

Abstract
Large greenhouse gas emissions occur via the release of carbon dioxide (CO2) and methane (CH4) from the surface layer of lakes. Such emissions are modeled from the air–water gas concentration gradient and the gas transfer velocity (k). The links between k and the physical properties of the gas and water have led to the development of methods to convert k between gases through Schmidt number normalization. However, recent observations have found that such normalization of apparent k estimates from field measurements can yield different results for CH4 and CO2. We estimated k for CO2 and CH4 from measurements of concentration gradients and fluxes in four contrasting lakes and found consistently higher (on an average 1.7 times) normalized apparent k values for CO2 than CH4. From these results, we infer that several gas-specific factors, including chemical and biological processes within the water surface microlayer, can influence apparent k estimates. We highlight the importance of accurately measuring relevant air–water gas concentration gradients and considering gas-specific processes when estimating k.
Keywords: carbon dioxide, methane, lake, gas transfer, greenhouse gas, piston velocity
Short abstract
Higher gas transfer velocities for CO2 than CH4 in lakes challenge previous results and commonly made assumptions and highlight the importance of gas-specific transport in aquatic greenhouse gas exchange.
1. Introduction
Lakes cover less than 2% of the terrestrial surface area1 and are estimated to emit carbon dioxide (CO2) and methane (CH4) in notable amounts with respect to the continental greenhouse gas exchange.2,3 Transport of dissolved gases across the water surface to the atmosphere constitutes the main pathway for lake emission of CO2 and is also a major flux pathway for CH4. The exchange of dissolved gases (Flux (F); mol m–2 d–1; units provided here and below are examples) between air and water depends on the concentration gradient expressed as the difference between the surface water gas concentration (Cw; mol m–3) and the gas concentration in equilibrium with the air (Cair; mol m–3), and the gas transfer velocity (k; m d–1), according to eq 1,
| 1 |
Flux measurements by floating chambers (FCs) or eddy covariance have been used together with measurements of Cw and Cair to calculate k from eq 1, yielding local apparent k estimates.4−7 There has also been several attempts to develop general k models, predicting k from external drivers.8−11 A common assumption is that k can be converted between gases of interest if the key thermodynamic properties of the dissolved gases (i.e., the ratio of the kinematic viscosity and mass diffusivity, expressed through the “Schmidt number”) are considered.10 Usually, k-values are normalized to a Schmidt number of 600 (k600), corresponding to k for CO2 at 20 °C. This Schmidt normalization procedure was derived for ideal well-defined k values considered predictable from fundamental physical principles. However, the apparent k used for estimating gas fluxes in nature is influenced by many factors,12 such as turbulence within the water column,9,11 physical mechanisms,13 biological and chemical mechanisms,14,15 or methodological uncertainties,16,17 that all determine how well-defined and accurate the apparent k becomes.
Recently, mismatches between apparent k600 for CO2 and CH4 in lakes and other aquatic systems have been observed.4,5,7,13,18−20 In some cases, higher k600 were found for CH4 than for CO2, attributed to CH4 microbubble formation and transport.13,18 This explanation is linked to the assumption that gases with low solubility form or enter microbubbles that move faster across the water surface boundary layer than dissolved gases, but its importance for CH4 depends on circumstantial indications. In one other case, higher k600 for CO2 than for CH4 were observed, which could be attributed to hydrodynamic and processes in the surface boundary layer that favors production and transport of CO2,7 but the importance and general validity of this explanation for the k600 difference between CO2 and CH4 is also unclear. Further, for both these types of observations, there may be other, not yet widely discussed, potential explanations (details in the Discussion).
The incompatibility between apparent k600 for CO2 and CH4 generates questions regarding the assumption that apparent k determined for one gas can be easily converted to k for another gas via physical properties. If apparent k estimates are not directly comparable across gases, the general use of models to determine k beyond the physicochemical domains and the specific gas(es) represented by the measurements behind the model can be questioned. All of this could undermine the global estimates of gas emissions which are dependent on such models, and the interpretation of apparent k and comparability of apparent k600 among gases are challenged by the above discrepancies. In this study, we investigate differences in apparent k600 for CO2 and CH4 in four lakes, with different nutrient and dissolved organic carbon concentrations and discuss possible mechanisms for gas-specific variations in apparent k600. We hypothesized contrasting k600 for CO2 and CH4 and that the k600 ratio between CO2 and CH4 would differ depending on lake characteristics (trophic status).
2. Materials and Methods
2.1. Study Areas
Empirical data were collected in four lakes (Table 1): Övre Björntjärn (OBJ; Aug. 21–24, 2012), Sörsjön (SOR; Oct. 31, 2019), Bolen (BOL; Nov. 1, 2019), and Södra Teden (SOD; Nov. 1, 2019). OBJ is a small boreal and humic lake located in northern Sweden with an inlet draining a catchment of mires and coniferous forests.11,21 SOR, BOL, and SOD are oligo-, meso-, and eutrophic lakes, respectively, located in southern Sweden. Their catchments consist of coniferous and deciduous forests, and BOL and SOD are adjacent to small urban areas (about 500 inhabitants).
Table 1. Lake Characteristics for OBJ, SOR, BOL, and SODa.
| system | OBJ | SOR | BOL | SOD | ||||
|---|---|---|---|---|---|---|---|---|
| coordinates | N | E | N | E | N | E | N | E |
| 64.122 | 18.785 | 58.723 | 16.084 | 58.781 | 16.154 | 58.339 | 16.023 | |
| area (km2) | 0.05 | 0.21 | 0.48 | 0.69 | ||||
| average depth (m) | 4 | 4.4 | 7.2 | 1.8 | ||||
| maximum depth (m) | 9 | 9 | 14 | 3.4 | ||||
| pH | 4.0 | 6.8 | 7.2 | 7.8 | ||||
| DOC (mg L–1) | 22 | 21.5 | 10.9 | 14.7 | ||||
| TN (mg L–1) | 0.5 | 0.95 | 0.6 | 1.5 | ||||
| TP (μg L–1) | 19 | 10 | 12 | 323 | ||||
| trophic/humic status | mesotrophic/humic | oligotrophic/humic | mesotrophic | eutrophic | ||||
Water chemistry data for OBJ from Klaus et al.21 and MacIntyre et al.11 Water chemistry for SOR, BOL, and SOD was sampled near the surface (0.1–0.5 m depth) during the ice-free season in 2020. DOC, TN, and TP denote dissolved organic carbon, total nitrogen, and total phosphorous, respectively. Trophic status is based on calculations from Carlson,22 using TP.
2.2. Measurements
2.2.1. Gas Flux
Gas fluxes were measured using FCs, similar to those used in Natchimuthu et al.23 The FCs consisted of plastic buckets with an opening of 0.062–0.075 m2 facing the water surface and volumes of 5.4–8.6 L, depending on the model used. FCs were covered with reflective aluminum tape to minimize internal heating and had floats attached to the outer edges such that the edges penetrated 2–3 cm below the water surface. In previous studies, this FC design yielded flux estimates similar to other methods that do not interfere with the water surface.24,25
In OBJ, 5–6 FCs were deployed simultaneously for 15–30 min. FC deployments were made 1–3 times per day over 4 days, usually between 11:00 and 14:00, and one day between 20:45 and 21:00, yielding 37 individual FC deployments. Sampling of 30 mL gas from inside the chamber was made every 10–30 min during 30-minute-long deployments by syringe sampling via a 50 cm long polyurethane tube (inner and outer diameter of 3 and 5 mm, respectively) at the top of each chamber. This resulted in 2–4 samples for each FC deployment (13 deployments with two samples, 11 deployments with three samples, and 8 deployments with four samples). Samples were analyzed within 24 h on a greenhouse gas analyzer (LGR DLT 100, Los Gatos Research Inc. USA) equipped with a custom-made syringe injection system. Regressions between FC headspace concentrations and time generated the rate of gas accumulation in each FC (ppm d–1). Data were discarded when the regression R2 for gas accumulation of either CO2 or CH4 was below 0.9 or if the data indicated leakage or sample handling errors (5 out of 37 measurements were discarded in total). After processing the data, 32 pairs of CO2 and CH4 accumulation rates were obtained.
In SOR, BOL, and SOD, one FC was deployed for repeated continuous measurements of gas flux for 10–16 min. Measurements were made by connecting the top of the FC to two polyurethane tubes, which were inserted to the inlet and outlet of an ultra-portable greenhouse gas analyzer (UGGA; Los Gatos Research Inc. USA). The FC connected to the UGGA was left floating freely on the water surface, see Figure S1. Between each measurement, the FC was lifted for ventilating it, allowing concentrations of CO2 and CH4 to reach background atmospheric concentrations. This provided data for a total of 16 individual FC deployments: seven in SOR, four in BOL, and five in SOD (no measurements were discarded). At least the first 60 s of gas accumulation was removed to allow time for mixing between the FC headspace and the UGGA measurement cell. Ten-second (0.1 Hz) data were used in regressions for gas accumulation rates (ppm d–1) for a time interval set as the minimum time needed for a gas concentration increase rate with an R2 > 0.7 to be established (between 40 and 100 s of data; R2 threshold slightly lower than for OBJ above to account for the random noise in gas spectrometry). Gas fluxes were calculated from the gas accumulation rates in the chambers and the ideal gas law according to Rudberg et al.26 (See Supporting Information, Text S1 for detailed information.)
2.2.2. Dissolved CO2 Concentration
In SOR, BOL, and SOD, CwCO2 was sampled through a headspace extraction technique (e.g., Cole et al.27). Near-surface water was collected with a Ruttner sampler deployed horizontally at ∼0.1 m. The outlet tube of the Ruttner sampler was inserted at the bottom of a 1.2 L plastic bottle and water was transferred, overflowing the bottle with at least two times the bottle volume. Then, the bottle was capped with a rubber stopper pierced by a long and a short polyurethane tube (inner and outer diameters of 3 and 5 mm), reaching the bottom of the bottle and the end of the stopper, respectively, each connected to closed 3-way luer-lock valves at the outer end. After connecting a 60 mL syringe (Becton–Dickinson) filled with atmospheric air to the short tube and connecting an empty 60 mL syringe to the long tube, air was pushed into the bottle via the short tube, while the added pressure pushed out a similar amount of water through the long tube, filling the empty syringe. The bottle was shaken for two minutes to equilibrate gases between the headspace and the water. The equilibrated headspace air was then extracted by the inverse procedure and transferred to evacuated vials. Separate air samples were collected to correct for initial CO2 in the headspace.
For 21 of the flux measurements in OBJ, surface water gas concentrations of CO2 (CwCO2) were derived from separate chambers (here termed as equilibration chambers) equipped with CO2 sensors (Senseair K33 ELG, Sweden) according to Bastviken et al.28 Equilibration chambers were deployed for 24 h prior to flux measurements, allowing CO2 concentrations in the chamber headspace to equilibrate with the water underneath. Equilibration chambers were placed next to the FCs used for flux measurements to minimize differences in spatial variability between measurements of CO2 flux and CwCO2. To make sure that the equilibration chambers provided reliable measures for CwCO2, snapshot samples for CwCO2 were collected using a headspace extraction technique described above. Parallel measurements with the equilibrated chambers and the headspace extraction technique of water samples differed by less than 5%.11 For the additional 11 flux measurements in OBJ, CO2 concentrations were measured with a common syringe headspace extraction technique by extracting 40 mL of water at ∼0.1 m depth and 20 mL of air with a 60 mL syringe. The sample was equilibrated by shaking, and the resulting headspace gas was transferred to a dry syringe and analyzed within 120 min. Separate air samples were collected to correct for initial CO2 in the headspace.
2.2.3. Dissolved CH4 Concentration
Dissolved CH4 (CwCH4) was sampled using two approaches: for SOR, BOL, SOD, and for 21 of the flux measurements in OBJ, water was sampled next to the FCs at ∼0.1 m depth using a 10 mL plastic syringe (Becton–Dickinson). The sampled volume was adjusted to 5 mL and transferred to a 22 mL N2-filled glass vial (Agilent) containing 0.1 mL 85% H3PO4 and sealed with a butyl rubber stopper and aluminum crimp. The initial overpressure of N2 was removed prior to injection to adjust the vial pressure to ambient conditions. The acid preserved the sample and allowed the storage of CH4 until analysis.
For the remaining 11 flux measurements in OBJ, water concentrations were sampled similarly to what was described in Section 2.2.2 for the additional 11 dissolved CO2 concentration samples, with a common syringe headspace extraction technique.
2.2.4. Atmospheric Pressure and Wind Speed
At OBJ, atmospheric pressure and wind speed were measured at 10 m height from a meteorological station located 300 m north-east of the lake using an Onset S-WCA-M003 and an Onset S-BPB-CM50, respectively.18 For SOR, BOL, and SOD, atmospheric pressure and wind speed at 10 m were obtained from the Swedish Meteorological and Hydrological Institute (SMHI) MESAN model. The model interpolates measurements from nearby weather stations combined with a meteorological model to estimate hourly means on a 2.5 × 2.5 km grid.29 Although MESAN estimates wind speed at 10 m height, previous results from two lakes showed reasonable agreement between hourly MESAN values and wind speed measured at lake level (R2 = 0.65–0.74).26
2.3. Analysis of Dissolved Gas
The samples in vials from all lakes were analyzed by gas chromatography (Agilent 7890) with a Poropak Q column and FID detection connected to a headspace autosampler (Agilent 7697 headspace sampler). Samples in syringes from OBJ were analyzed in the field on the Los Gatos DLT100 greenhouse gas analyzer as described above. Water concentrations were calculated using the partial pressures (the measured mixing ratio times the barometric pressure) combined with (i) the ideal gas law to calculate the amount of the gas in the headspace (nh; mol) and (ii) the temperature adjusted Henry’s law30 to calculate the residual gas in the water within the vial/syringe (naq; mol). The sum of nh and naq, after subtracting the background target gas amount in the gas forming the headspace (N2 in vials and atmospheric air in the syringe extractions), divided with the volume of the water sample extracted yielded target gas concentrations. For CO2, we accounted for shifts in the carbonic acid equilibrium during equilibration according to Koschorreck et al.17 and Rudberg et al.26
2.4. Calculating k600
We used paired gas flux and concentration measurements of CO2 and CH4 to derive k from eq 1. These values were then converted to a standardized k with the same Schmidt number of 600 (k600), i.e., the relationship between the kinematic viscosity of water divided by the diffusion coefficient of gas normalized to CO2 at 20 °C,10 to allow comparison of gases12 according to eq 2:
| 2 |
where kis and Scis are k and Schmidt numbers in situ, 600 is the reference Schmidt number, and n is a variable that is linked to the roughness of the water surface. Similar to other studies4−7 and according to Jähne et al.12 and Liss and Merlivat,31 we have used a value for n of 2/3 and 1/2 in conditions when wind speeds, either measured at 10 m or corrected to a height of 10 m, were below and above 3.6 m s–1, respectively.
2.5. Statistical Analysis
Parameters and R2, adjusted to the number of observations (n) and predictor variables (p), according to eq 3, were derived from Ordinary Least Squares regression for linear relationships and from direct curve-fitting in Python v.3.7 (scipy.optimize.curve_fit) for exponential relationships, where the latter estimates the parameters without log-transformation to not bias large x-values.
| 3 |
Adjusted R2 values according to eq 3 are in the following sections, including text and figures, referred to as R2. Since k600 data violated terms for parametric tests by not being normally distributed, the non-parametric Wilcoxon signed-rank (S-R) test and the Kruskal–Wallis analysis of variance rank test were performed in IBM SPSS statistics 28 to test for differences in k600 values for CO2 and CH4. We considered p-values below 0.05 as statistically significant to reject null-hypotheses in statistical tests and regressions.
3. Results
All surface waters were supersaturated with CO2 and CH4, with water to air concentration ratios (Cw/Cair) ranging from 3.0 to 5.5 for CO2 and from 95 to 335 for CH4. CwCO2 and CwCH4 ranged from 70 to 118 and from 0.39 to 1.38 μmol L–1, respectively. This variability was mostly due to differences between lakes whereas within-lake variability in CwCH4 and CwCO2 was small (within-lake CV: 2–31% and 2–11% for CwCH4 and CwCO2, respectively; Figure S2a). The measured fluxes ranged from 4 to 132 mmol m–2 d–1 and 0.02 to 0.98 mmol m–2 d–1 for CO2 and CH4, respectively, with the highest fluxes observed in OBJ (Figure S2b). Fluxes of CO2 and CH4 were highly related (p < 0.001, R2 = 0.91; Figure S2b). The relationship was mainly driven by OBJ data where a greater flux range was observed, but it was also significant when OBJ data were excluded (p < 0.05, R2 = 0.43).
Values of k600 ranged between 0.12 and 2.24 m d–1 for CO2 and between 0.08 and 1.84 m d–1 for CH4 (Figure 1), with an overall mean and median k600 ratio (k600CO2/k600CH4) of 1.68 ± 0.77 (mean ± 1 standard deviation) and 1.37, respectively, with only small differences in mean values between the lakes (OBJ = 1.67 ± 0.75 m d–1, SOR = 1.72 ± 0.76 m d–1, BOL = 1.52 ± 0.24 m d–1, and SOD = 1.76 ± 1.07 m d–1). This confirms the first part of our hypothesis that k600 for CO2 and CH4 would differ (p < 0.001). However, the second part of our hypothesis, that lake trophic status would influence the CO2/CH4k600 ratio, was not verified as this ratio was similar among the studied lakes (p > 0.05).
Figure 1.
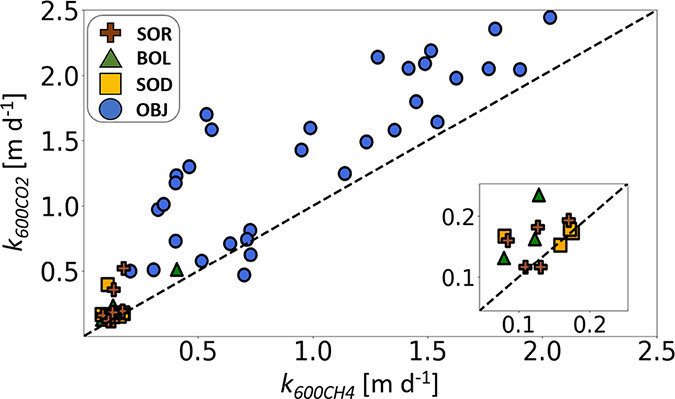
Plot of apparent gas transfer velocities normalized to a Schmidt number of 600, derived for CO2 (k600CO2) and CH4 (k600CH4) for lakes OBJ (blue circles), BOL (green triangles), SOD (orange squares), and SOR (brown crosses). The dashed line shows a 1:1 relation, where values above and below this line denote higher k600 for CO2 and CH4, respectively. The inset panel shows the smallest k-values and the dashed 1:1 line for clarity.
We observed that k600CO2 and k600CH4 were exponentially related to wind speed, which ranged from <0.4 to 3.9 m s–1 in OBJ and from 1.2 to 1.7 m s–1 elsewhere (R2 = 0.69 to 0.75; Figure 2a; the relationship is primarily generated by OBJ data, where the wind speed range was greatest). There was no clear unidirectional relationship between wind speed and the k600 ratio for CO2 and CH4 (Figure 2b). The k600 ratio was negatively and positively linked to CO2 and CH4 saturation (gas concentration in the water divided by the theoretical concentration in equilibrium with the atmospheric partial pressure), respectively (Figure S3). Such trends were weak, yet significant considering all lakes combined (CO2; p < 0.05, R2 = 0.12, CH4; p < 0.01, R2 = 0.20), and driven mainly by the OBJ lake data (CO2; p < 0.001, R2 = 0.32, CH4; p < 0.001, R2 = 0.64).
Figure 2.
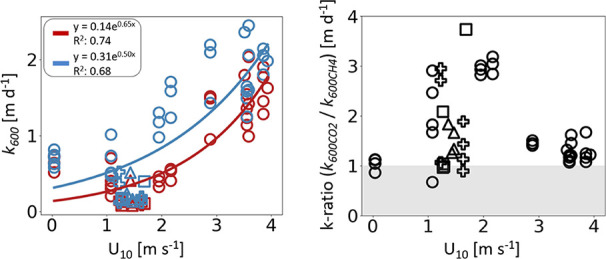
Regressions between wind speed at 10 m height (U10) and (a) apparent gas transfer velocities normalized to Schmidt number 600 for CH4 (k600CH4; red) and CO2 (k600CO2; blue), and (b) apparent k600-ratio (k600CO2/k600CH4). Different symbols represent different lakes: OBJ (circles), BOL (triangles), SOD (squares), and SOR (crosses). Gray area highlights where k600CO2 < k600CH4.
4. Discussion
4.1. Comparisons with Previous Studies
Our mean and median apparent k600 ratios for lakes, showing higher k600CO2 by a factor of 1.7 and 1.4 compared to k600CH4, are consistent with one other study, suggesting that apparent k600CO2 exceeded k600CH4.7 More specifically, Rosentreter et al.7 observed mean and median values for k600 ∼ 2.8 and 1.6 times greater for CO2 compared to CH4 in a mangrove estuary. In contrast, observations from some other studies report higher apparent k600 for CH4 than for CO2 (Table 2). Prairie and del Giorgio,13 McGinnis et al.,18 and Rantakari et al.19 found higher k600 for CH4 in 90–100% of their measurements, compared to 10 and 17% of the measurements in our study and Rosentreter et al.,7 respectively. These mixed results indicate variability in k600 between gases, among systems and conditions for reasons not yet understood.
Table 2. Examples of Results from Studies Comparing k600 for CO2 and CH4 in Order of Lower to Higher k600CO2/k600CH4 Ratiosa.
| source | systems |
Caq method |
k600 (m d–1) |
|||||
|---|---|---|---|---|---|---|---|---|
| CO2 | CH4 | CO2 | CH4 | CO2/CH4 | ||||
| min | max | min | max | |||||
| McGinnis et al.18 | temperate lake | Eq | Eq | 1.97 | 7.2 | 3.7 | 22 | 0.4 |
| Paranaíba et al.6 | three tropical reservoirs | Eq | Eq | 0.1 | 7.9 | 0.2 | 19.1 | 0.4 |
| Prairie and del Giorgio13 | boreal reservoir and lakes | Eq | Hs | 0.1 | 9.3 | 0.1 | 25 | 0.43 |
| Rantakari et al.19 | two boreal lakes | Hs | Hs | 0.5 | 3.4 | 1.1 | 14.5 | 0.56 |
| Rosentreter et al.20 | six mangrove estuaries | Eq | Eq | 1 | 24 | 1 | 28 | 0.83 |
| Beaulieu et al.5 | temperate river | Hs | Hs | 0.2 | 13.7 | 0.4 | 16.8 | < 1 |
| Guérin et al.4 | tropical reservoir | Hs | Hs | 0.05 | 1.9 | 0.1 | 2.2 | 1.16 |
| This study | four boreal lakes | Hs, Eqb | Hs | 0.1 | 2.4 | 0.1 | 2 | 1.68 |
| Rosentreter et al.7 | mangrove river estuary | Eq | Hs | 0.3 | 48 | 0.02 | 16 | 2.78 |
Eq and Hs denote measurements with flow through equilibrator and headspace extraction techniques, respectively, for surface water concentrations. The equilibrated chambers technique includes headspace extraction in floating chambers allowed to equilibrate.
Equilibrated chambers were used in 21 of the samples from OBJ, and headspace extraction was used in the remaining 11 samples from OBJ and in SOR, BOL, and SOD.
The results seem to indicate a hump-shaped relationship between the k600 ratio and wind speed (Figure 2b). The results indicate that differences in k600 between CO2 and CH4 can be substantial even at low wind speeds in both estuaries7 and lakes and could be larger at intermediate wind speeds for unknown reasons. If correct, it can be speculated that some of the mechanisms behind differences in k600 for CO2 and CH4 could have more effect at intermediate wind and intermediate k, while at higher wind speeds other factors become more important for gas flux and reduce the k600 differences. It is worth noting that other processes and factors other than wind influence k(10,11) and that the relationships shown in (Figure 2) should not be considered generally valid.
4.2. Possible Explanations for Differences in k600 among Gases
The mixed results from previous studies, where some observe higher k600CO2 and others higher k600CH4, can be due to mechanisms specific to either CH4, CO2, or to both gases. To highlight how specific mechanisms may influence k for CH4 and CO2, we discuss these mechanisms separately in the subsections below. Mechanisms related to the sampling methods are discussed in the section “Methodological reasons for gas k600 differences”. A summary of the mechanisms discussed can be found in Figure 3 and Table 3.
Figure 3.
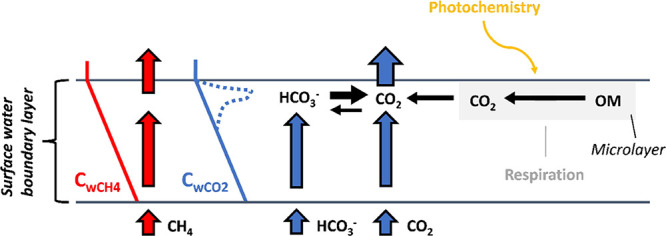
Conceptual figure for potential effect of chemical reactivity and degradation processes in the surface microlayer on concentration gradients of CO2 (blue dashed line) in relation to assumed concentration gradients for CO2 or CH4 without CO2 reactivity (solid lines in blue and red, respectively) in waters supersaturated with both CO2 and CH4. Please note that our intention with the figure is to present processes that contributes to the increase of apparent k600 for CO2 relative to CH4, hence relating to the findings from our empirical measurements. Therefore, we chose not to include processes in the figure that have the opposite effect, i.e., enhancing apparent k600 for CH4, e.g., by the contribution of microbubbles or other processes that may be the result of sampling bias.
Table 3. Overview of Potential Processes for Different Apparent k600 (Referred to as app. k600 in Table) Values for CH4 and CO2a.
| no. | process | effect | explanation | study |
|---|---|---|---|---|
| 1 | microbubble flux | app. k600CH4 > app. k600CO2 | based on hypothesis that microbubbles form that move faster than dissolved gases across the water surface boundary layer and that CH4 enters these bubbles to a greater extent than CO2. The apparent difference in k would in this case result because of a combination of different flux processes combined. | (5,13,18,20) |
| 2 | oxic surface water CH4 production | app. k600CH4 > app. k600CO2 | if there is oxic surface CH4 production above the depth where surface water CH4 and CO2 concentrations are measured, the true concentration gradient of CH4 is underestimated and apparent kCH4 will be overestimated. | |
| 3 | high surface primary production | app. k600CH4 > app. k600CO2 | if there is high primary productivity above the depth where surface water CH4 and CO2 concentrations are measured, the true concentration gradient of CO2 is overestimated and apparent kCO2 will be underestimated. | |
| 4 | chemical reactivity | app. k600CH4 < app. k600CO2 | chemical enhancement of CO2 due to equilibration reactions between CO2 and bicarbonate can alter the near-surface CO2 gradient. In cases for which this process is important, the k600CO2 should truly exceed k600CH4. | (7) |
| 5 | surface microfilm respiration processes | app. k600CH4 < app. k600CO2 | for cases with surface films enriched with organic matter where microbial or photochemical processes generate greater respiration and greater CO2 concentrations above the depth where surface water CH4 and CO2 concentrations are measured, the true concentration gradient of CO2 is underestimated and apparent kCO2 will be overestimated. | |
| 6 | possible biased gas concentration measurements using equilibrators | app. k600CH4 > app. k600CO2 | if gas concentration measurements do not fully account for the slower equilibration times for CH4, relative to CO2, between water and a gas headspace, the CH4 concentration will be underestimated and k600CH4 will be overestimated. | (6,18,20) |
| 7 | headspace extraction in waters where CwCO2 is undersaturated relative to CO2 concentrations in the atmosphere | app. k600CH4 < app. k600CO2 | if not accounting for the chemical equilibration of the carbonate system inside the water sample when calculating CwCO2 in undersaturated waters, CwCO2 will be underestimated relative to CwCH4. This in turn will overestimate apparent k600CO2. | |
| 8 | headspace extraction in waters where CwCO2 is supersaturated relative to CO2 concentrations in the atmosphere | app. k600CH4 > app. k600CO2 | if not accounting for the chemical equilibration of the carbonate system inside the water sample when calculating CwCO2 in supersaturated waters, CwCO2 will be overestimated relative to CwCH4. This in turn will underestimate apparent k600CO2. |
Here k600 represents the gas transfer velocity normalized to the Schmidt number 600. The explanations are considering processes when k600 is estimated from combined flux and surface water concentration measurements. Each process in the table is discussed further in the separate paragraphs in Section 4.2. The last column, study, is an attempt trying to link possible effects on apparent k600 from each specific mechanism presented in Section 4.2 to the studies that are shown in Table 2.
4.2.1. Potential Mechanisms Leading to Higher Estimated k600 for CH4 than for CO2
4.2.1.1. Microbubble Flux
Several studies reporting higher k600CH4 than k600CO2 considered the possibility of microbubble flux.5,13,18 Due to the buoyancy of bubbles, allowing faster transport of dissolved gases, microbubbles could favor the transport of gases with low water solubility, resulting in higher apparent k for low-compared to high-soluble gases.
Microbubble flux, enhancing k for CH4, has also been suggested to be positively linked to the level of CH4 supersaturation.13 Although dissolved CH4 concentrations in freshwater systems are usually supersaturated relative to atmospheric partial pressures (in the order of 2 μatm), the surface water concentrations are far from supersaturated relative to pure CH4 (1 atm). Therefore, it is not realistic that the CH4 itself should form microbubbles in surface water. However, microbubbles based on other gases can form due to entrainment of air in breaking waves in turbulent aquatic environments32 and can remain entrained for several days.33 Breaking waves and whitecap formation at wind speeds of 2–3 m s–1 has been suggested even at large-fetch systems.34 The studies conducted by McGinnis et al.18 and Rosentreter et al.20 are, to our knowledge, the only freshwater studies that identified relations between potential freshwater microbubble flux and wind speed or current velocity. In contrast, studies conducted by Rantakari et al.19 and Prairie and del Giorgio13 that suggest higher k600CH4 than k600CO2, did not observe such patterns. Studies by Beaulieu et al.5 and Paranaíba et al.,6 also suggesting higher k600CH4 than k600CO2, did not test for the above relationships. When it comes to our study, we did not experience any conditions with breaking waves or whitecap formation while sampling, thus limiting the potential of microbubble flux contribution.
Microbubbles, regardless of the formation mechanism, could transport all supersaturated gases, i.e., not only CH4 but also CO2 and other gases. The relative microbubble transport rates could be solubility dependent as suggested (favoring the transport of CH4 over CO2), but given the higher transfer rates of supersaturated CO2 from water into a headspace (Figure S4), significant transport of CO2 or other soluble gases via microbubbles cannot be excluded. Moreover, Beaulieu et al.5 observed no significant differences between k for CH4 and nitrous oxide (N2O) (t-test, p = 0.52), although solubility of N2O is similar to CO2 and would be expected to have lower k than CH4 if microbubble flux was occurring and was solubility dependent.
Our results may indicate that k600CH4 relative to k600CO2 decrease with CH4 supersaturation (Section 3; Figure S2b), and our calculations on the potential microbubble flux (Fmb), according to Prairie and del Giorgio13 (Text S2), show negligible effects on the enhancement of k for CH4. This does not support previous suggestions on microbubble formation and gas transport in boreal lakes13 and shows that the relationship between microbubble flux and enhanced k for CH4 needs further consideration. Based on the above-mentioned discussion, it is clear that the microbubble hypothesis, as an explanation for enhanced k for CH4 relative to k for CO2, is not generally applicable and instead context dependent. The extent to which microbubbles influence apparent k600 for different gases is therefore still an open question. Some of the alternative explanations outlined below may in many situations be more likely when explaining differences in k600 among gases.
4.2.1.2. Oxic Surface Water CH4 Production
CH4 can be produced in waters under oxic conditions,35,36 and there is support for such production associated with photosynthesis by cyanobacteria and other mechanisms. The extent of this surface water CH4 production is debated, but if it occurs close to the water surface, it could contribute to CH4 being formed in the surface boundary layer, making the real concentration gradient steeper than measured from deeper sampling, and leading to overestimating the apparent k600CH4 when estimating k from concentration measurements below the boundary layer.
4.2.1.3. High Surface Primary Production
Under some conditions, e.g., severe light limitation caused by algal blooms, it is possible (albeit unlikely—see “Surface microfilm processes” below) that the majority of the primary production may be restricted to the uppermost (few centimeters) of the water, resulting in limited productivity below this layer.37 This in turn could lead to elevated CO2 consumption near the water surface, and CwCO2 sampled some cm or deeper below the water surface may not represent the real CwCO2 driving the CO2 gas exchange. In such cases, CwCO2 could be overestimated yielding underestimation of k600CO2, resulting in a higher apparent k600CH4 than k600CO2. This explanation illustrates a case of method bias if gas concentration measurements do not represent the actual concentration gradient shaping the gas fluxes.
4.2.2. Potential Mechanisms Leading to Higher k600 for CO2 than for CH4
4.2.2.1. Chemical Reactivity
In contrast to dissolved CH4, CO2 has two ways to pass the water-side boundary layer at the water-atmosphere interface. One way, which is shared with other gases, is via molecular diffusion. In addition, CO2 can react with the water and form bicarbonate or carbonate ions. Accordingly, a part of the CO2 can diffuse through the water boundary layer as bicarbonate (Figure 3). This generates a second way for CO2 to cross the water-atmosphere interface that is not shared by dissolved CH4. At high pH, which is common in lakes with high primary productivity and low inorganic carbon concentrations, this is well acknowledged as chemical enhancement.15,21 However, at low pH, when the carbonic acid equilibrium system favors a dominance of CO2 as the net result of equilibrium reactions, there is still a continuous formation of bicarbonate that could potentially contribute to the transport of CO2 across the surficial boundary layer. As a part of this mechanism and in the case with CO2-supersaturated waters, loss of CO2 through emissions to the atmosphere will shift the inorganic carbon equilibrium balance in the surface water boundary layer to convert more bicarbonate to CO2, which leads to a higher resupply of CO2 in the layer where the actual loss to the atmosphere occurs. This dual mechanism for passage across the diffusive boundary layer differs from gases not reacting with the water (such as CH4). Thereby, water samples taken below the boundary layer may underestimate the concentration gradient of CO2 and in turn overestimate k600 for CO2 in relation to gases such as CH4 (Figure 3). A higher reactivity for CO2 compared to CH4 in the surficial boundary layer, that is not limited to conditions of high pH, might explain the higher apparent k-values of CO2 that we observed during conditions of CO2 supersaturation, and similar observations being consistent with this explanation have also been obtained in CO2-supersaturated parts of estuaries.
4.2.2.2. Surface Microfilm Processes Stimulating CO2 Production
The surface microfilm at the air–water interface of water bodies is often enriched in nutrients, particulate and dissolved carbon, phytoplankton, and microbes.38−40 This is a zone where photochemical processes can degrade dissolved organic matter to CO2 and labile compounds, providing additional substrate for microbes41−44 (Figure 3). This is also a zone where photoinhibition may reduce primary production37,45,46 and where buoyant particles aggregate and are respired, which enhances net production of CO2 (Figure 3). Hence, CwCO2 measurements a few centimeters into the water may underestimate the CwCO2 and in such cases k600CO2 will be overestimated and the apparent result would be that k600CO2 is greater than k600CH4.
Biological and chemical processes in ocean surface microlayers have been suggested to influence k600,14,47−49 but to our knowledge their effect on k600 in inland waters is largely unknown. A study using FCs in the tropical Atlantic Ocean found mismatches in k600 between CH4, carbon monoxide (CO), N2O, and hydrogen (H2) and concluded that microbial gas consumption within the surface layer film was the only plausible explanation for such differences in k600.14 Frost47 came to the same conclusion when he identified a mismatch with up to 8% higher k600 for CH4 compared to sulfur hexafluoride (SF6) in the North Sea, and results were corroborated by Upstill-Goddard et al.,50 who replicated similar patterns in a controlled experiment by adding methanotrophs to the surface microlayer film. In the subtropical Atlantic, Calleja et al.51 found that k600 for CO2 was controlled by microbial metabolism in the uppermost (<2 cm) water layer, with sevenfold and tenfold differences in respiration and gross primary production, respectively, compared to the mixed photic layer below. If the above findings in ocean systems are valid for lakes, microbial communities within the uppermost water layer could potentially alter the concentration of CH4 or CO2 in lakes and bias our k determination method. It might explain the contrasting patterns found in different systems, where some studies indicate enhanced k600CH4 and some show enhancement of k600CO2.
4.2.3. Methodological Reasons for Gas k600 Differences
Discrete sample headspace analysis is associated with a risk of introducing systematic error into calculations if chemical equilibration of the carbonate system inside the sample vial is not accounted for. Koschorreck et al.17 found that such bias could lead to error, underestimating CwCO2 by a factor of 3 in highly undersaturated waters when using atmospheric air for the equilibration, but overestimating CwCO2 by less than 5% in supersaturated (>1000 μatm) waters. Bias is reduced if a high water to air volume ratio is used in the sampling syringe. We accounted for this error when calculating CwCO2, but bias would otherwise only be ∼2% due to high supersaturation in our lakes and a water to air volume ratio of ∼19 when sampling. Nevertheless, it is possible that the findings from Koschorreck et al.17 could influence the k600-ratios (CO2/CH4) presented in Table 2 if not considered in all studies.
When using flow-through equilibrators, a mismatch between k for CO2 and CH4 could increase if differences in gas equilibration times are not accounted for.16 The equilibration time for CH4 is considerably longer than for CO2 due to the greater supersaturation of CH4 in water and its lower solubility, and thereby the greater proportional mass transport of CH4 is needed from the water to the gas phase to reach equilibrium (Figure S4). Accordingly, if measurements of CwCH4 are not fully equilibrated, this will underestimate CwCH4 and consequently result in overestimation of k600 for CH4.
Regardless of choosing equilibrator or headspace extraction approaches, the depth of the water sampling is critical, as highlighted above. Potential explanations for the differences between apparent k600 for CO2 and CH4 could be attributed to methodological inconsistency regarding the depth where water samples are taken to measure the gas concentration due to gradients in the very near-surface water. From this perspective, it could be argued that a more accurate way to estimate the surface water gas concentrations driving the flux may be to use long-term FC equilibrations, designed to make in situ chamber headspace gas concentrations reflect the gas concentrations in the uppermost water layer. Further exploration of such chambers designed for quickest possible equilibration rates (e.g., large surface area to volume ratio and constant water renewal under the chamber28) and evaluating their pros and cons would be of interest to improve future k measurements.
Another methodological aspect regards the common temporal mismatch between flux measurements (integrating gas transport over 10–30 min) and instantaneous snap-shot concentration measurements or from the delay in the equilibration of chambers. This contributes uncertainty to k-estimates as the flux and concentration data used to calculate k represents different time periods. The direction of this bias depends on how gas concentrations in the water change during the flux measurement. This time-mismatch may therefore not cause a systematic bias on k600 differences among gases if considering many measurement periods and systems. However, reducing the related k uncertainty needs further consideration by, e.g., combining flux measurements with repeated or continuous concentration measurements during the whole flux measurement period or optimized FCs for rapid equilibration.
4.3. Future Considerations and Implications
It is imperative to understand controls on k600 to accurately estimate gas fluxes from surface waters. Our results from four boreal lakes add to the growing literature which indicates that current assumptions made when calculating k600 from measurements designed for other gases, do not hold when tested in situ. Additional mechanisms than those outlined here may be possible. For example, variability in k values for CO2 and CH4 may also depend on conditions within the water column, and exploring potential patterns in k600CH4 to k600CO2 versus, e.g., local hydrodynamics influenced by heating, cooling, and wind speed and direction would be of interest. Clearly, several explanations to observed differences in k600 among gases exist that can interact and differ in relative importance among studies. This is a matter of concern as k-dependent gas exchange models are widely used in regional studies due to the perceived simplicity of collecting water samples to estimate gas concentration and modeling k to derive fluxes in comparison with making actual flux measurements. Many such studies, incorporating k-dependent gas exchange models, are used in global upscaling of fluxes, and the discrepancy-ranges observed in k600 seem large enough to substantially affect aquatic greenhouse gas emissions estimates. Future studies should be designed to address potential biases in gas concentration measurements and account for the possible mechanisms that may affect k differently for CO2 and CH4. Before these challenges are addressed, attempts to convert k from one gas to another based solely on physical properties may not be reliable beyond controlled laboratory conditions, and in situ empirical assessments of k for each gas of interest remain important for accurate flux assessments.
Acknowledgments
This work was funded by the European Research Council (ERC), European Union’s Horizon 2020 research and innovation program (grant agreement No. 725546), the Knut and Alice Wallenberg Foundation (grant agreement No. 2016.0083), the Swedish Research Council (VR; grant agreement No. 2016-04829), and the Swedish Research Council for Sustainable Development (FORMAS; grant agreement No. 2018-01794). Support and development for manuscript review was also provided by the US National Science Foundation (Division of Environmental Biology, grant number 1753856). Many thanks to Balathandayuthabani Panneer Selvam and Sivakiruthika Balathandayuthabani for contributions made in the empirical collection phase, lab analysis, or discussions regarding the experimental setup and data handling.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.est.2c09230.
Gas calculations for CO2 and CH4 in lake systems SOR, BOL, and SOD (Text S1) and supplemental method description of microbubble flux calculations (Text S2), with figures showing the in situ sampling setup using UGGA and flux chamber for measuring fluxes of CH4 and CO2 (Figure S1), the relationship between measured, lake dissolved CO2 and CH4 concentrations, and lake CO2 and CH4 fluxes (Figure S2), the measured k600 ratio (CO2/CH4) and CO2 saturation, and CH4 saturation (Figure S3), and gas specific equilibration time measurements for CO2 and CH4 in an aquatic environment (Figure S4) (PDF)
Source data contain data needed to reproduce results in the manuscript and include in situ measurements of the apparent k600 for CO2 and CH4, wind speeds approximated at 10 m height (U10), apparent k600 ratio (CO2/CH4), dissolved CO2 and CH4, gas fluxes of CO2 and CH4, and apparent ratio of dissolved gases in the surface waters versus the above lying atmosphere (Cw/Cair) for CO2 and CH4, respectively (XLSX)
Author Contributions
D.B., G.P., D.R., and M.G. designed the study and the experimental setup. D.B. was supervising the study and secured resources for it. D.B., G.P., and M.G. performed most of the empirical data collection, with assistance from the other authors. Lab analysis of the empirical data collected was done by G.P., J.S., M.G., I.S., and D.B. Calculating and structuring the data was done by G.P. and D.R. with help from D.B. and M.G., G.P. and D.R. led the development of the manuscript, including writing the first draft of the manuscript with input from D.B. All authors contributed to the development of the manuscript and have approved the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Messager M. L.; Lehner B.; Grill G.; Nedeva I.; Schmitt O. Estimating the volume and age of water stored in global lakes using a geo-statistical approach. Nat. Commun. 2016, 7, 13603. 10.1038/ncomms13603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond P. A.; Hartmann J.; Lauerwald R.; Sobek S.; McDonald C.; Hoover M.; Butman D.; Striegl R.; Mayorga E.; Humborg C.; Kortelainen P.; Dürr H.; Meybeck M.; Ciais P.; Guth P. Global carbon dioxide emissions from inland waters. Nature 2013, 503, 355–359. 10.1038/nature12760. [DOI] [PubMed] [Google Scholar]
- Johnson M. S.; Matthews E.; Du J.; Genovese V.; Bastviken D. Methane Emission From Global Lakes: New Spatiotemporal Data and Observation-Driven Modeling of Methane Dynamics Indicates Lower Emissions. J. Geophys. Res.: Biogeosci. 2022, 127, e2022JG006793 10.1029/2022JG006793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guérin F.; Abril G.; Serça D.; Delon C.; Richard S.; Delmas R.; Tremblay A.; Varfalvy L. Gas transfer velocities of CO2 and CH4 in a tropical reservoir and its river downstream. J. Mar. Syst. 2007, 66, 161–172. 10.1016/j.jmarsys.2006.03.019. [DOI] [Google Scholar]
- Beaulieu J. J.; Shuster W. D.; Rebholz J. A. Controls on gas transfer velocities in a large river. J. Geophys. Res.: Biogeosci. 2012, 117, 2007. 10.1029/2011JG001794. [DOI] [Google Scholar]
- Paranaíba J. R.; Barros N.; Mendonça R.; Linkhorst A.; Isidorova A.; Roland F. B.; Almeida R. M.; Sobek S. Spatially Resolved Measurements of CO2 and CH4 Concentration and Gas-Exchange Velocity Highly Influence Carbon-Emission Estimates of Reservoirs. Environ. Sci. Technol. 2018, 52, 607–615. 10.1021/acs.est.7b05138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosentreter J. A.; Wells N. S.; Ulseth A. J.; Eyre B. D. Divergent Gas Transfer Velocities of CO2, CH4 and N2O Over Spatial and Temporal Gradients in a Subtropical Estuary. J. Geophys. Res.: Biogeosci. 2021, 126, e2021JG006270 10.1029/2021JG006270. [DOI] [Google Scholar]
- MacIntyre S., Wanninkhof R., Chanton J. P., Trace gas exchange across the air-sea interface in fresh water and coastal marine environments; Blackwell Science: Oxford, 1995; pp 52–97. [Google Scholar]
- Zappa C. J.; McGillis W. R.; Raymond P. A.; Edson J. B.; Hintsa E. J.; Zemmelink H. J.; Dacey J. W.; Ho D. T. Environmental turbulent mixing controls on air-water gas exchange in marine and aquatic systems. Geophys. Res. Lett. 2007, 34, L10601. 10.1029/2006GL028790. [DOI] [Google Scholar]
- Wanninkhof R. Relationship between wind speed and gas exchange over the ocean revisited. Limnol. Oceanogr.: Methods 2014, 12, 351–362. 10.4319/lom.2014.12.351. [DOI] [Google Scholar]
- MacIntyre S.; Bastviken D.; Arneborg L.; Crowe A. T.; Karlsson J.; Andersson A.; Gålfalk M.; Rutgersson A.; Podgrajsek E.; Melack J. M. Turbulence in a small boreal lake: Consequences for air–water gas exchange. Limnol. Oceanogr. 2021, 66, 827–854. 10.1002/lno.11645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jähne B.; Münnich K. O.; Bösinger R.; Dutzi A.; Huber W.; Libner P. On the parameters influencing air-water gas exchange. J. Geophys. Res. 1987, 92, 1937. 10.1029/JC092iC02p01937. [DOI] [Google Scholar]
- Prairie Y.; del Giorgio P. A new pathway of freshwater methane emissions and the putative importance of microbubbles. Inland Waters 2013, 3, 311–320. 10.5268/IW-3.3.542. [DOI] [Google Scholar]
- Conrad R.; Seiler W. Influence of the surface microlayer on the flux of nonconservative trace gases (CO, H2, CH4, N2O) across the ocean-atmosphere interface. J. Atmos. Chem. 1988, 6, 83–94. 10.1007/BF00048333. [DOI] [Google Scholar]
- Wanninkhof R.; Knox M. Chemical enhancement of CO2 exchange in natural waters. Limnol. Oceanogr. 1996, 41, 689–697. 10.4319/lo.1996.41.4.0689. [DOI] [Google Scholar]
- Webb J. R.; Maher D. T.; Santos I. R. Automated, in situ measurements of dissolved CO2, CH4, and δ13C values using cavity enhanced laser absorption spectrometry: Comparing response times of air-water equilibrators. Limnol. Oceanogr.: Methods 2016, 14, 323–337. 10.1002/lom3.10092. [DOI] [Google Scholar]
- Koschorreck M.; Prairie Y. T.; Kim J.; Marcé R. CO2 is not like CH4–limits of and corrections to the headspace method to analyse pCO2 in fresh water. Biogeosciences 2021, 18, 1619–1627. 10.5194/bg-18-1619-2021. [DOI] [Google Scholar]
- McGinnis D. F.; Kirillin G.; Tang K. W.; Flury S.; Bodmer P.; Engelhardt C.; Casper P.; Grossart H.-P. Enhancing surface methane fluxes from an oligotrophic lake: exploring the microbubble hypothesis. Environ. Sci. Technol. 2015, 49, 873–880. 10.1021/es503385d. [DOI] [PubMed] [Google Scholar]
- Rantakari M.; Heiskanen J.; Mammarella I.; Tulonen T.; Linnaluoma J.; Kankaala P.; Ojala A. Different apparent gas exchange coefficients for CO2 and CH4: Comparing a brown-water and a clear-water lake in the boreal zone during the whole growing season. Environ. Sci. Technol. 2015, 49, 11388–11394. 10.1021/acs.est.5b01261. [DOI] [PubMed] [Google Scholar]
- Rosentreter J.; Maher D. T.; Ho D.; Call M.; Barr J.; Eyre B. D. Spatial and temporal variability of CO2 and CH4 gas transfer velocities and quantification of the CH4 microbubble flux in mangrove dominated estuaries. Limnol. Oceanogr. 2017, 62, 561–578. 10.1002/lno.10444. [DOI] [Google Scholar]
- Klaus M.; Geibrink E.; Jonsson A.; Bergström A.-K.; Bastviken D.; Laudon H.; Klaminder J.; Karlsson J. Greenhouse gas emissions from boreal inland waters unchanged after forest harvesting. Biogeosciences 2018, 15, 5575–5594. 10.5194/bg-15-5575-2018. [DOI] [Google Scholar]
- Carlson R. E. A trophic state index for lakes 1. Limnol. Oceanogr. 1977, 22, 361–369. 10.4319/lo.1977.22.2.0361. [DOI] [Google Scholar]
- Natchimuthu S.; Sundgren I.; Gålfalk M.; Klemedtsson L.; Bastviken D. Spatiotemporal variability of lake pCO2 and CO2 fluxes in a hemiboreal catchment. J. Geophys. Res.: Biogeosci. 2017, 122, 30–49. 10.1002/2016JG003449. [DOI] [Google Scholar]
- Cole J. J.; Bade D. L.; Bastviken D.; Pace M. L.; Van de Bogert M. Multiple approaches to estimating air-water gas exchange in small lakes. Limnol. Oceanogr.: Methods 2010, 8, 285–293. 10.4319/lom.2010.8.285. [DOI] [Google Scholar]
- Gålfalk M.; Bastviken D.; Fredriksson S.; Arneborg L. Determination of the piston velocity for water-air interfaces using flux chambers, acoustic Doppler velocimetry, and IR imaging of the water surface. J. Geophys. Res.: Biogeosci. 2013, 118, 770–782. 10.1002/jgrg.20064. [DOI] [Google Scholar]
- Rudberg D.; Duc N.; Schenk J.; Sieczko A.; Pajala G.; Sawakuchi H.; Verheijen H.; Melack J.; MacIntyre S.; Karlsson J. Diel Variability of CO2 Emissions from Northern Lakes. J. Geophys. Res.: Biogeosci. 2021, 126, e2021JG006246 10.1029/2021JG006246. [DOI] [Google Scholar]
- Cole J. J.; Caraco N. F.; Kling G. W.; Kratz T. K. Carbon dioxide supersaturation in the surface waters of lakes. Science 1994, 265, 1568–1570. 10.1126/science.265.5178.1568. [DOI] [PubMed] [Google Scholar]
- Bastviken D.; Sundgren I.; Natchimuthu S.; Reyier H.; Gålfalk M. Technical Note: Cost-efficient approaches to measure carbon dioxide (CO2) fluxes and concentrations in terrestrial and aquatic environments using mini loggers. Biogeosciences 2015, 12, 3849–3859. 10.5194/bg-12-3849-2015. [DOI] [Google Scholar]
- Häggmark L.; Ivarsson K.-I.; Gollvik S.; Olofsson P.-O. Mesan, an operational mesoscale analysis system. Tellus A 2000, 52, 2–20. 10.3402/tellusa.v52i1.12250. [DOI] [Google Scholar]
- Sander R.; Acree W. E. Jr.; De Visscher A.; Schwartz S. E.; Wallington T. J. Henry’s law constants (IUPAC Recommendations 2021). Pure Appl. Chem. 2022, 94, 71–85. 10.1515/pac-2020-0302. [DOI] [Google Scholar]
- Liss P. S.; Merlivat L., Air-sea gas exchange rates: Introduction and synthesis. In The role of air-sea exchange in geochemical cycling; Springer: 1986; pp 113–127. [Google Scholar]
- Woolf D. K.; Thorpe S. Bubbles and the air-sea exchange of gases in near-saturation conditions. J. Mar. Res. 1991, 49, 435–466. 10.1357/002224091784995765. [DOI] [Google Scholar]
- Turner W. Microbubble persistence in fresh water. J. Acoust. Soc. Am. 1961, 33, 1223–1233. 10.1121/1.1908960. [DOI] [Google Scholar]
- Monahan E. C.; Muircheartaigh I. Optimal power-law description of oceanic whitecap coverage dependence on wind speed. J. Phys. Oceanogr. 1980, 10, 2094–2099. . [DOI] [Google Scholar]
- Grossart H. P.; Frindte K.; Dziallas C.; Eckert W.; Tang K. W. Microbial methane production in oxygenated water column of an oligotrophic lake. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 19657–19661. 10.1073/pnas.1110716108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bižić M.; Klintzsch T.; Ionescu D.; Hindiyeh M.; Günthel M.; Muro-Pastor A. M.; Eckert W.; Urich T.; Keppler F.; Grossart H.-P. Aquatic and terrestrial cyanobacteria produce methane. Sci. Adv. 2020, 6, eaax5343 10.1126/sciadv.aax5343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staehr P. A.; Brighenti L. S.; Honti M.; Christensen J.; Rose K. C. Global patterns of light saturation and photoinhibition of lake primary production. Inland Waters 2016, 6, 593–607. 10.1080/IW-6.4.888. [DOI] [Google Scholar]
- Danos S. C.; Maki J. S.; Remsen C. C. Stratification of microorganisms and nutrients in the surface microlayer of small freshwater ponds. Hydrobiologia 1983, 98, 193–202. 10.1007/BF00021021. [DOI] [Google Scholar]
- Hillbricht-Ilkowska A.; Kostrzewska-Szlakowska I. Surface microlayer in lakes of different trophic status: nutrients concentration and accumulation. Pol. J. Ecol. 2004, 52, 461–478. [Google Scholar]
- Hörtnagl P.; Perez M. T.; Zeder M.; Sommaruga R. The bacterial community composition of the surface microlayer in a high mountain lake. FEMS Microbiol. Ecol. 2010, 73, 458–467. 10.1111/j.1574-6941.2010.00904.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X.; Mopper K. Photochemical production of low-molecular-weight carbonyl compounds in seawater and surface microlayer and their air-sea exchange. Mar. Chem. 1997, 56, 201–213. 10.1016/S0304-4203(96)00076-X. [DOI] [Google Scholar]
- Bertilsson S.; Stefan L. J. Photochemically produced carboxylic acids as substrates for freshwater bacterioplankton. Limnol. Oceanogr. 1998, 43, 885–895. 10.4319/lo.1998.43.5.0885. [DOI] [Google Scholar]
- Bertilsson S.; Tranvik L. J. Photochemical transformation of dissolved organic matter in lakes. Limnol. Oceanogr. 2000, 45, 753–762. 10.4319/lo.2000.45.4.0753. [DOI] [Google Scholar]
- Anesio A. M.; Granéli W. Increased photoreactivity of DOC by acidification: Implications for the carbon cycle in humic lakes. Limnol. Oceanogr. 2003, 48, 735–744. 10.4319/lo.2003.48.2.0735. [DOI] [Google Scholar]
- Patterson J. C. Modelling the effects of motion on primary production in the mixed layer of lakes. Aquat. Sci. 1991, 53, 218–238. 10.1007/BF00877060. [DOI] [Google Scholar]
- Karentz D.; Bothwell M.; Coffin R.; Hanson A.; Herndl G.; Kilham S.; Lesser M.; Lindell M.; Moeller R.; Morris D. Impact of UV-B radiation on pelagic freshwater ecosystems: report of working group on bacteria and phytoplankton. Ergeb. Limnol. 1994, 43, 31–69. [Google Scholar]
- Frost T.Environmental controls of air-water gas exchange; University of Newcastle upon Tyne, 1999. [Google Scholar]
- Cunliffe M.; Upstill-Goddard R. C.; Murrell J. C. Microbiology of aquatic surface microlayers. FEMS Microbiol. Rev. 2011, 35, 233–246. 10.1111/j.1574-6976.2010.00246.x. [DOI] [PubMed] [Google Scholar]
- Cunliffe M.; Engel A.; Frka S.; Gašparović B.; Guitart C.; Murrell J. C.; Salter M.; Stolle C.; Upstill-Goddard R.; Wurl O. Sea surface microlayers: A unified physicochemical and biological perspective of the air–ocean interface. Prog. Oceanogr. 2013, 109, 104–116. 10.1016/j.pocean.2012.08.004. [DOI] [Google Scholar]
- Upstill-Goddard R. C.; Frost T.; Henry G. R.; Franklin M.; Murrell J. C.; Owens N. J. Bacterioneuston control of air-water methane exchange determined with a laboratory gas exchange tank. Global Biogeochem. Cycles 2003, 17, 19-1. 10.1029/2003GB002043. [DOI] [Google Scholar]
- Calleja M. L.; Duarte C. M.; Navarro N.; Agusti S. Control of air-sea CO2 disequilibria in the subtropical NE Atlantic by planktonic metabolism under the ocean skin. Geophys. Res. Lett. 2005, 32, L08606. 10.1029/2004GL022120. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.