

Abstract
The fungal strain Fusarium gramineanim FM1010 was isolated from a shallow-water volcanic rock known as “live rock” at the Carl Smith Beach, Hilo, Hawaii. Eleven specialised metabolites, including two undescribed diketopiperazines, three undescribed polyketides, and one undescribed isochromanone, along with five known fusarielin derivatives were obtained from F. gramineanim FM1010. The structures of the six undescribed compounds were elucidated by extensive analysis of NMR spectroscopy, HRESIMS, chemical reactions, and electronic circular dichroism (ECD) data. Kaneoheoic acids G-I showed mild inhibitory activity against S. aureus with the MIC values in the range of 20–40 μg/mL when assayed in combination with chloramphenicol (half of the MIC, 1 μg/mL), an FDA approved antibiotic. Kaneoheoic acid I exhibited both anti-proliferative activity against ovarian cancer cell line A2780 and TNF-α induced NF-κB inhibitory activity with the IC50 values of 18.52 and 15.86 μM, respectively.
Keywords: Fusarium graminearum, Nectriaceae, Polyketides, Diketopiperazines, Isochromanone, NMR, Antibacterial activity, NF-κB, Antiproliferative
Graphical Abstract
1. Introduction
Natural product drug discovery from relatively new or distinctive natural sources is considered a reasonable strategy to overcome the current huge demand for new drugs for the large global population. One approach is to uncover new biologically active molecules from these natural sources as hits or leads, which have the potential to be developed further into drugs. Fungi, as a unique ecological niche, produce a vast number of biologically active specialised metabolites, some of which have been transformed into clinically significant drugs such as beta-lactam antibiotics like penicillin and cephalosporin, as well as the immunosuppressant cyclosporine and cholesterol-lowering agents, compactin and lovastatin [Aly et al., 2011; Manzoni and Rollini, 2002]. The fungal kingdom is very diverse but only a small fraction of fungi has been explored for the biologically active specialised metabolites. Hence, there is an enormous possibility of getting novel biologically active compounds from fungi.
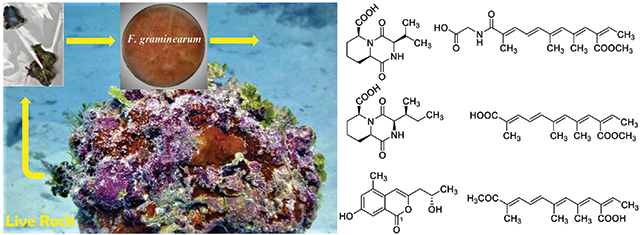
Due to the geological location in the central Pacific Ocean, Hawaii has its own ecologically rich marine habitats that support a wide variety of marine life, where fungi play a dynamic role. In our continuing search for biologically active compounds from fungi collected in Hawaii [Zaman et al., 2021a, 2021b, 2020a, 2020b; Wang et al., 2020, 2019; Li et al., 2019, 2018a, 2018b, 2017a, 2017b, 2017c, 2017d, 2016a, 2016b, 2015a, 2015b; Fei-Zhang et al., 2016; Huang et al., 2017], we isolated a fungal strain Fusarium graminearum Schwabe FM1010 (Family: Nectriaceae) (Genbank accession # MW703613) from a shallow-water volcanic rock, locally known as ‘live rock’ collected at the Carl Smith Beach, Big Island, Hawaii. Marine life is visibly attached or affixed to live rock. The living part of the rock is not the rock itself, but rather the small invertebrates, algae, bacteria and fungi that live in and on it. Fusarium species is well-known to produce numerous specialised metabolites such as fusaproliferin, fumonisins, fusaric acid, moniliformin, trichothecenes, and enniatins, with unique structures including terpenoids, alkaloids, and polyketides [Wei and Wu, 2020]. These types of compounds are interesting due to a broad spectrum of biological properties, for example, antifungal, antibacterial, insecticidal, and cytotoxic activities [Song et al., 2015]. Recently, a crude methanolic extract of F. graminearum FM1010 isolated from a live rock was found active against gram-positive bacteria Staphylococcus aureus at 80 μg/mL. From F. graminearum FM1010, we have isolated eleven specialized metabolites, including two unique diketopiperazines (1 and 2), three polyketides (3–5), and one isochromanone (6), along with five known fusarielin derivatives (7–11). Herein, we report the isolation and structural elucidation by HR-ESIMS, NMR spectral interpretation, chemical reactions and ECD analysis as well as the biological evaluation of compounds 1–11 from F. graminearum FM1010.
2. Results and discussion
2.1. Structural elucidation of the undescribed compounds
Compound 1 was isolated as a light yellow powder. Its molecular formula, C12H18N2O4, was determined by HRESIMS, requiring five degrees of unsaturation. Comprehensive analysis of the 1D and 2D NMR data of 1 (Table 1 and Figures S2–6) indicated the presence of two methyl groups, three methylenes, four methines including three nitrogenated ones, and three nonprotonated carbons. The chemical shifts (δH/δC) of the three methines (-CH-) groups at 3-, 6- and 10-positions were 3.67/59.8, 4.11/54.3, and 4.95/52.9, respectively, indicating that each of these three methines must connect to a nitrogen atom, not an oxygen atom otherwise their carbon chemical shifts must be more than 60 ppm. The last methine (-CH-) group at 11-position appeared at relatively higher field at 2.18/34.0 (δH/δC), indicating that this methine group must connect to three carbon atoms. Also, HMBC data showed that there were three carbonyl groups (-CON-, or -COO-) (δC 166.9, 168.1, and 173.2) in compound 1. Based on the molecular formula of compound 1 (C12H18N2O4), there must be amide and acid bonds in the molecule. The chemical shifts of the three methines (-CH-) groups at 3-, 6- and 10-positions are in the range of chemical shifts of alpha-position (-N-CHR-CON-) of amino acids in peptides, which further confirmed the presence of three nitrogenated methines in compound 1. Two spin systems, –CH–CH2-CH2–CH2–CH–, and –CH–-CH(CH3)2 (Figure 2), were established based on the analysis of the COSY spectrum. In the HMBC spectrum, H-3 showed correlations to C-2, C-5, C-11, C-13, and C-14, and H-6 correlated to C-5, C-7, C-8, and C-10. Also, H-10 exhibited HMBC correlations to C-2, C-6, C-8, C-9, and C-15 (Figure 2). All these HMBC correlations along with the two identified COSY spin systems indicated the planar structure of compound 1 as shown in Figure 2.
Table 1.
1H and 13C NMR Spectroscopic Data for Compounds 1 and 2 in DMSO-d6
| 1 |
2 |
|||
|---|---|---|---|---|
| no. | δC | δH mult. J (Hz) | δC | δH mult. J (Hz) |
|
|
|
|||
| 2 | 166.9 | 167.2 | ||
| 3 | 59.8 | 3.67, t (3) | 57.9 | 3.80, t (3) |
| 5 | 168.1 | 167.7 | ||
| 6 | 54.3 | 4.11, dd (4, 12) | 54.3 | 4.08, dd (4, 12) |
| 7 | 29.7 | 1.30, m | 29.8 | 1.31, m |
| 2.20, m | 2.19, m | |||
| 8 | 20.7 | 1.31, m | 20.8 | 1.31, m |
| 1.68, m | 1.69, m | |||
| 9 | 26.6 | 1.48, m | 26.6 | 1.49, m |
| 2.17, m | 2.17, m | |||
| 10 | 52.9 | 4.95, d (5) | 52.7 | 4.99, d (5) |
| 11 | 34.0 | 2.18, m | 40.7 | 1.92 |
| 12 | 25.9 | 1.24, m; 1.38, m | ||
| 13 | 19.3 | 0.94, d (7) | 12.3 | 0.86, t (7) |
| 14 | 16.6 | 0.88, d (7) | 13.9 | 0.85, d (7) |
| 15 | 173.2 | 172.9 | ||
Fig. 2.
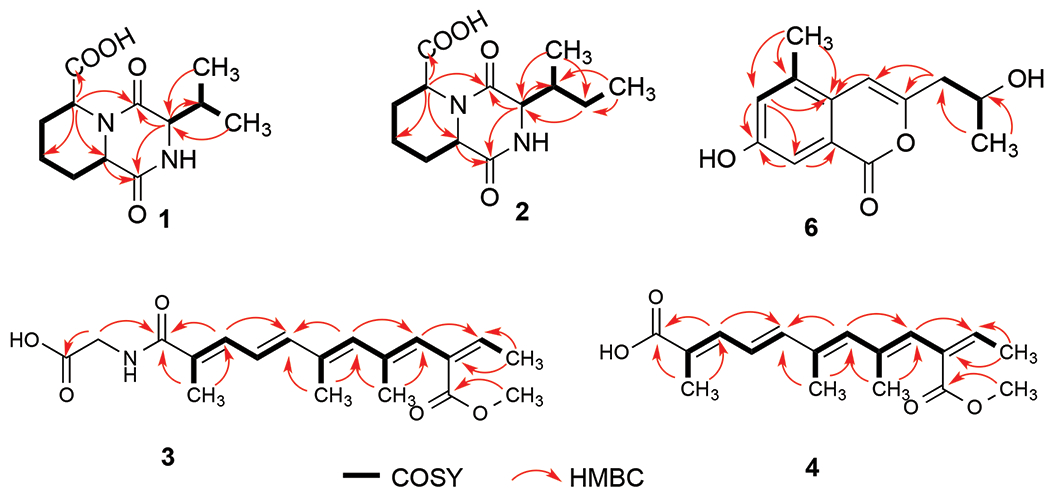
Key COSY (bolds) and HMBC (red arrows) correlations of compound 1-4, and 6
In the ROESY spectrum (Figure S7) of 1, H-6 and H-10 showed correlation, indicating a cis relationship between H-6 and H-10. As there was no NOE between H-3 and H-6/H-10, it was inconclusive whether H-3 and H-6/H-10 were cis or trans. To determine the relationship of H-3 and H-6/H-10, we carried out DFT calculations of NMR chemical shifts of 1-SRS(3,6,10) and 1-RRS(3,6,10) coupled with DP4+ analysis [Marcarino et al., 2021]. After a preliminary conformational search using the MMFF force field, all conformations found within a 5 kcal/mol window were fully optimized at the B3LYP/6-31G* level. The GIAO isotropic shielding constants were computed at the PCM/mPW1PW91/6-31+G** level, and where Boltzmann averaged with the relative energies refined at the SMD/M06-2X/6-31G* level [Zanardi et al., 2020]. The DP4+ calculations identified 1-RRS(3,6,10) as the most likely structure (DP4+ >99%, Figure S8) [Grimblat et al., 2015], suggesting that the relative configuration should be 3R*,6R*,10S*. Once the relative configuration was established, we next undertook TDDFT-ECD calculations for (3R*,6R*,10S*)-1 to determine its absolute configuration [Pescitelli and Bruhn 2016]. Each B3LYP/6-31G* optimized structure was submitted to TDDFT-ECD calculations at the PBE0/def2-SVP level, and the resulting spectra were Boltzmann-averaged using the relative energies refined at the SMD/M06-2X/6-31G* level. Our calculations (Figure S10) showed that the (3R,6R,10S)-1 isomer showed good agreement with the experimental ECD collected for 1. The 3R configuration was further confirmed by the hydrolysis of 1 using Marfey’s reagent [Hess, 2019], as amino acid analysis revealed the presence of D-valine ((R)-valine) (Figure S9). Therefore, the absolute configuration of 1 was determined as shown in Figure 1, and it was given a trivial name gramipiperazine A ((3R,6S,9aR)-3-isopropyl-1,4-dioxooctahydro-2H-pyrido[1,2-a]pyrazine-6-carboxylic acid).
Fig. 1.
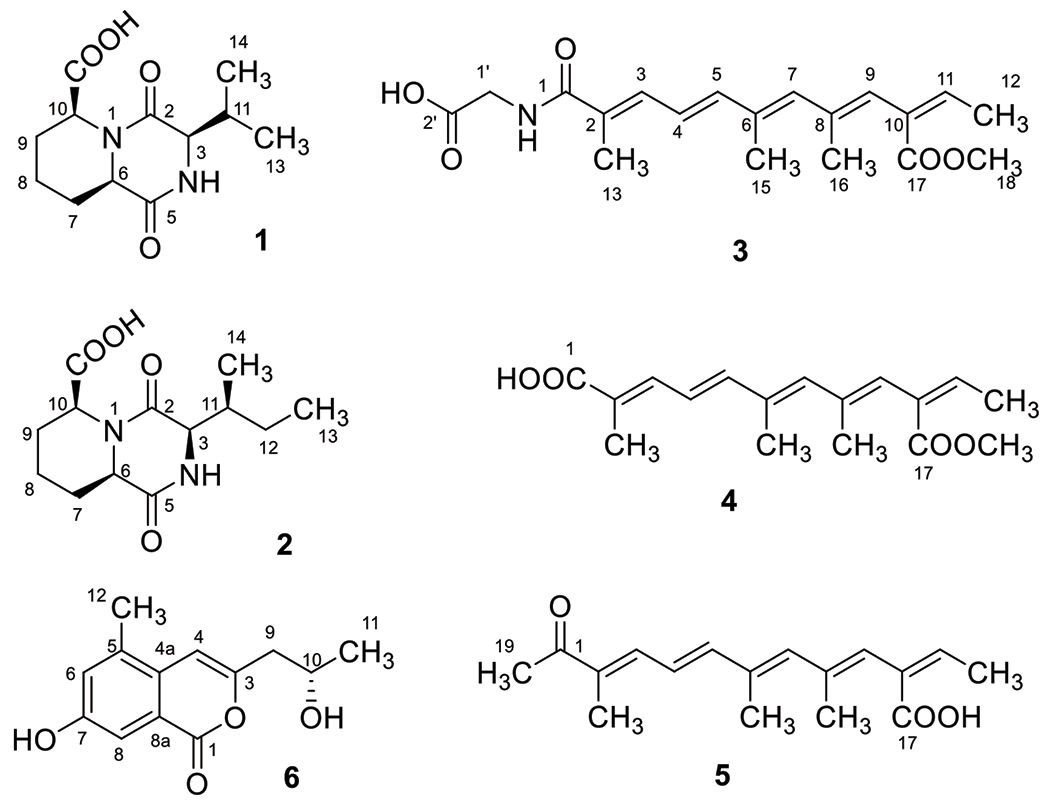
Chemical structures of compounds 1–6
The molecular formula of compound 2 was determined as C13H20N2O4 by HR-ESIMS analysis, with five degrees of unsaturation which has one methylene group more than compound 1. Detailed 1D and 2D NMR analysis (Table 1 and Figures S13–17) confirm the presence of one more methylene group between 11- and 13-positions, which meant that the substitute at 3-position in 2 was an isobutyl group rather than an isopropyl group as in 1. This substitution was confirmed by the HMBC correlations of H3-13 to C-11 and C-12, and H3-14 to C-3, C-11, and C-12 (Figure 2). Hence, the planer structure of compound 2 was established as shown in Figure 2.
In the ROESY spectrum (Figure S18) of 2, like 1, H-6 and H-10 showed correlation to each other, indicating a cis relationship between them. To determine the relative configuration at C-3 and C-11 we computed the NMR chemical shifts of 2-SRSS(3,6,10,11), 2-SRSR(3,6,10,11), 2-RRSS(3,6,10,11), and 2-RRSR(3,6,10,11) using the same computational procedure described for 1. The DP4+ calculations strongly supported 2-RRSS(3,6,10,11) as the most likely isomer (DP4+ > 99.9%, Figure S19). Hence, the relative configuration should be 3R*,6R*,10S*,11S* TDDFT-ECD calculations suggested that the absolute configuration of 2 was (3R,6R,10S,11S) (Figure S21), in clear agreement with the results obtained for 1. The 3R and 11S absolute configurations were further confirmed by hydrolysis of 2 with the Marfey’s reagent [Hess, 2019], revealing the presence of allo-D-isoleucine ((2R,3S)-2-amino-3-methylpentanoic acid) (Figure S20). Therefore, the absolute configuration of 2 was determined as shown in Figure 1, and it was given a trivial name gramipiperazine B ((3R,6S,9aR)-3-((S)-sec-butyl)-1,4-dioxooctahydro-2H-pyrido[1,2-a]pyrazine-6-carboxylic acid).
Compound 3 was obtained as brownish powder and its molecular formula was determined as C19H25NO5 by HRESIMS, requiring eight degrees of unsaturation. Comprehensive analysis of the 1H, DEPTQ and HSQC NMR indicated the presence of 19 carbons including five methyl groups (5 × CH3), one methylene (1 × CH2), six olefinic methines (6 × CH=C), and seven non-protonated carbons including three carboxyl/amide groups (3 × -COO−/−CONH-) (Table 2 and Figures S24–28). 1H–1H COSY spectrum established three spin systems, –C(CH3)=CH–CH=CH–, –C(CH3)=CH–C(CH3)=CH- and -C=CH-CH3 (Figure 2). HMBC correlations (Figure 2) from H3-15 to C-5, C-6, and C-7, and H3-16 to C-7, C-8, and C-9, confirmed the presence of the first two spin systems. In the HMBC spectrum of 3, H-9 correlated to C-11, which enabled us to connect the three spin systems together, as –C(CH3)=CH–CH=CH–C(CH3)=CH–C(CH3)=CH-C=CH-CH3. Also, both H-9 and H3-18 correlated to C-17 (a carbonyl), indicating the formation of a 2,6,8,10-tetrasubstituted straight chain with five conjugated double bonds at 2-, 4-, 6-, 8-, and 10-positions [-C(CH3)=CH–CH=CH–C(CH3)=CH–C(CH3)=CH–C(COO–CH3)=CH–CH3]. HMBC correlations from H3-13 to C-1 (a carbonyl), C-2 and C-3, and H2-1′ to C-1 and C-2′ established the planar structure of compound 3 as HOOC–CH2–NH–CO–C(CH3)=CH–CH=CH–C(CH3)=CH–C(CH3)=CH–C(COO–CH3)=CH–CH3 with a glycine moiety connecting to the polyketide through an amide bond. The conjugated double bonds at 2-, 4-, 6-, 8, and 10-positions were determined to be trans based on ROESY correlations (Figure S29) and comparison of chemical shifts and coupling constants with the literature [Vesonder 1996; Alecio et al., 1998]. Hence, the structure of compound 3 was determined as shown, which was given a trivial name kaneoheoic acid G (((2E,4E,6E,8E,10Z)-10-(methoxycarbonyl)-2,6,8-trimethyldodeca-2,4,6,8,10-pentaenoyl)glycine).
Table 2.
1H and 13C NMR Spectroscopic Data for Compounds 3-5 in CD3OD
| 3 |
4 |
5 |
||||
|---|---|---|---|---|---|---|
| no. | δC | δH mult. J (Hz) | δC* | δH mult. J (Hz) | δC* | δH mult. J (Hz) |
|
|
|
|
||||
| 1 | 170.3 | 171.1 | 200.9 | |||
| 2 | 128.8 | 126.8 | 134.7 | |||
| 3 | 134.4 | 7.06, d (9) | 138.1 | 7.32, d (9) | 141.3 | 7.35, d (10) |
| 4 | 123.0 | 6.63, dd (9,15) | 123.0 | 6.67 dd (9,15) | 123.8 | 6.69, dd (10,15) |
| 5 | 143.5 | 6.65, d(15) | 144.1 | 6.68, d (15) | 146.6 | 6.96, d (14) |
| 6 | 135.0 | 135.0 | 134.7 | |||
| 7 | 136.9 | 6.21, s | 137.3 | 6.25, s | 139.1 | 6.33, s |
| 8 | 137.5 | 137.5 | 136.8 | |||
| 9 | 124.0 | 6.00, s | 124.1 | 6.03, s | 126.0 | 6.10, s |
| 10 | 130.3 | 130.3 | 130.2 | |||
| 11 | 139.6 | 6.96, m | 139.3 | 7.00, m | 138.7 | 6.93 |
| 12 | 14.7 | 1.81, d (7) | 14.5 | 1.81, d (7) | 15.0 | 1.81, d (7) |
| 13 | 11.7 | 2.05, s | 11.4 | 1.99, s | 10.3 | 1.93, s |
| 15 | 13.0 | 2.10, s | 12.8 | 2.10, s | 13.2 | 2.13, s |
| 16 | 17.8 | 1.71, s | 17.5 | 1.72, s | 17.9 | 1.76, s |
| 17 | 168.0 | 167.8 | 169.9 | |||
| 18 | 50.9 | 3.75, s | 50.6 | 3.70, s | ||
| 19 | 24.4 | 2.38, s | ||||
| 1′ | 42.1 | 3.93, s | ||||
| 2′ | 173.4 | |||||
13C NMR shifts were obtained from HSQC and HMBC NMR
The molecular formula of compound 4 was determined as C17H22O4 by HR-ESIMS analysis, with seven degrees of unsaturation. A comprehensive analysis of the 1H, 13C and HSQC NMR spectra indicated the presence of 17 carbons including five methyl groups (5× CH3), six olefinic methines (6× CH=C), and six non-protonated carbons including two carboxyl groups (2×-COO-) (Table 2 and Figures S32–35). 1H-1H COSY spectrum establishes three spin systems, -C(CH3)=CH-CH=CH-, -C(CH3)=CH-C(CH3)=CH-, and-C=CH-CH3 (Figure 2). HMBC correlations (Figure 2) from H3-15 to C5, C6, and C7 enabled us to connect the first two spin systems together, as -C(CH3)=CH-CH=CH-C(CH3)=CH-C(CH3)=CH-. In the HMBC spectrum of 4, H-9 correlated to C-11 and both H-9 and H3-18 correlated to C-17 (a carbonyl), indicating the formation of a 2,6,8,10-tetrasubstituted straight chain with five conjugated double bonds at 2-, 4-, 6-, 8-, and 10-positions [-C(CH3)=CH–CH=CH–C(CH3)=CH–C(CH3)=CH–C(COO–CH3)=CH–CH3]. Compound 4 was almost the same as compound 3 except for the absence of amide linkage to a glycine moiety at 1-position. Like compound 3, the conjugated double bonds at 2-, 4-, 6-, 8, and 10-positions were confirmed to be trans based on ROESY correlations (Figure S36) and comparison of chemical shifts and coupling constants with the literature [Vesonder 1996; Alecio et al., 1998]. Hence, the structure of compound 4 is established as shown, given a trivial name kaneoheoic acid H ((2E,4E,6E,10Z)-10-(methoxycarbonyl)-2,6,8-trimethyldodeca-2,4,6,8,10-pentaenoic acid).
Compound 5 was obtained as a yellow powder and its molecular formula was determined as C17H22O3 by HRESIMS, requiring seven degrees of unsaturation. Comprehensive analysis of the 1D and 2D NMR (HSQC, COSY, and HMBC) data indicated the similarity between compounds 4 and 5 (Table 2 and Figures S39–42). The main differences were the substitution of methyl ester group (–COOCH3) in 4 by a carboxyl group (–COOH) in 5 at 10-position, and also the presence of acetyl group (–COCH3) in 5 instead of the carboxyl group (–COOH) in 4 at 1-position. Like compound 3 and 4, the conjugated double bonds were determined to be trans based on ROESY correlations (Figure S43) and comparison of chemical shifts and coupling constants with the literature [Vesonder 1996; Alecio et al., 1998]. Therefore, the structure of compound 5 is expressed as shown, given a trivial name kaneoheoic acid I ((2Z,3E,5E,7E,9E)-2-ethylidene-4,6,10-trimethyl-11-oxododeca-3,5,7,9-tetraenoic acid).
Compound 6 was isolated as a brownish powder with a molecular formula of C13H14O4, which was determined by HRESIMS, requiring seven degrees of unsaturation. Comprehensive analysis of the 1H, DEPTQ, and HSQC NMR (Tables S1 and Figures S46–50) indicated the presence of two methyl groups, one methylene, four methines including one oxygenated, a pair of meta-coupled aromatic protons and an olefinic proton, and six nonprotonated carbons. Two spin systems, –CH2–CH–CH3, and =CH–C(CH3)= (Figure 2), were established by the COSY spectrum. The HMBC correlations from H-4 to C-3, C-4a, C-5, and C-8a, from H- 6 to C-4a, C-5, C-7, and C-8, from H-8 to C-4a, C-6, C-7, and C-8a, as well as the molecular formula of 6, established an isocoumarin skeleton. HMBC correlations from H3-11 to C-9 and C-10, together with the 1H and 13C NMR signals (δH/δC) at 9- (2.71/42.9) and 10-positions (4.21/64.9), confirmed the presence of a 2-hydroxyl-propanyl group. The substituted position of the 2-hydroxyl-propanyl group was determined to be at C-3 based on the HMBC correlations from H2-9 to C-3 and C-4 and H-4 to C-9. One methyl group and one hydroxyl group were attached to C-5 and C-7, respectively, by HMBC correlations from methyl protons H3-12 to C-4a, C-5, and C-6 and 13C NMR signal at C-7 (δc 161.0). Based on the above analysis, the planar structure of 6 was determined to be 7-hydroxy-3-(2-hydroxy-propyl)-5-methyl-isochromen-1-one which was the same as the previously isolated compound 7-hydroxy-3-(2-hydroxy-propyl)-5-methyl-isochromen-1-one with an R configuration (Tables S1) [Wang et al., 2012].
However, the absolute configuration at C-10 was determined to be S by comparison of its optical rotation value ([α]25d-13.3 (c 0.3, MeOH)) with that of previously isolated compound ([α]25d+17.8 (c 0.3, MeOH)) with an R configuration [Wang et al., 2012] and (R)-orthosporin ([α]22 d+61.8, MeOH) [Hallock et al., 1988; Ichihara et al., 1989]. This was further confirmed by TDDFT-ECD calculations at the PBE0/def2-SVP//B3LYP/6-31G* level, with the (S)-6 isomer showing clear agreement with the experimental ECD data obtained for the natural product (Figure 3).
Fig. 3.
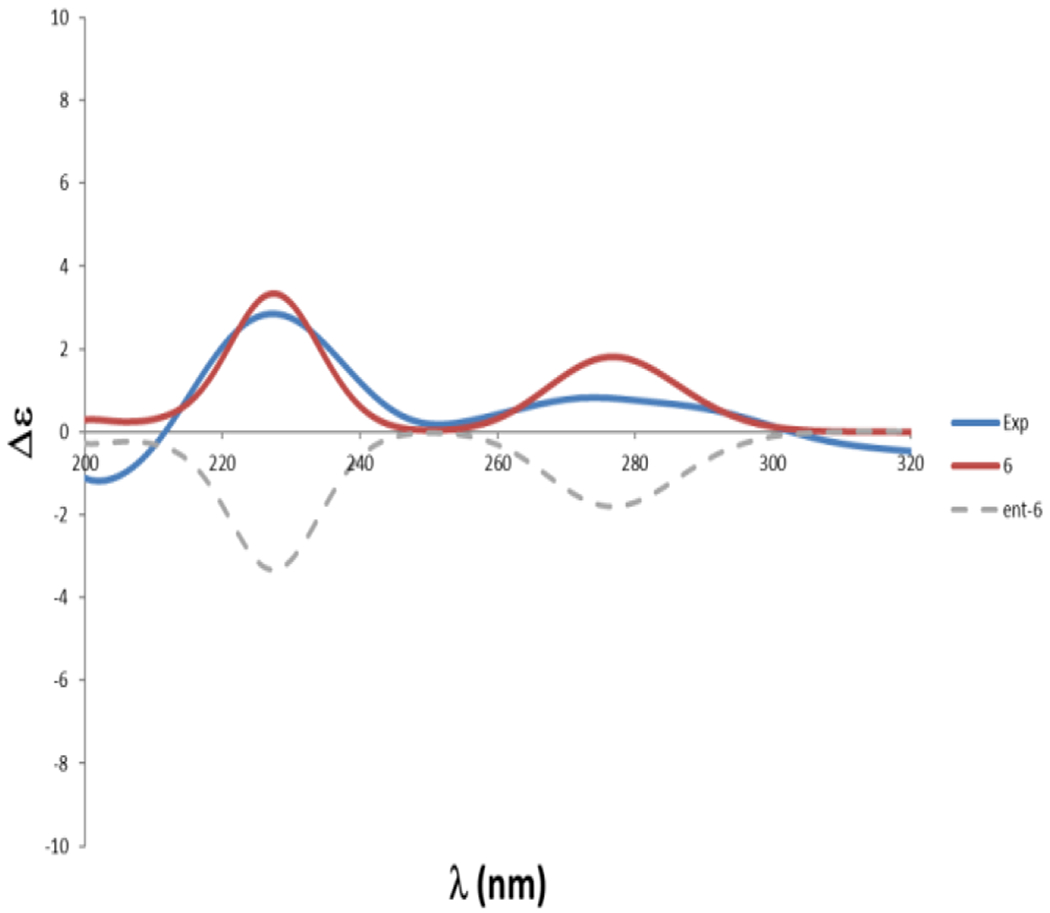
Experimental and calculated ECD of compound 6
In addition to compounds 1–6, five known fusareilins, fusarielin F (7) [Sorensen et al., 2012], fusarielin G (8) [Sorensen et al., 2012], fusarielin H (9) [Sorensen et al., 2012], fusarielin M (10) [Chen et al., 2021], and 3-O-methyl fusarielin (11) [Tchoukoua et al., 2018] (Figure S52) were also isolated. The structures of these compounds were determined based on comparisons of 1D and 2D NMR and HRESIMS data with previously reported ones.
2.2. Antibacterial and anti-proliferative activity of 1-11
Compounds 1–11 were evaluated for their antibacterial activity against S. aureus, methicillin-resistant S. aureus, and E. coli (Table 3). Although compounds 2–5, 7, 9, and 11 showed weak activity against S. aureus and methicillin-resistant S. aureus with the MIC values ranging from 80 to 160 μg/mL when assayed alone, compounds 3–5 showed significantly increased antibacterial activity with decreased MIC values by two to four folds in the presence of chloramphenicol (1 μg/mL, half of its MIC value (2-8 μg/mL)), None of the isolated compounds showed any activity against gram-negative bacteria E. coli.
Table 3.
Activities of compounds 1–11 against S. aureus (ATCC® 12600™) and methicillin-resistant S. aureus (ATCC®43300™) in the presence and absence of chloramphenicol (1 μg/mL)
| Compounds | MIC [μg/mL] |
|||
|---|---|---|---|---|
|
S.
aureus
|
Methicillin-resistant
S. aureus |
|||
| Compound alone | Compound + Chloramphenicol [1 μg/mL] | Compound alone | Compound + Chloramphenicol [1 μg/mL] | |
| 2 | 160 | 160 | NA | NA |
| 3 | 80 | 40 | NA | NA |
| 4 | 80 | 20 | NA | NA |
| 5 | 160 | 40 | NA | NA |
| 7 | 80 | 80 | 160 | 80 |
| 9 | 80 | 80 | 160 | 80 |
| 11 | 80 | 80 | 80 | 80 |
NA→ Not active
Compounds 1–11 were further evaluated for their antiproliferative activity against A2780 human ovarian cancer cells, only compound 5 showed activity with an IC50 value of 18.52 μM. When evaluated in a mammalian cell-based assay designed to monitor TNF-α-induced NF-κB activity, compound 5 were found to inhibit NF-κB with an IC50 value of 15.86 μM. Compounds 1–11 were not toxic to the human embryonic kidney cells 293 (HEK 293) at 50 μM. Thus, in the absence of cytotoxic response, inhibition of TNF-α-induced NF-κB activity suggests the potential of mediating a cancer chemopreventive response.
3. Conclusion
Diketopiperazines are a common class of specialised metabolites from various natural sources. 2,5-Diketopiperazines are cyclodipeptides usually cyclized through condensation of two α-amino acids. Compounds 1 and 2 are atypical 2,5-diketopiperazines because biogenetically they could be derived from 2-amino-5-hydroxyadipic acid and valine (for 1), and 2-amino-5-hydroxyadipic acid and isoleucine (for 2), respectively. As we know, no natural product containing 1,4-dioxooctahydro-2H-pyrido[1,2-a]pyrazine-6-carboxylic acid (a compound without the isopropyl group in 1) has been reported before. Also, diketopiperazine rarely have a core structure of hexahydro-4H-pyrido[1,2-a]pyrazine-1,4(6H)-dione (a compound without the carboxylic acid and the isopropyl group in 1) like nocarazepine A [Zhou et al., 2017], Compounds 3-5 from F. graminearum FM1010 showed antibacterial activities against S. aureus and methicillin-resistant S. aureus when combined with the FDA-approved antibiotic chloramphenicol. The findings from the present study warrant future research on the mechanism of action responsible for these synergistic activities.
4. Experimental
4.1. General experimental procedure
Optical rotations, CD, and FT-IR spectra were measured with a Rudolph Research analytical autoPol automatic polarimeter, JASCO J-815 CD, and Thermo Scientific Nicolet iS10 IR spectrometer, respectively. 1D and 2D NMR spectra were recorded on a Bruker AM-400 spectrometer. The 3.31 and 49.1 ppm resonances for CD3OD and 2.50 and 39.5 ppm resonances for DMSO-d6 were used as the internal references for 1H and 13C NMR spectra, respectively. An Agilent 6530 Accurate-Mass Q-TOF LC-MS spectrometer was used to record high-resolution mass spectra. Preparative HPLC was carried out on an Ultimate 3000 chromatographic system with a Phenomenex preparative column (Phenyl-Hexyl, 5 μ, 100 × 21.2 mm) and semipreparative HPLC on an Ultimate 3000 chromatographic system with a Phenomenex semipreparative column (C8, 5 μ, 250 × 10 mm), a Dionex Ultimate 3000 DAD detector, and a Dionex Ultimate 3000 automated fraction collector; all solvents were HPLC grade. Diaion HP-20 was used to run open-column chromatography.
4.2. Strain isolation and identification
The study material Fusarium graminearum Schwabe (Nectriaceae) strain FM1010 was isolated from a shallow-water volcanic rock, locally known as ‘live rock’ collected at the Carl Smith Beach, Big Island, Hawaii (latitude 19.7342° N; longitude 155.0261° W), in September 2019. The sequence of the 18S rDNA gene of FM1010 was obtained, identified, and aligned against previously reported sequences available in the GeneBank database using the BLAST tool to identify the similarity score as well as to calculate the statistical significance of the matches (http://www.blast.ncbi.nlm.nih.gov/Blast). Based on the obtained result the strain was confirmed as Fusarium graminearum (Family: Nectriaceae) and deposited in GenBank under accession no. MW703613. The strain was deposited in a −80°C freezer at Daniel K. Inouye College of Pharmacy, University of Hawaii at Hilo, HI, USA.
4.3. Fermentation, extraction, and isolation
The strain was grown on PDA plates at 28 °C for 5 days; then it was cut into small pieces and inoculated into 20 L of autoclaved sterilized liquid medium [mannitol 20 g, glucose 10 g, monosodium glutamate 5 g, KH2PO4 (0.5 g), MgSO4·7H2O 0.3 g, and yeast extract 3 g in 1 L of distilled water; pH 6.5 prior to sterilization] containing 15% sea salt for fermentation at 24 °C for 28 days. After 28 days, the mycelia of FM1010 were filtered and extracted with acetone under ultrasonic conditions (1 L × 3 times), followed by removal of acetone under reduced pressure to afford an aqueous solution. After combining the aqueous mycelia extraction and supernatant solution, it was subjected to an HP-20 column eluted with MeOH–H2O (10, 50, 90, and 100%) to afford four fractions (Fr.1–4). Fraction 3 (8.4 g) was separated by preparative HPLC (Phenyl-Hexyl, 5 μ, 100 × 21.2 mm; 8 mL/min) eluted with 60–100% MeOH–H2O in 20 min to yield subfractions SFr 3–1-20. Also, Fraction 2 (2.4 g) was separated by preparative HPLC (Phenyl-Hexyl, 5 μ, 100 × 21.2 mm; 8 mL/min) eluted with 30% isocratic in 10 min to yield subfractions SFr 2-1-10. SFr 2-4 was purified by semipreparative HPLC (25% isocratic of MeOH–H2O with 0.1% formic acid for 20 min; 3 mL/ min) to afford compound 1 (1.2 mg, tR 18.5 min). Compounds 2 (1.8 mg, tR 19 min) was separated from SFr 3-5 by using the same HPLC (40% isocratic of MeOH–H2O with 0.1% formic acid for 20 min). Compounds 5 (0.9 mg, tR 32 min) was isolated from SFr 3-12 by using 40% isocratic of ACN–H2O with 0.1% formic acid for 40 min (3 mL/min). Compounds 4 (0.8 mg, tR 20 min) was separated from SFr 3-15 by using the same HPLC (70% isocratic of MeOH–H2O with 0.1% formic acid for 20 min). Compounds 5 (0.9 mg, tR 19 min) and 7 (1.3 mg, tR 22.5 min) were separated from SFr 3-14 by using the same HPLC (68% isocratic of MeOH–H2O with 0.1% formic acid for 30 min). Compound 6 (1.6 mg, tR 23 min) was isolated from SFr 3-6 with the elution of a 40% isocratic of MeOH–H2O with 0.1% formic acid for 30 min (3 mL/min). Compound 8 (0.8 mg, tR 27 min) was isolated from SFr 3-18 with the elution of a 75% isocratic of MeOH–H2O (0.1% formic acid) for 30 min (3 mL/min). Compound 9 (1.9 mg, tR 27.5 min) was isolated from SFr 3-19 with the elution of a 40% isocratic of acetonitrile–H2O (0.1% formic acid) for 30 min (3 mL/min). SFr 3-22 and 3-20 were purified by semipreparative HPLC (80% isocratic of acetonitrile–H2O with 0.1% formic acid for 20 min; 3 mL/min) to afford compound 10 (1.2 mg, tR 18.5 min) and compound 11 (1.7 mg, tR 16.5 min), respectively.
4.4. Spectroscopic data of compounds 1-6
Gramipiperazine A (1):
Light yellow powder; −12.3 (c 0.002, MeOH); UV (MeOH) λmax (log ε) = 202 (3.8) nm; ECD (c 0.01, MeOH) λmax +232; IR (MeOH) vmax 3306, 2942, 2834, 1650, 1451, 1403, 1109, 1014 cm−1; 1H and 13C NMR data, Table 1; HRESIMS m/z 253.1190 [M - H]− (calcd for C12H17N2O4, 253.11883).
Gramipiperazine B (2):
Light yellowish powder; −8.6 (c 0.002, MeOH); UV (MeOH) λmax (log ε) = 206 (3.8) nm; ECD (c 0.01, MeOH) λmax +232, −206; IR (MeOH) vmax 3327, 2945, 2835, 1652, 1453, 1404, 1114, 1016 cm−1; 1H and 13C NMR data, Table 1; HRESIMS m/z 269.14887 [M + H]+ (calcd for C13H21N2O4, 269.15013).
Kaneoheoic acid G (3):
Brownish powder; UV (MeOH) λmax (log ε) = 332 (3.8) nm; IR (MeOH) vmax 3302, 2948, 2832, 1661, 1450, 1407, 1114, 1016 cm−1; 1H and 13C NMR data, Table 2; HRESIMS m/z 348.17069 [M + H]+(calcd for C19H26NO5, 348.18110).
Kaneoheoic acid H (4):
Brownish powder; UV (MeOH) λmax (log ε) = 335 (3.6) nm; IR (MeOH) vmax 3296, 2945, 2829, 1646, 1453, 1398, 1111, 1016 cm−1; 1H and 13C NMR data, Table 2; HRESIMS m/z 291.15148 [M + H]+ (calcd for C17H23O4, 291.15963).
Kaneoheoic acid I (5):
Brownish powder; UV (MeOH) λmax (log ε) = 347 (3.8) nm; IR (MeOH) vmax 3299, 2945, 2832, 1649, 1450, 1404, 1114, 1016 cm−1; 1H and 13C NMR data, Table 2; HRESIMS m/z 275.15700 [M + H]+ (calcd for C17H23O3, 275.16472), 297.13959 [M + Na]+ (calcd for C17H22NaO3, 297.1467).
7-Hydroxy-3-(2-hydroxy-propyl)-5-methyl-epiisochromen-1-one (6):
Brownish powder; −13.3 (c 0.3, MeOH; UV (MeOH) λmax (log ε) = 223 (3.8), 248 (3.4), 293 (2.9) nm; ECD (c 0.01, MeOH) λmax +226, +274,−202; IR (MeOH) vmax 3299, 2948, 2826, 1646, 1450, 1407, 1114, 1016 cm−1; 1H and 13C NMR data, Table S1; HRESIMS m/z 235.09584 [M + H]+ (calcd for C13H15O4, 235.09703).
4.5. Hydrolysis of gramipiperazine A (1) and B (2) and Marfey reactions [Hess, 2019]
Gramipiperazine A (1) and B (2) (0.1 mg each) were hydrolyzed separately with 6N HCl at 110°C for 14 h, respectively. The hydrolysate was concentrated to dryness under vacuum, followed by 3 times wash with H2O to remove the residual acid. The residue was dissolved in 100 μL 1N NaHCO3, and the solution was treated with 50 μL of Marfey reagent (1-fluoro-2,4-dinitrophenyl-5-L-alanine amide, 10 mg/mL in acetone). The reaction mixture was heated at 80°C for 5 min, which was followed by adding 50 μL 2N HCl to quench the solution. Aqueous 50% CH3CN (50 μL) was added before LC-MS analysis.
4.6. Computational section
All the quantum mechanical calculations were performed using Gaussian 09 [Frisch et al., 2009]. Systematic conformational searches were done for each compound in the gas phase using the MMFF force field, implemented in Macromodel [MacroModel, 2018] (mixed torsional/low-mode sampling protocol) using an energy cutoff of 5 kcal/mol. The choice for the 5 kcal/mol of cutoff was set as a balance between reducing the overall CPU calculation time and minimizing the possibility of losing further contributing conformers. All conformers were kept for full geometry optimization at the B3LYP/6-31G* level in the gas phase. Frequency calculations were done at the same level to determine the nature of the stationary points found. The magnetic shielding constants (σ) were computed using the gauge including atomic orbitals (GIAO) [Wolinski et al., 1990] method at PCM/mPW1PW91/6-31+G** level of theory, the recommended for DP4+ calculations [Grimblat et al., 2015]. The unscaled chemical shifts (δu) were computed using TMS as reference standard according to δu = σ0 - σx, where σx is the Boltzmann averaged shielding tensor (over all significantly populated conformations) and σ0 is the shielding tensor of TMS computed at the same level of theory employed for σx. The Boltzmann averaging was done according to eq 1:
| (eq. 1) |
where σi x is the shielding constant for nucleus x in conformer i, R is the molar gas constant (8.3145 J K−1 mol−1), T is the temperature (298 K), and Ei is the energy of conformer i (relative to the lowest energy conformer), obtained at the SMD/M06-2X/6-31G* level of theory. where σi x is the shielding constant for nucleus x in conformer i, R is the molar gas constant (8.3145 J K−1 mol−1), T is the temperature (298 K), and Ei is the energy of conformer i (relative to the lowest energy conformer), obtained at the SMD/M06-2X/6-31G* level of theory. The scaled chemical shifts (δs) were computed as δs = (δu – b)/m, where m and b are the slope and intercept, respectively, resulting from a linear regression calculation on a plot of δu against δexp. The DP4+ values were computed using the Excel spreadsheet provided in https://sarotti-nmr.weebly.com. The ECD calculations were carried out using the B3LYP/6-31G* optimized geometries. The excitation energies (nm) and rotatory strength (R) in dipole velocity (Rvel) of the first forty singlet excitations were calculated using TDDFT implemented in Gaussian 09 at the PBE0/def2-SVP level from all significantly populated conformers, which were averaged using Boltzmann weighting. The Boltzmann amplitudes obtained by refining the Gibbs free energies of all compounds at the SMD/M06-2X/6-31G* level. The calculated rotatory strength were simulated into the ECD curve as the sum of Gaussians with 0.3 eV width at half-heights (σ), which were UV-corrected and scaled [Pescitelli and Bruhn 2016].
4.7. Antibacterial assay
Antibacterial assay was conducted by using the previously described method with slight modifications [Zaman et al., 2020a]. Bacteria were grown on agar plates [Tryptic Soy Agar (TSA) or Luria–Bertani Agar (LBA)] for 1 day at 37°C and then added to a liquid medium (TSB for S. aureus, and methicillin-resistant S. aureus and LB for E. coli). After incubation at 37°C for 20 h, the cultures were diluted with TSB or LB media to obtain an OD600 value of approx. 0.1. One hundred microliters of fresh media with samples at the desired concentration of 160 μg/mL (dissolved in DMSO) was put in the first well and then a two-fold dilution continued to the lowest concentration. The bacterium-containing media (100 μL) were then added to each well of 96-well plates. Additionally, samples were tested in combination with the FDA-approved antibiotic chloramphenicol at 1 μg/mL. DMSO (5%) and chloramphenicol (1 μg/mL) were used as negative controls in these sets of experiments. Chloramphenicol, which is active against S. aureus, methicillin-resistant S. aureus, and E. coli at MIC values of 6.25 μg/ml, 6.25 μg/ml, and 3.2 μg/ml, respectively, was employed as a positive control.
4.8. Anti-proliferative Assay
The viability of A2780 human ovarian cancer cells was determined using the CyQuant assay according to the manufacturer’s instructions (Life Technologies, CA, USA). Briefly, cells were cultured in 96-well plates at 1000 cells per well for 24 h and subsequently treated with compounds (20 μg/mL) for 72 h and analyzed. The relative viability of the treated cells was normalized to the DMSO-treated control cells. Cisplatin was used as a positive control, which had an IC50 value of 0.36 μM. All experiments were performed in triplicate.
4.9. NF-κB Assay
We employed HEK 293 from Panomics for monitoring changes occurring along the NF-κB pathway.32 Stable constructed cells were seeded into 96-well plates at 20 × 103 cells per well. Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen Co.), supplemented with 10% fetal bovine serum (FBS), 100 units/mL penicillin, 100 μg/mL streptomycin, and 2 mM L-glutamine. After 48 h of incubation, the medium was replaced and the cells were treated with various concentrations of test substances. TNF-α (human, recombinant, E. coli, Calbiochem) was used as an activator at a concentration of 2 ng/mL (0.14 nM). The plate was incubated for 6 h. Spent medium was discarded, and the cells were washed once with PBS. Cells were lysed using 50 μL (for 96-well plate) of reporter lysis buffer from Promega, by incubating for 5 min on a shaker, and stored at −80 °C. The luciferase assay was performed using the Luc assay system from Promega. The gene product, luciferase enzyme, reacts with luciferase substrate, emitting light, which was detected using a luminometer (LUMIstar Galaxy BMG). Data for NF-κB inhibition are expressed as IC50 values (i.e., the concentration required to inhibit TNF-α-induced NF-κB activity by 50%). As positive controls, two known NF-κB inhibitors were used, TPCK (Nα-tosyl-L-phenylalanine chloromethyl ketone) and BAY-11- 7082 (which selectively and irreversibly inhibits NF-κB activation by blocking TNF-α-induced phosphorylation of IκB-α without affecting constitutive IκB-α phosphorylation), yielding IC50 values of 5.3 ± 0.9 and 11 ± 1.8 μM, respectively. All experiments were performed in triplicate.
4.10. SRB Assay
To assess the potential of mediating a cytotoxic response, the cells were treated under the same experimental conditions with each test compound at a concentration of 50 μM, and cell survival was determined by the sulforhodamine B (SRB) assays. After incubation of HEK 293 cells with test compounds, cells were fixed with 10% trichloroacetic acid solution for 30 min and stained with 0.4% SRB in 1% acetic acid solution for 30 min. Protein-bound SRB was dissolved in 10 mM Tris buffer (pH 10.0), and the absorbance was measured at 515 nm. The effect of compounds on cell survival was demonstrated as percentage survival in comparison with vehicle (DMSO)-treated control cells.
Supplementary Material
Highlights.
Six undescribed compounds including 2 uncommon diketopiperazines were identified.
Absolute configurations were confirmed by ECD calculation and Marfey’s reaction.
Compounds 2-5 showed antibacterial activities when combined with chloramphenicol.
Compounds 5 exhibited anti-proliferative activity against ovarian cancer cell line.
Compounds 5 also showed TNF-α induced NF-κB inhibitory activity.
Acknowledgement
This work was financially supported by the Victoria S. and Bradley L. Geist Foundation (15ADVC-74420, 17CON-86295, and 20CON-102163) (to SC). Funding for this work was also supported by Hawaii IDeA Network for Biomedical Research Excellence III and IV (INBRE-III and INBRE-IV) project: NIGMS Grant 5P20GM103466 (to SC). We would also like to express our gratitude to Mr. Justin Reinicke for his help with HRMS, NMR, optical rotation and ECD data collection.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Appendix A. Supplementary material
The Supporting Information is available online at https://doi.org/XXX
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Alécio AC, V da Silva Bolzani, Young MCM, Kato MJ, Furlan M, 1998. Antifungal Amide from Leaves of Piper hispidam. J. Nat. Prod 61, 637–639. 10.1021/np9703656 [DOI] [PubMed] [Google Scholar]
- Aly AH, Debbab A, Proksch P, 2011. Fungal endophytes: unique plant inhabitants with great promises. App. Micro. Biotech 90, 1829–1845. 10.1007/s00253-011-3270-y [DOI] [PubMed] [Google Scholar]
- Chen D, Liu L, Lu Y Chen S, 2021. Identification of fusarielin M as a novel inhibitor of Mycobacterium tuberculosis protein tyrosine phosphatase B (MptpB). Bioorg. Chem 106, 104495. 10.1016/j.bioorg.2020.104495 [DOI] [PubMed] [Google Scholar]
- Fei-Zhang DJ, Li C, Cao S, 2016. Hawaii natural compounds are promising to reduce ovarian cancer deaths. Cancer Biol. Ther 17,709–712. 10.1080/15384047.2016.1178428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr., Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ Gaussian 09, Gaussian, Inc.: Wallingford, CT, 2009. [Google Scholar]
- Grimblat N, Zanardi MM, Sarotti AM, 2015. Beyond DP4: an improved probability for the stereochemical assignment of isomeric compounds using quantum chemical calculations of NMR shifts. J. Org. Chem 80, 12526–12534. 10.1021/acs.joc.5b02396 [DOI] [PubMed] [Google Scholar]
- Hallock YF, Clardy J, Kenfield DS, Strobel G, 1988. De-O-methyldiaporthin, a phytotoxin from Drechslera sicccms. Phytochemistry 27, 3123. 10.1016/0031-9422(88)80012-8 [DOI] [Google Scholar]
- Hess S, 2019. A Universal HPLC-MS Method to Determine the Stereochemistry of Common and Unusual Amino Acids. Amino Acid Analysis 263–275. 10.1007/978-1-61779-445-2_7 [DOI] [PubMed] [Google Scholar]
- Huang P, Li C, Sarotti AM, Turkson J, Cao S, 2017. Sphaerialactonam, a γ-lactam–isochromanone from the Hawaiian endophytic fungus Paraphaeosphaeria sp. FT462. Tetrahedron Lett. 58, 1330–1333. 10.1016/j.tetlet.2017.02.052 [DOI] [Google Scholar]
- Ichihara A, Hashimoto M, Hirai T, 1989. Structure, Synthesis, and Stereochemistry of (+)-Orthosporin, a Phytotoxic Metabolite of Rhynchosporium Orthosporum. Chem. Lett 18, 1495. 10.1246/cl.1989.1495 [DOI] [Google Scholar]
- Li C, Ding Y, Yang B, Hoffman N, Yin H-Q, Mahmud T, Turkson J, Cao S, 2016. Eremophilane sesquiterpenes from Hawaiian endophytic fungus Chaetoconis sp. FT087. Phytochemistry 126, 41–46. 10.1016/j.phytochem.2016.03.005 [DOI] [PubMed] [Google Scholar]
- Li C, Ding Y, Yang B, Miklossy G, Yin H, Walker LA, Turkson J, Cao S, 2015. A New Metabolite with a Unique 4-Pyranone–γ-Lactam–1,4-Thiazine Moiety from a Hawaiian-Plant Associated Fungus.Org. Lett 17, 3556–3559. 10.1021/acs.orglett.5b01650 [DOI] [PubMed] [Google Scholar]
- Li C, Hu Z, Liu Q, Wu X, Cao S, 2018. Two new tricycloalternarenes from Hawaiian endophytic fungus Didymella sp. FT433. Tetrahedron Lett. 59, 3381–3383. 10.1016/j.tetlet.2018.07.061 [DOI] [Google Scholar]
- Li C, Ren G, Yang B, Miklossy G, Turkson J, Fei P, Ding Y, Walker LA, Cao S, 2016. Meroterpenoids with Antiproliferative Activity from a Hawaiian-Plant Associated Fungus Peyronellaea coffeae-arabicae FT238. Org. Lett 18, 2335–2338. 10.1021/acs.orglett.6b00685 [DOI] [PubMed] [Google Scholar]
- Li C, Sarotti AM, Huang P, Dang UT, Hurdle JG, Kondratyuk TP, Pezzuto JM, Turkson J, Cao S, 2017. NF-κB inhibitors, unique γ-pyranol-γ-lactams with sulfide and sulfoxide moieties from Hawaiian plant Lycopodiella cernua derived fungus Paraphaeosphaeria neglecta FT462. Sci. Rep 7, 10424. 10.1038/s41598-017-10537-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Sarotti AM, Turkson J, Cao S, 2017. Verbenanone, an octahydro-5H-chromen-5-one from a Hawaiian-Plant Associated Fungus FT431. Tetrahedron Lett. 58, 2290–2293. 10.1016/j.tetlet.2017.04.096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Sarotti AM, Wu X, Yang B, Turkson J, Chen Y, Liu Q, Cao S, 2019. An unusual benzoisoquinoline-9-one derivative and other related compounds with antiproliferative activity from Hawaiian endophytic fungus Peyronellaea sp. FT431. Molecules 24, pii:E196. 10.3390/molecules24010196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Sarotti AM, Yang B, Turkson J, Cao S, 2017. A New N-methoxypyridone from the Co-Cultivation of Hawaiian Endophytic Fungi Camporesia sambuci FT1061 and Epicoccum sorghinum FT1062. Molecules 22, pii:E1166. 10.3390/molecules22071166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Sarotti AM, Yoshida W, Cao S, 2018. Two new polyketides from Hawaiian endophytic fungus Pestalotiopsis sp. FT172. Tetrahedron Lett. 59, 42–45. 10.1016/j.tetlet.2017.11.045 [DOI] [Google Scholar]
- Li C, Yang B, Fenstemacher R, Turkson J, Cao S, 2015. Lycopodiellactone, an unusual δ-lactone-isochromanone from a Hawaiian plant-associated fungus Paraphaeosphaeria neglecta FT462. Tetrahedron Lett. 56, 1724–1727. 10.1016/j.tetlet.2015.02.076 [DOI] [Google Scholar]
- Li C, Yang B, Turkson J, Cao S, 2017. Anti-proliferative ambuic acid derivatives from Hawaiian endophytic fungus Pestalotiopsis sp. FT172. Phytochemistry 140, 77–82. 10.1016/j.phytochem.2017.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacroModel Schrodinger release 2018-3, Schrodinger LLC: New York, 2018. [Google Scholar]
- Manzoni M, Rollini M, 2002. Biosynthesis and biotechnological production of statins by filamentous fungi and application of these cholesterol-lowering drugs. App. Micro. Biotech 58, 555–564. 10.1007/s00253-002-0932-9 [DOI] [PubMed] [Google Scholar]
- Marcarino MO, Cicetti S, Zanardi MM, Sarotti AM, 2021. A critical review on the use of DP4+ in the structural elucidation of natural products: the good, the bad and the ugly. A practical guide. Nat. Prod. Rep 10.1039/D1NP00030F. [DOI] [PubMed] [Google Scholar]
- Pescitelli G, Bruhn T, 2016. Good computational practice in the assignment of absolute configurations by TDDFT calculations of ECD spectra. Chirality 28, 466–474. 10.1002/chir.22600 [DOI] [PubMed] [Google Scholar]
- Song D, Zhou X, Peng Q, Chen T, Zhang F, Huang T, Zhang T, Li A, Huang D, Wu Q, He H, Tang Y, 2015. Newly Emerged Porcine Deltacoronavirus Associated With Diarrhoea in Swine in China: Identification, Prevalence and Full-Length Genome Sequence Analysis. Trans. Emerg. Dis 62(6), 575–580. 10.1111/tbed.12399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen JL, Hansen FT, Sondergaard TE, Straek D, Lee TV, Wimmer R, Klitgaard LG, Purup S, Giese H, Frandsen RJN, 2012. Production of novel fusarielins by ectopic activation of the polyketide synthase 9 cluster in Fusarium graminearum. Environ. Microb 14(5), 1159–1170. 10.1111/j.1462-2920.2011.02696.x [DOI] [PubMed] [Google Scholar]
- Tchoukoua A, Hasegawa R, Hendracipta KA, Sato S, Koseki T, Shiono Y, 2018. Structure elucidation of new fusarielins from Fusarium sp. and their antimicrobial activity. Magn. Reson. Chem 56, 32–36. 10.1002/mrc.4662 [DOI] [PubMed] [Google Scholar]
- Vesonder RF, 1996. (all trans)-2,4,6,8,10,12-Tetradecahexene-1,14-dial, a New Pigment from Conidiobolus paulns. J. Nat. Prod 59, 441–442. 10.1021/np9602271 [DOI] [PubMed] [Google Scholar]
- Wang C, Wu X, Bai H, Zaman KA, Hou S, Saito J, Wongwiwatthananukit S, Kim KS, Cao S, 2020. Antibacterial and NF-κB Inhibitory Lumazine Peptides, Aspochalasin, γ-Butyrolactone Derivatives, and Cyclic Peptides from a Hawaiian Aspergillus flavipes. J. Nat. Prod 83, 2233–2240. 10.1021/acs.jnatprod.0c00344 [DOI] [PubMed] [Google Scholar]
- Wang F, Hu Z, Li C, Wu X, Cao S, 2019. Circumdatin M, a new benzodiazepine alkaloid with a unique pyrimidone-4-pyrone moiety from a Hawaiian marine fungus Aspergillus sp. FM242. Tetrahedron Lett. 60, 1724–1726. 10.1016/j.tetlet.2019.05.061 [DOI] [Google Scholar]
- Wang Q, Bao L, Yang X, Guo H, Yang R, Ren B, Zhang L, Dai H, Guo L, Liu H, 2012. Polyketides with antimicrobial activity from the solid culture of an endolichenic fungus Ulocladium sp. Fitoterapia. 83, 209–214. 10.1016/j.fitote.2011.10.013 [DOI] [PubMed] [Google Scholar]
- Wei J, Wu B 2020. Chemistry and bioactivities of secondary metabolites from the genus Fusarium. Fitoterapia. 146, 104638. 10.1016/j.fitote.2020.104638 [DOI] [PubMed] [Google Scholar]
- Wolinski K, Hinton JF, Pulay P, 1990. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc 112, 8251. 10.1021/ja00179a005 [DOI] [Google Scholar]
- Zaman KA, Park JH, Devine L, Hu Z, Wu X, Kim HS, Cao S, 2021. Secondary Metabolites from the Leather Coral-Derived Fungal Strain Xylaria sp. FM1005 and Their Glycoprotein IIb/IIIa Inhibitory Activity. J. Nat. Prod 84, 2, 466–473. 10.1021/acs.jnatprod.0c01330 [DOI] [PubMed] [Google Scholar]
- Zaman KA, Wu X, Hu Z, Cao S, 2020. Tryptoquivalines W and X, two new compounds from a Hawaiian fungal strain and their biological activities. Tetrahedron Lett. 61(14), e151730. 10.1016/j.tetlet.2020.151730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman KA, Wu X, Hu Z, Cao S, 2021. Antibacterial kaneoheoic acids AF from a Hawaiian fungus Fusarium sp. FM701. Phytochemistry 181, 112545. 10.1016/j.phytochem.2020.112545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman KA, Wu X, Hu Z, Hou S, Saito J, Kondratyuk TP, Pezzuto JM, Cao S, 2020. NF-κB Inhibitory and Antibacterial Helvolic and Fumagillin Derivatives from Aspergillus terreus. J. Nat. Prod 83, 730–737, 10.1021/acs.jnatprod.9b01190 [DOI] [PubMed] [Google Scholar]
- Zanardi MM, Marcarino MO, Sarotti AM, 2020. Redefining the impact of Boltzmann analysis in the stereochemical assignment of polar and flexible molecules by NMR calculations. Org. Lett 22, 52–56, 10.1021/acs.orglett.9b03866 [DOI] [PubMed] [Google Scholar]
- Zhou H, Yang X, Li F, Yi X, Yu L, Gao C, Huang R, 2017. A New Diketopiperazine of Nocardiopsis alba Isolated from Anthogorgia caerulea. Chem. Nat. Comp 53, 338–340, 10.1007/s10600-017-1983-6 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.