


Abstract
The influence of chromatin structure on cisplatin DNA damage was investigated in intact human cells. The epsilon-globin gene promoter was utilised as the target DNA sequence and the terminal transferase-dependent PCR technique was employed to examine adduct formation at base pair resolution. It was found that cisplatin preferentially damaged at runs of consecutive guanine bases in intact cells. By comparing the relative intensity of adduct formation in intact cells and in purified genomic DNA, it was possible to assess the influence of chromatin proteins on the extent of cisplatin DNA damage. Enhanced damage in intact cells was found at the CACC site where a member of the Sp1 family of proteins is thought to bind. It is postulated that protein binding at this site bends the DNA double-helix so that enhanced cisplatin binding occurs. The altered DNA binding of cisplatin in the presence of chromatin proteins could be important in the properties of cisplatin as an anti-tumour drug.
INTRODUCTION
Cisplatin is widely used in the clinic as a cancer chemotherapeutic drug (1). It forms covalent adducts with DNA mainly at the N-7 of guanine (2,3). Intrastrand DNA crosslinks are the main damage lesion although interstrand DNA crosslinks, monofunctional DNA lesions and protein–DNA crosslinks are also formed. DNA is thought to be the biological target for the anti-tumour activity of cisplatin. There have been several studies examining the sequence specificity of cisplatin damage in plasmid DNA sequences (4–7) and in intact human cells (8,9). Runs of consecutive Gs are the preferred damage sites for cisplatin with lesser damage at GA, AG and GC sequences. There has been a previous study where the sequence specificity of cisplatin damage has been determined in a single-copy gene (9). However, no study has examined the effect of chromatin structure on cisplatin DNA damage in a single-copy gene in intact human cells.
The influence of chromatin proteins bound to DNA inside the cell is expected to be profound on the binding of cisplatin to DNA. Protection of certain sequences by bound proteins is a likely outcome. The binding of proteins can distort the DNA double-helix and novel cisplatin adducts could form at these distorted DNA sequences. This latter point could be extremely important as novel adducts could evade the repair process and be crucial in the anti-tumour activity of cisplatin. There is a report (9) of a novel cisplatin adduct forming in cells at 5′-TACT-3′ sequences. The precise details of the molecular events that give rise to these novel cisplatin adducts are yet to be clarified; however, a likely explanation is distortion of the DNA double-helix by binding of chromatin proteins.
There have been several studies on the interactions of proteins with cisplatin and DNA in vitro (2,10,11). This work has shown that certain proteins (especially those containing an HMG box) preferentially interact with cisplatin-modified DNA. The bending of DNA by cisplatin is thought to be crucial in this interaction. The bending of DNA by a cisplatin adduct, such as a GG intrastrand crosslink, is recognised by proteins containing an HMG box. It has been postulated that the binding of proteins to cisplatin-modified DNA, is important in the anti-tumour activity of cisplatin. This could be caused either by prevention of repair due to the binding of protein or by sequestration of crucial transcription factors on cisplatin-damaged DNA (11). There are also direct interactions between proteins and cisplatin. This usually involves sulphydryl groups forming coordination complexes with cisplatin (2).
The aim of this work was to examine the effect of chromatin structure on cisplatin damage in intact human cells. We utilised the human epsilon-globin promoter region as a DNA target in this study. The human epsilon-globin promoter region has previously been studied using several other DNA damaging agents (see below) (12). These previous studies have used DNA damaging agents such as bleomycin, DMS and nitrogen mustard analogues. This damage can be analysed using the ligation-mediated PCR (LMPCR) technique at base pair resolution in a human single-copy gene. Because these agents lead to cleavage of the phosphodiester backbone of DNA, the LMPCR technique can be used. However, cisplatin damage cannot be examined directly by LMPCR because it does not cleave DNA but forms stable covalent adducts with DNA. An alternative technique must be used and we employed terminal transferase-dependent PCR (TDPCR) to analyse cisplatin damage at base pair resolution in a human single-copy gene.
The TDPCR technique (13) involves extension of a gene-specific oligonucleotide primer up to the site of adduct formation by linear amplification; the addition of approximately three guanine ribo-nucleotides (14) by terminal transferase to the 3′-end of these amplified products; ligation of a double-stranded linker containing a three base cytosine 3′-overhang; PCR with a second gene-specific oligonucleotide and an oligonucleotide complementary to the ligated linker. The second gene-specific oligonucleotide is labelled with 32P and the PCR products are analysed on a DNA sequencing gel.
The epsilon-globin promoter region has been shown to contain a number of transcription factor binding motifs that can bind their cognate transcription factors. Previous studies with other DNA damaging agents (DMS, bleomycin and nitrogen mustard analogues) in intact human cells revealed several transcription factor binding site footprints at the CCAAT, CACC, GATA-1, ɛF1 and the TATA binding motifs (12). In our experiments we employed two human tissue culture cell lines: K562 cells are derived from an erythroid lineage and express epsilon- and gamma-globin genes; HeLa cells were used as controls and do not express any globin genes. Cellular cisplatin DNA damage was compared to purified cisplatin damage in order to reveal any effects of cellular chromatin proteins on cisplatin damage. These effects were quantified by densitometry.
MATERIALS AND METHODS
HeLa and K562 cells were maintained and subcultured by conventional techniques in RPMI 1640 media. Cisplatin was dissolved in dimethyl formamide at a stock concentration of 5 mM. Cells were treated at 37°C for 18 h at various concentrations of cisplatin in RPMI 1640 medium containing no serum. After incubation the cells were washed twice with phosphate buffered saline and genomic DNA was purified. All experiments included controls containing no cisplatin. DNA was purified from human cells as previously described (15). Briefly this involved cell lysis in SDS, proteinase K digestion, phenol extraction and ethanol precipitation. DNA was dissolved in 10 mM Tris–HCl, pH 8.8, 0.1 mM EDTA to give an approximate DNA concentration of 1 mg/ml.
Purified genomic human DNA was also treated with varying concentrations of cisplatin for 18 h at 37°C in 10 mM HEPES, pH 7.5, 10 mM NaCl. After ethanol precipitation and two ethanol washes, the genomic DNA was also dissolved in 10 mM Tris–HCl, pH 8.8, 0.1 mM EDTA to give an approximate DNA concentration of 1 mg/ml.
The TDPCR procedure used in this experiment was modified from the method of Komura and Riggs (13) and is described in detail elsewhere (N.P.Davies and V.Murray, submitted for publication). Briefly genomic DNA was linearly amplified with an epsilon-globin gene specific primer (5′-AAATGGAAATTTGTGTTGCAGATAG-3′) by Taq DNA polymerase in a 10 µl reaction with 25 cycles of 30 s at 95°C, 60°C for 60 s, and 72°C for 90 s and an additional 5 min incubation at 72°C. RiboG tailing was accomplished by adding 2 µl of the LA products to a terminal deoxynucleotidyl transferase (total volume 20 µl) reaction and incubating at 37°C for 15 min. After ethanol precipitation, the products were dissolved in 10 µl of TE (1 mM Tris, 0.1 mM EDTA) and 2.4 µl was ligated to the linker γ (5′-GCGGTGACCCGGGAGATCTGAATTCCC-3′ and 5′-AATTCAGATCTCCCGGGTCACCGC-3′) (13) for 16 h at 17°C in a total volume of 6 µl. All 6 µl were then subjected to a PCR reaction (in a total volume of 10 µl) with a second nested 32P-labelled epsilon-globin primer (5′-AGGAGCCAACAAAAAAGAGCCTCAG-3′) and an oligonucleotide (5′-GCGGTGACCCGGGAGATCTGAATTC-3′) complementary to the linker utilising Taq DNA polymerase and 25 thermal cycles of 30 s at 95°C, 60°C for 60 s, and 72°C for 90 s and an additional 5 min incubation at 72°C. After electrophoresis, the DNA sequencing gel was dried on 3MM paper and scanned with a Molecular Dynamics PhosphorImager. Quantitation of damage band intensity in the digital image was achieved at each position by integration of peak areas with ImageQuaNT software. After subtraction of background and normalisation of lane intensity, the relative band intensity in cellular and purified DNA samples was calculated. Chemical and enzymatic sequencing fragments run in parallel provided a sequence reference to the exact position of damage sites. The primer used for ‘long oligonucleotide’ sequencing (16) was 5′-GGGAATTCAGATCTCCCGGGTCACCGCAGGAGCCAACAAAAAAGAGCCTCAG-3′.
The G+A chemical sequencing protocol was derived from that of Belikov and Wieslander (17). Purified human DNA (10–20 µg) was incubated with 10 µl of 3% diphenylamine in formic acid at 20°C for 3–5 min and the reaction stopped by the addition of 0.3 M sodium acetate (pH 5.5). The mixture was then extracted four times with water-saturated ether and the tube left for 5 min to remove traces of ether. The DNA was precipitated by the addition of 2.5 volumes of ethanol.
LMPCR was performed as previously described (12,18,19) using the same epsilon-globin specific primers as above. The type 1 LMPCR used the following two annealed oligonucleotides 5′-GAAGAGAAGGT-3′ and 5′-CCAAAACGCCATTTCCACCTTCTCTTC-3′ as linker while type 2 used 5′-GCGGTGACCCGGGAGATCTGAATTCCC-3′ and 5′-GGGAATTCAGATCTCCCGGGTCACCGC-3′ as linker.
RESULTS
The technique used to determine the DNA sequence specificity of cisplatin in the epsilon-globin promoter region was TDPCR. This technique uses controlled ribonucleotide tailing by terminal deoxynucleotidyl transferase at the 3′-ends of primer extension products to give a suitable target for cohesive-end ligation of a common linker.
After cisplatin damage, DNA fragments generated by primer extension to the adduct sites were subjected to TDPCR and resolved on a DNA sequencing gel. The sequence specificity was determined by direct alignment with LMPCR-amplified Maxam and Gilbert sequencing fragments, and with linear amplification products using a ‘long oligonucleotide’ on PCR product (see below) (16). This demonstrated that the ‘long oligonucleotide’ sequencing method can be used on PCR products as well as the plasmid DNA used by Cairns and Murray (16). Maxam and Gilbert sequencing fragments subjected to TDPCR did not produce useful sequencing information; this is in agreement with the observation of Komura and Riggs (13), that chemically cleaved DNA is not a preferred substrate for TDPCR.
Figures 1 and 2 are phosphorimages derived from sequencing gels that display the effects of cisplatin adduct formation in non-erythroid HeLa cells, erythroid K562 cells and in purified DNA. Undamaged DNA was also included to indicate the level of background noise in the DNA samples. The sequence specificity of cisplatin was similar to that previously observed, with damage occurring preferentially at runs of Gs, with lesser damage also occurring at GA dinucleotides.
Figure 1.
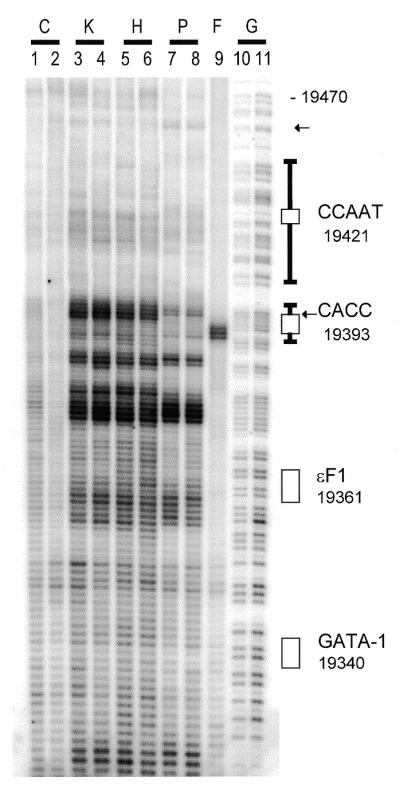
PhosphorImage of a DNA sequencing gel comparing cisplatin damage in purified DNA and in intact human cells using TDPCR in the human epsilon-gene promoter region. Lanes 1–9 were subjected to the TDPCR technique while lanes 10 and 11 (G) are G+A reactions on genomic human DNA with LMPCR type 1. Lanes 1 and 2 (C) are control samples from undamaged DNA. Lanes 3–8 are derived from cisplatin-treated samples: K562 cells (K), 100 µM (lane 3); K562 cells, 60 µM (lane 4); HeLa cells (H), 50 µM (lane 5); HeLa cells, 25 µM (lane 6); purified DNA (P), 1 µM (lane 7); purified DNA, 0.3 µM (lane 8). Lane 9 (F) is from HinfI-digested human genomic DNA. Rectangular boxes (and T-bar extensions) indicate previously observed footprints (12). Arrows indicate differences between cellular and purified DNA samples (see text). The base pair positions on the side of the gel have been aligned with the TDPCR bands by subtracting 1.25 bases from the LMPCR type 1 G+A sequencing (see text). These base pair positions indicate the 5′-end of the binding motifs. The CACC box is 111 bp upstream from the start of transcription at bp 19 504.
Figure 2.

PhosphorImage of a DNA sequencing gel examining cisplatin damage and three methods of DNA sequencing analysis. Lanes 1, 4, 7, 8 and 14 employed LMPCR type 1 and lanes 2, 5, 9 and 15 used LMPCR type 2 while lanes 3, 6, and 16–24 used TDPCR. Lanes 1–3 (A) are from an AluI digest of human genomic DNA while lanes 4–6 and 16 (F) are from a HinfI digest of human genomic DNA. Lanes 7–9 and 14–15 (G) are derived from a G+A reaction on human genomic DNA. ‘Long oligonucleotide’ DNA sequencing (LO) is shown for dideoxy C (lane 10), T (lane 11), A (lane 12) and G (lane 13). Lanes 17–22 are derived from cisplatin-damaged samples. Lanes 17 (0.3 µM) and 18 (1 µM) (P) are from purified human DNA, lanes 19 (25 µM) and 20 (50 µM) (H) are from HeLa cells, lanes 21 (60 µM) and 22 (100 µM) (K) are from K562 cells. Lanes 23 and 24 (C) are undamaged control samples. The base pair positions on the side of the gel have been aligned with the TDPCR bands by subtracting 1.25 bases from the LMPCR type 1 G+A sequencing (see text).
Visual inspection of cellular and purified DNA cisplatin damage in Figures 1 and 2 indicated that relatively enhanced cellular damage was occurring at the CACC site (bp 19 396). In order to confirm this, a densitometric comparison of cellular and purified DNA was carried out. The densitometric comparison indicated that cellular enhancement was indeed occurring at the CACC site (bp 19 396) for both K562 and HeLa cells (the CACC site extends from bp 19 393 to 19 396). This enhancement was between 50 and 100%. It should be noted that because TDPCR produces multiple bands for each damage site, damage bands spanning ~3–6 bp were amalgamated for each damage site.
No significant protection or enhancement was observed at the GATA-1 or CCAAT motifs. There was a site of protection at bp 19 460 that does not correspond to any known transcription factor binding motif. It should be noted that the level of damage above bp 19 400 was low and densitometric analysis is likely to be inaccurate in this region.
In vitro DNase I footprinting was conducted to investigate the DNA binding factors present in a cellular extract from K562 and HeLa cells. The DNase I data showed that there was a footprint at the CACC site with both K562 and HeLa cell extracts (data not shown). There was also a major footprint at the CCAAT motif with both K562 and HeLa cell extracts.
It was important to establish an accurate method of DNA sequencing for use with TDPCR, in order to determine, for example, whether the enhancement observed at position 19 396 was centred on the CACC binding site, or on the periphery of this site. This sequencing was not, however, easy to obtain. It was not possible to use Maxam–Gilbert sequencing with the TDPCR protocol as, as already stated, this reaction does not provide a good substrate for TDPCR (13). Three types of DNA sequencing are shown in Figure 2 and diagramatically in Figure 3.
Figure 3.

Schematic diagram showing three methods of DNA sequencing analysis. The top single line represents DNA cleaved with HinfI (only one strand is shown). Using the HinfI digest as a DNA template, several methods for DNA sequencing are shown. Conventional linear amplification cycle sequencing will produce a product 27 bases shorter than TDPCR (because TDPCR involves ligation of a linker). The size of the expected HinfI product is shown in brackets after each sequencing method. Also in brackets is the type of DNA sequencing involved: either dideoxy enzymic sequencing or chemical G+A sequencing. The ‘long oligonucleotide’ method has a 27 base extension at the 5′-end of the primer. Both LMPCR methods and TDPCR use ligation of a linker during the experimental procedure and hence the PCR products are increased in size by 27 bp. The asterisk indicates the position of the 32P label.
(i) Lanes 7, 8 and 14 (Fig. 2) show Maxam–Gilbert sequencing which has been processed by LMPCR type 1 (18). The linker used in this procedure was 27 bp in length, and not 24 bp as in TDPCR. This was to allow for the average of three Gs which are added in the terminal transferase step of TDPCR. However, the linker used to produce this sequencing was of a different sequence to that used in TDPCR.
(ii) Lanes 9 and 15 show LMPCR type 2 Maxam–Gilbert sequencing using a 27 bp linker of the equivalent sequence to that used in TDPCR, necessitating inclusion of three Gs at the 3′-end of the linker. The LMPCR type 2 G+A sequencing runs 0.5–1 base faster than the LMPCR type 1 G+A sequencing (probably due to differences in the sequence composition of the linkers).
(iii) The last type of sequencing used was ‘long oligonucleotide’ dideoxy sequencing (lanes 10–13). This results in sequencing equivalent to TDPCR, except that the linker sequence (plus the three Gs) is at the 5′-end of the fragment rather than at the 3′-end of the DNA fragment. The ‘long oligonucleotide’ sequencing bands ran ~1 bp faster than the LMPCR type 1 G+A bands, and approximately the same as the LMPCR type 2 G+A bands.
The use of restriction enzyme fragments (HinfI and AluI) with TDPCR permitted the alignment of the cisplatin TDPCR damage with the three types of sequencing (Fig. 2). To align the LMPCR type 2 G+A sequencing (Fig. 2, lanes 9 and 15) with the cisplatin TDPCR products (Fig. 2, lanes 17–22), a 2 base difference was allowed; this assumes that all TDPCR cisplatin amplified damage contains an extra A added by Taq DNA polymerase (20) and destruction of the sugar (attached to the modified base) during Maxam–Gilbert G+A chemical sequencing (21). To align the LMPCR type 1 G+A sequencing (Fig. 1, lanes 10 and 11) with the cisplatin TDPCR products (Fig. 1, lanes 3–8), a 1–1.5 base difference was allowed. The ‘long oligonucleotide’ sequencing (lanes 10–13) ran with the same mobility as LMPCR type 2 G+A lane (lanes 9 and 15) in Figure 2.
Thus, in summary, the three types of sequencing give consistent results (to within a base pair) that permit base pair assignment of cisplatin damage bands with the TDPCR procedure. This indicated the enhancement of cisplatin damage, centred at bp 19 396, occurred at the 3′-end of of the CACC site (bp 19 393–19 396).
DISCUSSION
Sequence specificity of adduct formation
The damage caused by cisplatin in intact human cells was investigated at base pair resolution in a single-copy human gene using the TDPCR procedure. The epsilon-globin promoter region was utilised as the target sequence for cisplatin DNA damage. Runs of consecutive Gs were preferentially targeted by cisplatin in intact human cells. This is similar to the sequence specificity previously found for cisplatin in purified plasmid sequences and in intact cells (22). A previous study detected an unusual damage site at a 5′-TACT-3′ sequence (9). We did not observe any unusual adduct sites in this study. There were two 5′-TACT-3′ sequences in the sequence analysed (bp 19 434 and 19 468) but no significant damage was observed at these sequences in intact cells.
Several different methods were used to provide DNA sequencing lanes to determine the precise site of adduct formation. The TDPCR technique does not produce a satisfactory sequence with a Maxam–Gilbert G+A chemical sequencing reaction, as previously found (13). In this present study, three different sequencing methods were used: ligation-mediated PCR on G+A chemical reactions with two different oligonucleotide strategies; and dideoxy sequencing with the ‘long oligonucleotide’ technique (16). The three methods gave approximately the same result (to within a base pair) and hence we are confident of the base pair assignments of damage.
Effect of chromatin structure on cisplatin DNA damage
In this study we have investigated the effect of chromatin structure in intact human cells on cisplatin DNA damage. The targeted epsilon-globin promoter region contains a number of transcription factor binding motifs. Other studies have shown that certain transcription factors bind to these motifs as assessed by footprinting techniques. Footprinting in intact human cells has shown protection at the CCAAT, CACC, GATA-1, ɛF1 and the TATA motifs (12). Strong enhancement was also found at either end of the CCAAT binding motifs (12). In vitro DNase I footprinting has shown protection at the CCAAT and CACC motifs with cellular extracts from both HeLa and K562 cells (this study, data not shown) (23–25).
Our results showed enhanced cisplatin damage in intact cells at the CACC binding site. This site is thought to bind a member of the Sp1 transcription factor family (25). A footprint has been previously found at this site in intact cells using DMS and nitrogen mustard analogues as DNA damaging agents (12). In vitro footprinting with DNase I has also shown a footprint at this site using both K562 and HeLa cell extracts (this study, data not shown) (23,25).
Hence we have found enhanced damage in cells with cisplatin while other DNA damaging agents show protection at this region. This could be due to the different determinants that affect DNA damage. The precise mechanism for this enhancement is not clear. However, a likely explanation is that protein binding distorts the DNA double-helix and permits enhanced binding of cisplatin. There could be preferential binding of cisplatin to bent/distorted DNA.
Cisplatin intrastrand crosslinks are known to bend DNA (2). Certain proteins (e.g. those containing an HMG box) are known to preferentially bind to cisplatin-damaged DNA (10,11). DNA binding proteins can bend DNA. It is possible that the binding of a transcription factor to DNA bends DNA in such a way as to enhance cisplatin binding to DNA. In this manner the presence of transcription factor binding in cells leads, via bending of the DNA, to enhanced cisplatin damage in cells. To our knowledge the preferential binding of cisplatin to bent/distorted DNA in vitro has not been reported. This would not be unlikely as the preferential binding of cisplatin to DNA in a particular conformation is likely to be favoured.
It is not thought that a positioned nucleosome is present at this site in cells. Hence the distortion associated with wrapping DNA around the nucleosome core is probably not responsible for this effect. However, there is evidence that multi-protein complexes associated with the transcriptional machinery exist in the epsilon-globin promoter in intact cells that lead to great distortion of the DNA double-helix in this region (12).
There is also the possibility that repair is inhibited at the CACC site leading to a perception of enhanced damage at this site. The cisplatin DNA adducts at this site could be preferentially bound by proteins that prevent DNA repair (11).
The enhanced damage is also found in HeLa cells. Even though HeLa cells do not express beta-globin genes, it is common to observe transcription factor binding in the beta-globin cluster in these cells (12,18,19). It is likely that a member of the Sp1 family is binding at the CACC motif in HeLa cells.
No significant protection or enhancement was observed at the GATA-1 or CCAAT motifs. The lack of protection/enhancement at the CCAAT motif was probably due to the low intensity of damage in this region and the different DNA parameters recognised by cisplatin. There was evidence of protection at bp 19 460. However, no transcription factor is known to bind at this sequence. Evidence for protection at this site has also previously been obtained using nitrogen mustard analogues (12).
The damage caused by ultraviolet light has also been assessed in intact cells at the promoter of the c-jun gene (26). They found enhancement at the CCAAT site but protection at the CACC site. This is opposite to that found in this study at the CACC site but probably reflects the different determinants recognised by different DNA damaging agents.
The results presented in this paper have several implications for the action of cisplatin as an anti-tumour drug. When cisplatin is given to a cancer patient, the drug damages DNA in the form of chromatin. This study has shown that chromatin structure has a large impact on the degree of damage caused by cisplatin: the binding of a transcription factor resulted in enhancement of cisplatin damage. Hence the pattern of adducts produced in DNA complexed with chromatin proteins in intact human cells is very different to that observed in purified DNA. Protein-induced DNA distortion could lead to the production of novel adducts that could evade the repair pathway and be important in the anti-tumour activity of cisplatin. This influence of chromatin structure on cisplatin DNA damage should be taken into account during the design of new cisplatin analogues. In summary, this study has revealed a further layer of complexity in the mechanism of action of cisplatin as a cancer chemotherapeutic agent.
Acknowledgments
ACKNOWLEDGEMENTS
This research was supported by the National Health and Medical Council of Australia. N.P.D. and L.C.H. were supported by an Australian Postgraduate Award. This paper is dedicated to the memory of Leigh Hardman 1972–1999.
REFERENCES
- 1.Loeher P.J. and Einhorn,L.H. (1984) Ann. Intern. Med., 100, 704–713. [DOI] [PubMed] [Google Scholar]
- 2.Bruhn S.L., Toney,J.H. and Lippard,S.J. (1991) Prog. Inorg. Chem. Bioinorg. Chem., 38, 478–517. [Google Scholar]
- 3.Fichtinger Schepman A.M., van der Veer,J.L., den Hartog,J.H., Lohman,P.H. and Reedijk,J. (1985) Biochemistry, 24, 707–713. [DOI] [PubMed] [Google Scholar]
- 4.Royer Pokora B., Gordon,L.K. and Haseltine,W.A. (1981) Nucleic Acids Res., 9, 4595–4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pinto A.L. and Lippard,S.J. (1985) Proc. Natl Acad. Sci. USA, 82, 4616–4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ponti M., Forrow,S.M., Souhami,R.L., D’Incalci,M. and Hartley,J.A. (1991) Nucleic Acids Res., 19, 2929–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murray V., Motyka,H., England,P.R., Wickham,G., Lee,H.H., Denny,W.A. and McFadyen,W.D. (1992) J. Biol. Chem., 267, 18805–18809. [PubMed] [Google Scholar]
- 8.Murray V., Motyka,H., England,P.R., Wickham,G., Lee,H.H., Denny,W.A. and McFadyen,W.D. (1992) Biochemistry, 31, 11812–11817. [DOI] [PubMed] [Google Scholar]
- 9.Grimaldi K.A., McAdam,S.R., Souhami,R.L. and Hartley,J.A. (1994) Nucleic Acids Res., 22, 2311–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pil P.M. and Lippard,S.J. (1992) Science, 256, 234–237. [DOI] [PubMed] [Google Scholar]
- 11.Zamble D.B. and Lippard,S.J. (1995) Trends Biochem. Sci., 20, 435–439. [DOI] [PubMed] [Google Scholar]
- 12.Temple M.D., Cairns,M.J., Kim,A. and Murray,V. (1999) Biochim. Biophys. Acta, 1445, 245–256. [DOI] [PubMed] [Google Scholar]
- 13.Komura J. and Riggs,A.D. (1998) Nucleic Acids Res., 26, 1807–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmidt W.M. and Mueller,M.W. (1996) Nucleic Acids Res., 24, 1789–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murray V. and Martin,R.F. (1985) J. Biol. Chem., 260, 10389–10391. [PubMed] [Google Scholar]
- 16.Cairns M.J. and Murray,V. (1998) Biochem. Mol. Biol. Int., 46, 267–275. [DOI] [PubMed] [Google Scholar]
- 17.Belikov S. and Wieslander,L. (1995) Nucleic Acids Res., 23, 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cairns M.J. and Murray,V. (1996) Biochemistry, 35, 8753–8760. [DOI] [PubMed] [Google Scholar]
- 19.Temple M.D., Cairns,M.J., Denny,W.A. and Murray,V. (1997) Nucleic Acids Res., 25, 3255–3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clark J.M. (1988) Nucleic Acids Res., 16, 9677–9686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maxam A.M. and Gilbert,W. (1980) Methods Enzymol., 65, 499–560. [DOI] [PubMed] [Google Scholar]
- 22.Murray V. (2000) Prog. Nucleic Acid Res. Mol. Biol., 63, 367–415. [DOI] [PubMed] [Google Scholar]
- 23.Gong Q.H., Stern,J. and Dean,A. (1991) Mol. Cell Biol., 11, 2558–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ronchi A.E., Bottardi,S., Mazzucchelli,C., Ottolenghi,S. and Santoro,C. (1995) J. Biol. Chem., 270, 21934–21941. [DOI] [PubMed] [Google Scholar]
- 25.Yu C.Y., Motamed,K., Chen,J., Bailey,A.D. and Shen,C.K. (1991) J. Biol. Chem., 266, 8907–8915. [PubMed] [Google Scholar]
- 26.Tornaletti S. and Pfeifer,G.P. (1995) J. Mol. Biol., 249, 714–728. [DOI] [PubMed] [Google Scholar]