

Abstract
The polymerase delta interacting protein 2 (Poldip2) is a nuclear-encoded mitochondrial protein required for oxidative metabolism. Under hypoxia, Poldip2 expression is repressed by an unknown mechanism. Therefore, low levels of Poldip2 are required to maintain glycolytic metabolism.
The Cellular Communication Network Factor 2 (CCN2, Connective tissue growth factor, CTGF) is a profibrogenic molecule highly expressed in cancer and vascular inflammation in advanced atherosclerosis. Because CCN2 is upregulated under hypoxia and is associated with glycolytic metabolism, we hypothesize that Poldip2 downregulation is responsible for the upregulation of profibrotic signaling under hypoxia. Here, we report that Poldip2 is repressed under hypoxia by a mechanism that requires the activation of the enhancer of zeste homolog 2 repressive complex (EZH2) downstream from the Cyclin-Dependent Kinase 2 (CDK2). Importantly, we found that Poldip2 repression is required for CCN2 expression downstream of metabolic inhibition of the ubiquitin-proteasome system (UPS)-dependent stabilization of the serum response factor. Pharmacological or gene expression inhibition of CDK2 under hypoxia reverses Poldip2 downregulation, the inhibition of the UPS, and the expression of CCN2, collagen, and fibronectin. Thus, our findings connect cell cycle regulation and proteasome activity to mitochondrial function and fibrotic responses under hypoxia.
Keywords: Poldip2, mitochondria, hypoxia, proteasome, cell cycle, CCN2
INTRODUCTION
Hypoxia and pseudohypoxia are important pathophysiological signals in the cardiovascular system. Hypoxia triggers a secretory and proliferative phenotype in smooth muscle cells of various vascular beds, such as the lung [1] and the bladder [2]. Similarly, the stabilization of the hypoxia-inducible factor 1α in aortic smooth muscle cells is responsible for the increased deposition of extracellular matrix components such as collagen, contributing to Angiotensin II-induced vascular remodeling [3].
The polymerase delta interacting protein 2 (Poldip2) is a nuclear-encoded mitochondrial protein required for oxidative metabolism in various cell types, including highly glycolytic cancer cells [4]. We have previously reported that Poldip2 is repressed under hypoxia [4]. However, the molecular mechanism that regulates Poldip2 expression during hypoxia remains elusive.
The ubiquitin-proteasome system (UPS) is the primary site for regulated protein degradation. UPS function involves the breakdown of polyubiquitin-tagged proteins by the 26S proteasome, a multiprotein complex consisting of a 20S core particle associated with one or two 19S regulatory particles [5]. The 19S regulatory particle is responsible for UPS activation and serves to recognize, unfold, and translocate client proteins across the proteolytic chamber [6].
We recently reported that Poldip2 expression downregulation is responsible for inhibiting the UPS by a mechanism that involves glycosylation of one of the 19S components, the activator 26S proteasome regulatory subunit 4 (PSMC1) [7].
The cellular communication network factor 2 (CCN2, connective tissue growth factor, CTGF) is a profibrogenic protein overexpressed in fibrotic diseases and cancer [8–10]. CCN2 expression stimulates the synthesis of other connective proteins, including type I collagen, fibronectin, and elastin [11, 12]. Furthermore, CCN2 expression is upregulated by hypoxia [13, 14]. Regarding the vasculature, we and others have shown that CCN2 is upregulated in advanced atherosclerosis in humans [15] and during diabetes-induced vascular inflammation in mice [16], both of which are pathological states that have been linked to hypoxia and hypoxia-inducible factor 1 alpha (HIF1α) signaling.
Notably, several lines of evidence have established that glycolytic cells display high CCN2 expression [17–19]. This study aimed to elucidate the signaling pathway and metabolic reprogramming events leading to the upregulation of CCN2 and other profibrotic proteins in vascular smooth muscle cells during hypoxia.
Here, we report that under hypoxia, the expression of CCN2, CCN2-dependent collagen 1A, and fibronectin are upregulated by a mechanism that requires the metabolic inhibition of the ubiquitin-proteasome system downstream of Poldip2 repression. Additionally, we reveal that the signaling pathway leading to Poldip2 repression under hypoxia involves activation of the enhancer of zeste 2 (EZH2), a subunit of the polycomb repressive complex 2, downstream of cyclin-dependent kinase 2 (CDK2). Thus, our findings connect cell cycle regulation and proteasome activity to mitochondrial function and fibrotic responses under hypoxia.
RESULTS
CDK2/Cyclin A/EZH2-mediated repression of Poldip2 under hypoxia.
We have previously shown that Poldip2 expression is oxygen-dependent [4]. We first sought to investigate the mechanism of Poldip2 expression inhibition under hypoxic conditions since it is currently not known. Previous work showed that Poldip2 expression is required for normal cell cycle progression in mouse embryonic fibroblasts [20]. Consistent with this reported relationship, we observed that after thymidine-block release, the expression of Poldip2 in human aortic smooth muscle cells (HASMCs) follows a pattern that resembles the expression of B-type mitotic cyclins [21] (Supplementary Figure 1). This result suggests that Poldip2 expression is regulated by signaling pathways that control the cell cycle and that these pathways might be involved in the repression of Poldip2 expression during hypoxia. To investigate the possible role of cell cycle-related signaling pathways in inhibiting Poldip2 expression under hypoxia, we first characterized the effect of hypoxia on cell cycle signaling. We found that in HASMCs, hypoxia induces the expression of cyclin A2 (CCNA2) and the phosphorylation of cyclin-dependent kinase 2 (CDK2) at Threonine 160 (Figure 1), a posttranslational modification that is required for full activation of the CDK2−cyclin A dimer [22]. Thus, we posited that CDK2 activation is required for Poldip2 inhibition under hypoxia. Consistent with this hypothesis, downregulation of CDK2 (Figure 2A), or cyclin A (Figure 2B), reversed the hypoxia-induced downregulation of Poldip2. These results indicate that under hypoxia, Poldip2 expression is inhibited by a CDK2/Cyclin A-dependent mechanism.
Figure 1. Hypoxia induces the activation of CDK2 and EZH2 phosphorylation in HASMCs.
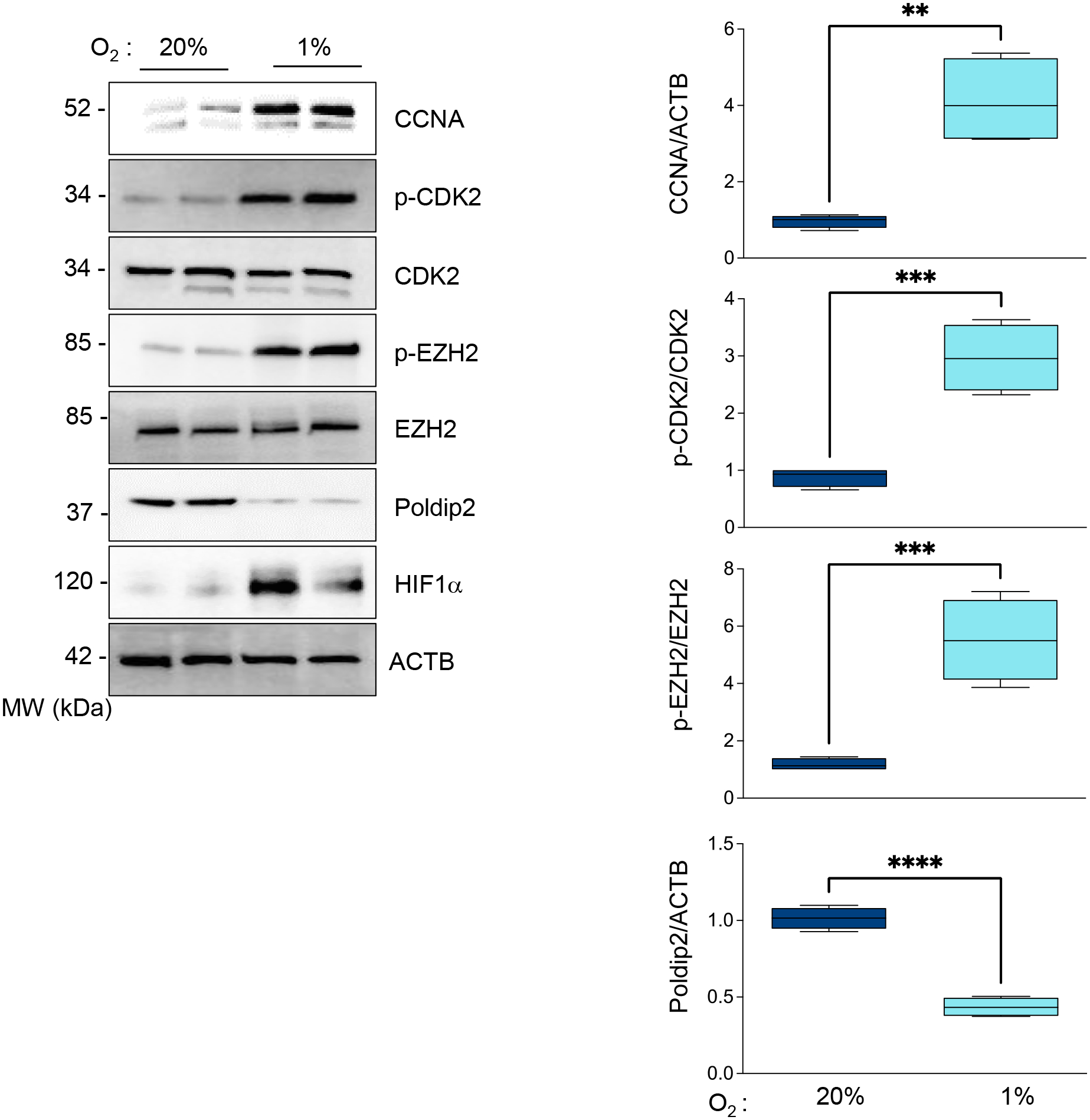
HASMCs were cultured under normoxia or hypoxia for 48 hours. Total lysates were prepared and analyzed by western blot using specific antibodies (Left). β-actin served as a loading control. Densitometric analysis of protein signals normalized to β-actin (Right). Bar graphs represent mean ± SE from 4 independent experiments.
Figure 2. A CDK2/CyclinA-dependent mechanism mediates EZH2 phosphorylation and Poldip2 downregulation under hypoxia.
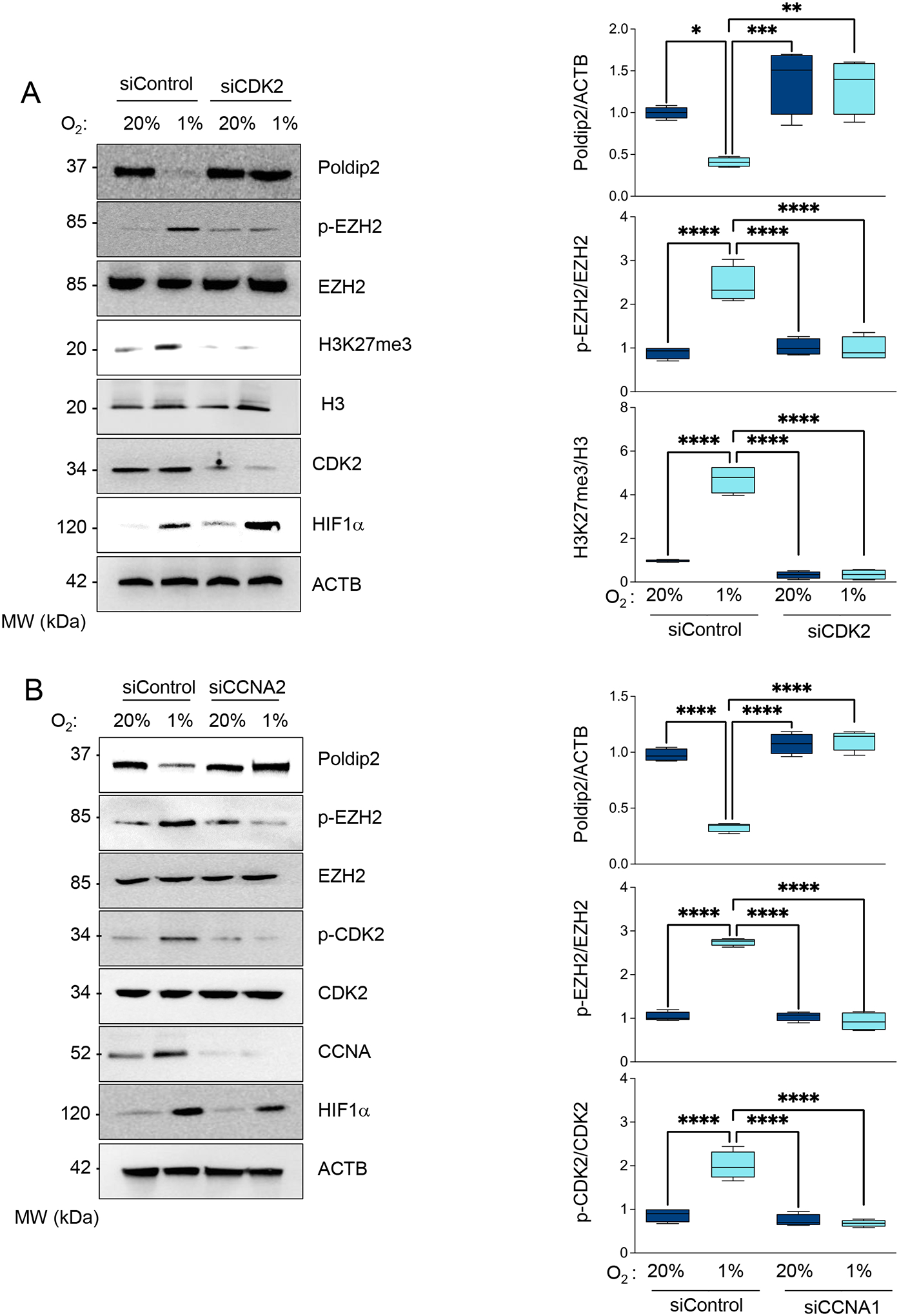
HASMCs were transfected with control siRNA (siControl) or siRNA against (A) CDK2 (siCDK2) or (B) Cyclin A2 (siCCNA2). HASMCs were cultured under normoxia or hypoxia for 48 h. Total lysates were prepared and analyzed by western blot using specific antibodies (Left). β-actin served as a loading control. Densitometric analysis of protein signals normalized to β-actin (Right). Bar graphs represent mean ± SE from 4 independent experiments.
The histone methyltransferase enhancer of zeste EZH2 is the catalytic subunit of the polycomb repressive complex 2, which methylates histone H3 to repress target genes by writing the inhibitory histone H3 lysine K27 di-and trimethylation (H3K27me2/3) [23]. Since it has been reported that CDK2-induced gene silencing proceeds through phosphorylation and activation of EZH2 [24, 25], we hypothesized that CDK2-mediated inhibition of Poldip2 under hypoxia occurs by an EZH2-dependent mechanism. Indeed, we observed that EZH2 is phosphorylated under hypoxia (Figure 1) in a CDK2 (Figure 2A) and Cyclin A-dependent manner (Figure 2B). Most importantly, hypoxia failed to downregulate Poldip2 in EZH2-deficient HASMCs (Figure 3 and Supplementary Figure 2) which overlaps with the inhibition of the hypoxia-induced, EZH2-dependent K27 trimethylation of Histone 3. Similar results are observed in human ventricular cardiac fibroblasts (Supplementary Figure 3). Consistent with our hypothesis, activation of the CDK2/Cyclin A/EZH2 pathway precedes H3 methylation and Poldip2 downregulation (Supplementary Figure 4). Together, these data indicate that hypoxia represses Poldip2 expression by a CDK2/EZH2-dependent mechanism.
Figure 3. Poldip2 is downregulated under hypoxia by an EZH2-dependent mechanism.
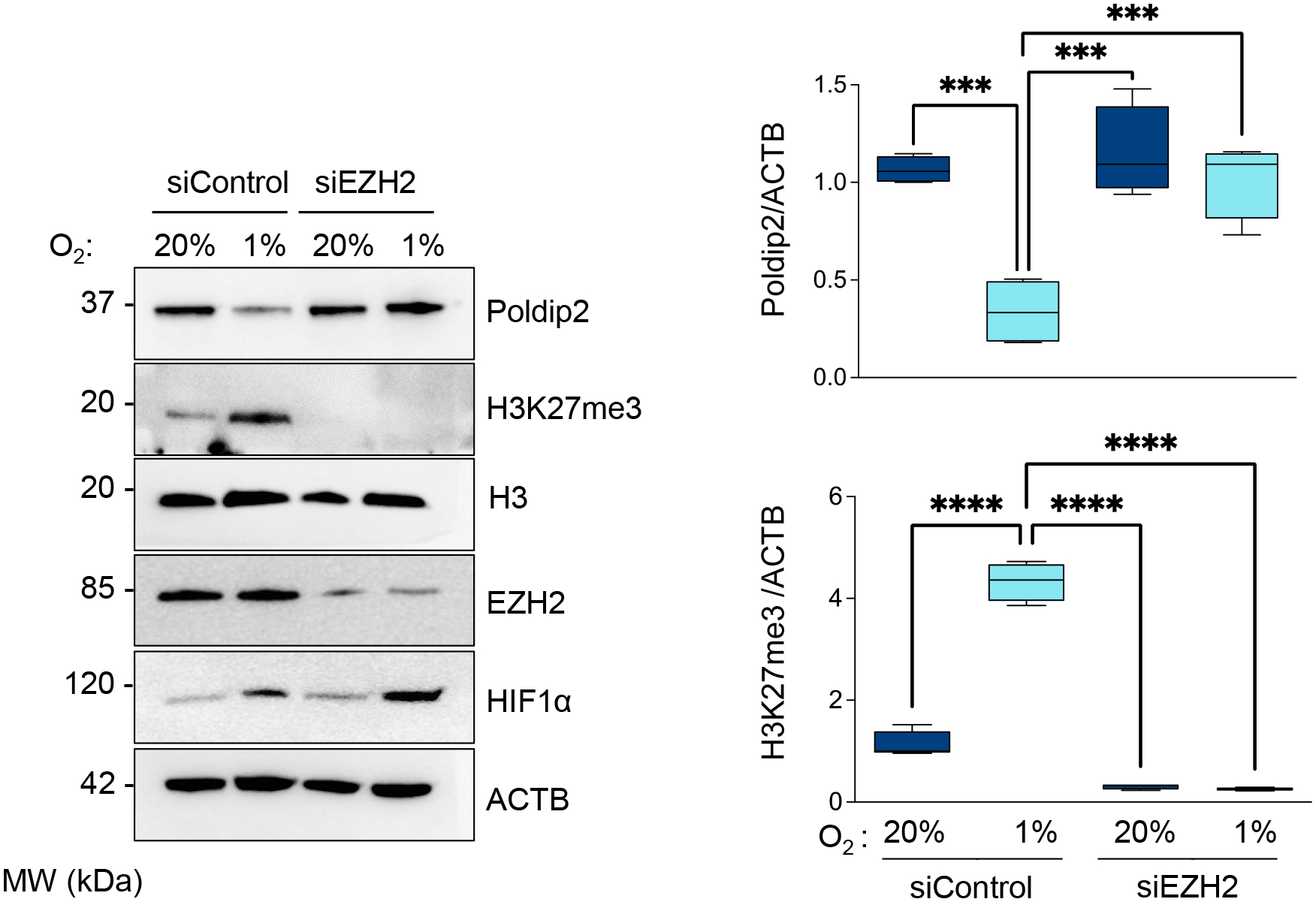
HASMCs were transfected with control siRNA (siControl) or siRNA against EZH2 (siEZH2). After 24 hours, cells were cultured under normoxia or hypoxia for 48 hours. Total lysates were prepared and analyzed by western blot using specific antibodies (Left). β-actin served as a loading control. Densitometric analysis of protein signals normalized to β-actin (Right). Bar graphs represent mean ± SE from 4 independent experiments.
Poldip2 repression is required for the metabolic inhibition of the Ubiquitin-Proteasome System under hypoxia in HASMCs.
Recent data from our laboratory showed that Poldip2-deficiency in normoxic cells inhibits the Krebs cycle [4] and leads to a metabolic reprogramming that causes the upregulation of the hexosamine pathways and the O-GlcNAc transferase (OGT)-dependent and glycosylation-mediated inhibition of the ubiquitin-proteasome system (UPS) [7]. Thus, we posited that repression of Poldip2 under hypoxia might participate in metabolic inhibition of the UPS.
To examine UPS activity under hypoxia, we tracked a fluorogenic UPS substrate’s degradation and observed that concomitant to Poldip2 repression, UPS activity is inhibited under hypoxia (Supplementary Figure 5A). Consistent with this result, hypoxic HASMCs accumulate poly-ubiquitinated aggregates and Ub-GFP (Supplementary Figure 5B).
Then, we use a forced expression of Poldip2 to restore its level under hypoxia. We confirmed that overexpressed Poldip2-myc was localized to the mitochondrion as endogenous Poldip2 (Supplementary Figure 6). Most importantly, we observed that Poldip2 expression rescued UPS activity (Figure 4A) and prevented the accumulation of poly-ubiquitinated aggregates or UPS activity reporters (Figure 4B). These experiments demonstrate that Poldip2 repression under hypoxia is required for hypoxia-induced UPS inhibition in HASMCs.
Figure 4. Proteasome inhibition under hypoxia requires Poldip2 downregulation.
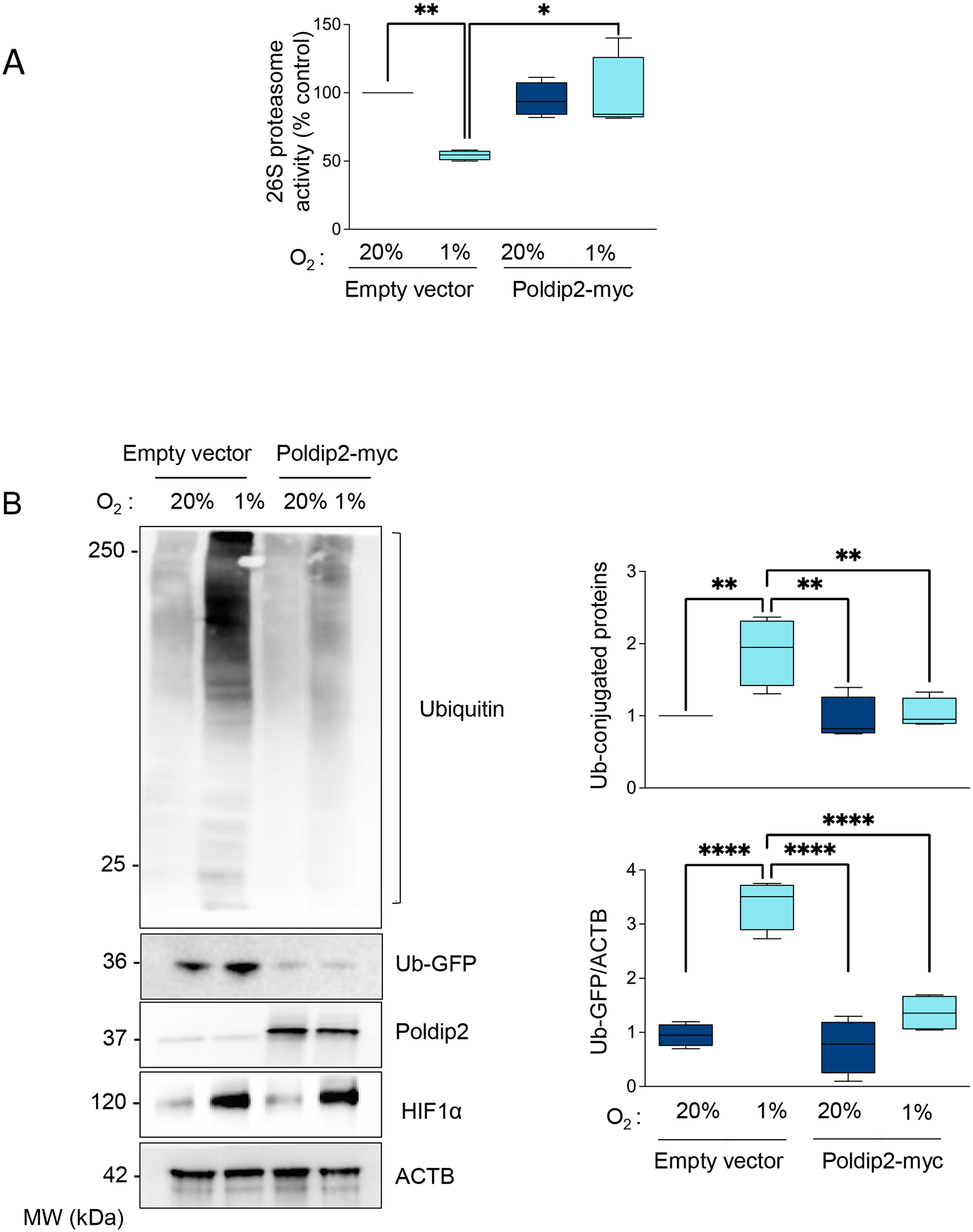
(A) HASMCs were transduced with empty vector or Poldip2-myc-expressing adenovirus and cultured under normoxia or hypoxia for 48 hours. Lysates were prepared, and UPS activity was measured using a fluorogenic assay described in the Methods section. Results were expressed as a percentage of the control. (B) Representative western blot to analyze the accumulation of Poly-ubiquitinated proteins and the reporter Ub-GFP signal. Bar graphs represent mean ± SE from 4 independent experiments.
Furthermore, regarding the upstream mechanism and consistent with the role of OGT-mediated glycosylation in hypoxia-induced UPS inhibition, we observed that hypoxia upregulated key enzymes of the hexosamine biosynthetic pathway (HBP). Hexokinase 2 (HK2), glutamine-fructose-6-phosphate transaminase 2 (GFPT2) (which catalyzes the HBP’s rate-limiting reaction), and OGT were significantly upregulated by hypoxia in HASMCs (Supplementary Figure 7), leading to the accumulation of O-GlcNAc-modified proteins in HASMCs (Supplementary Figure 7). This hypoxia-induced metabolic reprogramming event is mediated by Poldip2 repression since overexpression of Poldip2 reversed hypoxia-induced upregulation of HK2, GFPT2, and OGT, as well as the accumulation of O-linked glycosylated proteins (Figure 5).
Figure 5. Hypoxia-increased flux through the HBP and OGT activity requires Poldip2 downregulation.
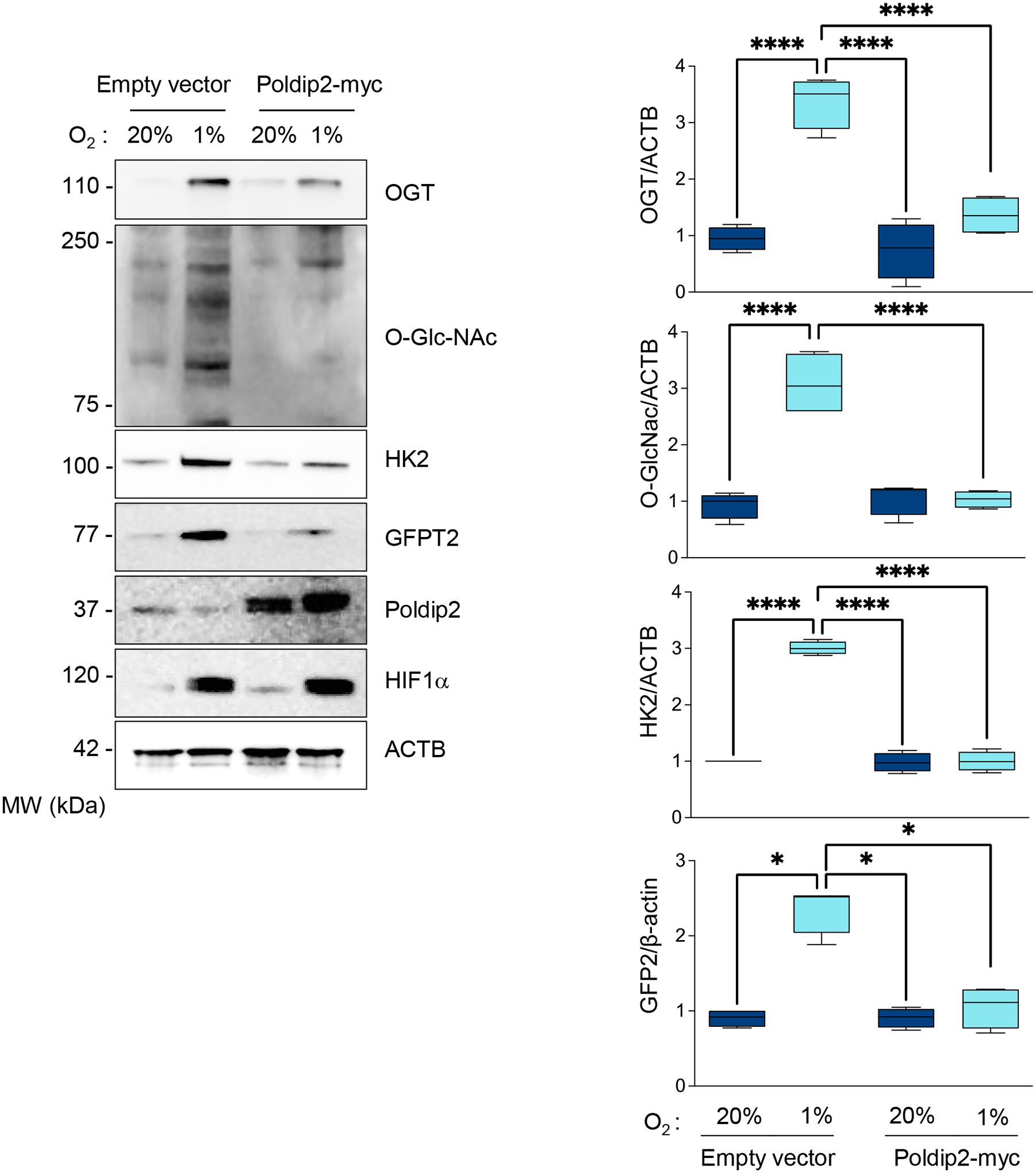
HASMCs were transduced with empty vector or Poldip2-myc-expressing adenovirus and cultured under normoxia or hypoxia for 48 hours. Lysates were prepared and analyzed by western blots using specific antibodies. Representative western blots show the expression of key components of the HBP and the accumulation of O-linked GlcNAcylated proteins. Bar graphs are presented as mean ± SE from 4 independent experiments.
Finally, and consistent with the role of OGT in UPS inhibition downstream of Poldip2 downregulation, we observed that UPS activity was restored, and the accumulation of ubiquitinated proteins and UPS activity reporters were reversed when OGT was downregulated in hypoxic HASMCs (Figures 6A and B). Together, these data demonstrate that the repression of Poldip2 under hypoxia inhibits UPS activity through upregulation of the HBP and OGT-mediated glycosylation.
Figure 6. UPS inhibition under hypoxia is OGT-dependent.
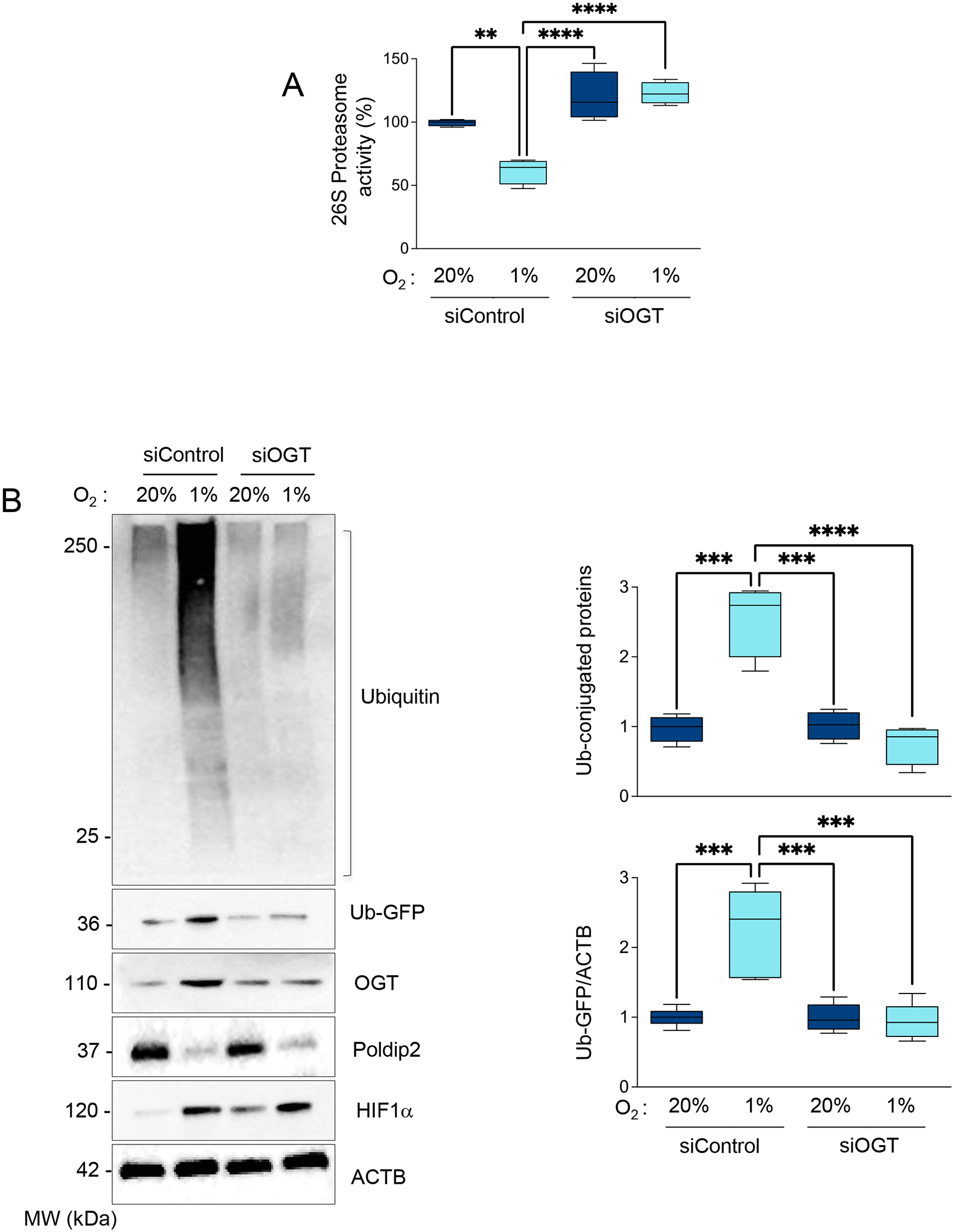
HASMCs were transfected with siRNA control (siControl) or siRNA against OGT (siOGT). After 24 hours, cells were cultured under normoxia or hypoxia for 48 hours. (A) Proteasome activity was measured using a fluorogenic assay described in the Methods section. (B) Representative western blot to analyze the accumulation of Poly-ubiquitinated proteins and the reporter Ub-GFP signal. Bar graphs represent mean ± SE from 4 independent experiments.
The CDK2/EZH2/Poldip2/OGT-dependent inhibition of the UPS is responsible for profibrotic gene expression under hypoxia.
CCN2 expression is upregulated by hypoxia in a variety of cell types [14, 26–29]. Furthermore, the rate of connective tissue deposition is accelerated in response to hypoxia [30] and the inhibition of the UPS [31]. Thus, we postulated that Poldip2-mediated UPS inhibition under hypoxia regulates fibrotic gene expression in HASMCs. First, we confirmed that hypoxia induces the expression of CCN2, Collagen 1A (COL1A), and Fibronectin 1 (FN1) in HASMCs (Supplemental Figure 8A). Our data also indicated that under hypoxia, the expression of CCN2 is upstream of COL1A and FN1 (Supplementary Figure 8B) in an upstream pathway that seems to be independent to the TGF-ß pathway, which displays no upregulation under hypoxic conditions (Supplementary Figure 8C). These experiments demonstrated that CCN2 is upregulated and drives the expression of COL1A and FN1 in HASMCs under hypoxia.
Importantly, we observed that forced expression of Poldip2 under hypoxia is sufficient to reverse the accumulation of CCN2, COL1A, and FN1 in HASMCs (Figure 7) and human ventricular cardiac fibroblasts (Supplementary Figure 9).
Figure 7. Hypoxia-induced profibrotic gene expression requires Poldip2 downregulation.
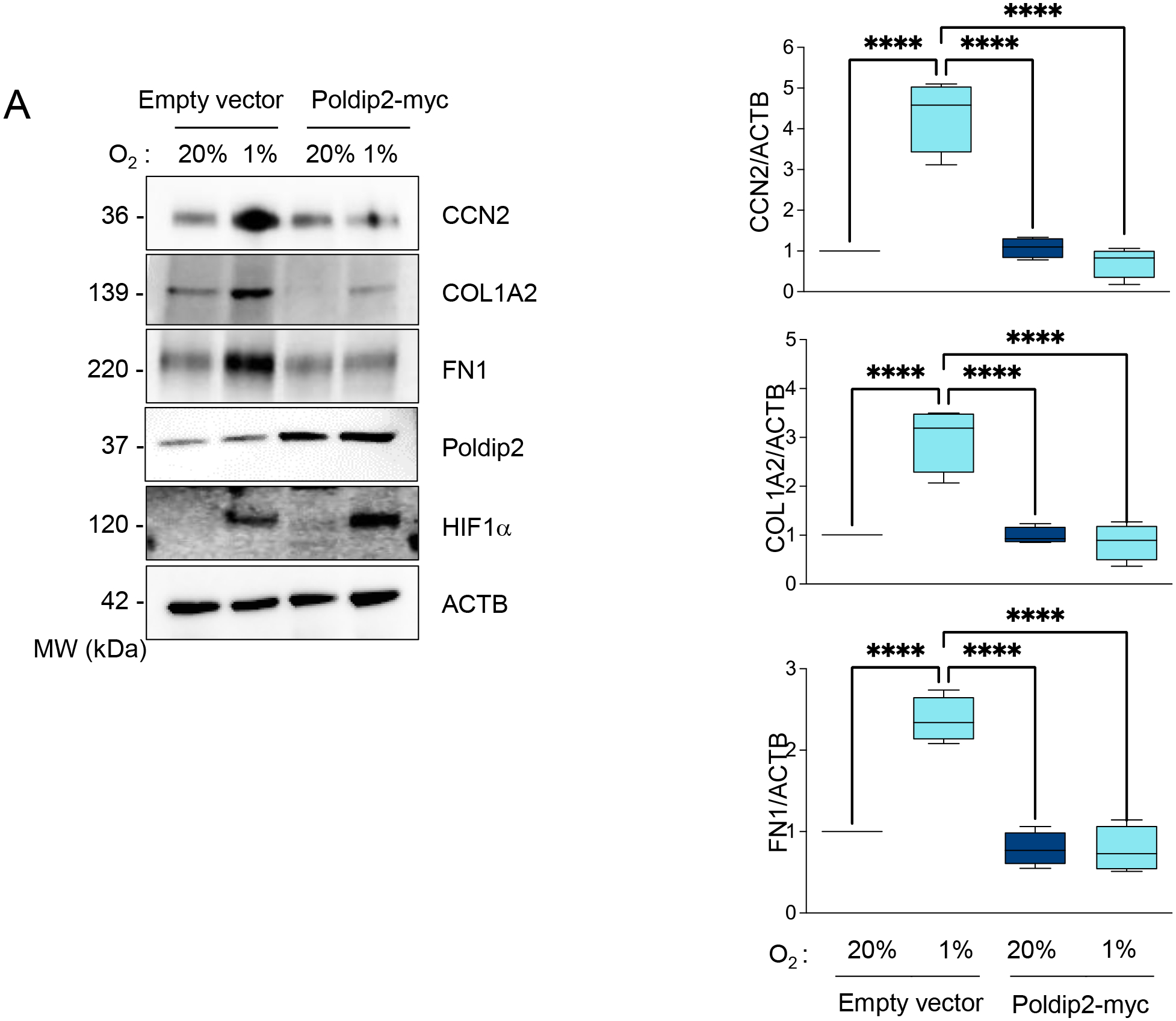
HASMCs were transduced with empty vector or Poldip2-myc-expressing adenovirus and cultured under normoxia or hypoxia for 48 hours. Lysates were prepared and analyzed by western blot using specific antibodies against profibrotic proteins (Left). β-actin served as a loading control. Densitometric analysis of protein signals normalized to β-actin (Right).
Angiotensin II strongly activates HIF1α in VSMCs [32–34]. Consistently, Poldip2 deficient mice infused with Angiotensin express higher levels of CCN2 in the media of coronary arteries (Figures 8A–B) and accumulation of interstitial collagen in the heart (Figure 8C) under this pathological condition which is prone to activate HIF1α. These experiments demonstrate a critical role for Poldip2 in fibrosis in vitro and in vivo.
Figure 8. The media of arteries of Poldip2 deficient animals display exacerbate expression of CCN2.
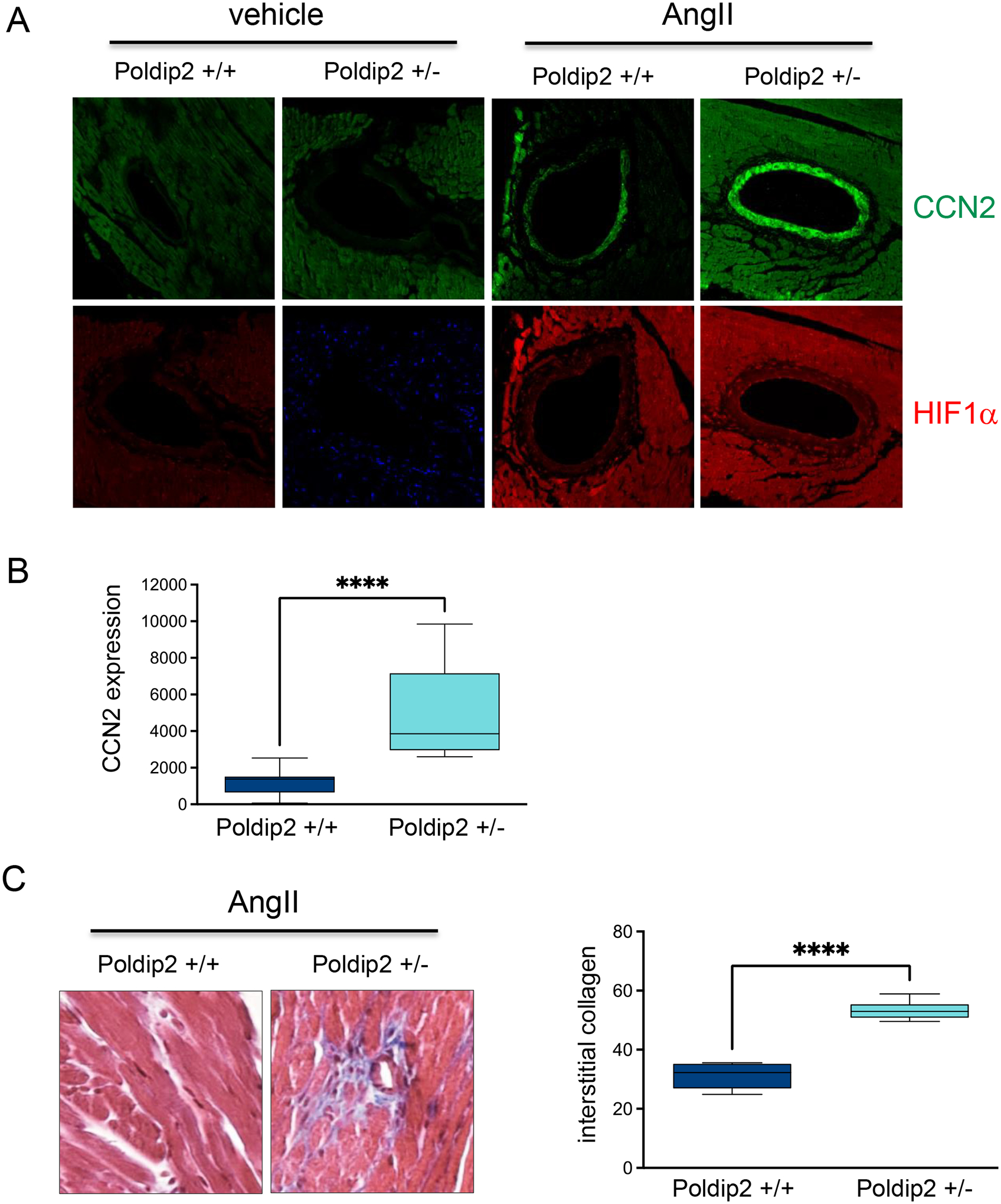
Wild type and Poldip2 deficient mice were treated with AngII for 30 days, hearts were harvested and process for histology. A. Expression of CCN2 and HIF1a in coronary arteries acquired by Confocal microcopy. B. Bar graphs of mean fluorescence intensity ± SE calculated using Image J software (NIH) from 10 independent experiments. C. Representative images of Masson’s Trichrome staining of heart tissue and Bar graphs of mean staining intensity ± SE calculated using Image J software (NIH) from 10 independent experiments.
In agreement with the role of OGT in mediating UPS inhibition in this pathway (Figure 6), we found that CCN2, COL1A, and FN1 upregulation under hypoxia is abrogated in OGT deficient HASMCs (Figure 9A).
Figure 9. Hypoxia-induced profibrotic gene expression requires OGT-mediated inhibition of the UPS.
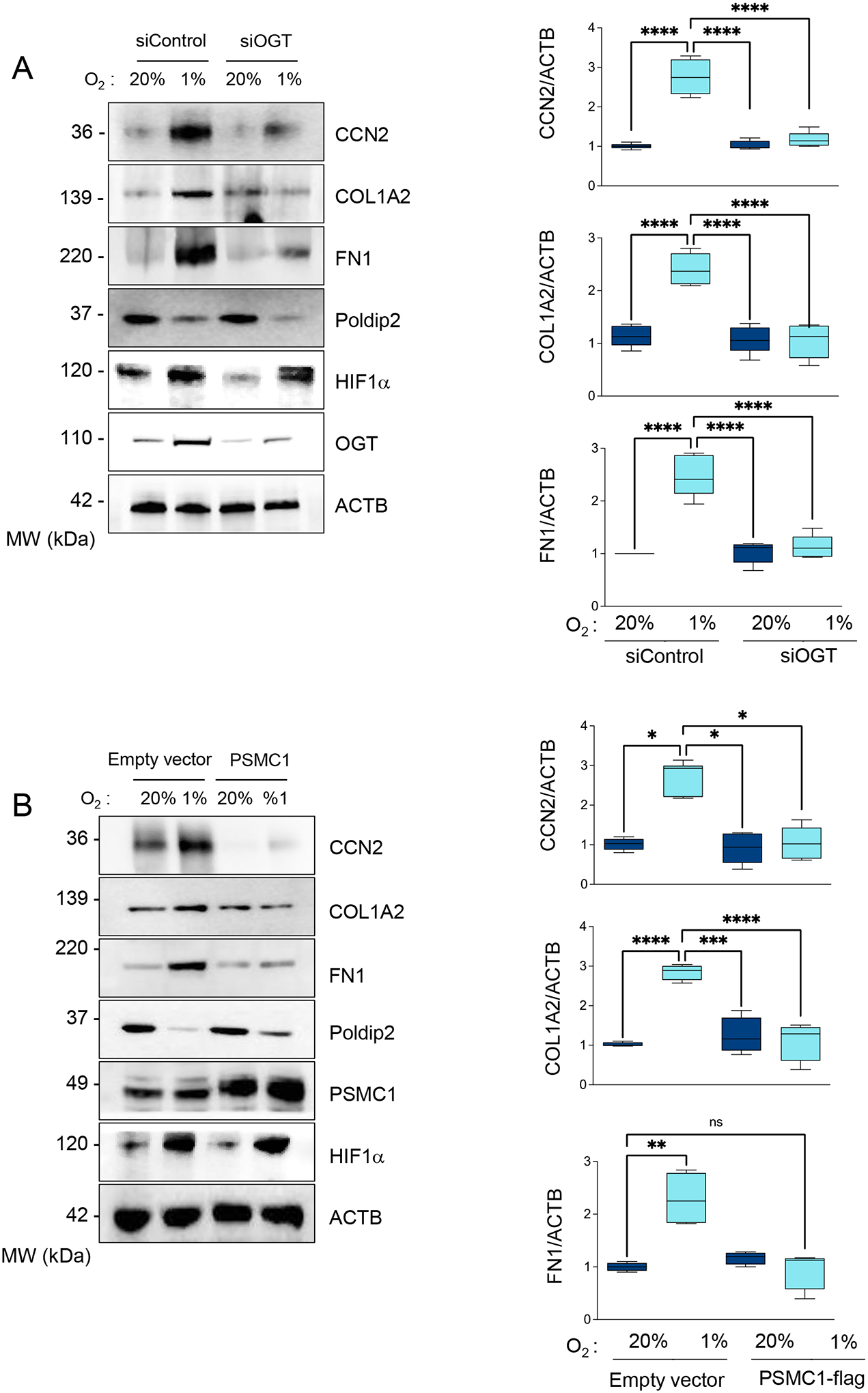
(A) HASMCs were transfected with control siRNA (siControl) or siRNA against OGT (siOGT) and cultured under normoxia or hypoxia for 48 hours. Lysates were prepared and analyzed by western blots using specific antibodies (Left). β-actin served as a loading control. Densitometric analysis of protein signals normalized to β-actin (Right). (B) HASMCs were transduced with an empty vector or adenovirus expressing PSMC1-myc and cultured under normoxia or hypoxia for 48 hours. Lysates were prepared and analyzed by western blots using specific antibodies (Left). β-actin served as a loading control. Densitometric analysis of protein signals normalized to β-actin (Right). Bar graphs are presented as mean ± SE from 4 independent experiments.
Previously, we identified the 19S component PSMC1 as the target of OGT-mediated UPS inhibition. OGT-mediated glycosylation impairs PSMC1 phosphorylation-induced activation [7]. Consistently, we found that forced expression of PSMC1 not only restores hypoxia-induced UPS inhibition (Supplemental Figures 5 A–B) but reverses the expression of profibrotic genes as well (Figure 9B). Finally, we confirmed that inhibition of CDK2 using siRNA (Figure 10) or pharmacological inhibitors (Supplementary Figures 10 A–B) and downregulation of EZH2 (Figure 11) are sufficient to block the expression of profibrotic genes under hypoxia and restore hypoxia-induced UPS inhibition. This series of experiments demonstrates that CCN2 and CCN2-induced expression of COL1A and FN1 under hypoxia depends on the Poldip2 repression by a CDK2/EZH2-dependent mechanism.
Figure 10. Hypoxia-induced profibrotic gene expression requires CDK2.
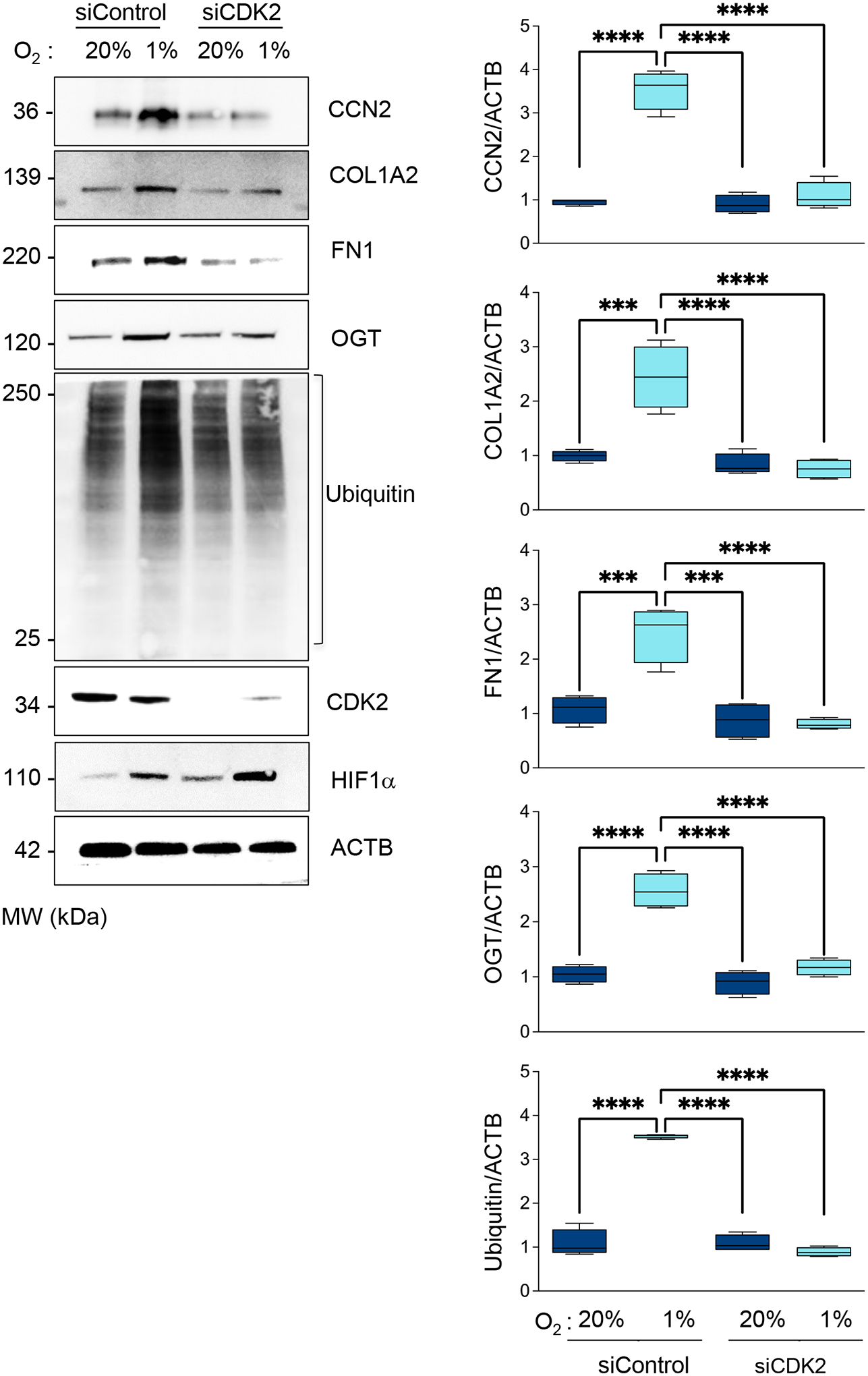
HASMCs were transfected with control siRNA (siControl) or siRNA against CDK2 (siCDK2). HASMCs were cultured under normoxia or hypoxia for 48 h. Total lysates were prepared and analyzed by western blot using specific antibodies (Left). β-actin served as a loading control. Densitometric analysis of protein signals normalized to β-actin (Right). Bar graphs are presented as mean ± SE from 4 independent experiments.
Figure 11. Hypoxia-induced profibrotic genes expression requires EZH2.
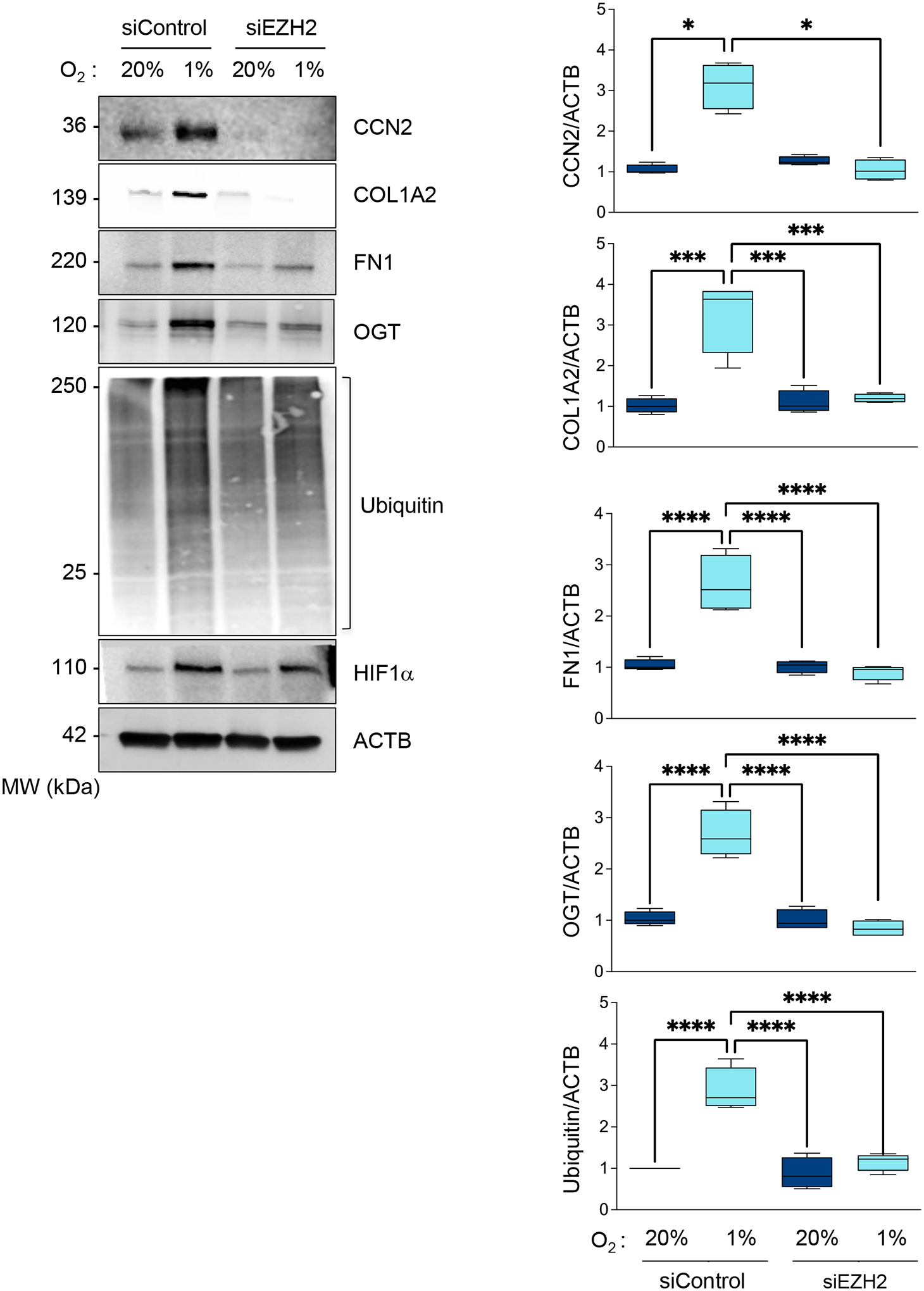
A. HASMCs were transfected with control siRNA (siControl) or siRNA against EZH2 (siEZH2) and cultured under normoxia or hypoxia for 48 hours. Lysates were prepared and analyzed by western blots using specific antibodies (Left). β-actin served as a loading control. Densitometric analysis of protein signals normalized to β-actin (Right). Bar graphs are presented as mean ± SE from 4 independent experiments.
The UPS inhibition-dependent stabilization of SRF is required for profibrotic gene expression under hypoxia.
factor (SRF) is known to induce the expression of CCN2 in a variety of cell types [35–37]. Importantly, we have demonstrated that inhibition of the proteasome downstream from Poldip2 downregulation induces the stabilization of SRF, leading to its increased transcriptional activity [7]. Thus, we posited that the stabilization of SRF under hypoxia downstream Poldip2 repression is responsible for the CCN2 upregulation and profibrotic signaling in HASMCs. In support of this notion, we found that SRF expression is increased under hypoxia in a Poldip2- and PSMC1-dependent manner (Figure 12A–B). In particular, SRF is required for the upregulation of CCN2, COL1A, and FN1 under hypoxia (Figure 12C). These data demonstrate that SRF stabilization under Poldip2 inhibition-mediated repression of the UPS is required for the expression of fibrogenic genes under hypoxia in HASMCs.
Figure 12. Stabilization of SRF by UPS inhibition downstream Poldip2 repression is required for profibrotic signaling.
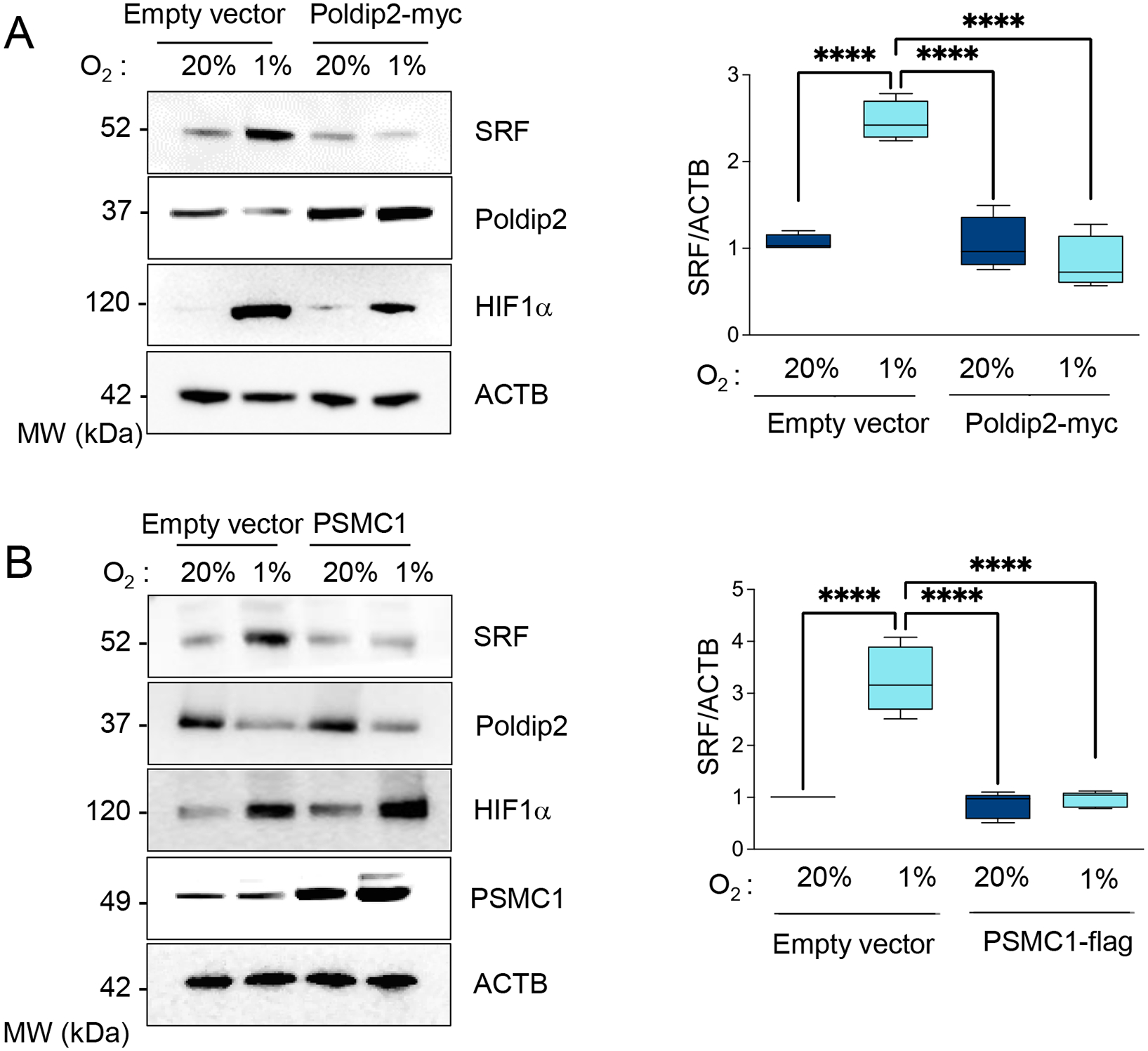
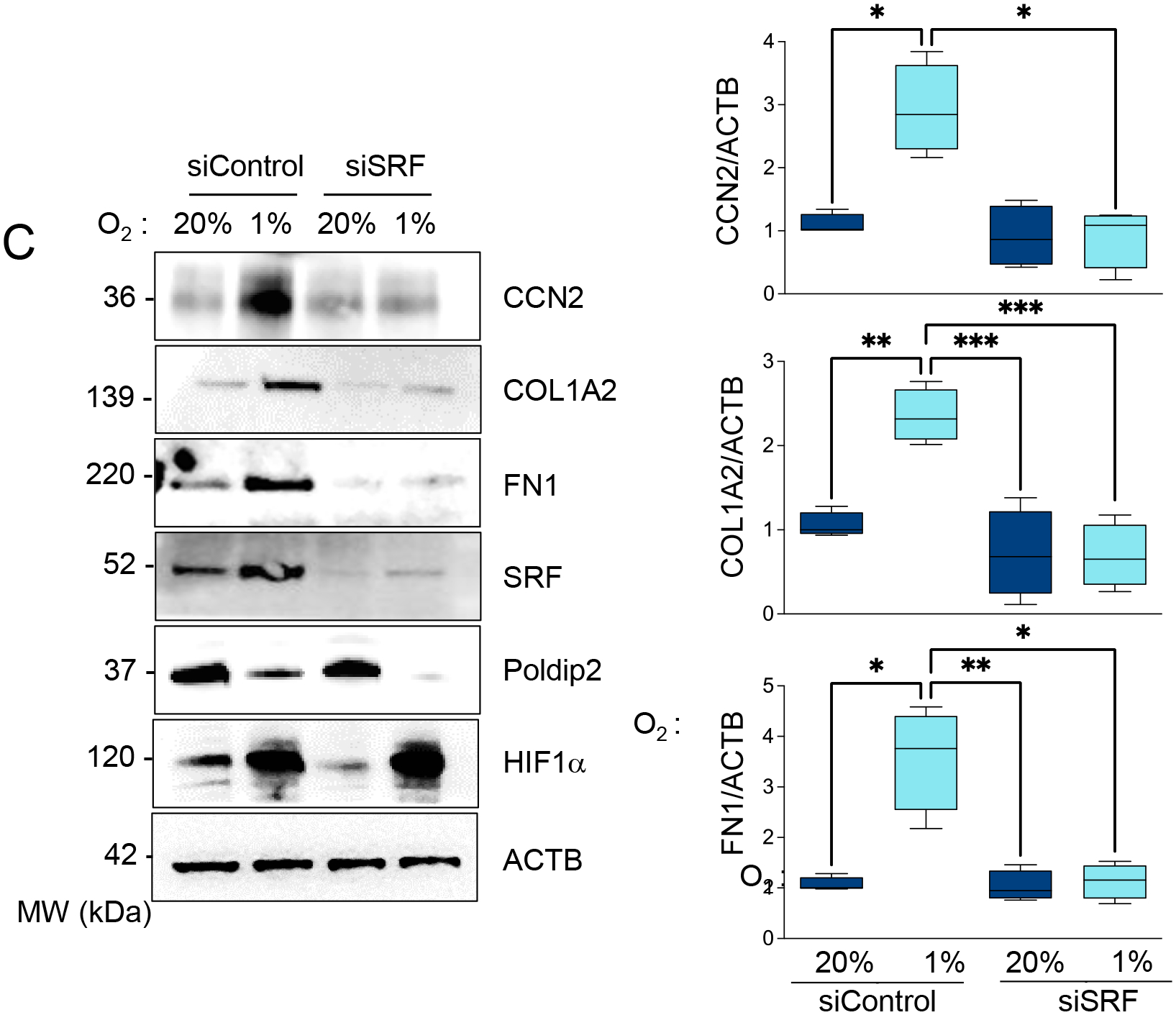
A. HASMCs were transduced with empty vector or Poldip2-myc-expressing adenovirus and cultured under normoxia or hypoxia for 48 hours. Lysates were prepared and analyzed by western blot using specific antibodies (Left). β-actin served as a loading control. Densitometric analysis of protein signals normalized to β-actin (Right). B. HASMCs were transduced with an empty vector or adenovirus expressing PSMC1-myc and cultured under normoxia or hypoxia for 48 hours. Lysates were prepared and analyzed by western blots using specific antibodies (Left). β-actin served as a loading control. Densitometric analysis of protein signals normalized to β-actin (Right). C. HASMCs were transfected with control siRNA (siControl) or siRNA against SRF (siSRF) and cultured under normoxia or hypoxia for 48 hours. Lysates were prepared and analyzed by western blots using specific antibodies (Left). β-actin served as a loading control. Densitometric analysis of protein signals normalized to β-actin (Right). Bar graphs are presented as mean ± SE from 4 independent experiments.
Based on our data, we propose a signaling pathway by which hypoxia leads to the upregulation of CCN2 and fibrosis (Figure 13). This mechanism leading to the expression of pro-fibrotic proteins links the cell cycle to the metabolic inhibition of the UPS. Importantly, we established that this mechanism required intraorganellar communication coordinated by the mitochondrial protein Poldip2.
Figure 13.
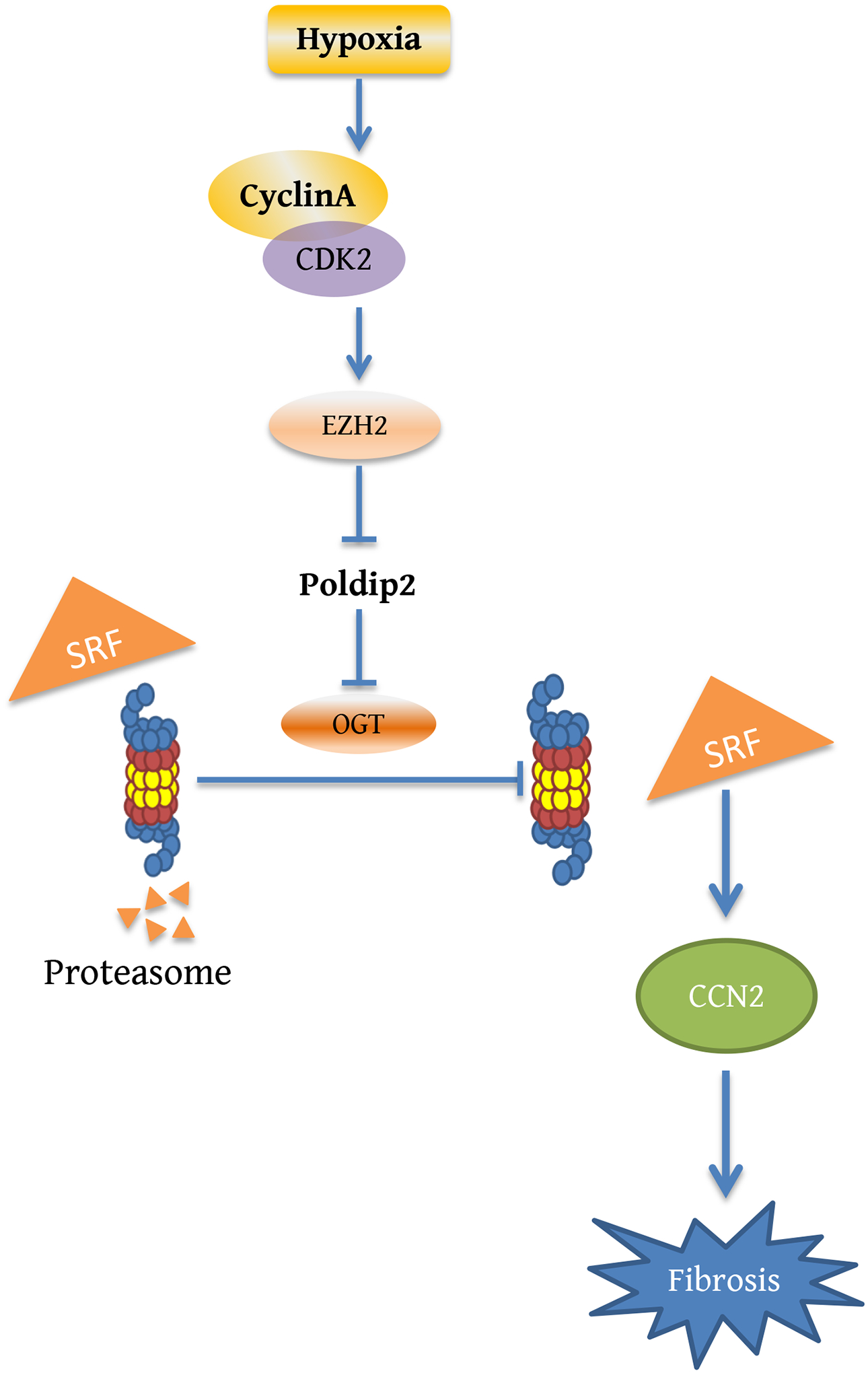
Proposed signaling pathway leading to increased expression of profibrotic genes under hypoxia.
DISCUSSION
Hypoxia is a significant environmental signal that initiates downstream physiological and pathological responses in the vasculature [38]. Our study demonstrates that hypoxia activates a particular metabolic reprogramming event in HASMCs, leading to inhibition of the UPS and induction of profibrotic signaling. We demonstrated that inhibition of the mitochondrial protein Poldip2 is critical for the metabolic reprogramming induced by hypoxia in HASMCs. Previously, we demonstrated that Poldip2 expression is required for oxidative metabolism and that its repression under hypoxia supports glycolytic metabolic shift [4]. Here, we expanded our observations and determined that under hypoxia, downregulation of Poldip2 activates the hexosamine pathways and increases OGT-mediated glycosylation. Similar metabolic reprogramming with increased OGT-mediated glycosylation has been reported in pancreatic ductal adenocarcinoma cancer cells [39]. Furthermore, since Poldip2 supports glycolytic reprogramming in triple-negative breast cancer cells [4], this mechanism may generally apply to metabolic adaptation to hypoxia in other cancer cells.
In cancer cells, hypoxia increases the expression of EZH2 by HIF-1α -dependent transcriptional activation (48). However, this seems cell type-dependent since we did not observe an induction of EZH2 expression by hypoxia.
Consistent with previous reports that show that HK2 is a direct target of HIF-1α [40], hypoxic cells displayed a significant increase in HK2 expression (Supplementary Figure 3). Interestingly, forced expression of Poldip2 reduced HK2 expression without affecting the level of HIF-1α (Figure 3). Thus, it is conceivable that post-translational modifications induced by changes in the metabolome affect the affinity of HIF-1α for hypoxia response elements (HRE) or for protein members of the PAS family to form heterodimers required for activity.
Our findings are consistent with earlier work showing the ability of hypoxia to activate CDK2 [41]. Additionally, previous studies have identified CDK2 as a positive regulator of glycolytic shift [42] and an upstream mediator of fibrosis [43]. In the current study, we identify Poldip2 as a target of CDK2-initiated signaling that mediates metabolic reprogramming, inhibition of the UPS, and the expression of fibrogenic factors.
CCN2 is a primary pathological marker of fibrosis since it is a mediator of TGF-ß1 signaling [44]. Recently, CCN2 expression has been linked to aerobic glycolysis [45]. Additionally, previous reports have shown that CCN2 expression is induced by hypoxia [28] via a HIF-1α-dependent mechanism [29]. Our results show that in addition to HIF-1α, the increased expression of CCN2, FN1, and COL1A under hypoxia requires the stabilization of SRF by a mechanism that involves Poldip2 downregulation and consequent inhibition of the UPS activity by OGT-mediated glycosylation.
Our data show that SRF is the specific target of UPS inhibition under hypoxia which is required to upregulate the expression of CCN2. This is consistent with previous reports showing that the stabilization of SRF by UPS inhibition increases its transcriptional activity [46] and with the fact that in different systems, SRF-mediated signaling is required for fibrosis [35–37].
In summary, we show that Poldip2-mediated glycolytic shift under hypoxia signals through an HBP/OGT-mediated signaling pathway inhibits the UPS activity that directly impacts fibrotic signaling. The reduction of UPS activity is likely to impact many cellular functions, and the relationship of this pathway to diseases associated with hypoxia or other metabolic switches to a more glycolytic phenotype warrants further investigation.
EXPERIMENTAL PROCEDURES
Cell Culture
Human aortic smooth muscle cells (HASMCs) and human mammary epithelial cells (HMECs) were purchased from Thermo-Fisher. Cells were grown as recommended by the vendor in base media with the addition of growth supplements. After cells reached ~ 80% confluence, growth supplements were removed 24h prior to experiments. For hypoxia experiments, cells were either exposed to normoxia or placed in 1% O2, 94% N2, 5% CO2 using a HypOxystation H35 (HypOxygen, MD, USA).
Animal studies and Histology
Poldip2+/− mice used were generated by a gene trap insertion in the first intron of Poldip2 (chromosome 11, NCBI Gene ID: 67811) at the Texas A&M Institute for Genomic Medicine (College Station, TX) and fully backcrossed with C57BL/6 as previously described [47]. Mice were anesthetized and osmotic mini-pumps (Azlet) containing [Asn1, Val5]-Ang II (Sigma) (750 μg/kg/day) or 0.9% saline were subcutaneously implanted into the mice, as previously described [48]. After 30 days, mice were euthanized using CO2, and tissues were pressure perfused with saline and fixed with 10% buffered formalin. Slices were prepared for histology and stained with Trichrome staining. For immunofluorescent imaging, sections were blocked and then incubated with CCN2 (Everest EB11760) or HIF1α (Bethyl, A300-286A) primary antibodies overnight at 4°C. Confocal micrographs were acquired with a Zeiss LSM 510 META Laser Scanning Confocal Microscope System using a 20x air objective lens and Zeiss ZEN acquisition software. When comparing sections from different experimental groups, confocal microscope image threshold settings remained constant. Mean fluorescence intensity and percent area were calculated using Image J software (NIH).
All procedures were approved by the Emory University Institutional Animal Care and Use Committee. Animal randomization and allocation concealment was performed for the analysis.
Inhibitors
CDK2 inhibitors were purchased from Cayman Chemical Company (CDK2 inhibitor II, Cat. 15154) and Sigma (GW8510, Cat. G7791). The inhibitors were prepared according to the supplier’s instructions at a stock concentration of 50mM for CDK2 inhibitor II and 10mM for GW8510. This stock solution was used directly to treat the cells at the concentration determined for each experiment.
G1/S Phase Synchronization using Double Thymidine Block
Thymidine synchronization was performed as previously described [49]. Briefly, HASMCs at 40% confluence were cultured in a growth medium supplemented with 2 mM thymidine (Sigma-Aldrich) for 18 hours (first block). After that, HASMCs were then washed with PBS to remove thymidine and cultured in a fresh growth medium for 9 hours. The second block of 16 hours was performed by adding 2 mM thymidine. After 16 hours, cells were washed with PBS, and a fresh medium was added.
Cell Transfection
Cells were transfected using Lipofectamine RNAiMAX reagent (Thermo Fisher Scientific) following the manufacturer’s suggested protocol. Sequences were: Poldip2 sense 5’-CGUGAGGUUUGAUCAGUAATT-3’; OGT sense 5’-TACGCGTGCCATCCAAATTAA-3’; EZH2 sense 5’GACUCUGAAUGCAGUUGCU3’; CCN2 sense 5’AAAGGTTAGTATCATCAGATA-3’; CDK2 sense 5’-GACGGAGCTTGTTATCGCAAA-3’; SRF sense 5’- AAGGAGCGGCCTCGCCATAAA-3’. Allstars Negative control (Qiagen Cat. 1027281) was used as a negative control in all experiments.
Cell Fractionation
Cells were washed with PBS and scraped in 1X buffer A provided by the cell fractionation kit (Abcam, ab109719). Cells were then fractionated according to the protocol provided by Abcam. The final cell fractions were sonicated twice for 10s, SDS loading buffer was added (2X), and samples were heated to 80°C for 5 min.
Western Blots
Protein samples were separated on Express-Plus Page Gels (GenScript) with Tris-MOPS buffer containing SDS and transferred onto an Immobilon-P membrane (Millipore, IPVH00010).
All primary antibodies were incubated for 24h at 4°C. Protein bands were visualized using ECL Western Blotting Substrate (Pierce, 32106) and imaged on a Kodak Camera System. Densitometric analyses of protein bands were performed using the software (Carestream and Image J). Densitometric values were normalized to housekeeping genes.
| Protein | Company | Catalog number |
|---|---|---|
| Poldip2 | Santa Cruz | sc-398591 |
| Poldip2 | Abcam | ab172435 |
| β-actin | Sigma | A5441 |
| OCT | Abcam | ab96718 |
| Ubiquitin | Cell Signaling | 3936S |
| COL1A | Bioss | Bs-10423R |
| Fibronectin | Millipore | AB2033 |
| HIF1A | Bethyl Laboratories | A300–286A |
| CCN2/CTGF | Santa Cruz | sc-14939 |
| GFP | Rockland | 600120215 |
| O-GlcNAc | Cell Signaling | 98755 |
| O-GlcNAc | Abcam | ab2739 |
| HK2 | Santa Cruz | sc-374091 |
| GFP2 | Cell Signaling | 98755 |
| p-CDK2 | Abcam | Ab183554 |
| CDK2 | Santa Cruz | sc-163 |
| p-EZH2 | Active Motif | 61241 |
| PSMC1 | Bethyl Laboratories | A303821A |
| SRF | Santa Cruz | sc-335 |
| Cyclin A | Santa Cruz | sc-53227 |
| EZH2 | Cell Signaling | 52465S |
Short-life reporter accumulation
HASMCs were transfected with siRNA control or against Poldip2 using RNAiMAX (Thermo-Fisher). After 24h, cells were transfected with GFP-Ub (Addgene#11928) using Lipofectamine 3000 (Thermo-Fisher) following the manufacturer’s recommendations. Total protein lysates were prepared after 24h, and the accumulation of fluorescent probe was quantified by immunoblot using a primary antibody recognizing GFP (Rockland#600-102-215).
Proteasome Activity
HASMCs were transfected with control siRNA or siRNA against Poldip2. After 48h, protein lysates were prepared, and 100μL of lysate was mixed with fusion protein for 48h at 25°C. After collection, 100μL of lysate was transferred to a 96-well plate, mixed with Bz-Val-Gly-Arg-Amc to a final concentration of 50 mM, and incubated at 37°C for 1h. Fluorescence readings were taken using filters at specific wavelengths (360/380 excitation/ 460 emission). The results were presented as % 26S proteasome activity compared to control activity. In some samples, 50 μM MG132 (Cayman Chemical, #10012628) was added 3h prior to samples collection as a positive control.
Adenoviral expression of Poldip2 and PSMC1
Human Poldip2 cDNA (Accession NP_056399) plus a C-terminal myc tag (EQKLISEEDL) was cloned into the pAdTrack-CMV vector, which allows for simultaneous expression of myc-tagged Poldip2 and GFP by two independent CMV promotors. The virus was packaged using the AdEasy Adenoviral Vector System. (Agilent Technologies) as previously described [50, 51].
The human PSMC1 cDNA (MGC24583) with C-terminal myc tag (EQKLISEEDL) and His tag was cloned into the pAdTrack-CMV vector, which allows for the expression of myc-tagged PSMC1 and His Tag by two independent CMV promoters. The adenovirus was obtained from Vigene Biosciences (Cat. VH827525).
Statistical Analysis
Data are presented as mean ± SEM from a minimum of 4 independent experiments. When samples passed the normality Shapiro Wilk test, significance was determined using a t-test for unpaired samples or one-way ANOVA followed by Dunnet’s post hoc test for multiple comparisons. When samples were not normally-distributed, the Mann-Whitney test for two groups or Kruskal Wallis for multiple group comparisons were used. GraphPad Prism 6 Software was used for statistical analyses. A threshold of P<0.05 was considered significant. * represents P < 0.05, ** represents P < 0.01, *** represents P < 0.001 and **** represents P < 0.0001.
Supplementary Material
Acknowledgment
This study was supported by The National Heart, Lung, and Blood Institute of the National Institutes of Health under awards HL095070 to ASM and by the American Heart Association under fellowship award POST3438089 to FP.
Footnotes
Conflict of interest: The authors declare that they have no conflicts of interest with the contents of this article.
References
- [1].Barman SA, Li X, Haigh S, Kondrikov D, Mahboubi K, Bordan Z, Stepp DW, Zhou J, Wang Y, Weintraub DS, Traber P, Snider W, Jonigk D, Sullivan J, Crislip GR, Butcher JT, Thompson J, Su Y, Chen F, Fulton DJR, Galectin-3 is expressed in vascular smooth muscle cells and promotes pulmonary hypertension through changes in proliferation, apoptosis, and fibrosis, Am J Physiol Lung Cell Mol Physiol 316(5) (2019) L784–L797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wiafe B, Adesida A, Churchill T, Adewuyi EE, Li Z, Metcalfe P, Hypoxia-increased expression of genes involved in inflammation, dedifferentiation, pro-fibrosis, and extracellular matrix remodeling of human bladder smooth muscle cells, In Vitro Cell Dev Biol Anim 53(1) (2017) 58–66. [DOI] [PubMed] [Google Scholar]
- [3].Imanishi M, Tomita S, Ishizawa K, Kihira Y, Ueno M, Izawa-Ishizawa Y, Ikeda Y, Yamano N, Tsuchiya K, Tamaki T, Smooth muscle cell-specific Hif-1alpha deficiency suppresses angiotensin II-induced vascular remodelling in mice, Cardiovasc Res 102(3) (2014) 460–8. [DOI] [PubMed] [Google Scholar]
- [4].Paredes F, Sheldon K, Lassegue B, Williams HC, Faidley EA, Benavides GA, Torres G, Sanhueza-Olivares F, Yeligar SM, Griendling KK, Darley-Usmar V, San Martin A, Poldip2 is an oxygen-sensitive protein that controls PDH and alphaKGDH lipoylation and activation to support metabolic adaptation in hypoxia and cancer, Proc Natl Acad Sci U S A 115(8) (2018) 1789–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Schmidt M, Finley D, Regulation of proteasome activity in health and disease, Biochim Biophys Acta 1843(1) (2014) 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tanaka K, The proteasome: overview of structure and functions, Proc Jpn Acad Ser B Phys Biol Sci 85(1) (2009) 12–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Paredes F, Williams HC, Quintana RA, San Martin A, Mitochondrial Protein Poldip2 (Polymerase Delta Interacting Protein 2) Controls Vascular Smooth Muscle Differentiated Phenotype by O-Linked GlcNAc (N-Acetylglucosamine) Transferase-Dependent Inhibition of a Ubiquitin Proteasome System, Circ Res 126(1) (2020) 41–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jun JI, Lau LF, Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets, Nat Rev Drug Discov 10(12) (2011) 945–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ramazani Y, Knops N, Elmonem MA, Nguyen TQ, Arcolino FO, van den Heuvel L, Levtchenko E, Kuypers D, Goldschmeding R, Connective tissue growth factor (CTGF) from basics to clinics, Matrix Biol 68–69 (2018) 44–66. [DOI] [PubMed] [Google Scholar]
- [10].Shen YW, Zhou YD, Chen HZ, Luan X, Zhang WD, Targeting CTGF in Cancer: An Emerging Therapeutic Opportunity, Trends Cancer (2020). [DOI] [PubMed] [Google Scholar]
- [11].Tache D, Bogdan F, Pisoschi C, Banita M, Stanciulescu C, Fusaru AM, Comanescu V, Evidence for the involvement of TGF-beta1-CTGF axis in liver fibrogenesis secondary to hepatic viral infection, Rom J Morphol Embryol 52(1 Suppl) (2011) 409–12. [PubMed] [Google Scholar]
- [12].Ferdoushi S, Paul D, Ghosh CK, Sultana T, Joarder AI, Islam MS, Islam MS, Mahmuduzzaman M, Rahman Q, Ahmed AN, Correlation of Connective Tissue Growth Factor (CTGF/CCN2) with Hepatic Fibrosis in Chronic Hepatitis B, Mymensingh Med J 24(3) (2015) 558–63. [PubMed] [Google Scholar]
- [13].De Francesco EM, Lappano R, Santolla MF, Marsico S, Caruso A, Maggiolini M, HIF-1alpha/GPER signaling mediates the expression of VEGF induced by hypoxia in breast cancer associated fibroblasts (CAFs), Breast Cancer Res 15(4) (2013) R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wu W, Li J, Zhao M, Liu X, HIF-1alpha mediates visfatin-induced CTGF expression in vascular endothelial cells, Cell Mol Biol (Noisy-le-grand) 63(4) (2017) 28–32. [DOI] [PubMed] [Google Scholar]
- [15].Oemar BS, Werner A, Garnier JM, Do DD, Godoy N, Nauck M, Marz W, Rupp J, Pech M, Luscher TF, Human connective tissue growth factor is expressed in advanced atherosclerotic lesions, Circulation 95(4) (1997) 831–9. [DOI] [PubMed] [Google Scholar]
- [16].San Martin A, Du P, Dikalova A, Lassegue B, Aleman M, Gongora MC, Brown K, Joseph G, Harrison DG, Taylor WR, Jo H, Griendling KK, Reactive oxygen species-selective regulation of aortic inflammatory gene expression in Type 2 diabetes, Am J Physiol Heart Circ Physiol 292(5) (2007) H2073–82. [DOI] [PubMed] [Google Scholar]
- [17].Akashi S, Nishida T, Mizukawa T, Kawata K, Takigawa M, Iida S, Kubota S, Regulation of cellular communication network factor 2 (CCN2) in breast cancer cells via the cell-type dependent interplay between CCN2 and glycolysis, J Oral Biosci 62(3) (2020) 280–288. [DOI] [PubMed] [Google Scholar]
- [18].Kim H, Son S, Ko Y, Shin I, CTGF regulates cell proliferation, migration, and glucose metabolism through activation of FAK signaling in triple-negative breast cancer, Oncogene 40(15) (2021) 2667–2681. [DOI] [PubMed] [Google Scholar]
- [19].Maeda-Uematsu A, Kubota S, Kawaki H, Kawata K, Miyake Y, Hattori T, Nishida T, Moritani N, Lyons KM, Iida S, Takigawa M, CCN2 as a novel molecule supporting energy metabolism of chondrocytes, J Cell Biochem 115(5) (2014) 854–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Brown DI, Lassegue B, Lee M, Zafari R, Long JS, Saavedra HI, Griendling KK, Poldip2 knockout results in perinatal lethality, reduced cellular growth and increased autophagy of mouse embryonic fibroblasts, PLoS One 9(5) (2014) e96657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tyers M, Fitch I, Tokiwa G, Dahmann C, Nash R, Linskens M, Futcher B, Characterization of G1 and mitotic cyclins of budding yeast, Cold Spring Harb Symp Quant Biol 56 (1991) 21–32. [DOI] [PubMed] [Google Scholar]
- [22].Stevenson LM, Deal MS, Hagopian JC, Lew J, Activation mechanism of CDK2: role of cyclin binding versus phosphorylation, Biochemistry 41(26) (2002) 8528–34. [DOI] [PubMed] [Google Scholar]
- [23].Sellers WR, Loda M, The EZH2 polycomb transcriptional repressor--a marker or mover of metastatic prostate cancer?, Cancer Cell 2(5) (2002) 349–50. [DOI] [PubMed] [Google Scholar]
- [24].Kaneko S, Li G, Son J, Xu CF, Margueron R, Neubert TA, Reinberg D, Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA, Genes Dev 24(23) (2010) 2615–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chen S, Bohrer LR, Rai AN, Pan Y, Gan L, Zhou X, Bagchi A, Simon JA, Huang H, Cyclin-dependent kinases regulate epigenetic gene silencing through phosphorylation of EZH2, Nat Cell Biol 12(11) (2010) 1108–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ren J, Guo H, Wu H, Tian T, Dong D, Zhang Y, Sui Y, Zhang Y, Zhao D, Wang S, Li Z, Zhang X, Liu R, Qian J, Wei H, Jiang W, Liu Y, Li Y, GPER in CAFs regulates hypoxia-driven breast cancer invasion in a CTGF-dependent manner, Oncol Rep 33(4) (2015) 1929–37. [DOI] [PubMed] [Google Scholar]
- [27].Kroening S, Neubauer E, Wullich B, Aten J, Goppelt-Struebe M, Characterization of connective tissue growth factor expression in primary cultures of human tubular epithelial cells: modulation by hypoxia, American journal of physiology. Renal physiology 298(3) (2010) F796–806. [DOI] [PubMed] [Google Scholar]
- [28].Hong KH, Yoo SA, Kang SS, Choi JJ, Kim WU, Cho CS, Hypoxia induces expression of connective tissue growth factor in scleroderma skin fibroblasts, Clin Exp Immunol 146(2) (2006) 362–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Higgins DF, Biju MP, Akai Y, Wutz A, Johnson RS, Haase VH, Hypoxic induction of Ctgf is directly mediated by Hif-1, American journal of physiology. Renal physiology 287(6) (2004) F1223–32. [DOI] [PubMed] [Google Scholar]
- [30].Xiong A, Liu Y, Targeting Hypoxia Inducible Factors-1alpha As a Novel Therapy in Fibrosis, Front Pharmacol 8 (2017) 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zhou Y, Zhang J, Li H, Huang T, Wong CC, Wu F, Wu M, Weng N, Liu L, Cheng ASL, Yu J, Wong N, Lo KW, Tang PMK, Kang W, To KF, AMOTL1 enhances YAP1 stability and promotes YAP1-driven gastric oncogenesis, Oncogene 39(22) (2020) 4375–4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lauzier MC, Page EL, Michaud MD, Richard DE, Differential regulation of hypoxia-inducible factor-1 through receptor tyrosine kinase transactivation in vascular smooth muscle cells, Endocrinology 148(8) (2007) 4023–31. [DOI] [PubMed] [Google Scholar]
- [33].Page EL, Robitaille GA, Pouyssegur J, Richard DE, Induction of hypoxia-inducible factor-1alpha by transcriptional and translational mechanisms, J Biol Chem 277(50) (2002) 48403–9. [DOI] [PubMed] [Google Scholar]
- [34].Richard DE, Berra E, Pouyssegur J, Nonhypoxic pathway mediates the induction of hypoxia-inducible factor 1alpha in vascular smooth muscle cells, J Biol Chem 275(35) (2000) 26765–71. [DOI] [PubMed] [Google Scholar]
- [35].Angelini A, Li Z, Mericskay M, Decaux JF, Regulation of Connective Tissue Growth Factor and Cardiac Fibrosis by an SRF/MicroRNA-133a Axis, PLoS One 10(10) (2015) e0139858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Plantier L, Renaud H, Respaud R, Marchand-Adam S, Crestani B, Transcriptome of Cultured Lung Fibroblasts in Idiopathic Pulmonary Fibrosis: Meta-Analysis of Publically Available Microarray Datasets Reveals Repression of Inflammation and Immunity Pathways, Int J Mol Sci 17(12) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sakai N, Chun J, Duffield JS, Lagares D, Wada T, Luster AD, Tager AM, Lysophosphatidic acid signaling through its receptor initiates profibrotic epithelial cell fibroblast communication mediated by epithelial cell derived connective tissue growth factor, Kidney Int 91(3) (2017) 628–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Marsch E, Sluimer JC, Daemen MJ, Hypoxia in atherosclerosis and inflammation, Curr Opin Lipidol 24(5) (2013) 393–400. [DOI] [PubMed] [Google Scholar]
- [39].Guillaumond F, Leca J, Olivares O, Lavaut MN, Vidal N, Berthezene P, Dusetti NJ, Loncle C, Calvo E, Turrini O, Iovanna JL, Tomasini R, Vasseur S, Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma, Proc Natl Acad Sci U S A 110(10) (2013) 3919–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kim JW, Gao P, Liu YC, Semenza GL, Dang CV, Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1, Mol Cell Biol 27(21) (2007) 7381–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Adachi S, Ito H, Tamamori-Adachi M, Ono Y, Nozato T, Abe S, Ikeda M, Marumo F, Hiroe M, Cyclin A/cdk2 activation is involved in hypoxia-induced apoptosis in cardiomyocytes, Circ Res 88(4) (2001) 408–14. [DOI] [PubMed] [Google Scholar]
- [42].Tang Z, Li L, Tang Y, Xie D, Wu K, Wei W, Xiao Q, CDK2 positively regulates aerobic glycolysis by suppressing SIRT5 in gastric cancer, Cancer Sci 109(8) (2018) 2590–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lee JG, Jung E, Heur M, Fibroblast growth factor 2 induces proliferation and fibrosis via SNAI1-mediated activation of CDK2 and ZEB1 in corneal endothelium, J Biol Chem 293(10) (2018) 3758–3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Song JJ, Aswad R, Kanaan RA, Rico MC, Owen TA, Barbe MF, Safadi FF, Popoff SN, Connective tissue growth factor (CTGF) acts as a downstream mediator of TGF-beta1 to induce mesenchymal cell condensation, J Cell Physiol 210(2) (2007) 398–410. [DOI] [PubMed] [Google Scholar]
- [45].Enzo E, Santinon G, Pocaterra A, Aragona M, Bresolin S, Forcato M, Grifoni D, Pession A, Zanconato F, Guzzo G, Bicciato S, Dupont S, Aerobic glycolysis tunes YAP/TAZ transcriptional activity, EMBO J 34(10) (2015) 1349–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sandbo N, Qin Y, Taurin S, Hogarth DK, Kreutz B, Dulin NO, Regulation of serum response factor-dependent gene expression by proteasome inhibitors, Mol Pharmacol 67(3) (2005) 789–97. [DOI] [PubMed] [Google Scholar]
- [47].Sutliff RL, Hilenski LL, Amanso AM, Parastatidis I, Dikalova AE, Hansen L, Datla SR, Long JS, El-Ali AM, Joseph G, Gleason RL Jr., Taylor WR, Hart CM, Griendling KK, Lassegue B, Polymerase delta interacting protein 2 sustains vascular structure and function, Arterioscler Thromb Vasc Biol 33(9) (2013) 2154–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Williams HC, Ma J, Weiss D, Lassegue B, Sutliff RL, San Martin A, The cofilin phosphatase slingshot homolog 1 restrains angiotensin II-induced vascular hypertrophy and fibrosis in vivo, Lab Invest 99(3) (2019) 399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bostock CJ, Prescott DM, Kirkpatrick JB, An evaluation of the double thymidine block for synchronizing mammalian cells at the G1-S border, Exp Cell Res 68(1) (1971) 163–8. [DOI] [PubMed] [Google Scholar]
- [50].He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B, A simplified system for generating recombinant adenoviruses, Proc Natl Acad Sci U S A 95(5) (1998) 2509–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Luo J, Deng ZL, Luo X, Tang N, Song WX, Chen J, Sharff KA, Luu HH, Haydon RC, Kinzler KW, Vogelstein B, He TC, A protocol for rapid generation of recombinant adenoviruses using the AdEasy system, Nat Protoc 2(5) (2007) 1236–47. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.