NCT05765812). This study deduced debio-0123 as an effective agent against glioblastoma in vitro. Furthermore, as a monotherapy, the oral administration of debio-0123 penetrated blood-brain barrier and inhibited tumor growth in vivo exceptionally well, thus highlighting debio-0123’s therapeutic potential.134
2.5.1. Synthesis of Pyrimidopyrimidine
Aryl-isocyanate is added to a mixture of the amine 98 and NaH in DMF at room temperature and stirred to produce a 6N-aryl pyrimidopyrimidine-2,4-dione moiety 99. In the first route, 99 is oxidized by m-CPBA, followed by reacting with an aryl amine in the presence of the base to give 102 via the formation of an intermediate 100. 4N-alkylation by an alkyl halide in diazabicycloundecene (DBU) gives Xa. In another route, 99 first undergoes 4N-alkylation to give 101, followed by oxidation with m-CPBA, then reacted with an aryl amine in the presence of DIPEA to produce Xa. Intermediate 101 can also be synthesized by reacting 20 with an alkyl amine in the basic environment, followed by reacting with aryl isocyanide (Scheme 16).135
Scheme 16. Synthesis of Pyrimidopyrimidine (Xa).

Adapted from Bamba et al.135
Analogues of 93 (Xb) can be synthesized by reacting 4-amino-2-chloropyrimidin-5-carbonitrile (103) with isocyanate in base (NaH). This reaction gives imino-pyridopyrimidine moiety 104, which undergoes 4N-alkylation by alkyl halide to generate a disubstituted intermediate 105, then undergoes amination by reacting with tosylate monohydrate to produce Xb (Scheme 17).136
Scheme 17. Synthesis of Pyrimidopyrimidine (Xb).

Adapted from Yoshizumi et al.136
4-Amino-2-methylsulfanyl-pyrimidin-5-carbonitrile (106) first reacts with an aryl amine to give 2°-aminopyrimidine 107, which is first oxidized by m-CPBA, then reacts with an aryl amine under basic conditions to give 2-aryl substituted pyrimidine 108. Xb is produced when 108 is allowed to react with isocyanate (Scheme 18).136
Scheme 18. Synthesis of Pyrimidopyrimidine.

Adapted from Yoshizumi et al.136
2.6. Pyrimidine-Based Tricyclics: De Novo Design and Optimization
However, de novo design and optimization of the pyrimidopyrimidine scaffold afforded the pyrimidine-based tricyclic (PBT) scaffold (XI,Figure 19). A 5-membered heterocyclic ring is fused to the pyrimidopyrimidine ring, which is confined between V313 and F433 like the pyridine ring of AZD1775, and these analogues (PBT) exhibit good potency, PK properties, and oral antitumor efficacy when tested in murine xenograft models.137 This series offered IMP7068 (95) from Impact Therapeutics, a potential phase-I clinical candidate against advanced solid tumors in monotherapy (NCT04768868). The trial so far recommends doses up to 160 mg to observe sound preliminary antitumor activities and pharmacodynamics; additionally, well-tolerated treatment-related toxicity profile with negligible dose-limiting toxicities was observed.138
Figure 19.
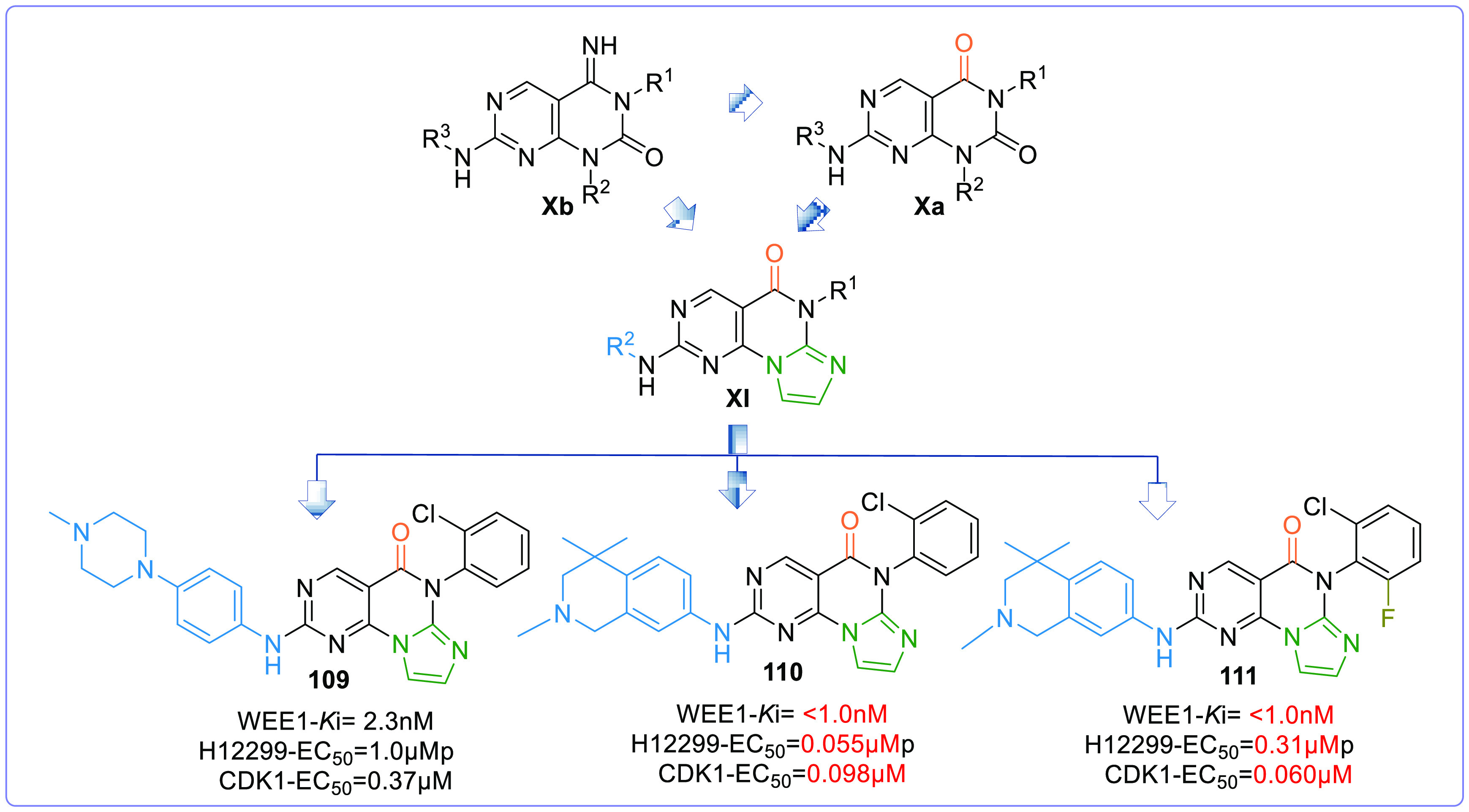
Optimization of PBT (adapted from Tong et al.137).
2.6.1. Structure–Activity Relationship and Optimization Approaches of Pyrimidine-Based Tricyclics
2-Amine and the 3N of the pyrimidine ring are associated with C379 through H-bonds. While the 5-carbonyl ‘O’ interacts with the N376-gatekeeper residue.137 The 2-amino substitutions extend out of the pocket and are solvent-exposed. The aryl substitution on the 2-amino group is very essential as it is sandwiched between I305 and G382 and forms π–σ interactions.137 The 6N-substituted benzene remains perpendicular to the tricyclic ring and fits into the lipophilic pocket beside the gatekeeper. Similar to pyrrolocarbazoles, these tricyclic compounds also prefer a 2′-Cl, but a 2′-Cl, 6′-F substitution resulted in increased potency (Figure 19).137 The substitutions to aniline must contain at least one “N”. However, as shown in Figure 19, tetrahydroisoquinoline with gem-dimethyl or cyclopropyl elevates the activity.137Figure 20 shows the overlap image of pyrimidopyrimidine and pyridine-based tricyclic analogues in the ATP pocket of WEE1.
Figure 20.

Pyrimidopyrimidine (91) and pyrimidine-based tricyclic scaffold (95) interactions with WEE1 kinase residues.
2.6.2. Synthesis of Pyrimidine-Based Tricyclics
The pyrimidine-based tricyclic core (XI) is synthesized from the key intermediate 99 when treated with phosphorus oxychloride (POCl3) in DIPEA, followed by a reaction with 2,2-dimethoxyethanamine (112) in acetonitrile at elevated temperature, then treated with acids like conc. HCl to give the key tricyclic intermediate 113. Another method of synthesizing 113 is by reacting 99 with either sodium azide at elevated temperature in DIPEA or by reaction with formyl hydrazine in acetonitrile. Moreover, reacting the ester 20 with 1-(2-aminoethyl)-3-alkyl urea 114 in the presence of DIPEA in acetonitrile and then treating with POCl3 also gives the tricyclic core 113. The prepared 113 is then oxidized with m-CPBA and then amination by reacting with an aryl amine in DIPEA to produce PBT analogues (XI) (Scheme 19).139,140
Scheme 19. Synthesis of Pyrimidine-Based Tricyclics (XI).

From 99 to 113 (adapted from Woods et al.139) and from 20 to 113 (adapted from Tian et al.140).
2.7. Other Scaffolds and Their Synthetic Schemes
Later in 2019, via ensemble docking of 20 WEE1 crystal structures, two new ligands 1 and 2 (Figure 5) were identified.141 In the same year, Hu et al. screened the ZINC database via ligand-based pharmacophore modeling and identified eight potential ligands with new scaffolds, which were further subjected to QSAR, ADMET, and binding free energy studies followed by molecular dynamics (MD) simulations and ultimately proposed 3 (Figure 5) as a potent WEE1i.142 Furthermore, vanillates (4) (Figure 5) have also exhibited micromolar WEE1 inhibition and were initially treated as an important class of WEE1i.143 Conversely, patent literature has also provided new scaffolds worth investigating. Shouyao Holdings’ SY-4835 (confidential structure) is a phase-I WEE1i being tested against advanced solid tumors to evaluate the pharmacokinetics, safety, tolerability, and anticancer activity (NCT05291182). Yang et al. were the first to identify the rhodium(III) complex 9, which induces mitotic catastrophe via induced DNA damage, as a potent single agent WEE1i in TP53 mutant (MDA-MB-231) cells.144 Olawale et al. performed in silico studies using FDA-approved drugs and proposed Dasatinib and Cangrelor as potential repurposed WEE1i.145
PF03814735 (6) is an Aurora kinase inhibitor in clinical trials. It showed nanomolar WEE1 inhibition (IC50 = 78.7 nM). It can be synthesized by reacting trifluoro-methylpyrimidine (115) with the amine 116 in the presence of ZnCl2 in ether, then treated with TEA in t-butanol and ethylene dichloride (DCE), followed by N-deprotection to give aminopyridine intermediate 117, which is first reacted with the carboxylic acid 118, then substituted with cyclopropylamine to produce PF03814735 (6) (Scheme 20).146
Scheme 20. Synthesis of PF03814735 (6).
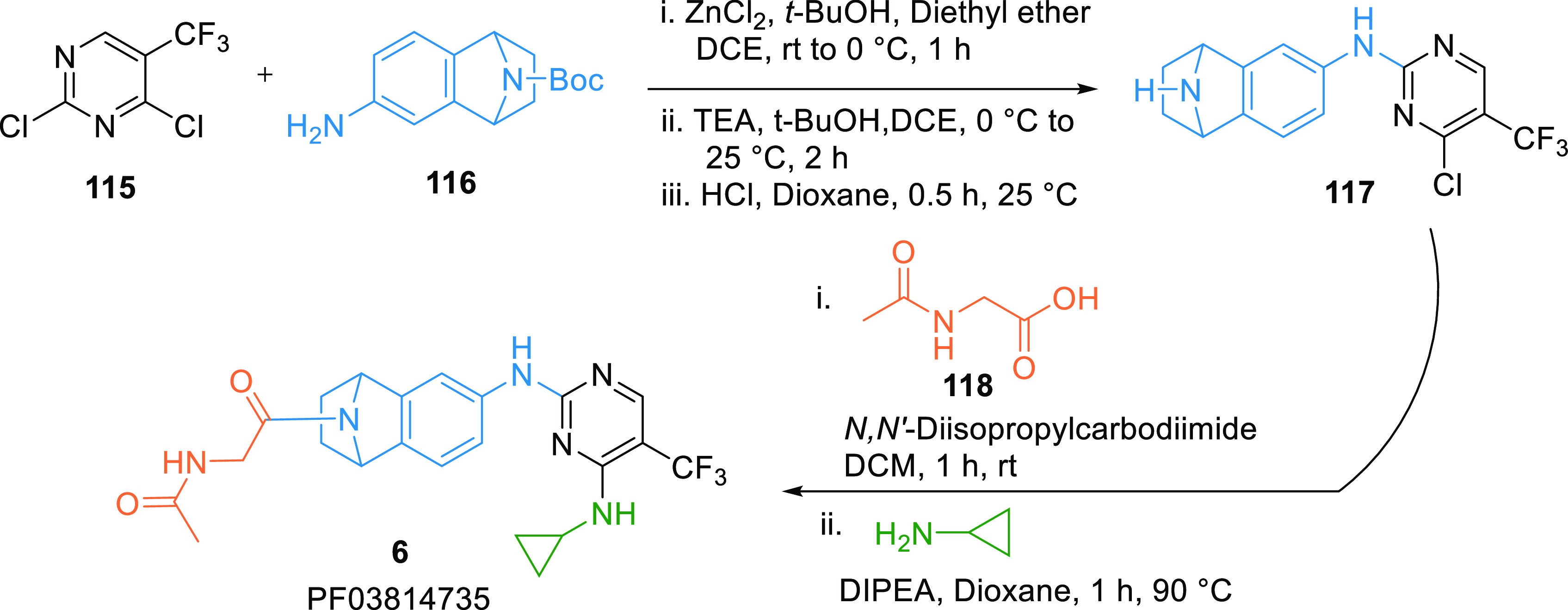
Adapted from Arcari et al.146
Bosutinib (7) is a tyrosine kinase (Abl and Src) inhibitor used for treating chronic myelogenous leuekemia. It showed IC50 = 644 ± 195 nM against WEE1 kinase. Its isomer (8) is also a dual Abl and Src inhibitor displayed IC50 = 54.8 ± 12 nM against WEE1.147 Both isomers can be synthesized by reacting chloroquinoline 119 with their respective dichlorophenylamine positional isomers 120 and 121 in pyrimidine hydrochloride and 2-methoxyethanol followed by reacting with N-methylpiperazine in KI (Scheme 21).148
Scheme 21. Synthesis of Bosutinib (7) and Its Isomer (8).
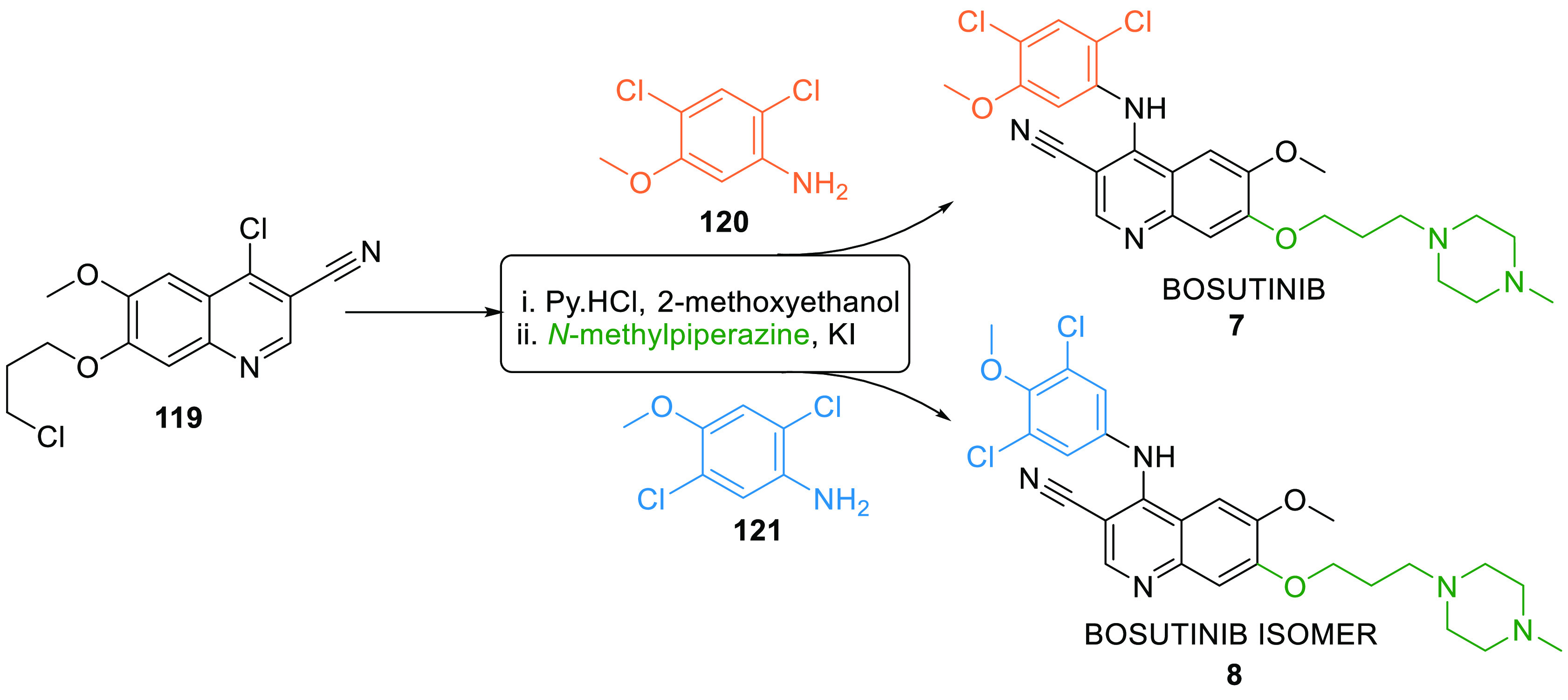
Adapted from Wang et al.148
An alkyl/aryl amine is substituted on pyrimidine 122 in the presence of a base like DIPEA to give 123, which in the presence of the palladium catalyst reacts with pyrazole 124 when exposed to microwaves giving pyrimidine-based inhibitor (I) (Scheme 22).149
Scheme 22. Synthesis of Pyrimidine-Based inhibitor (I).

Adapted from Reid et al.149
The reaction of imidazolyl pyrimidine 125 and an aryl amine is catalyzed by palladium catalyst and X-Phos, which generates the arylamine-substituted imidazoyl pyrimidine (II) (Scheme 23).150
Scheme 23. Synthesis of Pyrimidine Inhibitors (II).

Adapted from Choi et al.150
The amide 126 reacts with diiodomethane in a basic medium and undergoes cyclization to form an intermediate 127 which, when oxidized and reacted with an amine, gives the product pyrimidothiazenes (IXa) or pyrimidooxazines (IXb) (Scheme 24).151
Scheme 24. Synthesis of Pyrimidothiazenes (IXa) and Pyrimidooxazines (IXb).

Adapted from Kumar et al.151
Benzothiazineonedioxide 128 is initially reacted with N,N-dimethoxytrimethylamine and then reacted with S-methyl thiourea (initially treated with acetic acid) to produce pyrimidobenzothiazineonedioxide 129 which, when oxidized and reacted with an aryl-substituted amine, gives XIII (Scheme 25).152
Scheme 25. Synthesis of XIII.

Adapted from Miyashiro et al.152
Li et al. in 2018 performed pharmacophore modeling, docking, and MD simulations and identified a new scaffold (XVI) having an IC50 value of 22.32 μM against WEE1 and an IC50 value of 17.8 μM when tested against A-549 cell lines.153 Pyridothienopyrimidine (XVI) is synthesized by cyclization of pyridothiophene 130 with hydrazine, followed by reacting with bromophenylethanone (132) via forming an intermediate 131 (Scheme 26).154
Scheme 26. Synthesis of Pyridothienopyrimidine (XVI).

Adapted from Ahmed et al.154
2.7.1. Hybrid WEE1 Kinase Inhibitor and Its Synthesis
The hybrid set of new compounds (XV) comprises the essential structural and pharmacophore features of promising WEE1i (XI and IV) that were designed and synthesized (Figure 21). The kinase inhibition profile of hybrid analogues targeting WEE1 kinase provided a potential framework for developing of novel anticancer agents.
Figure 21.

Hypothetical design strategy of XV.
Aminoindazole 133 is substituted to ethyl 4-chloro-2-(methylthio)pyrimidine-5-carboxylate (134) in the presence of a base like DIPEA to give the amino indazole-substituted ethyl(methylthio)pyrimidine carboxylate (135), which is N-alkylated by bromo alkyl or aryl in basic medium to give 136. Base-catalyzed hydrolysis of 136 (an ester) gives an acid 137. Cyclization of obtained acid 137 catalyzed by hydroxybenzotriazole gives basic designed core 138. Oxidation followed by amination of 138 produces the hybrid scaffold XV (Scheme 27).155
Scheme 27. Synthesis of Hybrid Scaffold (XV).

Adapted from Takahashi et al.155
3. Degraders of WEE1 Kinase
WEE1i known so far are also off-target binders, i.e., apart from WEE1, they also interact with other kinases. Therefore, selective degradation of the protein using PROTACs (proteolysis targeting chimeras) is another strategy to explore.156 PROTACs are chemical chimera degraders that take over natural protein control systems. PROTACs contain three components. (1) The warhead or target binder agents that bind with the protein, (2) the E3 ligase binding moiety (EBM) that binds with a component of the protein control system (PCS), and (3) the linker that joins the warhead and the EBM.157 The PROTACs anchor the target and the component of PCS (E3 ligase), ease ubiquitination, and mark the protein for recognition by proteasomes or autophagosomes, which degenerate the target proteins.157 As PROTACs recruit the proteasomes, they are specific and selective, though the warhead is nonselective.158 Additionally, after degradation of the target protein, the released PROTACs continue degrading another target protein; thus, the PROTACs have relatively high potency. The efficiency of PROTACs is a product of inhibition and degradation of the target protein. The increase in selectivity and potency is also observed in WEE1 PROTACs.159 Li et al. constructed the first WEE1 PROTAC, ZNL-02-096 (142), by anchoring the E3 ubiquitin ligase recruiter pomalidomide (139), a cereblon binding ligand to AZD1775, which showed ten times more potency. Though it inhibits both WEE1 and PLK1 in vitro, it only degrades WEE1 in cells with maximal degradation at 100 nM.160
Aublette et al. linked VH032 (140) and pomalidomide analogues (139) that bind to VHL (Von Hippel-Lindau) E3 ligase and CRBN (Cereblon) E3 ligase, respectively, to AZD1775 using different linkers and proposed the SAR of WEE1 PROTAC degraders. According to him, the linker length and nature determine the efficiency of the degrader. He proposed using either shorter or longer linkers but not intermediate ones. The proposed degraders MA055 (143) and MA071 (145) have indeed shown increased selectivity but no apparent change in apoptosis or antiproliferative effects compared to AZD1775.156 Similar results were obtained by Zhu et al. in in vitro studies of 144, a PROTAC degrader constructed using AZD1775 and CRBN E3 ligase recruiter and in vivo studies using MV-4–11 cells showed good WEE1 degradation in the target site after 3 h treatment.159 Compound 144 has displayed sound pharmacokinetic properties in in vitro and in vivo studies. Moreover, one excess carbon in the linker of 144 contributed to an increase in WEE1 degradation compared to 142, accentuating the importance of linkers’ length to a PROTAC degrader.159 In contrast, compound libraries containing glutarimide-structural fragments were synthesized and screened for compounds promoting the ubiquitination of WEE1. WX106 (141), a CRBN competitive binder, induced the desired proteasome-dependent degradation of WEE1 and is effective against 18 cell lines tested.161 The toxicity of AZD1775 is a result of off-target and on-target inhibition, but the corresponding PROTACs offer ample selectivity and probably no off-target toxicity and tolerable on-target toxicity. Figure 22 depicts the components of PROTAC degraders and summarizes the PROTAC degraders of WEE1 kinase.
Figure 22.

Structure of WEE1 PROTACs containing AZD1775 as a warhead, E3 ligase binding moieties, and various linkers (adopted from Li et al.,160 Aublette et al.,156 and Zhu et al.)159.
3.1. Synthesis of E3 Ligase Recruiters and Their Corresponding Degraders
3.1.1. Synthesis of E3 Ligase Recruiters
Scheme 28 depicts the preparation of the VH032 analogue 150, a VHL E3 ligase recruiter. In the first step, p-bromobenzonitrile (147) reacted with 4-methylthiazole (148) in the presence of palladium acetate, KOAc, and dimethylacetamide (DMAc). Next, it is treated with NaBH4, cobalt(II)chloride in MeOH followed by reaction with Boc-Hyp-OH in DMF, then later DIPEA is added, followed by HATU to give 149. Deprotection of Boc by TFA and DCM followed by the addition of Boc-l-tert-leucine when treated with DIPEA, HATU in DMF generate 150 (Scheme 28).156
Scheme 28. Synthesis of VHL Recruiter 150.

Adapted from Aublette et al.156
On the other hand, pomalidomide analogue 153 is obtained by reacting 3-fluoro-phthalic anhydride (151) with 3-amino-2,6-piperidindionhydrobromide salt (152), when dissolved in acetic acid and heated with KOAc (Scheme 29).156
Scheme 29. Synthesis of Pomalidomide Analogue 153, a CRBN Recruiter.

Adapted from Aublette et al.156
3.1.2. Synthesis of Degraders
MA055 (143), a VHL-derived PROTAC, is produced when Boc deprotected VH032 analogue 150 undergoes amide formation with MK1775 acid analogue 154 (Scheme 30).156
Scheme 30. Synthesis of Degrader 143.

Adapted from Aublette et al.156
Li et al. in 2020 synthesized the first series of WEE1 PROTAC degraders where MK1775’s analogue 155 was coupled with Boc-linker 156 in basic medium to generate Boc-substituted MK1775’s analogue 157, when treated with TFA and DCM to remove Boc and give an amine 158. By substitution of the amine 158 to the pomalidomide analogue 153 in DIPEA and DMSO, alkyl-linked 142 and 144 can be synthesized (Scheme 31).160
Scheme 31. Synthesis of Degrader.

Moreover, CRBN PROTAC degrader is synthesized in two steps. First, the pomalidomide analogue 153 and diamine linker 159 are reacted in the presence of DIPEA and dimethylacetamide to generate the Boc-protected intermediate 160, which undergoes deprotection followed by base-catalyzed amide formation with 154 to produce MA071 (145) (Scheme 32).156
Scheme 32. Synthesis of Degrader 145.

Adapted from Aublette et al.156
CRBN recruiter 153 is substituted with an amine linker 161 and then hydrolyzed to acid 162 that is condensed with MK1775’s amine analogue 158 to give the degrader 146 containing a amide linker (Scheme 33).160
Scheme 33. Synthesis of Degrader.

Adapted from Li et al.160
4. Other Mechanisms or Strategies of WEE1 Downregulation
Modulation of WEE1 expression in tumor cells using miRNAs (microRNAs), siRNAs (small-interference or silencing RNAs), and lncRNAs (long noncoding RNAs) is another ingenious strategy. MicroRNAs (miR- or miRNAs) are small, noncoding single-stranded RNAs that target mRNA and prevent protein synthesis by recognizing and binding to the 3′UTR (3′ untranslated region).171 lncRNAs, on the other hand, act as miRNA sponges that regulate miRNA levels through actions indirectly regulating gene expression. Table 4 shows some of the lncRNAs and their target miRNAs that regulate WEE1, while miRNAs targeting WEE1 are tabulated in Table 5. Additionally, siRNAs targeting WEE1 are constructed.172 The silencing of WEE1 by siRNA has demonstrated increased chemosensitization in many cancers.70
Table 4. WEE1/miRNA/lncRNA Axis.
| S. no. | lncRNA | miRNA | refs |
|---|---|---|---|
| 1 | lncRNA DLX6-AS1 | miR-424-5p | (162) |
| 2 | lncRNA VIM-AS1 | miR-105-5p | (163) |
| 3 | lncRNA XIST | miR-16-5p | (164) |
| 4 | lncRNA FGD5-AS1 | miR-140-5p | (165) |
| 5 | lncRNA SNHG3 | miR-384 | (166) |
| 6 | lncRNA NEAT1_2 | miR-101-3p | (167) |
| 7 | circZNF91 | miR-1283 | (168) |
| 8 | lncRNA FOXD3-AS1 | miR-128-3p | (169) |
| 9 | cirSNAP47 | miR-625-5p | (170) |
Table 5. miRNA Regulating WEE1.
| S. no. | year | miRNA | cancer cells | refs |
|---|---|---|---|---|
| 1 | 2009 | miR-195 | human embryonic stem cells (hESCs) | (173) |
| 2 | 2010 | miR-516a-3p | pituitary adenomas | (174) |
| miR-128a | ||||
| miR-155 | ||||
| 3 | 2011 | miR-17 | unrestricted somatic stem cells from human cord blood (USSC) | (175) |
| miR-20a | ||||
| miR-106b | ||||
| 4 | 2011 | miR-128 | glioma cells | (176) |
| 5 | 2013 | miR-497 | neuroblastoma cells | (177) |
| 6 | 2013 | miR-424 | renal cancer cells | (178) |
| 7 | 2013 | miR-381 | ||
| 8 | 2015 | miR-17-92 cluster | leukemia | (179) |
| 9 | 2016 | miR-219-5p | RAW264.7 cells | (180) |
| 10 | 2017 | miR-503 | laryngeal carcinoma | (181) |
| 11 | 2017 | miR-194 | laryngeal squamous cell carcinoma (LSCC) | (182) |
| 12 | 2019 | miR-526b-3p | glioma cells | (183) |
| 13 | 2019 | miR-384 | laryngeal carcinoma cells | (166) |
| 14 | 2019 | miR-101-3P | HCC | (167) |
| 15 | 2020 | miR-140-5p | NSCLC | (165) |
| 16 | 2020 | miR-105-5p | glioma cells | (163) |
| 17 | 2021 | miR-16-5p | NSCLC | (184) |
| 18 | 2022 | miR-138-5P | glioma cells | (185) |
5. Adverse Events of Existing Clinical Candidates
The first clinical candidate AZD1775 has undergone many clinical trials as a single agent and in combination with radiotherapy and diverse chemotherapeutics. These trials unveiled AZD1775’s noxious toxicity profile.110 Most studies, be it monotherapy or combination therapy identified treatment emergent adverse events (TEAEs), drug-related adverse events (DRAEs), and dose-limiting toxicities (DLTs), including ≥ grade 3 myelosuppression and grade 1–3 gastrointestinal (GI) toxicity.110 Excruciating grade 4 or 3 myelosuppression is observed in almost all trials, limiting AZD1775’s potential as an ideal candidate.110 Initially, the toxicity profile of AZD1775 was inseparable from its off-target activity against PLK1, but recent studies and trials using more selective analogue ZN-c3 also showed treatment-related adverse events, including all grades of GI toxicities, ≥ grade 3 neutropenia, and other hematological toxicities.186,187 Additionally, a 2023 study to optimize selective AZD1775 analogues identified thrombocytopenia as an inevitable consequence of WEE1 inhibition and not PLK inhibition, asserting thrombocytopenia as an on-target toxicity.126 On the other hand, IMP7068 exhibited negligible DLTs and treatment-related adverse events (TRAEs), including grade 1/2 increased levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) and grade 1/2 diarrhea, which are tolerable.138 Other candidate Debio-0123 is currently tested against advanced solid tumors (NCT05109975, NCT03968653) in phase-I and glioblastoma (NCT05765812) phase-I, II in monotherapy and combination therapy with manageable toxicity profile.132 SY-4835 is also in phase-I trial against advanced solid tumor (NCT05291182), and its toxicity profile is not yet determined. Table 6 summarizes the adverse events exhibited by clinical candidates.
Table 6. Toxicity profile of clinical candidatesa.
| candidate | toxicities | grade |
|---|---|---|
| AZD1775 | myelosuppression | ≥3 |
| GI toxicities | 1—3 | |
| fatigue | 3 | |
| ZN-c3 | GI toxicities | all |
| neutropenia | ≥3 | |
| IMP7068 | increased ALT | 1 or 2 |
| increased AST | 1 or 2 | |
| diarrhea | 1 or 2 | |
| Debio-0123 | NA | NA |
| SY-4835 | NA | NA |
NA = not available.
6. Discussion
WEE1 is a CDK inhibition kinase with three family members: WEE1/WEE1A, WEE2/WEE1B, and MYT1/PKMYT1. This family of kinases inhibits CDKs by phosphorylating inhibitory P-sites (T14 and/or Y15), resulting in cell cycle arrest. WEE1, the mitotic regulator, mainly ensures the generation of mature daughter cells. Loss of WEE1 action results in abnormally small-sized daughter cells, ultimately leading to mitotic catastrophe. The somatic WEE1/WEE1A contains the classic kinase structure and is equipped with NLS, NES, AcM, and three PEST regions. It conserves the HxD, DxG, and AxE motifs and four RxL motifs and is furnished with P-sites at S642, T239, S123, S121, and S53, as well as other auto-P-sites, besides an acetylation site K177 enclosed within NES that regulates WEE1 activation. Additionally, binding consensus for 14-3-3, cyclin A-CDK complex, CRK, and CRM1 are present.
In addition to being a mitotic regulator, WEE1 is an epigenic modifier that inhibits H2B and blocks histone transcription. During the S and G2 phases, WEE1 is highly expressed and tightly shielded by the individual and combined efforts of 14-3-3, HSP90, and MIG6. In contrast, WEE1 is depressed during the initiation of the M-phase as it loses its shields and is subjected to hyperphosphorylation by PLK1, CDK1, and CK2 to generate phosphodegrons (PDs), which is then degraded by F-box proteins like SCF β-TrCP, Tome-1, Smurf1, and Pof1/3 during the M-phase. Additionally, WEE1 is subjected to inhibitory phosphorylation by NIM1/CDR1, BRSK1/2, and CDKs. WEE1 is highly expressed in many cancer types. Hence, suppressing WEE1 has a massive impact.
WEE1 inhibition sensitizes tumor cells to chemotherapy and radiotherapy while enhancing the antitumor activities of other kinase inhibitors. WEE1 suppression can be achieved by small molecule ATP competitive binders/WEE1i, PROTAC degraders, and noncoding RNAs. WEE1i are broadly classified as pyridopyrimidines, pyrrolocarbazoles, pyrazolopyrimidines, pyrimidopyrimidine, pyrimidine-based tricyclics (derived from pyrimidopyrimidine), and pyrrolopyrimidines, and their synthesis strategies are provided. Pyridopyrimidines and pyrrolocarbazoles exhibited a broad spectrum of kinase activity and were thus eliminated from the course of preclinical studies. The pyrazolopyrimidines series, however, offers two clinical candidates in phase-I, II, namely, AZD1775/MK1775/Adavosertib and ZN-c3/Azenosertib. Additionally, Debio-0123 (pyrimidopyrimidine), IMP7068 (PBT), and SY-4835 (confidential structure) are in phase-I clinical trials. All series bind to C379, N376, and F433 residues. 2-Amine substitutions are solvent-exposed (except pyrrolocarbazoles), and the other aromatic ring substitutions fill a small hydrophobic pocket near N376; this ring prefers 2′-Cl or 2′,6′-dichloro substitution (except pyrazolopyrimidines). The literature and patent study also unveiled other scaffolds showing WEE1 inhibition. On the other hand, PROTACs are selective degraders constructed using an AZD1775 analogue as the warhead, and the constructed PROTACs displayed better selectivity, potency, and anticancer activity than AZD1775. Hence, PROTACs may be a better option. Additionally, the ncRNAs like miRNAs, lncRNAs, and siRNAWEE1 also modulate WEE1 expression but absence of suitable vectors hampers their applications.
Many RNAi screening studies showed WEE1 as a better target to combat cancer, and the clinical candidate AZD1775, the most selective and most studied WEE1i, displayed adverse events in clinical studies, limiting its development. Additionally, some cancers acquired resistance against AZD1775 due to overexpression of other kinases like MYT1, YAP, etc. This resistance can be restricted by using a variety of kinase inhibitors alongside WEE1i as a form of combination therapy. However, ZN-c3, an analogue of AZD1775, is relatively selective for WEE1 and is in phase-I, II clinical trials. Nevertheless, it is in its early stages and cannot be considered a better alternative. Like ZN-c3, other clinical candidates, SY-4835, IMP7068, and Debio-0123, are in the early stages; thus, a relative best pick is not possible. Hence, further studies of existing WEE1 clinical candidates, optimization of WEE1i, and the design of new inhibitors may boost this strategy to new heights.
7. Challenges
WEE1 has become a promising anticancer target, and many inhibitors are being discovered and constructed. (i) WEE1, being a kinase, is a tricky target. (ii) WEE1i bind in the ATP binding pocket of WEE1. However, all kinases have ATP binding pockets; hence, WEE1i are less likely to be selective. (iii) WEE1i are also highly nonspecific (off-target). (iv) Acquired resistance against WEE1i due to mechanisms other than mutations in the WEE1 protein. (5) Eradication of on-target myelosuppression.
8. Opinion of Interest
WEE1 is one of the checkpoint kinases responsible for the mitotic delay. WEE1 has two functions: (1) to ensure the maturation of daughter cells before mitosis and (2) to ensure DNA repair before mitosis. Its absence causes mitotic catastrophe. In an account of its mitotic regulatory function, WEE1 is highly expressed in many types of cancers, displaying its role in cancer cell survival and growth. Therefore, targeting WEE1 is a promising strategy in the battle against cancer. Since the discovery of the first WEE1i PD0166285 in 2001, WEE1 inhibitory effects in cancer cells have been studied, and satisfactory anticancer effects have been observed. Many RNAi kinome screenings have also identified WEE1 as a better target. Moreover, AZD1775, the most studied clinical candidate, shows many adverse effects in monotherapy and combination therapy. Additionally, in monotherapy desensitization of cancers to AZD1775 is seen; thus, combination therapy is a better option.
Besides boosting anticancer activity, combination therapy also enhances potency that might reduce dose-limiting toxicities. In addition to DNA-damaging agents, WEE1 is also combined with PARP and ATR inhibitors. However, the G2-M checkpoint is not single-handedly regulated by WEE1, but MYT1 also plays a key role. Hence, combined WEE1 and MYT1 inhibition may be a better option, given MYT1’s overexpression in some WEE1i-resistant cancers. However, these kinases are equally important in normal cells with high replication; thus, their inhibition may be counterproductive as inhibition of WEE1 alone resulted in more severe myelosuppression. Additionally, WEE1i, along with mTOR, MYC, YAP, MET, and HDAC inhibition, offers an alternate method that is effective against the acquired resistance to WEE1i. Furthermore, simultaneous inhibition of WEE1 and HSP90/HDAC/ATR may be a good approach as they directly or indirectly regulate other checkpoint kinases, enhancing the cytotoxicity even in cancers resistant to WEE1i. Nevertheless, it is uncertain if these combinations result in a tolerable toxicity.
On the other hand, PROTACs may be a better alternative to inhibitors as they significantly enhance potency and selectivity, which might reduce off-target and dose-limiting toxicities. However, the studies have been limited in this direction. Additionally, gene-silencing therapies with RNAi are also effective, but ideal vectors are not available, thus limiting their application. To enhance WEE1 inhibition, compromising its stability is also an alternative. WEE1 stabilizers like MIG6 and 14-3-3, if inhibited, may contribute to the cause. Additionally, investigating the relationship between PD-L1 and WEE1 has opened a new path to treat SCLC, and ongoing clinical trials could afford possibilities. However, choosing precise WEE1i and PD-L1 inhibitors (either MAB or small molecule) as a combination therapy in the clinical investigation is a complicated task.
9. Future Perspectives
This review presents a diverse range of compounds exhibiting potent WEE1 inhibition. Among these molecules, certain compounds have been discontinued or advanced through clinical trials, while most of the inhibitors have not been progressed beyond initial testing. Consequently, there is a need for additional studies aimed at identifying and optimizing novel lead compounds from existing but not yet fully evaluated inhibitors. Notably, the extensively studied clinical candidate, AZD1775, was abandoned due to its dire toxicity profile. However, its more selective derivative, ZN-c3, demonstrated a superior preclinical profile. Nonetheless, its clinical data is sparse, including information on its toxicity profile. The other clinical candidates, IMP7068 and Debio-123, are also at the premium with limited data. A recent study however, identified the most alarming toxicity, myelosuppression as a consequence of WEE1 inhibition. However, adequate studies are to be conducted to authenticate this notion. Therefore, additional fugitive trials followed by comprehensive long-term studies are warranted. PROTACs are peerless in terms of their potency and selectivity, presenting an opportunity for WEE1 degradation with little or no off-target clinical toxicity. However, this potential needs to be confirmed. While miRNA therapy has shown promising results, further investigations in this direction are needed before a cancer therapeutic can be developed. Conversely, emerging concerns regarding WEE1i resistance have become apparent. Therefore, additional studies aiming to deepen our understanding of WEE1-associated pathways are needed. Furthermore, it has been substantiated that cancer types with TP53 mutations are more liable to WEE1 inhibition. However, other cancer types also exhibit sensitivity to WEE1 inhibition, while certain TP53 mutant types have developed resistance. Thus, an exhaustive analysis and systematic categorization of patient profiles are imperative to identify the beneficiary of this inhibition.
10. Conclusion
WEE1, the mitotic regulator and epigenetic modifier, is elected as a prime anticancer target. It is a typical kinase that is highly expressed and stabilized during the G2 and S-phases and suppressed during the M-phase, contributing to its mitotic regulatory function. Currently, WEE1i, in combination with PARP and ATM inhibitors, is in clinical trials. Other proteins like HDAC, HSP90, and CHK1 inhibitors, etc. are other proposed combinations to curb WEE1i resistance. Clinical studies on AZD1775, including monotherapy and combination therapy with DNA damaging agents, including radiation, showed adverse effects with remarkable anticancer activity. Hence, understanding the biology of WEE1 and concept of design and discovery of WEE1i may lead to the development of better candidates. Thus, this broad-spectrum multidisciplinary overview of WEE1i unveils benefits toward the design of new focused libraries against WEE1 kinase for further investigations.
Acknowledgments
V.J.A. thanks the CSIR-Indian Institute of Chemical Technology (CSIR-IICT), Hyderabad, for the Fellowship (Project: Design and Synthesis of Novel Kinase inhibitors; MLP-0102). S.S.J. thanks CSIR for providing the infrastructure facility. IICT/Pubs./2023/009.
Glossary
Abbreviations and Acronyms
- DDRs
DNA damage responses
- ATR
Ataxia-Telangiectasia mutated and Rad3-related, single-strand DNA breakage
- ATM
Ataxia-Telangiectasia mutated, double-strand DNA breakage
- CHK1/2
checkpoint kinase 1/2
- CDC kinases
cell division cycle kinase
- CDK1/2
cell division kinases 1/2
- S/T kinases
Serine/threonine kinases
- PDB
protein data bank
- NRD
N-terminal regulatory domain
- KD
central kinase domain
- CRD
C-terminal regulatory domain
- P-site
phosphorylation site
- PLK1
Polo-like kinase 1
- NLS
nuclear localization signal
- CK2
casein kinase 2
- NTD
N-terminal domain
- CTD
C-terminal domain
- BRSK1/2
BR serine/threonine kinase 1/2
- AKT
AKT kinase or Protein Kinase B
- CRM1
chromosomal maintenance 1 or exportin-1
- NES
nuclear export signal
- CRK
CRK adaptor protein
- AcM
acetylation motif
- MPF
mitosis promoting factor
- pCDK
phosphorylated CDK
- HSP90
heat shock protein 90
- SKP2
S-phase kinase associated protein 2
- FBXL1
F-box and leucine-rich repeat protein 1
- EME1
essential meiotic structure-specific endonuclease 1
- MIG6
mitogen-inducible gene 6
- c-Fos/AP1
c-Fos/Activator Protein 1
- NIM1
nonexpresser of PR genes 1
- CDR1
cerebellar degeneration Related Protein 1
- SCF
skp1/cul1/F-box
- β-TrCP
β-transduction repeat-containing protein
- Tome-1
trigger-of-mitotic-entry 1
- Smurf1
Smad ubiquitination regulatory factor 1
- Pof1/3
promoter of filamentation 1/3
- PIN1
peptidyl-prolyl cis–trans isomerase NIMA-interacting 1
- GCN5
general control non-depressible 5
- SIRT1
sirtuin 1
- γ-H2AX
histone family protein X
- CCNE1
cyclin E1
- NSCLC
nonsmall cell lung cancer
- DDAs
DNA damaging agents
- WEE1i
WEE1 inhibitors
- CIN
chromosomal instability
- BRCA
breast cancer gene
- RAS
rat sarcoma virus gene
- ATRX
ATP-dependent helicase ATRX, X-linked helicase II
- EGFR
epidermal growth factor receptor
- PARP
poly (ADP-ribose) polymerase
- PARPi
PARP inhibitors
- CHK1i
CHK1 inhibitors
- AML
acute myeloid leukemia
- TNBC
triple-negative breast cancer
- ATRi
ATR inhibitors
- HDAC
histone deacetylase
- HSP90i
HSP90 inhibitors
- c-MYC
cellular-myelocytomatosis gene
- PD-L1
programmed cell death ligand 1
- SCLC
small cell lung cancer
- c-Src
cellular-sarcoma gene
- THF
tetrahydrofuran
- DMF-DMA
Ν,Ν-dimethylformidedimethylacetal
- m-CPBA
m-chloroperoxybenzoic acid
- LAH
lithium aluminum hydride
- DMF
N,N-dimethylformamide
- HBD
hydrogen bond donor
- DDQ
2,3-dichloro-5,6-dicyano-1,4-benzoquinone
- TFA
trifluoroacetic acid
- PKC
protein kinase C
- FOXO
Forkhead box protein O
- FOXM1
Forkhead box protein M1
- APC/C
anaphase-promoting complex
- JAK2/3
Janus kinase 2/3
- MAP3K4
mitogen-activated protein kinase kinase kinase 4
- ABL1
tyrosine-protein kinase ABL1
- YES1
proto-oncogene tyrosine-protein kinase YES1
- AXL
AXL receptor tyrosine kinase
- mTOR
mechanistic target of Rapamycin kinase
- pS6
phosphorylated ribosomal protein S6
- MET
mesenchymal–epithelial transition kinase
- YAP
yes-associated protein
- FA
fanconi anemia
- CDDP
cis-diaminodichloroplatinum(II) or cis-platin
- AraC
cytarabine
- HNSCC
head and neck squamous cell carcinoma
- PK
pharmacokinetics
- MTD
maximum tolerant dose
- ADMET
absorption, distribution, metabolism, excretion, and toxicity
- CBDP
cannabidiphorol
- DIPEA
N,N-diisopropylethylamine
- PPMS
potassium peroxymonosulfate complex salt
- DBU
diazabicycloundecene
- PBT
pyrimidine-based tricyclics
- QSAR
quantitative structure–activity relationships
- MD simulations
molecular dynamics simulations
- DCE
dichloroethane
- PROTACs
proteolysis-targeting chimeras
- EBM
E3 ligase binding moiety
- PCS
protein control system
- VHL E3 ligase
Von Hippel-Lindau E3 ligase
- CRBN E3 ligase
Cereblon E3 ligase
- SAR
structure–activity relationship
- DMAc
dimethylacetamide
- HATU
hexafluorophosphate azabenzotriazole tetramethyl uronium
- DCM
dichloromethane
- DMSO
dimethyl sulfoxide
- miRNAs or miR
microRNAs
- siRNAs
small-interference or silencing RNAs
- lncRNAs
long noncoding RNAs
- 3′UTR
3′ untranslated region
- hESCs
human embryonic stem cells
- LSCC
laryngeal squamous cell carcinoma
- HCC
hepatocellular carcinoma
The authors declare no competing financial interest.
References
- Bukhari A. B.; Chan G. K.; Gamper A. M. Targeting the DNA Damage Response for Cancer Therapy by Inhibiting the Kinase Wee1. Front Oncol 2022, 12, 828684. 10.3389/fonc.2022.828684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheson C. J.; Backos D. S.; Reigan P. Targeting WEE1 Kinase in Cancer. Trends Pharmacol. Sci. 2016, 37 (10), 872–881. 10.1016/j.tips.2016.06.006. [DOI] [PubMed] [Google Scholar]
- Ha J.; Kang E.; Seo J.; Cho S. Phosphorylation Dynamics of JNK Signaling: Effects of Dual-Specificity Phosphatases (DUSPs) on the JNK Pathway. Int. J. Mol. Sci. 2019, 20 (24), 6157. 10.3390/ijms20246157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell M. J.; Raleigh J. M.; Verkade H. M.; Nurse P. Chk1 Is a Wee1 Kinase in the G2 DNA Damage Checkpoint Inhibiting Cdc2 by Y15 Phosphorylation. EMBO J. 1997, 16 (3), 545–554. 10.1093/emboj/16.3.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y.; Forbes K. C.; Wu Z.; Moreno S.; Piwnica-Worms H.; Enoch T. Replication Checkpoint Requires Phosphorylation of the Phosphatase Cdc25 by Cds1 or Chk1. Nature 1998, 395 (6701), 507–510. 10.1038/26766. [DOI] [PubMed] [Google Scholar]
- Furnari B.; Blasina A.; Boddy M. N.; McGowan C. H.; Russell P. Cdc25 Inhibited in Vivo and in Vitro by Checkpoint Kinases Cds1 and Chk1. Mol. Biol. Cell 1999, 10 (4), 833–845. 10.1091/mbc.10.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chehab N. H.; Malikzay A.; Appel M.; Halazonetis T. D. Chk2/HCds1 Functions as a DNA Damage Checkpoint in G1 by Stabilizing P53. Genes Dev. 2000, 14 (3), 278–288. 10.1101/gad.14.3.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirao A.; Kong Y.-Y.; Matsuoka S.; Wakeham A.; Ruland J.; Yoshida H.; Liu D.; Elledge S. J.; Mak T. W. DNA Damage-Induced Activation of P53 by the Checkpoint Kinase Chk2. Science (80-) 2000, 287 (5459), 1824–1827. 10.1126/science.287.5459.1824. [DOI] [PubMed] [Google Scholar]
- Russell P.; Nurse P. Negative Regulation of Mitosis by Wee1+, a Gene Encoding a Protein Kinase Homolog. Cell 1987, 49 (4), 559–567. 10.1016/0092-8674(87)90458-2. [DOI] [PubMed] [Google Scholar]
- Igarashi M.; Nagata A.; Jinno S.; Suto K.; Okayama H. Wee1+-like Gene in Human Cells. Nature 1991, 353 (6339), 80–83. 10.1038/353080a0. [DOI] [PubMed] [Google Scholar]
- Taviaux S. A.; Demaille J. G. Localization of Human Cell Cycle Regulatory Genes CDC25C to 5q31 and WEE1 to 11p15.3–11p15.1 by Fluorescence in Situ Hybridization. Genomics 1993, 15 (1), 194–196. 10.1006/geno.1993.1032. [DOI] [PubMed] [Google Scholar]
- Safran M.; Rosen N.; Twik M.; BarShir R.; Stein T. I.; Dahary D.; Fishilevich S.; Lancet D. The Genecards Suite. Pract Guid to life Sci. databases 2021, 27–56. 10.1007/978-981-16-5812-9_2. [DOI] [Google Scholar]
- McGowan C. H.; Russell P. Human Wee1 Kinase Inhibits Cell Division by Phosphorylating P34cdc2 Exclusively on Tyr15. EMBO J. 1993, 12 (1), 75–85. 10.1002/j.1460-2075.1993.tb05633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Den Haese G. J.; Walworth N.; Carr A. M.; Gould K. L. The Wee1 Protein Kinase Regulates T14 Phosphorylation of Fission Yeast Cdc2. Mol. Biol. Cell 1995, 6 (4), 371–385. 10.1091/mbc.6.4.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg D. R. Wee1-Dependent Mechanisms Required for Coordination of Cell Growth and Cell Division. J. Cell Sci. 2003, 116 (24), 4883–4890. 10.1242/jcs.00908. [DOI] [PubMed] [Google Scholar]
- Squire C. J.; Dickson J. M.; Ivanovic I.; Baker E. N. Structure and Inhibition of the Human Cell Cycle Checkpoint Kinase, Wee1A Kinase: An Atypical Tyrosine Kinase with a Key Role in CDK1 Regulation. Structure 2005, 13 (4), 541–550. 10.1016/j.str.2004.12.017. [DOI] [PubMed] [Google Scholar]
- Nakanishi M.; Ando H.; Watanabe N.; Kitamura K.; Ito K.; Okayama H.; Miyamoto T.; Agui T.; Sasaki M. Identification and Characterization of Human Wee1B, a New Member of the Wee1 Family of Cdk-Inhibitory Kinases. Genes to Cells 2000, 5 (10), 839–847. 10.1046/j.1365-2443.2000.00367.x. [DOI] [PubMed] [Google Scholar]
- Hanna C. B.; Yao S.; Patta M. C.; Jensen J. T.; Wu X. WEE2 Is an Oocyte-Specific Meiosis Inhibitor in Rhesus Macaque Monkeys. Biol. Reprod. 2010, 82 (6), 1190–1197. 10.1095/biolreprod.109.081984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solc P.; Schultz R. M.; Motlik J. Prophase I Arrest and Progression to Metaphase I in Mouse Oocytes: Comparison of Resumption of Meiosis and Recovery from G2-Arrest in Somatic Cells. Mol. Hum. Reprod. 2010, 16 (9), 654–664. 10.1093/molehr/gaq034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J. S.; Susor A.; Schindler K.; Schultz R. M.; Conti M. Cdc25A Activity Is Required for the Metaphase II Arrest in Mouse Oocytes. J. Cell Sci. 2013, 126 (5), 1081–1085. 10.1242/jcs.115592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller P. R.; Coleman T. R.; Kumagai A.; Dunphy W. G. Myt1: A Membrane-Associated Inhibitory Kinase That Phosphorylates Cdc2 on Both Threonine-14 and Tyrosine-15. Science (80-) 1995, 270, 86–90. 10.1126/science.270.5233.86. [DOI] [PubMed] [Google Scholar]
- Fattaey A.; Booher R. N. Myt1: A Wee1-Type Kinase That Phosphorylates Cdc2 on Residue Thr14. Prog. Cell Cycle Res. 1997, 3, 233–240. 10.1007/978-1-4615-5371-7_18. [DOI] [PubMed] [Google Scholar]
- Booher R. N.; Holman P. S.; Fattaey A. Human Myt1 Is a Cell Cycle-Regulated Kinase That Inhibits Cdc2 but Not Cdk2 Activity. J. Biol. Chem. 1997, 272 (35), 22300–22306. 10.1074/jbc.272.35.22300. [DOI] [PubMed] [Google Scholar]
- Liu F.; Stanton J. J.; Wu Z.; Piwnica-Worms H. The Human Myt1 Kinase Preferentially Phosphorylates Cdc2 on Threonine 14 and Localizes to the Endoplasmic Reticulum and Golgi Complex. Mol. Cell. Biol. 1997, 17 (2), 571–583. 10.1128/MCB.17.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Decker S. J.; Sebolt-Leopold J. Knockdown of Chk1, Wee1 and Myt1 by RNA Interference Abrogates G2 Checkpoint and Induces Apoptosis. Cancer Biol. Ther 2004, 3 (3), 305–313. 10.4161/cbt.3.3.697. [DOI] [PubMed] [Google Scholar]
- Watanabe N.; Broome M.; Hunter T. Regulation of the Human WEE1Hu CDK Tyrosine 15-Kinase during the Cell Cycle. EMBO J. 1995, 14 (9), 1878–1891. 10.1002/j.1460-2075.1995.tb07180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yde C. W.; Olsen B. B.; Meek D.; Watanabe N.; Guerra B. The Regulatory β-Subunit of Protein Kinase CK2 Regulates Cell-Cycle Progression at the Onset of Mitosis. Oncogene 2008, 27 (37), 4986–4997. 10.1038/onc.2008.146. [DOI] [PubMed] [Google Scholar]
- Li C.; Andrake M.; Dunbrack R.; Enders G. H. A Bifunctional Regulatory Element in Human Somatic Wee1Mediates Cyclin A/Cdk2 Binding and Crm1-Dependent Nuclear Export. Mol. Cell. Biol. 2010, 30 (1), 116–130. 10.1128/MCB.01876-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J. J.; Evans E. K.; Murakami M.; Moyer M. B.; Moseley M. A.; Vande Woude G.; Kornbluth S. Wee1-Regulated Apoptosis Mediated by the Crk Adaptor Protein in Xenopus Egg Extracts. J. Cell Biol. 2000, 151 (7), 1391–1400. 10.1083/jcb.151.7.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J. J.; Richardson D. A.; Kopf J.; Yoshida M.; Hollingsworth R. E.; Kornbluth S. Apoptotic Regulation by the Crk Adapter Protein Mediated by Interactions with Wee1 and Crm1/Exportin. Mol. Cell. Biol. 2002, 22 (5), 1412–1423. 10.1128/MCB.22.5.1412-1423.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X.; Su Q.; Xie H.; Song L.; Yang F.; Zhang D.; Wang B.; Lin S.; Huang J.; Wu M.; Liu T. SIRT1 Deacetylates WEE1 and Sensitizes Cancer Cells to WEE1 Inhibition. Nat. Chem. Biol. 2023, 19, 585. 10.1038/s41589-022-01240-y. [DOI] [PubMed] [Google Scholar]
- Parker L. L.; Atherton-Fessler S.; Piwnica-Worms H. P107wee1 Is a Dual-Specificity Kinase That Phosphorylates P34cdc2 on Tyrosine 15. Proc. Natl. Acad. Sci. 1992, 89 (7), 2917–2921. 10.1073/pnas.89.7.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollapour M.; Tsutsumi S.; Neckers L. Hsp90 Phosphorylation, Wee1 and the Cell Cycle. Cell Cycle 2010, 9 (12), 2310–2316. 10.4161/cc.9.12.12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollapour M.; Tsutsumi S.; Donnelly A. C.; Beebe K.; Tokita M. J.; Lee M.-J.; Lee S.; Morra G.; Bourboulia D.; Scroggins B. T.; Colombo G.; Blagg B. S.; Panaretou B.; Stetler-Stevenson W. G.; Trepel J. B.; Piper P. W.; Prodromou C.; Pearl L. H.; Neckers L. Swe1Wee1-Dependent Tyrosine Phosphorylation of Hsp90 Regulates Distinct Facets of Chaperone Function. Mol. Cell 2010, 37 (3), 333–343. 10.1016/j.molcel.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan K.; Fang B.; Koomen J. M.; Mahajan N. P. H2B Tyr37 Phosphorylation Suppresses Expression of Replication-Dependent Core Histone Genes. Nat. Struct Mol. Biol. 2012, 19 (9), 930–937. 10.1038/nsmb.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan K.; Mahajan N. P. WEE1 Tyrosine Kinase, a Novel Epigenetic Modifier. Trends Genet 2013, 29 (7), 394–402. 10.1016/j.tig.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan T.; Qin Q.; Nong C.; Gao S.; Wang L.; Cai B.; Zhang M.; Wu C.; Chen H.; Li T.; Xiong D.; Li G.; Wang S.; Yan S. A Novel WEE1 Pathway for Replication Stress Responses. Nat. plants 2021, 7 (2), 209–218. 10.1038/s41477-021-00855-8. [DOI] [PubMed] [Google Scholar]
- Elbaek C. R.; Petrosius V.; Benada J.; Erichsen L.; Damgaard R. B.; Sorensen C. S. WEE1 Kinase Protects the Stability of Stalled DNA Replication Forks by Limiting CDK2 Activity. Cell Rep 2022, 38 (3), 110261. 10.1016/j.celrep.2021.110261. [DOI] [PubMed] [Google Scholar]
- Martín Y.; Domínguez-Kelly R.; Freire R. Novel Insights into Maintaining Genomic Integrity: Wee1 Regulating Mus81/Eme1 2011, 2011 (6), 21. 10.1186/1747-1028-6-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domínguez-Kelly R.; Martín Y.; Koundrioukoff S.; Tanenbaum M. E.; Smits V. A. J.; Medema R. H.; Debatisse M.; Freire R. Wee1 Controls Genomic Stability during Replication by Regulating the Mus81-Eme1 Endonuclease. J. Cell Biol. 2011, 194 (4), 567–579. 10.1083/jcb.201101047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Jacobs C.; Hook K. E.; Duan H.; Booher R. N.; Sun Y. Binding of 14–3-3beta to the Carboxyl Terminus of Wee1 Increases Wee1 Stability, Kinase Activity, and G2-M Cell Population. Cell growth Differ Mol. Biol. J. Am. Assoc Cancer Res. 2000, 11 (4), 211–219. [PubMed] [Google Scholar]
- Lee J.; Kumagai A.; Dunphy W. G. Positive Regulation of Wee1 by Chk1 and 14–3-3 Proteins. Mol. Biol. Cell 2001, 12 (3), 551–563. 10.1091/mbc.12.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothblum-Oviatt C. J.; Ryan C. E.; Piwnica-Worms H. 14-3-3 Binding Regulates Catalytic Activity of Human Wee1 Kinase. Cell growth Differ Mol. Biol. J. Am. Assoc Cancer Res. 2001, 12 (12), 581–589. [PubMed] [Google Scholar]
- Lokeshwar V. B. Wee1-Hsp90 Inhibitor Combination Treatment: Molecular Therapy with Potentially Broad Applicability: Comment on: Iwai A, et Al. Cell Cycle 2012; 11:3649–55; PMID:22935698; Http://Dx.Doi.Org/10.4161/Cc.21926. Cell Cycle 2012, 11 (20), 3722–3723. 10.4161/cc.22119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman J. R.; Fukuda S.; Pelus L. M. Inhibition of Caspase-3 by Survivin Prevents Wee1 Kinase Degradation and Promotes Cell Survival by Maintaining Phosphorylation of P34Cdc2. Gene Ther Mol. Biol. 2009, 13, 264–273. [PMC free article] [PubMed] [Google Scholar]
- Sasaki M.; Terabayashi T.; Weiss S. M.; Ferby I. The Tumor Suppressor MIG6 Controls Mitotic Progression and the G2/M DNA Damage Checkpoint by Stabilizing the WEE1 Kinase. Cell Rep 2018, 24 (5), 1278–1289. 10.1016/j.celrep.2018.06.064. [DOI] [PubMed] [Google Scholar]
- Kawasaki H.; Komai K.; Ouyang Z.; Murata M.; Hikasa M.; Ohgiri M.; Shiozawa S. C-Fos/Activator Protein-1 Transactivates Wee1 Kinase at G(1)/S to Inhibit Premature Mitosis in Antigen-Specific Th1 Cells. EMBO J. 2001, 20 (16), 4618–4627. 10.1093/emboj/20.16.4618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier J.; Solomon M. J.; Booher R. N.; Bazan J. F.; Kirschner M. W. Cdc25 Is a Specific Tyrosine Phosphatase That Directly Activates P34cdc2. Cell 1991, 67 (1), 197–211. 10.1016/0092-8674(91)90583-K. [DOI] [PubMed] [Google Scholar]
- Sexl V.; Diehl J. A.; Sherr C. J.; Ashmun R.; Beach D.; Roussel M. F. A Rate Limiting Function of Cdc25A for S Phase Entry Inversely Correlates with Tyrosine Dephosphorylation of Cdk2. Oncogene 1999, 18 (3), 573–582. 10.1038/sj.onc.1202362. [DOI] [PubMed] [Google Scholar]
- Coleman T. R.; Tang Z.; Dunphy W. G. Negative Regulation of the Wee1 Protein Kinase by Direct Action of the Nim1/Cdr1Mitotic Inducer. Cell 1993, 72 (6), 919–929. 10.1016/0092-8674(93)90580-J. [DOI] [PubMed] [Google Scholar]
- Tang Z.; Coleman T. R.; Dunphy W. G. Two Distinct Mechanisms for Negative Regulation of the Wee1 Protein Kinase. EMBO J. 1993, 12 (9), 3427–3436. 10.1002/j.1460-2075.1993.tb06017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L.; Russell P. Nim1 Kinase Promotes Mitosis by Inactivating Wee1 Tyrosine Kinase. Nature 1993, 363 (6431), 738–741. 10.1038/363738a0. [DOI] [PubMed] [Google Scholar]
- Bright N. J.; Thornton C.; Carling D. The Regulation and Function of Mammalian AMPK-Related Kinases. Acta Physiol 2009, 196 (1), 15–26. 10.1111/j.1748-1716.2009.01971.x. [DOI] [PubMed] [Google Scholar]
- Müller M.; Lutter D.; Püschel A. W. Persistence of the Cell-Cycle Checkpoint Kinase Wee1 in SadA- and SadB-Deficient Neurons Disrupts Neuronal Polarity. J. Cell Sci. 2010, 123 (2), 286–294. 10.1242/jcs.058230. [DOI] [PubMed] [Google Scholar]
- Michael W. M.; Newport J. Coupling of Mitosis to the Completion of S Phase through Cdc34-Mediated Degradation of Wee 1. [Erratum to Document Cited in CA130:122527]. Science (Washington, D C) 1998, 282 (5395), 1886–1889. 10.1126/science.282.5395.1886. [DOI] [PubMed] [Google Scholar]
- Ayad N. G.; Rankin S.; Murakami M.; Jebanathirajah J.; Gygi S.; Kirschner M. W. Tome-1, a Trigger of Mitotic Entry, Is Degraded during G1 via the APC. Cell 2003, 113 (1), 101–113. 10.1016/S0092-8674(03)00232-0. [DOI] [PubMed] [Google Scholar]
- Lim H. H.; Surana U. Tome-1, Wee1, and the Onset of Mitosis: Coupled Destruction for Timely Entry. Mol. Cell 2003, 11 (4), 845–846. 10.1016/S1097-2765(03)00149-7. [DOI] [PubMed] [Google Scholar]
- Watanabe N.; Arai H.; Nishihara Y.; Taniguchi M.; Watanabe N.; Hunter T.; Osada H. M-Phase Kinases Induce Phospho-Dependent Ubiquitination of Somatic Wee1 by SCFbeta-TrCP. Proc. Natl. Acad. Sci. U. S. A. 2004, 101 (13), 4419–4424. 10.1073/pnas.0307700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei R.; Guo J.; Li M.; Yang X.; Zhu R.; Huang H.; Li K.; Zhang L.; Gao R. Smurf1 Controls S Phase Progression and Tumorigenesis through Wee1 Degradation. FEBS Lett. 2017, 591 (8), 1150–1158. 10.1002/1873-3468.12624. [DOI] [PubMed] [Google Scholar]
- Qiu C.; Yi Y.-Y.; Wu M.-J.; Sun J.-H.; Wang X.; Jin Q.-W.; Wang Y.; Lucena R. F-Box Proteins Pof3 and Pof1 Regulate Wee1 Degradation and Mitotic Entry in Fission Yeast. J. Cell Sci. 2018, 131 (3), jcs202895. 10.1242/jcs.202895. [DOI] [PubMed] [Google Scholar]
- Harvey S. L.; Charlet A.; Haas W.; Gygi S. P.; Kellogg D. R. Cdk1-Dependent Regulation of the Mitotic Inhibitor Wee1. Cell 2005, 122 (3), 407–420. 10.1016/j.cell.2005.05.029. [DOI] [PubMed] [Google Scholar]
- Olsen B. B.; Kreutzer J. N.; Watanabe N.; Holm T.; Guerra B. Mapping of the Interaction Sites between Wee1 Kinase and the Regulatory β-Subunit of Protein Kinase CK2. Int. J. Oncol. 2010, 36 (5), 1175–1182. 10.3892/ijo_00000600. [DOI] [PubMed] [Google Scholar]
- Katayama K.; Fujita N.; Tsuruo T. Akt/Protein Kinase B-Dependent Phosphorylation and Inactivation of WEE1Hu Promote Cell Cycle Progression at G2/M Transition. Mol. Cell. Biol. 2005, 25 (13), 5725–5737. 10.1128/MCB.25.13.5725-5737.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visconti R.; Palazzo L.; Della Monica R.; Grieco D. Fcp1-Dependent Dephosphorylation Is Required for M-Phase-Promoting Factor Inactivation at Mitosis Exit. Nat. Commun. 2012, 3, 894. 10.1038/ncomms1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K.; Sagata N. Mechanism for Inactivation of the Mitotic Inhibitory Kinase Wee1 at M Phase. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (10), 3753–3758. 10.1073/pnas.0607357104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo T.; Yamaguchi S.; Mitsui S.; Emi A.; Shimoda F.; Okamura H. Control Mechanism of the Circadian Clock for Timing of Cell Division in Vivo. Science (80-) 2003, 302, 255–259. 10.1126/science.1086271. [DOI] [PubMed] [Google Scholar]
- Vriend L. E. M.; De Witt Hamer P. C.; Van Noorden C. J. F.; Würdinger T. WEE1 Inhibition and Genomic Instability in Cancer. Biochim. Biophys. Acta 2013, 1836 (2), 227–235. 10.1016/j.bbcan.2013.05.002. [DOI] [PubMed] [Google Scholar]
- Hauge S.; Macurek L.; Syljuasen R. G. P21 Limits S Phase DNA Damage Caused by the Wee1 Inhibitor MK1775. Cell Cycle 2019, 18 (8), 834–847. 10.1080/15384101.2019.1593649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Jamaei A. H.; de Visscher J. G. A. M.; Subramanyam V. R.; Forouzanfar T.; Sminia P.; Doulabi B. Z.; Helder M. N. WEE1 Kinase Inhibitor MK-1775 Sensitizes Oral Tongue Squamous Cell Carcinoma Cells to Radiation Irrespective of TP53 Status. Oral Dis 2022, 10.1111/odi.14269. [DOI] [PubMed] [Google Scholar]
- Esposito F.; Giuffrida R.; Raciti G.; Puglisi C.; Forte S. Wee1 Kinase: A Potential Target to Overcome Tumor Resistance to Therapy. Int. J. Mol. Sci. 2021, 22 (19), 10689. 10.3390/ijms221910689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Witt Hamer P. C.; Mir S. E.; Noske D.; Van Noorden C. J. F.; Würdinger T. WEE1 Kinase Targeting Combined with DNA-Damaging Cancer Therapy Catalyzes Mitotic Catastrophe. Clin cancer Res. an Off J. Am. Assoc Cancer Res. 2011, 17 (13), 4200–4207. 10.1158/1078-0432.CCR-10-2537. [DOI] [PubMed] [Google Scholar]
- Seligmann J. F.; Fisher D. J.; Brown L. C.; Adams R. A.; Graham J.; Quirke P.; Richman S. D.; Butler R.; Domingo E.; Blake A.; Yates E.; Braun M.; Collinson F.; Jones R.; Brown E.; de Winton E.; Humphrey T. C.; Parmar M.; Kaplan R.; Wilson R. H.; Seymour M.; Maughan T. S. Inhibition of WEE1 Is Effective in TP53- and RAS-Mutant Metastatic Colorectal Cancer: A Randomized Trial (FOCUS4-C) Comparing Adavosertib (AZD1775) With Active Monitoring. J. Clin Oncol 2021, 39 (33), 3705–3715. 10.1200/JCO.21.01435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Yang D.; Carey J. P. W.; Karakas C.; Albarracin C.; Sahin A. A.; Arun B. K.; Guray Durak M.; Li M.; Kohansal M.; Bui T. N.; Ha M.-J.; Hunt K. K.; Keyomarsi K. Targeting Replicative Stress and DNA Repair by Combining PARP and Wee1 Kinase Inhibitors Is Synergistic in Triple Negative Breast Cancers with Cyclin E or BRCA1 Alteration. Cancers (Basel) 2021, 13 (7), 1656. 10.3390/cancers13071656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole K. A. Targeting ATRX Loss through Inhibition of the Cell-Cycle Checkpoint Mediator WEE1. Cancer Res. 2020, 80 (3), 375–376. 10.1158/0008-5472.CAN-19-3587. [DOI] [PubMed] [Google Scholar]
- Liang J.; Zhao H.; Diplas B. H.; Liu S.; Liu J.; Wang D.; Lu Y.; Zhu Q.; Wu J.; Wang W.; Yan H.; Zeng Y.-X.; Wang X.; Jiao Y. Genome-Wide CRISPR-Cas9 Screen Reveals Selective Vulnerability of ATRX-Mutant Cancers to WEE1 Inhibition. Cancer Res. 2020, 80 (3), 510–523. 10.1158/0008-5472.CAN-18-3374. [DOI] [PubMed] [Google Scholar]
- Ha D.-H.; Min A.; Kim S.; Jang H.; Kim S. H.; Kim H.-J.; Ryu H. S.; Ku J.-L.; Lee K.-H.; Im S.-A. Antitumor Effect of a WEE1 Inhibitor and Potentiation of Olaparib Sensitivity by DNA Damage Response Modulation in Triple-Negative Breast Cancer. Sci. Rep 2020, 10 (1), 9930. 10.1038/s41598-020-66018-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cetin M. H.; Rieckmann T.; Hoffer K.; Riepen B.; Christiansen S.; Gatzemeier F.; Feyerabend S.; Schoof M.; Schüller U.; Petersen C.; Mynarek M.; Rothkamm K.; Kriegs M.; Struve N. G2 Checkpoint Targeting via Wee1 Inhibition Radiosensitizes EGFRvIII-Positive Glioblastoma Cells. Radiat Oncol 2023, 18 (1), 19. 10.1186/s13014-023-02210-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S.; Yao S.; Yuan Y.; Previs R. A.; Elias A. D.; Carvajal R. D.; George T. J.; Yuan Y.; Yu L.; Westin S. N.; Xing Y.; Dumbrava E. E.; Karp D. D.; Piha-Paul S. A.; Tsimberidou A. M.; Ahnert J. R.; Takebe N.; Lu K.; Keyomarsi K.; Meric-Bernstam F. Multicenter Phase II Trial of the WEE1 Inhibitor Adavosertib in Refractory Solid Tumors Harboring CCNE1 Amplification. J. Clin Oncol Off J. Am. Soc. Clin Oncol 2023, 41 (9), 1725–1734. 10.1200/JCO.22.00830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta N.; Huang T.-T.; Horibata S.; Lee J.-M. Cell Cycle Checkpoints and beyond: Exploiting the ATR/CHK1/WEE1 Pathway for the Treatment of PARP Inhibitor–Resistant Cancer. Pharmacol. Res. 2022, 178, 106162. 10.1016/j.phrs.2022.106162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra V.; Wang A. T.; Castroviejo-Bermejo M.; Polanska U. M.; Palafox M.; Herencia-Ropero A.; Jones G. N.; Lai Z.; Armenia J.; Michopoulos F.; Llop-Guevara A.; Brough R.; Gulati A.; Pettitt S. J.; Bulusu K. C.; Nikkilä J.; Wilson Z.; Hughes A.; Wijnhoven P. W. G.; Ahmed A.; Bruna A.; Gris-Oliver A.; Guzman M.; Rodríguez O.; Grueso J.; Arribas J.; Cortés J.; Saura C.; Lau A.; Critchlow S.; Dougherty B.; Caldas C.; Mills G. B.; Barrett J. C.; Forment J. V.; Cadogan E.; Lord C. J.; Cruz C.; Balmaña J.; O’Connor M. J. Identification of a Molecularly-Defined Subset of Breast and Ovarian Cancer Models That Respond to WEE1 or ATR Inhibition, Overcoming PARP Inhibitor Resistance. Clin cancer Res. an Off J. Am. Assoc Cancer Res. 2022, 28 (20), 4536–4550. 10.1158/1078-0432.CCR-22-0568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauge S.; Naucke C.; Hasvold G.; Joel M.; Rodland G. E.; Juzenas P.; Stokke T.; Syljuasen R. G. Combined Inhibition of Wee1 and Chk1 Gives Synergistic DNA Damage in S-Phase Due to Distinct Regulation of CDK Activity and CDC45 Loading. Oncotarget 2017, 8 (7), 10966–10979. 10.18632/oncotarget.14089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X.; Kim I.-K.; Kallakury B.; Chahine J. J.; Iwama E.; Pierobon M.; Petricoin E.; McCutcheon J. N.; Zhang Y.-W.; Umemura S.; Chen V.; Wang C.; Giaccone G. Acquired Small Cell Lung Cancer Resistance to Chk1 Inhibitors Involves Wee1 Up-Regulation. Mol. Oncol 2021, 15 (4), 1130–1145. 10.1002/1878-0261.12882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukhari A. B.; Lewis C. W.; Pearce J. J.; Luong D.; Chan G. K.; Gamper A. M. Inhibiting Wee1 and ATR Kinases Produces Tumor-Selective Synthetic Lethality and Suppresses Metastasis. J. Clin Invest 2019, 129 (3), 1329–1344. 10.1172/JCI122622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi W.; Xu X.; Wang M.; Li X.; Wang C.; Sun L.; Zhao D.; Sun L. Inhibition of Wee1 Sensitizes AML Cells to ATR Inhibitor VE-822-Induced DNA Damage and Apoptosis. Biochem Pharmacol (Amsterdam, Netherlands) 2019, 164, 273–282. 10.1016/j.bcp.2019.04.022. [DOI] [PubMed] [Google Scholar]
- Jin J.; Fang H.; Yang F.; Ji W.; Guan N.; Sun Z.; Shi Y.; Zhou G.; Guan X. Combined Inhibition of ATR and WEE1 as a Novel Therapeutic Strategy in Triple-Negative Breast Cancer. Neoplasia (New York, NY, United States) 2018, 20 (5), 478–488. 10.1016/j.neo.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rødland G. E.; Hauge S.; Hasvold G.; Bay L. T. E.; Raabe T. T. H.; Joel M.; Syljuåsen R. G. Differential Effects of Combined ATR/WEE1 Inhibition in Cancer Cells. Cancers (Basel) 2021, 13 (15), 3790. 10.3390/cancers13153790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindemann A.; Patel A. A.; Tang L.; Tanaka N.; Gleber-Netto F. O.; Bartels M. D.; Wang L.; McGrail D. J.; Lin S.-Y.; Frank S. J.; Frederick M. J.; Myers J. N.; Osman A. A. Combined Inhibition of Rad51 and Wee1 Enhances Cell Killing in HNSCC through Induction of Apoptosis Associated with Excessive DNA Damage and Replication Stress. Mol. Cancer Ther 2021, 20 (7), 1257–1269. 10.1158/1535-7163.MCT-20-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L.; Zhang Y.; Chen S.; Kmieciak M.; Leng Y.; Lin H.; Rizzo K. A.; Dumur C. I.; Ferreira-Gonzalez A.; Dai Y.; Grant S. A Regimen Combining the Wee1 Inhibitor AZD1775 with HDAC Inhibitors Targets Human Acute Myeloid Leukemia Cells Harboring Various Genetic Mutations. Leukemia 2015, 29 (4), 807–818. 10.1038/leu.2014.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N.; Patel A. A.; Tang L.; Silver N. L.; Lindemann A.; Takahashi H.; Jaksik R.; Rao X.; Kalu N. N.; Chen T.-C.; Wang J.; Frederick M. J.; Johnson F.; Gleber-Netto F. O.; Fu S.; Kimmel M.; Wang J.; Hittelman W. N.; Pickering C. R.; Myers J. N.; Osman A. A. Replication Stress Leading to Apoptosis within the S-Phase Contributes to Synergism between Vorinostat and AZD1775 in HNSCC Harboring High-Risk TP53 Mutation. Clin. Cancer Res. 2017, 23 (21), 6541–6554. 10.1158/1078-0432.CCR-17-0947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy M. J.; O’Grady S.; Tang M.; Crown J. MYC as a Target for Cancer Treatment. Cancer Treat Rev. 2021, 94, 102154. 10.1016/j.ctrv.2021.102154. [DOI] [PubMed] [Google Scholar]
- Lourenco C.; Resetca D.; Redel C.; Lin P.; MacDonald A. S.; Ciaccio R.; Kenney T. M. G.; Wei Y.; Andrews D. W.; Sunnerhagen M.; Arrowsmith C. H.; Raught B.; Penn L. Z. MYC Protein Interactors in Gene Transcription and Cancer. Nat. Rev. Cancer 2021, 21 (9), 579–591. 10.1038/s41568-021-00367-9. [DOI] [PubMed] [Google Scholar]
- Iwai A.; Bourboulia D.; Mollapour M.; Jensen-Taubman S.; Lee S.; Donnelly A. C.; Yoshida S.; Miyajima N.; Tsutsumi S.; Smith A. K.; Sun D.; Wu X.; Blagg B. S.; Trepel J. B.; Stetler-Stevenson W. G.; Neckers L. Combined Inhibition of Wee1 and Hsp90 Activates Intrinsic Apoptosis in Cancer Cells. Cell Cycle 2012, 11 (19), 3649–3655. 10.4161/cc.21926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi H.; Caeser R.; Chavan S. S.; Zhan Y. A.; Chow A.; Manoj P.; Uddin F.; Kitai H.; Qu R.; Hayatt O.; Shah N. S.; Quintanal Villalonga A.; Allaj V.; Nguyen E. M.; Chan J.; Michel A. O.; Mukae H.; de Stanchina E.; Rudin C. M.; Sen T. WEE1 Inhibition Enhances the Antitumor Immune Response to PD-L1 Blockade by the Concomitant Activation of STING and STAT1 Pathways in SCLC. Cell Rep 2022, 39 (7), 110814. 10.1016/j.celrep.2022.110814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA . Approved Drug Products with Therapeutic Equivalence Evaluations. https://www.accessdata.fda.gov/scripts/cder/ob/search_product.cfm (accessed on 26-07-2022).
- Wang Y.; Li J.; Booher R. N.; Kraker A.; Lawrence T.; Leopold W. R.; Sun Y. Radiosensitization of P53 Mutant Cells by PD0166285, a Novel G2 Checkpoint Abrogator. Cancer Res. 2001, 61 (22), 8211–8217. [PubMed] [Google Scholar]
- Mizenina O. A.; Moasser M. M. S-Phase Inhibition of Cell Cycle Progression by a Novel Class of Pyridopyrimidine Tyrosine Kinase Inhibitors. Cell Cycle 2004, 3 (6), 794–801. 10.4161/cc.3.6.899. [DOI] [PubMed] [Google Scholar]
- Zeng G.; Wu W.; Zhang R.; Sun J.; Xie W.; Shen Y. 3D-QSAR and Docking Studies of Pyrido[2,3-d]Pyrimidine Derivatives as Wee1 Inhibitors. Chinese J. Chem. Phys. 2012, 25 (3), 297–307. 10.1088/1674-0068/25/03/297-307. [DOI] [Google Scholar]
- Palmer B. D.; Smaill J. B.; Rewcastle G. W.; Dobrusin E. M.; Kraker A.; Moore C. W.; Steinkampf R. W.; Denny W. A. Structure-Activity Relationships for 2-Anilino-6-Phenylpyrido[2,3-d]Pyrimidin-7(8H)-Ones as Inhibitors of the Cellular Checkpoint Kinase Wee1. Bioorg. Med. Chem. Lett. 2005, 15 (7), 1931–1935. 10.1016/j.bmcl.2005.01.079. [DOI] [PubMed] [Google Scholar]
- Mastracchio A.; Lai C.; Torrent M.; Bromberg K.; Buchanan F. G.; Ferguson D.; Bontcheva V.; Johnson E. F.; Lasko L.; Maag D.; Shoemaker A. R.; Penning T. D. Investigation of Biaryl Heterocycles as Inhibitors of Wee1 Kinase. Bioorg. Med. Chem. Lett. 2019, 29 (12), 1481–1486. 10.1016/j.bmcl.2019.04.017. [DOI] [PubMed] [Google Scholar]
- Mastracchio A.; Chunqiu Lai T. D. P.. Pyridopyrimidinone Inhibitors of Kinases. WO 2013059485 A1, 2013.
- Klutchko S. R.; Hamby J. M.; Boschelli D. H.; Wu Z.; Kraker A. J.; Amar A. M.; Hartl B. G.; Shen C.; Klohs W. D.; Steinkampf R. W.; Driscoll D. L.; Nelson J. M.; Elliott W. L.; Roberts B. J.; Stoner C. L.; Vincent P. W.; Dykes D. J.; Panek R. L.; Lu G. H.; Major T. C.; Dahring T. K.; Hallak H.; Bradford L. A.; Showalter H. D.; Doherty A. M. 2-Substituted Aminopyrido[2,3-d]Pyrimidin-7(8H)-Ones. Structure-Activity Relationships against Selected Tyrosine Kinases and in Vitro and in Vivo Anticancer Activity. J. Med. Chem. 1998, 41 (17), 3276–3292. 10.1021/jm9802259. [DOI] [PubMed] [Google Scholar]
- Woods K.; Mastracchio A.; Lai C.; Gandhi V. B.; Penning T.. Preparation of Pyridopyrimidinone as Inhibitors of Wee-1 Kinase for Cancer Therapy. WO 2013126656 A1, 2013.
- Booth R. J.; Denny W. A.; Dobrusin E. M.; Kraker A. J.; Mitchell L. H.; Smaill J. B.; Thompson A. M.; Lee H. H.; Mccarthy J.; Oliver F.; Palmer B. D.. Inhibitors of Checkpoint Kinases (Wee1 and Chk1). WO 2003091255 A1, 2003.
- Palmer B. D.; Thompson A. M.; Booth R. J.; Dobrusin E. M.; Kraker A. J.; Lee H. H.; Lunney E. A.; Mitchell L. H.; Ortwine D. F.; Smaill J. B.; Swan L. M.; Denny W. A. 4-Phenylpyrrolo[3,4-c]Carbazole-1,3(2H,6H)-Dione Inhibitors of the Checkpoint Kinase Wee1. Structure-Activity Relationships for Chromophore Modification and Phenyl Ring Substitution. J. Med. Chem. 2006, 49 (16), 4896–4911. 10.1021/jm0512591. [DOI] [PubMed] [Google Scholar]
- Smaill J. B.; Baker E. N.; Booth R. J.; Bridges A. J.; Dickson J. M.; Dobrusin E. M.; Ivanovic I.; Kraker A. J.; Lee H. H.; Lunney E. A.; Ortwine D. F.; Palmer B. D.; Quin J. III; Squire C. J.; Thompson A. M.; Denny W. A. Synthesis and Structure-Activity Relationships of N-6 Substituted Analogues of 9-Hydroxy-4-Phenylpyrrolo[3,4-c]Carbazole-1,3(2H,6H)-Diones as Inhibitors of Wee1 and Chk1 Checkpoint Kinases. Eur. J. Med. Chem. 2008, 43 (6), 1276–1296. 10.1016/j.ejmech.2007.07.016. [DOI] [PubMed] [Google Scholar]
- Smaill J. B.; Lee H. H.; Palmer B. D.; Thompson A. M.; Squire C. J.; Baker E. N.; Booth R. J.; Kraker A.; Hook K.; Denny W. A. Synthesis and Structure-Activity Relationships of Soluble 8-Substituted 4-(2-Chlorophenyl)-9-Hydroxypyrrolo[3,4-c]Carbazole-1,3(2H,6H)-Diones as Inhibitors of the Wee1 and Chk1 Checkpoint Kinases. Bioorg. Med. Chem. Lett. 2008, 18 (3), 929–933. 10.1016/j.bmcl.2007.12.046. [DOI] [PubMed] [Google Scholar]
- Sagara T.; Otsuki S.; Sunami S.; Sakamoto T.; Niiyama K.; Yamamoto F.; Yoshizumi T.; Furuyama H.; Goto Y.; Bamba M.; Takahashi K.; Hirai H.; Nishibata T.. Dihydropyrazolopyrimidinone Derivatives. US 20070254892 A1, 2007.
- Hirai H.; Iwasawa Y.; Okada M.; Arai T.; Nishibata T.; Kobayashi M.; Kimura T.; Kaneko N.; Ohtani J.; Yamanaka K.; Itadani H.; Takahashi-Suzuki I.; Fukasawa K.; Oki H.; Nambu T.; Jiang J.; Sakai T.; Arakawa H.; Sakamoto T.; Sagara T.; Yoshizumi T.; Mizuarai S.; Kotani H. Small-Molecule Inhibition of Wee1 Kinase by MK-1775 Selectively Sensitizes P53-Deficient Tumor Cells to DNA-Damaging Agents. Mol. Cancer Ther 2009, 8 (11), 2992–3000. 10.1158/1535-7163.MCT-09-0463. [DOI] [PubMed] [Google Scholar]
- Leijen S.; van Geel R. M. J. M.; Sonke G. S.; de Jong D.; Rosenberg E. H.; Marchetti S.; Pluim D.; van Werkhoven E.; Rose S.; Lee M. A.; Freshwater T.; Beijnen J. H.; Schellens J. H. M. Phase II Study of WEE1 Inhibitor AZD1775 plus Carboplatin in Patients with TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy within 3 Months. J. Clin Oncol 2016, 34 (36), 4354–4363. 10.1200/JCO.2016.67.5942. [DOI] [PubMed] [Google Scholar]
- Kong A.; Mehanna H. WEE1 Inhibitor: Clinical Development. Curr. Oncol Rep 2021, 23 (9), 107. 10.1007/s11912-021-01098-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do K.; Doroshow J. H.; Kummar S. Wee1 Kinase as a Target for Cancer Therapy. Cell Cycle 2013, 12 (19), 3348–3353. 10.4161/cc.26062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin A. D.; Li J.; Liu Y.; Hurd M. S.; Schuller A. G.; Long B.; Hirsch H. A.; Feldman I.; Benita Y.; Toniatti C.; Zawel L.; Fawell S. E.; Gilliland D. G.; Shumway S. D. Preclinical Evaluation of the WEE1 Inhibitor MK-1775 as Single-Agent Anticancer Therapy. Mol. Cancer Ther 2013, 12 (8), 1442–1452. 10.1158/1535-7163.MCT-13-0025. [DOI] [PubMed] [Google Scholar]
- Do K.; Wilsker D.; Ji J.; Zlott J.; Freshwater T.; Kinders R. J.; Collins J.; Chen A. P.; Doroshow J. H.; Kummar S. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients with Refractory Solid Tumors. J. Clin Oncol 2015, 33 (30), 3409–3415. 10.1200/JCO.2014.60.4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright G.; Golubeva V.; Remsing Rix L. L.; Berndt N.; Luo Y.; Ward G. A.; Gray J. E.; Schonbrunn E.; Lawrence H. R.; Monteiro A. N. A.; Rix U. Dual Targeting of WEE1 and PLK1 by AZD1775 Elicits Single Agent Cellular Anticancer Activity. ACS Chem. Biol. 2017, 12 (7), 1883–1892. 10.1021/acschembio.7b00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen T.; Tong P.; Diao L.; Li L.; Fan Y.; Hoff J.; Heymach J. V.; Wang J.; Byers L. A. Targeting AXL and MTOR Pathway Overcomes Primary and Acquired Resistance to WEE1 Inhibition in Small-Cell Lung Cancer. Clin. Cancer Res. 2017, 23 (20), 6239–6253. 10.1158/1078-0432.CCR-17-1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F.; Guo E.; Huang J.; Lu F.; Yang B.; Xiao R.; Liu C.; Wu X.; Fu Y.; Wang Z.; Peng S.; Lei Y.; Guo Z.; Li L.; Xi L.; Sun C.; Liu S.; Chen G. MTOR Inhibition Overcomes Primary and Acquired Resistance to Wee1 Inhibition by Augmenting Replication Stress in Epithelial Ovarian Cancers. Am. J. Cancer Res. 2020, 10 (3), 908–924. [PMC free article] [PubMed] [Google Scholar]
- Lewis C. W.; Bukhari A. B.; Xiao E. J.; Choi W.-S.; Smith J. D.; Homola E.; Mackey J. R.; Campbell S. D.; Gamper A. M.; Chan G. K. Upregulation of Myt1 Promotes Acquired Resistance of Cancer Cells to Wee1 Inhibition. Cancer Res. 2019, 79 (23), 5971–5985. 10.1158/0008-5472.CAN-19-1961. [DOI] [PubMed] [Google Scholar]
- Garcia T. B.; Uluisik R. C.; van Linden A. A.; Jones K. L.; Venkataraman S.; Vibhakar R.; Porter C. C. Increased HDAC Activity and C-MYC Expression Mediate Acquired Resistance to WEE1 Inhibition in Acute Leukemia. Front Oncol 2020, 10, 296. 10.3389/fonc.2020.00296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oku Y.; Nishiya N.; Tazawa T.; Kobayashi T.; Umezawa N.; Sugawara Y.; Uehara Y. Augmentation of the Therapeutic Efficacy of WEE1 Kinase Inhibitor AZD1775 by Inhibiting the YAP-E2F1-DNA Damage Response Pathway Axis. FEBS Open Bio 2018, 8 (6), 1001–1012. 10.1002/2211-5463.12440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J.; Liu W.; Chung H.; Izadi H.; DeJong P.; Harismendy O.; li J.; Doñate F.; Samatar A.; Lackner M.; Escoubet L.; Bunker K.; Kim D.; Jameson N.; Ozmen T. Y.; Jeong K.; Zhang D.; Pan W.-A.; Mills G. Abstract 2153: Cyclin E1 Protein Overexpression Sensitizes Ovarian Cancer Cells to ZN-C3, a Novel, Selective and Oral Bioavailable Inhibitor of Wee1. Cancer Res. 2023, 83, 2153. 10.1158/1538-7445.AM2023-2153. [DOI] [Google Scholar]
- Yang C.; Li Z.; Li Q.; Xia Y.; Chan C.-C.; Yuan X.; Wang Y.; Chen S.; Qian W.. Preclinical Evaluation of SC0191, a Small Molecule Inhibitor of Wee1 Kinase; American Society of Clinical Oncology, 2020. [Google Scholar]
- Zhu J.-Y.; Cuellar R. A.; Berndt N.; Lee H. E.; Olesen S. H.; Martin M. P.; Jensen J. T.; Georg G. I.; Schonbrunn E. Structural Basis of Wee Kinases Functionality and Inactivation by Diverse Small Molecule Inhibitors. J. Med. Chem. 2017, 60 (18), 7863–7875. 10.1021/acs.jmedchem.7b00996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheson C. J.; Casalvieri K. A.; Backos D. S.; Reigan P. Development of Potent Pyrazolopyrimidinone-Based WEE1 Inhibitors with Limited Single-Agent Cytotoxicity for Cancer Therapy. ChemMedChem. 2018, 13 (16), 1681–1694. 10.1002/cmdc.201800188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheson C. J.; Venkataraman S.; Amani V.; Harris P. S.; Backos D. S.; Donson A. M.; Wempe M. F.; Foreman N. K.; Vibhakar R.; Reigan P. A WEE1 Inhibitor Analog of AZD1775 Maintains Synergy with Cisplatin and Demonstrates Reduced Single-Agent Cytotoxicity in Medulloblastoma Cells [Erratum to Document Cited in CA164:432708]. ACS Chem. Biol. 2016, 11 (7), 2066–2067. 10.1021/acschembio.6b00466. [DOI] [PubMed] [Google Scholar]
- Huang P. Q.; Boren B. C.; Hegde S. G.; Liu H.; Unni A. K.; Abraham S.; Hopkins C. D.; Paliwal S.; Samatar A. A.; Li J.; Bunker K. D. Discovery of ZN-C3, a Highly Potent and Selective Wee1 Inhibitor Undergoing Evaluation in Clinical Trials for the Treatment of Cancer. J. Med. Chem. 2021, 64 (17), 13004–13024. 10.1021/acs.jmedchem.1c01121. [DOI] [PubMed] [Google Scholar]
- Guler S.; DiPoto M. C.; Crespo A.; Caldwell R.; Doerfel B.; Grossmann N.; Ho K.; Huck B.; Jones C. C. V.; Lan R.; Musil D.; Potnick J.; Schilke H.; Sherer B.; Simon S.; Sirrenberg C.; Zhang Z.; Liu-Bujalski L. Selective Wee1 Inhibitors Led to Antitumor Activity In Vitro and Correlated with Myelosuppression. ACS Med. Chem. Lett. 2023, 14, 566. 10.1021/acsmedchemlett.2c00481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh Pham S.; Kankanala J.; Chakravarty S.; Pujala B.; Kumar V.. Heterocyclic Compounds and Uses Thereof. WO 2022082174 A1, 2022.
- Yu Y.; Liu S.; Fan Z.. Novel Chemical Synthesis Method of Wee1 Protein Kinase Inhibitor Avaposertib. CN 113880844 A, 2022.
- Qian W.; Yang C.; Li Z.; Li J.; Li J.. Crystal Form of WEE1 Inhibitor Compound and Use Thereof. WO 2020221358 A1, 2020.
- Chen C.; Wang Y.; Hu M.-Q.; Li H.; Chen X.; Qiang G.; Sun Y.; Zhu Y.; Li B. Discovery of Pyrrolo[2,3-d]Pyrimidine-Based Molecules as a Wee1 Inhibitor Template. Bioorg. Med. Chem. Lett. 2022, 75, 128973. 10.1016/j.bmcl.2022.128973. [DOI] [PubMed] [Google Scholar]
- Bamba M.; Furuyama H.; Niiyama K.; Sakamoto T.; Sunami S.; Takahash K.; Yamamoto F.; Yoshizumi T.. Bicycloaniline Derivative. WO 2008153207 A1, 2008.
- O’Dowd C.; Gavory G.; Burkamp F.; Treder A.; Boyd C.; Harrison T.; Massière F.; Glück A.; Chardonnens C.; Zaffalon A.; Rigotti S.; Mader R.; Vuagniaux G.; Chessex A. V. Abstract 4423: Antitumor Activity of the Novel Oral Highly Selective Wee1 Inhibitor Debio 0123. Cancer Res. 2019, 79, 4423. 10.1158/1538-7445.AM2019-4423. [DOI] [Google Scholar]
- Piggott L.; Vaslin-Chessex A.; Luong N.; Tschumi B.; Vuagniaux G. Abstract 2303: The WEE1 Inhibitor Debio 0123 Enhances the Efficacy of Standard of Care DNA Damaging Agents in Lung Cancer Models. Cancer Res. 2022, 82, 2303. 10.1158/1538-7445.AM2022-2303. [DOI] [Google Scholar]
- Piggott L.; Luong N.; Massiere F.; Kunze A.; Chardonnens C.; Vaslin A. Abstract 6185: Debio 0123 Is a Selective WEE1 Inhibitor That Effectively Penetrates the Brain and Demonstrates Anti-Tumor Activity in Preclinical Models of Glioblastoma. Cancer Res. 2023, 83, 6185. 10.1158/1538-7445.AM2023-6185. [DOI] [Google Scholar]
- Bamba M.; Sunami S.. Dihydropyrimidopyrimidine Derivatives. WO 2010067888 A1, 2010.
- Bamba M.; Furuyama H.; Sakamoto T.; Sunami S.; Takahashi K.; Yamamoto F.; Yoshizumi T.. Dihydropyrimidopyrimidine Derivative. WO 2010067886 A1, 2010.
- Tong Y.; Torrent M.; Florjancic A. S.; Bromberg K. D.; Buchanan F. G.; Ferguson D. C.; Johnson E. F.; Lasko L. M.; Maag D.; Merta P. J.; Olson A. M.; Osterling D. J.; Soni N.; Shoemaker A. R.; Penning T. D. Pyrimidine-Based Tricyclic Molecules as Potent and Orally Efficacious Inhibitors of Wee1 Kinase. ACS Med. Chem. Lett. 2015, 6 (1), 58–62. 10.1021/ml5002745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.-C.; Grewal J. S.; Sommerhalder D.; Strauss J. F.; Bai L.-Y.; Shen L.; Yeh Y.-M.; Hsieh C.-Y.; Cai S. X.; Tian Y. E.; Xia H.; Zhang C.; Li B.; Zhang M.; Ma N.; Zhao L.; Ma J.; Shi H.; Yu Z. A Phase 1 Dose-Escalation and -Expansion Study of IMP7068, a WEE1 Inhibitor, in Patients with Advanced Solid Tumors. J. Clin Oncol 2022, 40, e15052 10.1200/JCO.2022.40.16_suppl.e15052. [DOI] [Google Scholar]
- Tong Y.; Penning T. D.; Florjancic A. S.; Miyashiro J.; Woods K. W.. Tricyclic Inhibitors of Kinases. US 20120220572 A1, 2012.
- Cai S. X.; Tian Y. E.. 8,9-Dihydroimidazole[1,2-a]Pyrimido[5,4-e]Pyrimidine-5(6h)-Ketone Compound. WO 2018090939 A1, 2018.
- Li Y.; Pu Y.; Liu H.; Zhang L.; Liu X.; Li Y.; Zuo Z. Discovery of Novel Wee1 Inhibitors via Structure-Based Virtual Screening and Biological Evaluation. J. Comput. Aided Mol. Des 2018, 32 (9), 901–915. 10.1007/s10822-018-0122-1. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Zhou L.; Zhu X.; Dai D.; Bao Y.; Qiu Y. Pharmacophore Modeling, Multiple Docking, and Molecular Dynamics Studies on Wee1 Kinase Inhibitors. J. Biomol Struct Dyn 2019, 37 (10), 2703–2715. 10.1080/07391102.2018.1495576. [DOI] [PubMed] [Google Scholar]
- Lamoral-Theys D.; Pottier L.; Kerff F.; Dufrasne F.; Proutière F.; Wauthoz N.; Neven P.; Ingrassia L.; Antwerpen P. Van; Lefranc F.; Gelbcke M.; Pirotte B.; Kraus J.-L.; Nève J.; Kornienko A.; Kiss R.; Dubois J. Simple Di- and Trivanillates Exhibit Cytostatic Properties toward Cancer Cells Resistant to pro-Apoptotic Stimuli. Bioorg. Med. Chem. 2010, 18 (11), 3823–3833. 10.1016/j.bmc.2010.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G.-J.; Zhong H.-J.; Ko C.-N.; Wong S.-Y.; Vellaisamy K.; Ye M.; Ma D.-L.; Leung C.-H. Identification of a Rhodium(III) Complex as a Wee1 Inhibitor against TP53-Mutated Triple-Negative Breast Cancer Cells. Chem. Commun. (Cambridge, United Kingdom) 2018, 54 (20), 2463–2466. 10.1039/C7CC09384E. [DOI] [PubMed] [Google Scholar]
- Olawale F.; Ogunyemi O.; Folorunso I. M. Repurposing Clinically Approved Drugs as Wee1 Checkpoint Kinase Inhibitors: An in Silico Investigation Integrating Molecular Docking, Ensemble QSAR Modelling and Molecular Dynamics Simulation. Mol. Simul 2022, 48 (16), 1490–1512. 10.1080/08927022.2022.2101673. [DOI] [Google Scholar]
- Arcari J. T.; Bhattacharya S. K.; Brosius A. D.; Luzzio M. J.; Nelson K. L.; Pan G.; Southers J. A. Jr.; Wishka D. G.; Xiao J.. Pyrimidine Derivatives for the Treatment of Abnormal Cell Growth and Their Preparation. WO 2007072158 A2, 2007.
- Beeharry N.; Banina E.; Hittle J.; Skobeleva N.; Khazak V.; Deacon S.; Andrake M.; Egleston B. L.; Peterson J. R.; Astsaturov I.; Yen T. J. Re-Purposing Clinical Kinase Inhibitors to Enhance Chemosensitivity by Overriding Checkpoints. Cell Cycle 2014, 13 (14), 2172–2191. 10.4161/cc.29214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y.; Shen Y.; Wang H.; Zhu C.; Zhu G.; Tian X. A New and Practical Synthesis of Bosutinib. Heterocycles an Int. J. Rev. Commun. Heterocycl Chem. 2014, 89 (12), 2806–2813. 10.3987/COM-14-13117. [DOI] [Google Scholar]
- Reid P.; Drewry D. H.; Deanda F. Jr.. Chemical Compounds. WO 2008115742 A1, 2008.
- Choi H. G.; Ko E.; Kim N. D.; Kim H.. Novel Imidazolyl Pyrimidine Derivative, Method for Preparing Same, and Pharmaceutical Composition Comprising Same as Active Ingredient for Prevention or Treatment of Cancer. WO 2018056621 A1, 2018.
- Chakravarty S.; Pham S. M.; Kankanala J.; Pujala B.; Kumar V.. Heterocyclic Compounds and Uses Thereof. WO 2020210377 A1, 2020.
- Woods K. W.; Lai C.; Penning T. D.; Miyashiro J. M.. New Chemical Entities To Be Used For Wee1 Inhibition For The Treatment Of Cancer. US 20130018045 A1, 2013.
- Li Y.; Pu Y.; Liu H.; Zhang L.; Liu X.; Li Y.; Zuo Z. Discovery of Novel Wee1 Inhibitors via Structure-Based Virtual Screening and Biological Evaluation. J. Comput. Aided Mol. Des 2018, 32 (9), 901–915. 10.1007/s10822-018-0122-1. [DOI] [PubMed] [Google Scholar]
- Ahmed E. Synthesis of New Pyrido[4″,3″:4″,5″]Thieno[2″,3″:4,5]Pyrimido[2,1-b][1,3,4]Thiadiazine Derivatives. Phosphorus Sulfur Silicon Relat Elem - PHOSPHOR SULFUR SILICON 2002, 177, 989–1000. 10.1080/10426500210671. [DOI] [Google Scholar]
- Goto Y.; Niiyama K.; Sunami S.; Takahashi K.. Pyrimidopyrimidoindazole Derivative. WO 2010098367 A1, 2010.
- Aublette M. C.; Harrison T. A.; Thorpe E. J.; Gadd M. S. Selective Wee1 Degradation by PROTAC Degraders Recruiting VHL and CRBN E3 Ubiquitin Ligases. Bioorg. Med. Chem. Lett. 2022, 64, 128636. 10.1016/j.bmcl.2022.128636. [DOI] [PubMed] [Google Scholar]
- Yin L.; Hu Q. Chimera Induced Protein Degradation: PROTACs and Beyond. Eur. J. Med. Chem. 2020, 206, 112494. 10.1016/j.ejmech.2020.112494. [DOI] [PubMed] [Google Scholar]
- Jiang B.; Wang E. S.; Donovan K. A.; Liang Y.; Fischer E. S.; Zhang T.; Gray N. S. Development of Dual and Selective Degraders of Cyclin-Dependent Kinases 4 and 6. Angew. Chemie Int. Ed 2019, 58 (19), 6321–6326. 10.1002/anie.201901336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S.; Liu J.; Xiao D.; Wang P.; Ma J.; Hu X.; Fu J.; Zhou Y.; Li J.; Lu W. Design, Synthesis, and Biological Evaluation of Wee1 Kinase Degraders. Eur. J. Med. Chem. 2022, 243, 114786. 10.1016/j.ejmech.2022.114786. [DOI] [PubMed] [Google Scholar]
- Li Z.; Pinch B. J.; Olson C. M.; Donovan K. A.; Nowak R. P.; Mills C. E.; Scott D. A.; Doctor Z. M.; Eleuteri N. A.; Chung M.; Sorger P. K.; Fischer E. S.; Gray N. S. Development and Characterization of a Wee1 Kinase Degrader. Cell Chem. Biol. 2020, 27 (1), 57–65.e9. 10.1016/j.chembiol.2019.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y.; Cang Y.; Peng B.; Lei M.; Xu Y.; Chen S.. Fused Cyclic Compound Capable of Degrading Protein and Use Thereof. WO 2021047627 A1, 2021.
- Li D.; Tang X.; Li M.; Zheng Y. Long Noncoding RNA DLX6-AS1 Promotes Liver Cancer by Increasing the Expression of WEE1 via Targeting MiR-424–5p. J. Cell Biochem 2019, 120 (8), 12290–12299. 10.1002/jcb.28493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suo S.-T.; Gong P.; Peng X.-J.; Niu D.; Guo Y.-T. Knockdown of Long Non-Coding RNA VIM-AS1 Inhibits Glioma Cell Proliferation and Migration, and Increases the Cell Apoptosis via Modulation of WEE1 Targeted by MiR-105–5p. Eur. Rev. Med. Pharmacol Sci. 2020, 24 (12), 6834–6847. 10.26355/eurrev_202006_21673. [DOI] [PubMed] [Google Scholar]
- Du R.; Jiang F.; Yin Y.; Xu J.; Li X.; Hu L.; Wang X. Knockdown of LncRNA X Inactive Specific Transcript (XIST) Radiosensitizes Non-Small Cell Lung Cancer (NSCLC) Cells through Regulation of MiR-16–5p/WEE1 G2 Checkpoint Kinase (WEE1) Axis. Int. J. Immunopathol Pharmacol 2021, 35, 205873842096608. 10.1177/2058738420966087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J.; Cai H.; Wu Y.; Fang S.; Wang D. Elevation of FGD5-AS1 Contributes to Cell Progression by Improving Cisplatin Resistance against Non-Small Cell Lung Cancer Cells through Regulating MiR-140–5p/WEE1 Axis. Gene 2020, 755, 144886. 10.1016/j.gene.2020.144886. [DOI] [PubMed] [Google Scholar]
- Wang L.; Su K.; Wu H.; Li J.; Song D. LncRNA SNHG3 Regulates Laryngeal Carcinoma Proliferation and Migration by Modulating the MiR-384/WEE1 Axis. Life Sci. 2019, 232, 116597. 10.1016/j.lfs.2019.116597. [DOI] [PubMed] [Google Scholar]
- Chen X.; Zhang N. Downregulation of LncRNA NEAT1_2 Radiosensitizes Hepatocellular Carcinoma Cells through Regulation of MiR-101–3p/WEE1 Axis. Cell Biol. Int. 2019, 43 (1), 44–55. 10.1002/cbin.11077. [DOI] [PubMed] [Google Scholar]
- Li S.; Chen J.; Fan Y.; Xu X.; Xiong M.; Qi Y.; Wu W.; Zhao Y. CircZNF91 Promotes the Malignant Phenotype of Chronic Lymphocytic Leukemia Cells by Targeting the MiR-1283/WEE1 Axis. Biomed Res. Int. 2022 2022, 2022, 2855394. 10.1155/2022/2855394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling Z.; Zhang J.; Liu Q. Oncogenic Forkhead Box D3 Antisense RNA 1 Promotes Cell Survival and Confers Temozolomide Resistance in Glioblastoma Cells through the MiR-128–3p/WEE1 G2 Checkpoint Kinase Axis. Bioengineered 2022, 13 (3), 6012–6023. 10.1080/21655979.2022.2042133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou C.; Rong F.; Zeng Y.; Zeng J.; Wei R.; Wei D. Circ-SNAP47 (Hsa_circ_0016760) and MiR-625–5p Are Regulators of WEE1 in Regulation of Chemoresistance, Growth and Invasion of DDP-Tolerant NSCLC Cells via CeRNA Pathway. Environ. Toxicol 2022, 37 (2), 224–236. 10.1002/tox.23391. [DOI] [PubMed] [Google Scholar]
- Kaikkonen M. U.; Lam M. T. Y.; Glass C. K. Non-Coding RNAs as Regulators of Gene Expression and Epigenetics. Cardiovasc. Res. 2011, 90 (3), 430–440. 10.1093/cvr/cvr097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khvorova A.; Reynolds A.; Leake D.; Marshall W.; Read S.; Scaringe S.. SiRNA Targeting WEE1 Homolog (WEE1). US 20070249819 A1, 2007.
- Qi J.; Yu J.-Y.; Shcherbata H. R.; Mathieu J.; Wang A. J.; Seal S.; Zhou W.; Stadler B. M.; Bourgin D.; Wang L.; Nelson A.; Ware C.; Raymond C.; Lim L. P.; Magnus J.; Ivanovska I.; Diaz R.; Ball A.; Cleary M. A.; Ruohola-Baker H. MicroRNAs Regulate Human Embryonic Stem Cell Division. Cell Cycle 2009, 8 (22), 3729–3741. 10.4161/cc.8.22.10033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butz H.; Likó I.; Czirják S.; Igaz P.; Khan M. M.; Zivkovic V.; Bálint K.; Korbonits M.; Rácz K.; Patócs A. Down-Regulation of Wee1 Kinase by a Specific Subset of MicroRNA in Human Sporadic Pituitary Adenomas. J. Clin Endocrinol Metab 2010, 95 (10), E181–91. 10.1210/jc.2010-0581. [DOI] [PubMed] [Google Scholar]
- Trompeter H.-I.; Abbad H.; Iwaniuk K. M.; Hafner M.; Renwick N.; Tuschl T.; Schira J.; Müller H. W.; Wernet P. MicroRNAs MiR-17, MiR-20a, and MiR-106b Act in Concert to Modulate E2F Activity on Cell Cycle Arrest during Neuronal Lineage Differentiation of USSC. PLoS One 2011, 6 (1), e16138 10.1371/journal.pone.0016138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuchty S.; Arjona D.; Li A.; Kotliarov Y.; Walling J.; Ahn S.; Zhang A.; Maric D.; Anolik R.; Zenklusen J. C.; Fine H. A. Prediction of Associations between MicroRNAs and Gene Expression in Glioma Biology. PLoS One 2011, 6 (2), e14681 10.1371/journal.pone.0014681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creevey L.; Ryan J.; Harvey H.; Bray I. M.; Meehan M.; Khan A. R.; Stallings R. L. MicroRNA-497 Increases Apoptosis in MYCN Amplified Neuroblastoma Cells by Targeting the Key Cell Cycle Regulator WEE1. Mol. Cancer 2013, 12, 23. 10.1186/1476-4598-12-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.; Duan L.; Yin G.; Tan J.; Jiang X. Simultaneously Expressed MiR-424 and MiR-381 Synergistically Suppress the Proliferation and Survival of Renal Cancer Cells---Cdc2 Activity Is up-Regulated by Targeting WEE1. Clinics (Sao Paulo) 2013, 68 (6), 825–833. 10.6061/clinics/2013(06)17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockway S.; Zeleznik-Le N. J. WEE1 Is a Validated Target of the MicroRNA MiR-17–92 Cluster in Leukemia. Cancer Genet 2015, 208 (5), 279–287. 10.1016/j.cancergen.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou W.; Zhang X.; Hu X.-Y.; Hu A.-R. MicroRNA-219–5p Inhibits Morphine-Induced Apoptosis by Targeting Key Cell Cycle Regulator WEE1. Med. Sci. Monit Int. Med. J. Exp Clin Res. 2016, 22, 1872–1879. 10.12659/MSM.895439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H.; Lian R.; Wu Z.; Li X.; Yu W.; Shang Y.; Guo X. MiR-503 Enhances the Radiosensitivity of Laryngeal Carcinoma Cells via the Inhibition of WEE1. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2017, 39 (10), 1010428317706224. 10.1177/1010428317706224. [DOI] [PubMed] [Google Scholar]
- Li P.; Yang Y.; Liu H.; Yang A.-K.; Di J.-M.; Tan G.-M.; Wang H.-F.; Qiu J.-G.; Zhang W.-J.; Jiang Q.-W.; Zheng D.-W.; Chen Y.; Wei M.-N.; Huang J.-R.; Wang K.; Shi Z.; Ye J. MiR-194 Functions as a Tumor Suppressor in Laryngeal Squamous Cell Carcinoma by Targeting Wee1. Journal of hematology & oncology 2017, 10, 32. 10.1186/s13045-017-0402-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M.; Li X.; Liu Q.; Xie Y.; Yuan J.; Wanggou S. MiR-526b-3p Serves as a Prognostic Factor and Regulates the Proliferation, Invasion, and Migration of Glioma through Targeting WEE1. Cancer Manag Res. 2019, 11, 3099–3110. 10.2147/CMAR.S192361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.; Chen Y.; Lu H.; Wang H.; Feng H.; Xu J.; Zhang B. Quercetin Radiosensitizes Non-Small Cell Lung Cancer Cells through the Regulation of MiR-16–5p/WEE1 Axis. IUBMB Life 2020, 72 (5), 1012–1022. 10.1002/iub.2242. [DOI] [PubMed] [Google Scholar]
- Gong J.; Tang Z.; Yu Z.; Deng Z.; Liu Y.; Ren N.; Wang L.; He Z. MiR-138–5p Inhibits the Growth and Invasion of Glioma Cells by Regulating WEE1. Anal Cell Pathol (Amst) 2022 2022, 2022, 7809882. 10.1155/2022/7809882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore K.Zentalis Pharmaceuticals Announces Positive Initial Clinical Data on Zn-C3, Its Wee1 Inhibitor. In Patients With Advanced OVARIAN CANCER AT AACR; Zentalis Pharmaceuticals, Inc, 2022.
- Mauro G.ZN-c3 Shows Preliminary Efficacy. Safety in Recurrent/Advanced Uterine Serous Carcinoma; OncLive, 2022.