

Abstract
Canonical histone messenger RNAs (mRNAs) are transcribed during S phase and do not terminate with a poly(A) tail at the 3′ end. Instead, the histone mRNAs display a stem-loop structure at their 3-end. Stem-loop-binding protein (SLBP) binds the stem-loop and regulates canonical histone mRNA metabolism. We previously demonstrated that exposure to arsenic, an environmental carcinogen, induces polyadenylation of canonical histone H3.1 mRNA, causing transformation of human cells in vitro. Arsenic decreased cellular levels of SLBP by inducing its proteasomal degradation and inhibiting SLBP transcription via epigenetic mechanisms. Similarly, we also reported that nickel and arsenic have similar effects on canonical histone mRNA transcription and translation. Most recently, we further demonstrated that bisphenols’ exposure increased polyadenylation of canonical histone H3.1 mRNA possibly through down-regulation of SLBP expression. This facilitates the abnormal stability of at least one canonical histone isoform (H3.1), and also increases H3 protein levels. Excess expression of canonical histones have been shown to increase sensitivity to DNA damage as well as increase the frequency of missing chromosomes and induce genomic instability. Thus, polyadenylation of canonical histone mRNA following arsenic, nickel and bisphenols exposure may contribute to metal and bisphenol-induced carcinogenesis.
1. Introduction
1.1. Regulation and function of stem-loop binding protein (SLBP)
Canonical histones, such as H3.1 are replication dependent and are the only genes in eukaryotes with mRNAs ending in a stem loop structure at the 3′ end and not in a poly(A) tail. Canonical histone pre-mRNAs have a 3’UTR end that consists of a conserved sequence of 25–26 nucleotides, including a sequence of 5 nucleotides that is located before the stem-loop, followed by the 16-nucleotide stem-loop sequence and ending with a 4–5 nucleotides after the stem-loop. In metazoan, the stem-loop sequence is evolutionary conserved (Marzluff & Koreski, 2017; Marzluff, Wagner, & Duronio, 2008). The formation of the 3′ end of the mature histone mRNA is mediated by an endonucleolytic cleavage, and this is the only processing step necessary. In this reaction both the stem-loop and a downstream element that binds to U7 small nuclear ribonucleoprotein particle (U7 snRNP) is required (Lanzotti, Kaygun, Yang, Duronio, & Marzluff, 2002). The 3′ end of histone mRNAs is a cis-acting element that coordinates the regulation of histone mRNAs post-transcriptionally (Hanson, Sun, Willis, & Marzluff, 1996; Zheng et al., 2003).
This 26-nucleotide sequence is the binding site for the stem loop binding protein (SLBP). SLPB is a 31-kDa protein that has a distinct small 73 amino acid RNA-binding domain (RBD) and no other RNA-binding protein has this particular RBD (Marzluff et al., 2008). In mammals, there is only one SLBP and SLBP binding to the stem-loop is a required step for the processing of canonical histone mRNA during the S phase of the cell cycle (Sanchez & Marzluff, 2002). SLBP is involved in all aspects of histone mRNA metabolism including processing and regulation in both the nucleus and the cytoplasm (E. Sullivan et al., 2001). SLBP and U7 snRNP work together forming a complex to recruit another protein complex that carries out the pre-mRNA processing reaction, including the heat-labile factor and hypothetical cleavage factor (Lanzotti et al., 2002). SLBP accompanies the mature histone mRNA to the cytoplasm, where it stimulates histone mRNA translation as part of the histone messenger ribonucleoprotein particle (mRNP). In the cytoplasm, SLBP initiates translation of histone mRNA when the N-terminal domain of the bound SLBP interacts with the Slo-interacting protein 1 (SIP1), thus coordinating regulation of histone mRNAs (Brocato et al., 2014). SLBP is stoichiometrically required for accumulation of histone mRNA (Zheng et al., 2003). However, SLBP and histone mRNA are regulated by different signals and mechanisms. Histone pre-mRNA processing requires the binding of SLBP with histone pre-mRNA. After binding, SLBP stabilizes the binding of U7 snRNP to the histone downstream element (Zheng et al., 2003). The study of Zheng et al. (2003), showed that SLBP is a cell cycle regulated protein. In the nucleus, SLBP is present as a free protein (Whitfield et al., 2000).
Canonical histone gene (replication- dependent) expression is limited to the S phase of the cell cycle, since large amounts of histone protein synthesis are required during DNA replication. The canonical histone proteins are mainly synthesized during the S-phase and their function is packaging the newly replicated DNA into compact nucleosomes. Canonical histone mRNAs are tightly regulated in the cell cycle and the mRNA of all five histone proteins are coordinately regulated post-transcriptionally (Zheng et al., 2003). It is important that histone mRNA accumulation is limited to the S-phase for proper chromatin function during development (Zheng et al., 2003).
SLBP mRNA is present throughout the cell cycle. SLBP protein concentrations increase rapidly during the S phase of the cell cycle. Its translation to protein occurs during the latter part of S phase of the cell cycle. During that time, free and active SLPB protein levels increase dramatically just before the maximal accumulation of histone mRNA (Whitfield et al., 2000). Cell cycle signals, regulate SLBP levels post-translationally, regulating its expression throughout the cell cycle, causing a 10–20-fold increase in the late G1 phase to early S phase, followed by its rapid degradation as cell progress to the late S phase/early G2 phase. This accumulation of SLBP in the late G1 phase suggest a possible connection of this process to cyclin E and the rapid degradation at the end of S phase/start of G2 phase is likely due to a second cell cycle signal (Whitfield et al., 2000). This rapid proteasomal degradation of SLBP in the late S/early G2 phase is initiated by the phosphorylation of two threonine residues, Thr61 and Thr60. Thr61 is phosphorylated by cyclin A/Cdk1 at the end of S-phase, priming Thr60 for phosphorylation by Casein Kinase 2 (CK2) (Smith et al., 2018). Following that, dephosphorylation of a phospho-threonine in a conserved region of the RNA binding domain of SLBP, mediated by the co-operation of Peptidyl-prolyl cis/trans isomerase (Pin1) and protein phosphatase 2 (PP2A), will release SLBP from the histone mRNA in the cytoplasm (Brocato et al., 2014). Free SLBP in the cytoplasm will then be targeted by Pin1 for polyubiquitination via an interaction with Ser20 and Ser23 in the N-terminal of SLBP thus resulting in its degradation (Brocato et al., 2014, 2015).
SLBP mRNA is readily detectable in all adult tissues as well, suggesting that SLBP protein synthesis is also pos-transcriptionally regulated in normal somatic cells (Zheng et al., 2003). Histone RNA increases and decreases in a similar manner as SLBP. In the S-phase for example, histone mRNA levels increase approximately 35-fold. Accumulation of histone mRNAs without the presence of SLBP protein is not possible as SLBP is a component of the histone mRNP. The process of synthesis and export of histone mRNA from the nucleus to the cytoplasm is very rapid, thus activation of SLBP mRNA translation results in the rapid accumulation of histone mRNA in the cytoplasm.
Overall, the translation and proteasomal degradation regulation of SLBP has as a goal to work co-operatively to maintain very low SLBP levels in the G1 phase. The proteasomal degradation pathway in G1 phase is independent of the proteasomal degradation pathway of SLBP at the S/G2 phase transition (Whitfield et al., 2000). SLBP must be synthesized in each cell cycle to allow histone RNA accumulation and proper processing of the newly replicated DNA into nucleosomes. Degradation of SLBP at the end of S phase, will inhibit histone pre-mRNA processing further and function to prevent more accumulation of histone mRNA prior to entering the next cell cycle (Zheng et al., 2003). Finally, a secondary mechanism for stopping histone mRNA biosynthesis at the end of S phase suggested by Lanzotti et al. (2002), is the higher percentage of stable SLBP accumulating in the cytoplasm at the end of S-phase.
1.2. Arsenic causes loss of SLBP resulting in polyadenylation of all canonical histones and increases their protein levels outside the S-phase of the cell cycle
Arsenic is a very toxic metal, but it is not a mutagen, instead it causes its carcinogenic effects via epigenetic mechanisms. Arsenic ranks as number one at the ATSDR substance priority list and it’s a big concern with regards to the toxic and carcinogenetic effects it exerts in humans today. Arsenic is present in water sources, it is used as a wood preservative, and it’s in certain herbicides, as such it impacts hundreds of millions of people around the world. Exposure to arsenic can happen via the dermal route, for example from pressure treated wood found in kids’ playgrounds or other preserved woods, like telephone poles. Arsenic can also be inhaled through many industrial processes and can also be ingested via contaminated food (such as seafood and rice) or water, as apart of its anthropogenic release. It is also naturally occurring and ubiquitous in the environment. Arsenic water contamination has been a global issue with varied severity from country to country. There are different forms of arsenic in the environment. Most of the arsenic is found as inorganic arsenate, which is the pentavalent form of arsenic (Mendez Jr. et al., 2017). Arsenic can also be found in the environment in its more toxic inorganic trivalent form (arsenite) and that usually happens under anaerobic conditions in the drinking water (Mendez Jr. et al., 2017). In fish and seafood, arsenic is found in its organic form, such as arsenobetaine and arsenocholine; both are considered non-toxic forms, especially when compared to the inorganic forms of arsenic. Arsenite in the TCA cycle, can inhibit pyruvate dehydrogenase (PDH) by interacting with lipoic acid, thus interfering with cellular metabolism (Mendez Jr. et al., 2017). On the other hand, the lesser evil arsenate mimics phosphate thus, can also interfere in mitochondrial respiration and energy metabolism. In general principle, arsenic can interfere with the normal cell metabolism by displacing Zinc and Selenium from their binding site and can also cause oxidative stress by producing nitric oxides, superoxide, and hydroxyl radical via indirect mechanisms, resulting in damaged organelles as well as DNA, protein, and lipids (Mendez Jr. et al., 2017).
Acute exposure to arsenic can be very toxic or even fatal, but that is avoidable nowadays, due to all the implemented controls by regulatory agencies. The biggest issue with arsenic exposure today, is low dose, chronic exposure and that is because even though arsenic is a well-established carcinogen in humans, the mechanisms by which arsenic mediated carcinogenesis epigenetically are yet to be understood. What we know for certain thus far, is that inorganic arsenic epigenetically regulates carcinogenesis and not via direct DNA interaction. Furthermore, it has been correlated with skin, lung, bladder, prostate, liver, and kidney cancer in humans based on epidemiological studies, though the association with liver, prostate, and kidney cancer is weak and limited according to Zhou and Xi (2018), cited by Costa (2019). However, all these epidemiological studies that have made these associations in humans are mainly done in populations that were exposed to very high arsenic levels, as such there is a need for epidemiological studies looking at the health effects and cancer development associations in populations exposed to low doses of arsenic chronically (Mendez Jr. et al., 2017). In vivo studies have been conducted with hopes to induce tumors following arsenic exposure and uncover the mechanisms of arsenic induced carcinogenesis in experimental animals. As does not induce tumors easily in experimental animals, tumors have been induced in whole animals following whole life exposure, which includes exposure during development and during the entire lifetime of the animal (Waalkes, Qu, Tokar, Kissling, & Dixon, 2014). As a result, we are still unclear today as to why arsenic is so carcinogenic in humans but not in animals (Mendez Jr. et al., 2017). Some recent studies however have demonstrated that arsenic can cause loss of SLBP, thus resulting in the default polyadenylation of canonical histone H3.1 and a dramatic increase in the H3.1 protein levels, present outside the S-phase of the cell cycle (Lanzotti et al., 2002; Marzluff et al., 2008; Mendez Jr. et al., 2017; Wang, Xie, Lin, & Zhang, 2014). As mentioned previously in this review, canonical histone genes like H3.1, are replication- dependent histone genes, and these genes are the only genes in metazoan that do not end with a poly (A) tail, instead at their 3′ end they have a stem loop structure. The presence of SLBP is critical in the translation process of these histone genes and its absence during the S phase of the cell cycle can cause significant effects in the package of newly replicated DNA into nucleosomes, as well as result in genetic instability. SLBP levels dramatically increase during the G1/S phase transition and then degrade at the end of the S phase, prior to entering the G2 phase. On the other hand, variant histones such as H3.3 are replication independent genes that are polyadenylated and expressed throughout the cell cycle as they play an important role in active promoters and enhancers of genes, and they are constitutively expressed in quiescent cells (Mendez Jr. et al., 2017; Wang et al., 2014). Nucleosomes containing H3.3 can perform specialized functions that depends on their location (Szenker, Ray-Gallet, & Almouzni, 2011). H3.3 in the promoter region of genes, can activate chromatin complexes and allow transcriptional machinery to bind there, thus activating transcription (Szenker et al., 2011). On the other hand, H3.3 present at the telomeres, has the opposite effect, resulting in gene silencing (Goldberg et al., 2010). If a variant histone like H3.3 gets replaced by a canonical histone within the nucleosomes, the structure, stability, and dynamics of the chromatin structure will change, resulting in altered gene expression, impaired cell cycle control and genomic instability (Mendez Jr. et al., 2017). It is worth noting here that H3.3. and H3.1 proteins only differ by 5 amino acids thus, it is very easy to replace one another (Mendez Jr. et al., 2017). Finally, the study of Chen et al. (2020) showed that the knockdown of the variant H3.3 can induce cell transformation, as such the replacement of it by H3.1, could facilitate this process, and this was shown later by Chen et al. (2020). On the other hand, the study of Chen et al. (2020), showed that cells could potentially be rescued from arsenic toxicity and cell transformation by overexpressing H3.3. The study of Bush et al. (2013) and Jang, Shibata, Starmer, Yee, and Magnuson (2015) showed that H3.3 knockout mice were embryonically lethal, due to the severe chromosomal aberrations (Bush et al., 2013; Jang et al., 2015). As such, the displacement of H3.3 by H3.1 seems to be very important to the tumorigenic effects of arsenic. As of today, the mechanisms of the displacement of H3.3 by H3.1 is still unclear. One suggested mechanism by Chen et al. (2020) that was based on their findings, was that excess H3.1 protein can potentially increase the association of H3.1 with CAF-1 complex including the histone chaperones p60 and p48, resulting in shortage of available chaperons and decreasing the interaction between H3.3 and HIRA complex (an H3.3 specific histone chaperone) and compromising its assembly. Another potential mechanism could be that during the M phase of the cell cycle, H3.1 protein directly competes with H3.3 variant (Wang et al., 2014).
The study of Brocato et al. (2014) found that in several different cell lines, an increase of H3.1 polyadenylation occurred following exposure to arsenic. As mentioned previously, Brocato et al. (2014) showed that arsenic can silence the SLBP gene transcription via epigenetic regulation and enhance SLBP proteasomal degradation. Once SLBP is lost due to arsenic, there is less binding occurring at the stem loop structure of the H3.1 mRNA (Marzluff et al., 2008). This results in the acquisition of a poly (A) tail by activation of downstream poly (A) signal sequence that will then provide enhanced mRNA stability by increasing its half-life (Marzluff et al., 2008). The increased half-life of the polyadenylated H3.1 mRNA is due to the poly (A) tail’s ability to act as a protective wall against degradation enzymes in the cytosol (Marzluff et al., 2008). The poly (A) tail can also enhance the translation and transcription process, by guiding the mRNA to the ribosome, thus promoting the initiation of translation (Marzluff et al., 2008). This process will then result in increased H3.1 translation to protein that is now also present outside of the S phase (Mendez Jr. et al., 2017). Arsenic-induced Polyadenylated H3.1 mRNA is not susceptible to normal degradation at the end of the S phase (Marzluff et al., 2008). As a result, H3.1 protein is now also present outside the S phase and can interfere with the function of variant histone H3.3 during the cell cycle, by displacing it (Mendez Jr. et al., 2017). That will then lead to the disruption of the delicate balance that exists during normal physiological conditions between the H3.1 and H3.3, changing the landscape and function of chromatin (Mendez Jr. et al., 2017). This displacement and increased expression of canonical histone H3.1 protein, will then result in an increased sensitivity to DNA damage and genomic instability (Marzluff et al., 2008). The study of Brocato et al. (2015) showed that ectopic expression of polyadenylated H3.1 caused by arsenic, resulted in anchorage-independent cell growth suggesting arsenic-induced cell transformation via this mechanism.
Arsenic can therefore induce changes in gene expression via the alteration of post-translational histone modifications levels occurring in gene promoters. Arsenic is also believed to cause alterations in the post-translational level of the histone genes themselves, as in the study of Brocato et al. (2014), they showed that arsenic treated PBMCs (peripheral blood mononuclear cells) had 61 histone genes that were remarkably increased. This alterations in the stoichiometry of histones that are caused by arsenic exposure, contribute to the indirect arsenic-induced genomic instability (Marzluff et al., 2008). In the study of Brocato et al. (2014), they found that arsenic can increase histone cluster gene expression at both the translational and transcriptional level. Furthermore, the study of Brocato et al. (2014) suggested that the arsenic-induced increase of the polyadenylated H3.1 mRNA, could be the result of increased gene transcription or the increased stability of the polyadenylated H3.1 mRNA. This increase of the polyadenylated H3.1 mRNA induced by arsenic, was indirectly caused by the depletion of SLBP that consequently results in the aberrant pre-mRNA processing and accumulation of the polyadenylated mRNA outside of the S-phase of the cell cycle (Marzluff et al., 2008). Thus, having a potentially significant impact on cellular processes due to the inability of the cells to suppress canonical histone expression, resulting in abnormal chromosomal segregation during mitosis, where the levels of the polyadenylated H3.1 mRNA increase 3-fold following arsenic exposure and this was highly toxic (Marzluff et al., 2008).
Arsenic has been shown in the past to have the ability to alter DNA methylation at the promoters of genes, leading to gene silencing, and this is one possible mechanism by which it depletes the cells of SLBP, via silencing of its transcription (Marzluff et al., 2008). SLBP plays a critical role in the arsenic induced H3.1 mRNA polyadenylation according to Brocato et al., 2014, as also in the study of Chen et al. (2020), where they didn’t find any changes in any other genes involved in canonical histone mRNA processing (such as U7 snRNP or CPSF2), suggesting SLBP being the sole target. Arsenic also depletes SLBP at the translational level, preventing its translation to protein via epigenetic changes. Even though arsenic resulted in the addition of a poly (A) tail to H3.1 histone, not all H3.1 was polyadenylated, there was still some H3.1 with intact stem-loop structure, as 1μM of arsenic exposure on BEAS-2B (human bronchial epithelial) cells only partially depleted SLBP at the protein level. This finding of Brocato et al. (2014) suggests that H3.1 mRNA disability and translation are still regulated by SLBP to some extent, despite arsenic exposure (Marzluff et al., 2008). Though, the arsenic induced partial SLBP depletion and polyadenylated H3.1 mRNA increase that is dose-dependent, still has a significant impact on the cell. The mechanisms by which a poly (A) tail is formed following arsenic induced SLBP depletion is still unclear and further studies need to shed more light into this mechanism. Following the decrease of nuclear SLBP levels, the stem-loop structure is thought to become unbound and degraded via an unknown mechanism, thus allowing the formation of a poly (A) tail. One proposed mechanism by Brocato et al., that hasn’t been explored yet, is that the enzyme 3′hExo, which is part of the SLBP complex, is recruited by the altered conformation of the stem loop and degrades it prior to polyadenylation (Marzluff et al., 2008). Another alternative mechanism that has been proposed but hasn’t been explored yet is the possibility that post-transcriptionally, the stem-loop structure is spliced out of the H3.1 mRNA. According to Brocato et al., arsenic also has the tendency to affect proteins’ methyl, phospho and acetyl groups, thus this mechanism can potentially be mediating the arsenic induced SLBP degradation. Some of the epigenetic mechanisms by which arsenic was found to affect SLBP mRNA expression were via induction of histone deacetylase and DNA methyltransferase activity, both of which result in silencing of genes, but the specific mechanisms by which arsenic does this is still unclear (Marzluff et al., 2008). H3.1 mRNA polyadenylation induced by arsenic exposure and displacement of H3.3 by H3.1 in the nucleosome are critical in arsenic induced cell transformation, but the key is SLBP depletion, which is the prime reason for these consequential events such as polyadenylation and aberrant expression of H3.1 outside the S phase that then competes and displaces H3.3 in the nucleosome leading to arsenic induced toxicity and carcinogenicity (Marzluff et al., 2008).
2. Stem loop binding protein loss and polyadenylation of H3.1 (H3.2?) induce carcinogenesis
2.1. Importance of the loss of SLBP and H3.1 upregulation
As per Brocato et al., depletion of SLBP and increase in polyadenylated (polyA) histone 3.1 (H3.1) mRNA is observed following arsenic exposures in human bronchial epithelial BEAS-2B cells. These data suggest that arsenic regulate the polyadenylation of canonical histone mRNAs. Arsenic may have similar effects on canonical histone mRNA transcription and translation as other heavy metal such as nickel and cadmium (Brocato et al., 2014, 2015; Jordan et al., 2017). These results are significant because previous reports on a carcinogenic metal, arsenic indicated that depletion of SLBP, increases in H3.1 mRNA levels (total and poly A) and can potentiate carcinogenic transformation in vitro, Fig. 1 (courtesy iScience) (Brocato et al., 2014, 2015). The increased poly(A) H3.1 mRNA suggests that nickel and arsenic may have similar signaling pathways of carcinogenicity. Previous investigations have indicated that in the absence of SLBP, canonical H3.1 histone mRNA acquires an otherwise absent poly-adenylated tail (Brocato et al., 2014, 2015). As cells enter the S phase the synthesis of SLBP is activated by increasing the translation rate of the SLBP mRNA. SLBP is rapidly degraded at the end of S phase as a result of its phosphorylation (Whitfield et al., 2000; Zheng et al., 2003). Previous study showed that the SLBP stimulates translation of reporter mRNAs ending in the stem–loop both in vivo and in vitro (Sanchez & Marzluff, 2002). Histone mRNAs are the only eukaryotic cellular mRNAs that are not polyadenylated. Synthesis of mature histone mRNA requires only a single processing reaction: an endonucleolytic cleavage between a conserved stem–loop and a purine-rich downstream element to form the 3′ end.The SLBP is required for processing, and following processing, histone mRNA is transported to the cytoplasm, where SLBP participates in translation of the histone mRNA and is also involved in regulation of histone mRNA degradation (K. D. Sullivan, Mullen, Marzluff, & Wagner, 2009).
Fig. 1.
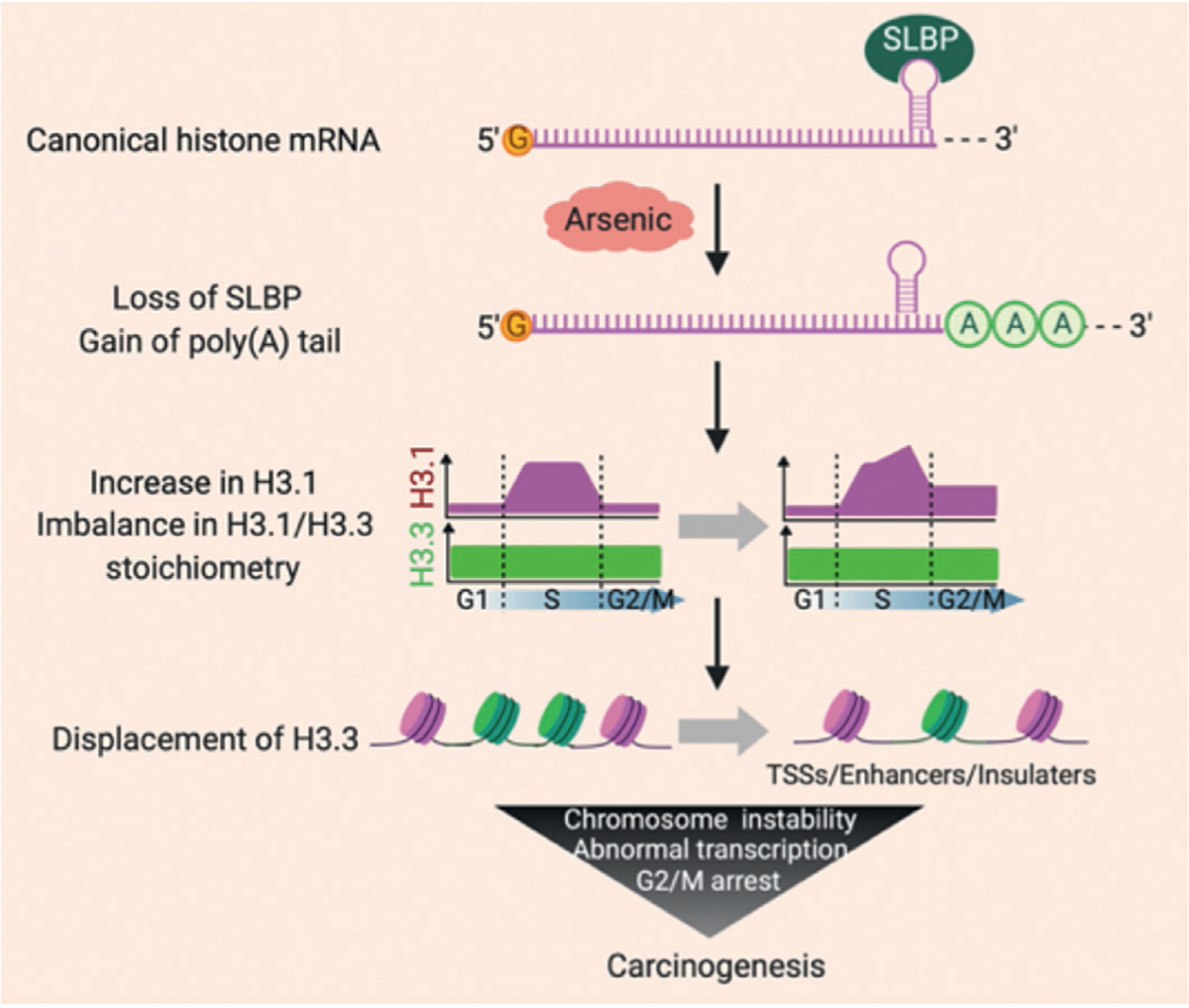
Loss of SLBP and gain of poly(A) tail and increase of H3.1 causes carcinogenesis.
2.2. The significance of increased polyadenylated H3.1 mRNA
Canonical histone mRNAs are rapidly degraded at the end of S phase, since they exhibit less stability because of the absence of a poly(A) tail (Harris et al., 1991; Whitfield et al., 2000). The absence of the poly(A) tail is vital for maintaining the steady state levels of canonical histone mRNAs during S phase. Disruption of pre-mRNA processing by the loss of SLBP results in polyadenylation of canonical histone mRNAs, increasing their stability and elevating the level of H3.1 protein (Lanzotti et al., 2002; Sullivan et al., 2001). The studies demonstrated that the 3′ end of canonical histone H3.1 mRNA can be polyadenylated following exposure to arsenic (Brocato et al., 2014). Arsenic, a confirmed human carcinogen, induces lung, bladder, and skin cancers (Mendez Jr. et al., 2017) and is linked to cancers of the kidney, liver, and prostate (Roh et al., 2017; Smith et al., 2018; W. Wang et al., 2014). Overexpression of polyadenylated H3.1 mRNA enhanced anchorage-independent growth of human bronchial epithelial BEAS-2B cells, demonstrating that polyadenylation of H3.1 mRNA induces cell transformation (Brocato et al., 2015). Polyadenylated canonical histone H3.1 mRNA could induce genomic instability and cell transformation by disrupting the balance between canonical and variant histones. Canonical replication dependent histone expression is tightly linked to the cell cycle and is absolutely required for cell survival. Previous studies have implicated abnormal overexpression of histones in cellular transformation (Brocato et al., 2015). Recent studies suggests that bisphenols’ exposure facilitates the abnormal stability of at least one canonical histone isoform (H3.1) (Veerappan, Zhang, & Costa, 2022a, 2022b). A review of recent findings that arsenic induced polyadenylation of H3.1 caused enhanced levels of H3.1 protein displacing H3.3 protein from its cellular binding sites, since the two proteins differ by only 5 amino acids. Knockdown of H3.3 alone can induce carcinogenesis, and therefore displacement of functional H3.3 protein by increased H3.1 protein, is likely a mechanism of arsenic and/or bisphenol carcinogenesis (Costa, 2019). Replication-dependent canonical histone mRNAs do not terminate with a poly(A) tail at the 3′ end (Marzluff et al., 2008). We previously demonstrated that exposure to arsenic induces polyA of canonical histone H3.1 mRNA, causing transformation of human cells in vitro. Further, we reported that polyA of H3.1 mRNA increases H3.1 protein, resulting in displacement of histone variant H3.3 at active promoters, enhancers, and insulator regions, leading to transcriptional deregulation, G2/M cell-cycle arrest, chromosome aneuploidy, and aberrations. In support of these observations, knocking down the expression of H3.3 induced cell transformation, whereas ectopic expression of H3.3 attenuated arsenic-induced cell transformation. Notably, arsenic exposure also resulted in displacement of H3.3 from active promoters, enhancers, and insulator regions (Fig. 2) (courtesy iScience). These data suggest that H3.3 displacement might be central to carcinogenesis caused by polyadenylation of H3.1 mRNA upon arsenic exposure. Chen et al. findings illustrate the importance of proper histone stoichiometry in maintaining genome integrity (Chen et al., 2020).
Fig. 2.
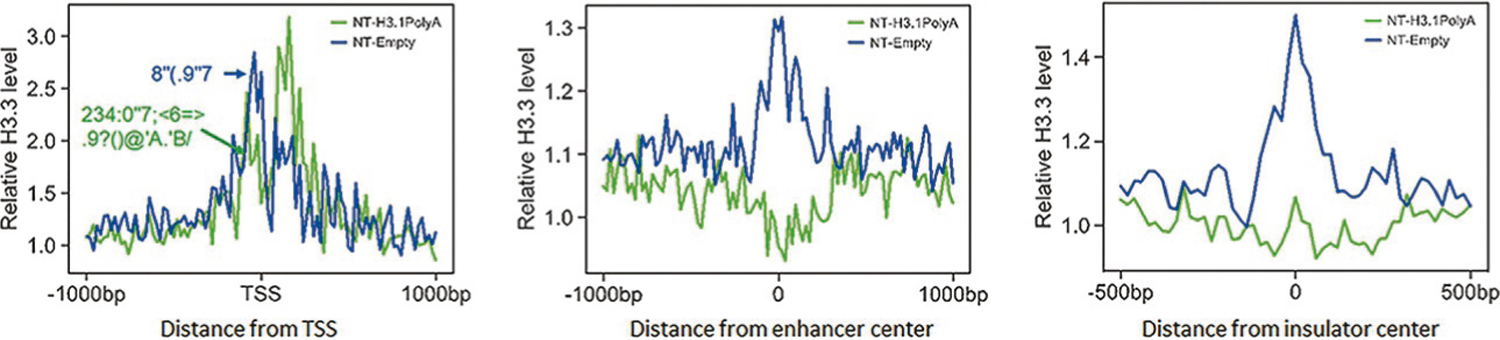
Overexpression of polyadenylated H3.1 mRNA displaces histone variant H3.3 from critical gene regulatory elements. Profile of endogenous H3.3-containg nucleosomes in untagged cells around the transcription start sites (TSSs) on the 2000 most highly expressed genes (active promoters) (left), DNase I hypersensitive sites (enhancers) (middle), or CTCF-binding sites (insulators) (right). H3.3-containing nucleosomes were isolated followed by ChIP-seq with the H3.3-specific antibody.
3. Carcinogens that cause the loss of SLBP and polyadenylation of H3.1
Loss of SLBP and polyadenylation of H3.1 were caused by carcinogens such as arsenic, nickel, cadmium and bisphenols, Table 1. Arsenic, nickel and cadmium may decrease SLBP mRNA expression by regulating SLBP promoter region, such as declining histone acetylation or increasing DNA methylation, then promote SLBP protein degradation, finally cause H3.1 polyadenylation (Fig. 3) (Jordan et al., 2017; Wang, Liu, & Jiang, 2021). Recent studies suggested that the polyadenylation of canonical histone mRNAs is targeted by bisphenols (Veerappan et al., 2022a, 2022b). This study investigates the mechanisms that underlie the tumorigenic effects of polyadenylated H3.1 mRNA followed by bisphenol exposure. We conclude that H3.3 displacement appears to be central to the tumorigenic responses caused by polyadenylated H3.1 mRNA following arsenic or bisphenols exposure. These findings add important insights not only into genomic instability induced by an imbalance in histone stoichiometry but also into the oncogenic role of H3.3 loss. Hence, we conclude that metals and endocrine disrupters such as bisphenols exposure causes human lung cancer mediated by loss of SLBP and polyadenylation of H3.1 by displacing non-canonical H3.3 from chromatin.
Table 1.
Carcinogens that cause the loss of SLBP and polyadenylation of H3.1.
| Arsenic |
Brocato et al. (2014)
Brocato et al. (2015) Chen et al. (2020) |
The Journal of Biological Chemistry, 289, 31751–31764 Biological Trace Element Research, 166, 72–81 iScience, 23, 101518, doi:https://doi.org/10.1016/j.isci.2020.101518 |
| Nickel | Jordan et al. (2017) | PLoS One, 12, e0173624, doi:https://doi.org/10.1371/journal.pone.0173624 |
| Cadmium | ||
| Bisphenols | Veerappan et al. (2022a, 2022b) | The Toxicologist, 186(S1), 108 Cancer Research 2022, 82(12 Suppl): 228 |
Fig. 3.
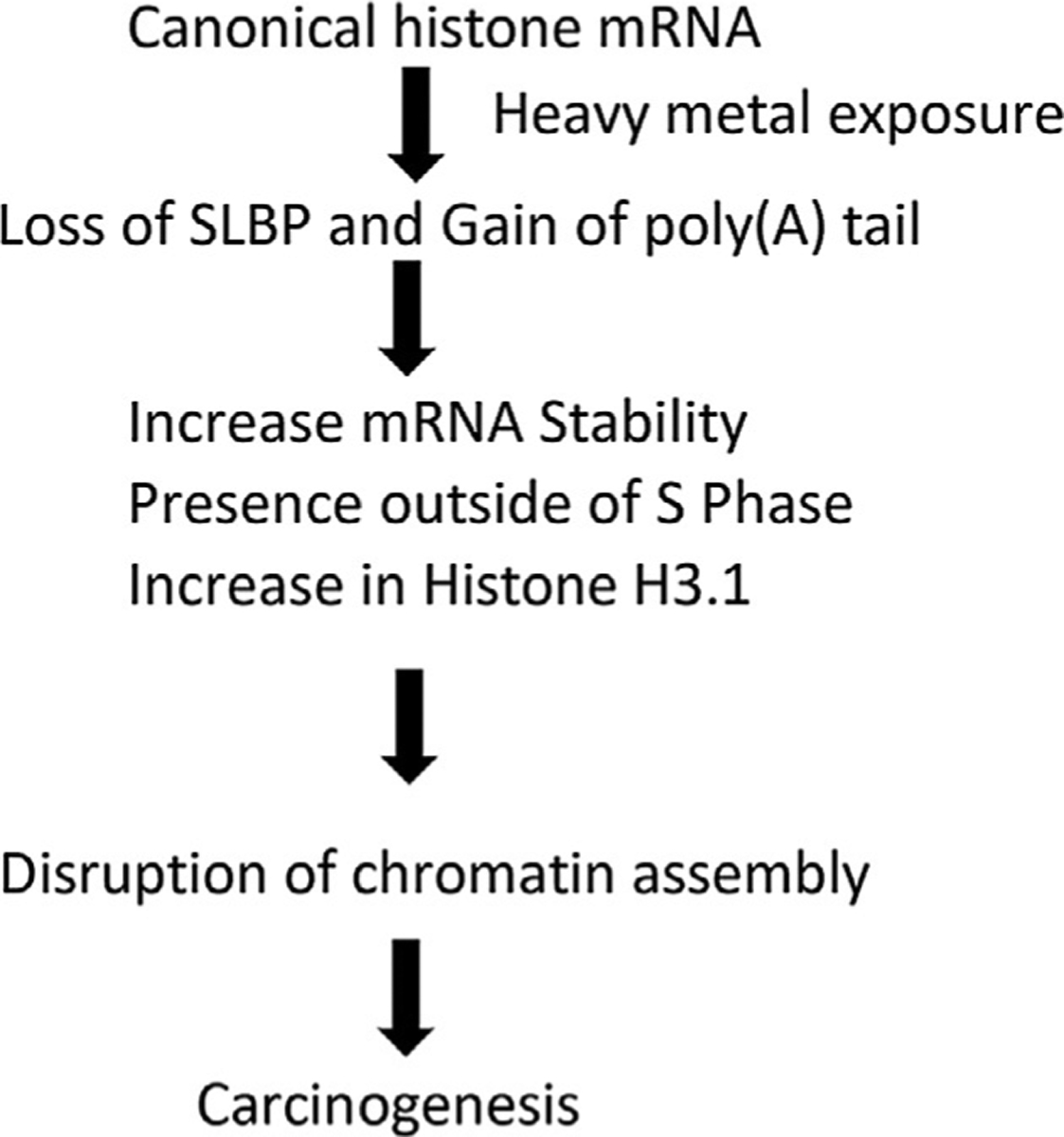
A schematic diagram of the regulation and functions of SLBP in the cells after exposed to heavy metals.
4. Conclusion
The polyadenylation of other canonical histones such as H4, H2B, and H2A does not seem to have the importance that polyadenylation of H3.1 has and this is likely due to the existence of a replacement histone H3.3 which is a noncanonical histone and is therefore polyadenylated and made through the cell cycle. Clearly the displacement of H3.3 by polyadenylation H3.1 at many chromatin loci is detrimental to cellular function and results in cancers.
Funding
This research was supported by the National Institute of Health (NIH) grants NIH/NIEHS R01ES030583 and ES030572-04 (M. Costa).
Abbreviation
- SLBP
stem-loop binding protein
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- Brocato J, Chen D, Liu J, Fang L, Jin C, & Costa M (2015). A potential new mechanism of arsenic carcinogenesis: Depletion of stem-loop binding protein and increase in polyadenylated canonical histone H3.1 mRNA. Biological Trace Element Research, 166(1), 72–81. 10.1007/s12011-015-0296-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocato J, Fang L, Chervona Y, Chen D, Kiok K, Sun H, et al. (2014). Arsenic induces polyadenylation of canonical histone mRNA by down-regulating stem-loop-binding protein gene expression. The Journal of Biological Chemistry, 289(46), 31751–31764. 10.1074/jbc.M114.591883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush KM, Yuen BT, Barrilleaux BL, Riggs JW, O’Geen H, Cotterman RF, et al. (2013). Endogenous mammalian histone H3.3 exhibits chromatin-related functions during development. Epigenetics & Chromatin, 6. 10.1186/1756-8935-6-7. Artn 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Chen QY, Wang Z, Zhu Y, Kluz T, Tan W, et al. (2020). Polyadenylation of histone H3.1 mRNA promotes cell transformation by displacing H3.3 from gene regulatory elements. iScience, 23(9), 101518. 10.1016/j.isci.2020.101518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa M (2019). Review of arsenic toxicity, speciation and polyadenylation of canonical histones. Toxicology and Applied Pharmacology, 375, 1–4. 10.1016/j.taap.2019.05.006. [DOI] [PubMed] [Google Scholar]
- Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S, et al. (2010). Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell, 140(5), 678–691. 10.1016/j.cell.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson RJ, Sun JH, Willis DG, & Marzluff WF (1996). Efficient extraction and partial purification of the polyribosome-associated stem-loop binding protein bound to the 3′ end of histone mRNA. Biochemistry, 35(7), 2146–2156. 10.1021/bi9521856. [DOI] [PubMed] [Google Scholar]
- Harris ME, Bohni R, Schneiderman MH, Ramamurthy L, Schumperli D, & Marzluff WF (1991). Regulation of histone mRNA in the unperturbed cell cycle: Evidence suggesting control at two posttranscriptional steps. Molecular and Cellular Biology, 11(5), 2416–2424. 10.1128/mcb.11.5.2416-2424.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang CW, Shibata Y, Starmer J, Yee D, & Magnuson T (2015). Histone H3.3 maintains genome integrity during mammalian development. Genes & Development, 29(13), 1377–1392. 10.1101/gad.264150.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan A, Zhang X, Li J, Laulicht-Glick F, Sun H, & Costa M (2017). Nickel and cadmium-induced SLBP depletion: A potential pathway to metal mediated cellular transformation. PLoS One, 12(3), e0173624. 10.1371/journal.pone.0173624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzotti DJ, Kaygun H, Yang X, Duronio RJ, & Marzluff WF (2002). Developmental control of histone mRNA and dSLBP synthesis during Drosophila embryogenesis and the role of dSLBP in histone mRNA 3′ end processing in vivo. Molecular and Cellular Biology, 22(7), 2267–2282. 10.1128/MCB.22.7.2267-2282.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzluff WF, & Koreski KP (2017). Birth and death of histone mRNAs. Trends in Genetics, 33(10), 745–759. 10.1016/j.tig.2017.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzluff WF, Wagner EJ, & Duronio RJ (2008). Metabolism and regulation of canonical histone mRNAs: Life without a poly(A) tail. Nature Reviews. Genetics, 9(11), 843–854. 10.1038/nrg2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez WM Jr., Eftim S, Cohen J, Warren I, Cowden J, Lee JS, et al. (2017). Relationships between arsenic concentrations in drinking water and lung and bladder cancer incidence in U.S. counties. Journal of Exposure Science & Environmental Epidemiology, 27(3), 235–243. 10.1038/jes.2016.58. [DOI] [PubMed] [Google Scholar]
- Roh T, Lynch CF, Weyer P, Wang K, Kelly KM, & Ludewig G (2017). Low-level arsenic exposure from drinking water is associated with prostate cancer in Iowa. Environmental Research, 159, 338–343. 10.1016/j.envres.2017.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez R, & Marzluff WF (2002). The stem-loop binding protein is required for efficient translation of histone mRNA in vivo and in vitro. Molecular and Cellular Biology, 22(20), 7093–7104. 10.1128/Mcb.22.20.7093-7104.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AH, Marshall G, Roh T, Ferreccio C, Liaw J, & Steinmaus C (2018). Lung, bladder, and kidney cancer mortality 40 years after arsenic exposure reduction. Journal of the National Cancer Institute, 110(3), 241–249. 10.1093/jnci/djx201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan KD, Mullen TE, Marzluff WF, & Wagner EJ (2009). Knockdown of SLBP results in nuclear retention of histone mRNA. RNA, 15(3), 459–472. 10.1261/rna.1205409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan E, Santiago C, Parker ED, Dominski Z, Yang X, Lanzotti DJ, et al. (2001). Drosophila stem loop binding protein coordinates accumulation of mature histone mRNA with cell cycle progression. Genes & Development, 15(2), 173–187. 10.1101/gad.862801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szenker E, Ray-Gallet D, & Almouzni G (2011). The double face of the histone variant H3.3. Cell Research, 21(3), 421–434. 10.1038/cr.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veerappan A, Zhang Z, & Costa M (2022a). Depletion of stem-loop binding protein and elevation of polyadenylation of histone 3.1 contribute to malignant cell transformation induced by bisphenol A. In Proceedings of the American association for cancer research annual meeting 2022; April 8–13. New Orleans (LA): AACR. Cancer Res; 2022;82 (Suppl): Abstract nr 1601 (in press). [Google Scholar]
- Veerappan A, Zhang Z, & Costa M (2022b). Exposure of bisphenol a and its substitutes to human lung epithelial cells causes the downregulation of stem-loop binding protein followed by polyadenylation of histone 3.1 and their role in bisphenols-induced carcinogenesis. The Toxicologist, 186(S1), 108. Suppl. to Toxicological Sciences. [Google Scholar]
- Waalkes MP, Qu W, Tokar EJ, Kissling GE, & Dixon D (2014). Lung tumors in mice induced by “whole-life” inorganic arsenic exposure at human-relevant doses. Archives of Toxicology, 88(8), 1619–1629. 10.1007/s00204-014-1305-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Liu LZ, & Jiang BH (2021). Dysregulation of microRNAs in metal-induced angiogenesis and carcinogenesis. Seminars in Cancer Biology, 76, 279–286. 10.1016/j.semcancer.2021.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Xie Z, Lin Y, & Zhang D (2014). Association of inorganic arsenic exposure with type 2 diabetes mellitus: A meta-analysis. Journal of Epidemiology and Community Health, 68(2), 176–184. 10.1136/jech-2013-203114. [DOI] [PubMed] [Google Scholar]
- Whitfield ML, Zheng LX, Baldwin A, Ohta T, Hurt MM, & Marzluff WF (2000). Stem-loop binding protein, the protein that binds the 3’ end of histone mRNA, is cell cycle regulated by both translational and posttranslational mechanisms. Molecular and Cellular Biology, 20(12), 4188–4198. 10.1128/Mcb.20.12.4188-4198.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng LX, Dominski Z, Yang XC, Elms P, Raska CS, Borchers CH, et al. (2003). Phosphorylation of stem-loop binding protein (SLBP) on two threonines triggers degradation of SLBP, the sole cell cycle-regulated factor required for regulation of histone mRNA processing, at the end of S phase. Molecular and Cellular Biology, 23(5), 1590–1601. 10.1128/Mcb.23.5.1590-1601.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, & Xi S (2018). A review on arsenic carcinogenesis: Epidemiology, metabolism, genotoxicity and epigenetic changes. Regul Toxicol Pharmacol, 99, 78–88. 10.1016/j.yrtph.2018.09.010. [DOI] [PubMed] [Google Scholar]