

ABSTRACT
We designed and developed a novel DNA topoisomerase I inhibitor MF-6, which was a more potent cytotoxin and a more potent inducer of immunogenic cell death compared with DXd. To utilize MF-6’s ability to induce antitumor immunity, a human epidermal growth factor receptor 2 (HER2)-targeted antibody–drug conjugate (ADC) trastuzumab-L6 that included a cleavable linker and MF-6 was developed. Different from traditional cytotoxic ADC, the antitumor activity of trastuzumab-L6 was assessed by inducing tumor cell immunogenic cell death, activating dendritic cells and cytotoxic CD8+ T cells to acquire durable adaptive immune memory. Tumor cells treated with trastuzumab-L6 were committed to immunogenic cell death, with upregulation of damage-associated molecular patterns and antigen presentation molecules. In a syngeneic tumor model with a mouse cell line that expressed human HER2, immunocompetent mice showed greater antitumor efficacy compared with nude mice. The trastuzumab-L6-cured immunocompetent mice acquired adaptive antitumor memory and rejected subsequent tumor cell challenge. The trastuzumab-L6 efficacy was abrogated when cytotoxic CD8+ T cells were depleted and enhanced when regulatory CD4+ T cells were depleted. The combination of trastuzumab-L6 with immune checkpoint inhibitors significantly increased antitumor efficacy. Enhanced T cell infiltration, dendritic cell activation, and decreased type M2 macrophages in tumor post trastuzumab-L6 administration confirmed the immune-activating responses. In conclusion, trastuzumab-L6 was considered to be an immunostimulatory agent, rather than a traditional cytotoxic ADC, and its antitumor efficacy was enhanced when combined with an anti-PD-L1 and anti-CTLA-4 antibody, which suggested a potential therapeutic strategy.
KEYWORDS: Adaptive antitumor immunity, antibody–drug conjugate, damage-associated molecular patterns, DNA topoisomerase I inhibitor, immunogenic cell death
Introduction
Substantial progress in the treatment of cancers has occurred due to the development of antibody-drug conjugates (ADC). Several ADCs are approved for treating hematological and solid tumors.1 Generally, ADCs are composed of an antibody that targets tumor cells and a cytotoxic compound; the two components are combined via a linker. Although the antibody itself is sometimes cytotoxic, e.g., trastuzumab, an ADC is primarily a chemotherapy drug that kills tumor cells via release of the cytotoxic component. Like most chemotherapeutics, the antitumor activities of ADC in vivo have two features. The antibody binds a target on the tumor cell surface, then endocytosis and toxin release by cathepsins digestion and killing tumor cell occur. Furthermore, the released toxin can also induce immunogenic cell death (ICD),2–5 followed by enhanced tumor antigen exposure, a boost in the release of tumor cell content, and immune cell infiltration.
At present, the toxins used in ADCs include mainly microtubule polymerization inhibitors, such as monomethyl auristatin E or F (MMAE, MMAF) and DM1, DNA alkylation agents such as pyrrolobenzodiazepines, and DNA topoisomerase I inhibitors, such as DXd and SN38.6 Most of these toxins induce tumor cell ICD, which could elicit or enhance an antitumor immune response.2,4,5 A common feature of tumor cell ICD is elevated expression or release of damage-associated molecular patterns (DAMPs), including calreticulin, heat-shock protein 70 and 90 (HSP70, HSP90), high-mobility group box 1 (HMGB1), and ATP. Released HMGB1 and ATP serve as chemoattractants to recruit the antigen-presenting cells (APCs), such as dendritic cells and macrophages.7–11 In addition, the translocation of calreticulin from endoplasmic reticulum to the plasma membrane, a feature of endoplasmic reticulum stress response, serves as “eat me” signaling to promote phagocytosis or efferocytosis by the APCs, which process and present the tumor antigens to T cells. Another endoplasmic reticulum stress response in tumor cells committed to ICD includes HSP70 and HSP90 translocation to membrane. In addition, tumor cells committed to ICD enhance antigen presentation via Major Histocompatibility Complex-class I (MHC-I) and MHC-II, which could be recognized by immune cells to enhance a tumor cell-specific response. The innate immune cells activated by ICD tumor cells secrete inflammatory cytokines and chemokines that promote dendritic cell maturation and T cell activation, which subsequently undergo proliferation and specifically attack tumor cells. Accordingly, the tumor cells committed to ICD function as an in situ vaccine by attracting APCs and inducing their activation and maturation to present tumor antigens to T cells. In addition to effector T cells, immunosuppressive T regulatory (Treg) cells are inhibited in the tumor microenvironment by ADCs treatment, which improves the antitumor activity of ADCs.2,12 In this way, ICD-inducing ADCs not only directly kill cancer cells, but they activate anticancer immunity that may enhance the therapeutic efficacy of immunotherapy.
Consideration of how to sufficiently make use of the immune-stimulation activity of ADCs in addition to cytotoxicity is critical when designing and developing an ADC. Systemic toxicity caused by drug release in circulation can limit the use and therapeutic window of ADCs and is another key factor to consider.13–16
In this study, we developed a camptothecin-derived compound, MF-6, as a drug in trastuzumab-L6 (TS-L6), coupled with a site-specific linker. The cytotoxicity and antitumor immunity of MF-6 and TS-L6 were investigated. We confirmed that MF-6 was 3–5 times stronger in DNA topoisomerase I inhibition and cytotoxicity compared with DXd. After the MF-6 was linked to HER2-targeted TS-L6 via a bridging and site-specific conjugating technology, its cytotoxicity was weakened due to a slow release rate, but the activity to induce tumor cell ICD was not affected. In an in vivo syngeneic tumor model with a mouse cell line that expressed human HER2 (CT26.WT-HER2), TS-L6 proved to be an immunomodulator, inducing dendritic cell activation and cytotoxic T cells infiltration. The mice cured by TS-L6 acquired adaptive antitumor memory. The tumor cells committed to ICD via MF-6 or TS-L6 treatment could be used as a tumor vaccine. When combined with an immune checkpoint inhibitor, such as anti- Programmed death-ligand 1 (PD-L1) or anti-cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) antibody or combined with an agent that depleted CD4+ Treg cells, the efficacy of TS-L6 was synergistically enhanced. We identified an immunostimulatory activity of TS-L6 distinct from the drug’s cytotoxic activity. Our findings suggest the possibility of developing ADCs with immunostimulatory activity in addition to the usual cytotoxicity effects.
Results
Trastuzumab-L6 structure and topoisomerase I inhibition activity of MF-6
To precisely deliver the highly potent toxin MF-6 into tumor cells, a HER2-targeted monoclonal antibody, trastuzumab (TS) was selected as vehicle to carry MF-6. Figure 1a shows the structure of trastuzumab-L6 (TS-L6), composed of the trastuzumab, a topoisomerase I inhibitor MF-6 as drug, and a maleimide valine-alanine dipeptide linker. The linker-drug was conjugated to two free sulfhydryl groups of the reduced antibody cysteine residues to form a bridge structure. After the ADC was internalized upon binding HER2 on the tumor surface and delivered to the lysosome, the dipeptide was digested by lysosomal enzymes, such as cathepsins B and L, which are highly expressed in tumor cells (Figure 1a). The released MF-6 drug inhibited DNA topoisomerase I, thereby specifically killing the tumor cells that express HER2. By using hydrophobic interaction chromatography, we determined that the drug–antibody ratio (DAR) of TS-L6 was approximately 4.0 with homogeneous drug distribution observed in the chromatography chart (Figure 1b).
Figure 1.
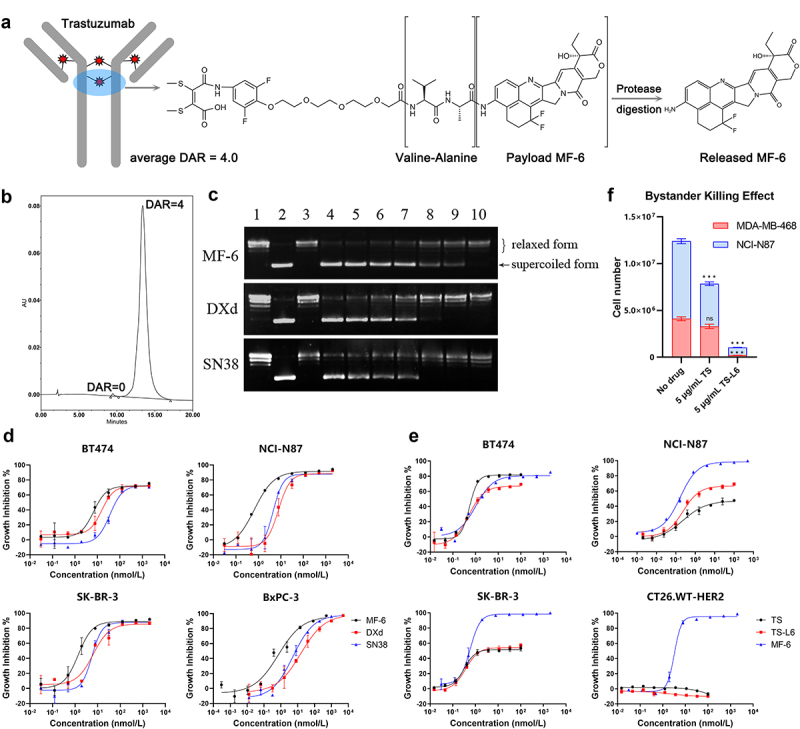
Structure and cytotoxicity of TS-L6.
a, Trastuzumab-L6 was composed of trastuzumab, a bridging valine-alanine linker between interchain disulfide bonds, and a topoisomerase I inhibitor MF-6, released by protease digestion.
b, The hydrophobic interaction chromatography chart of TS-L6 indicated a homogenous distribution with a drug–antibody ratio (DAR) of mostly 4.0.
c, The transformation of supercoiled plasmid into relaxed or nicked plasmid by DNA topoisomerase I in the presence of MF-6, DXd, and SN38 inhibitors was used to measure the activity of those three inhibitors. After the catalysis reaction was terminated, DNA agarose gel electrophoresis was used to separate the supercoiled plasmid with and nicked or relaxed plasmid; the supercoiled plasmid possessed higher migration rate due to tight conformation compared with nicked or relaxed plasmid. Lanes 1, 2, and 3 represent relaxed plasmid, supercoiled plasmid, and topoisomerase I only, respectively. Lanes 3 to 10 are a series of 2.5-fold dilutions of 100 μmol/L MF-6 (upper) and DXd (middle), and fivefold dilutions of 500 μmol/L SN38 (bottom).
d, Cytotoxicity of MF-6, DXd, and SN38 toward BT474, NCI-N87, SK-BR-3, and BxPC-3 cells was evaluated. Tumor cells were cultured with serially diluted drugs for 4 days. The cell growth inhibition ratio was calculated as the reduced cell viability signal in the drug-treated group compared with no drug treatment. The compound concentration and cell growth inhibition ratio were fitted with a four-parameter method to generate a dose–response curve.
e, Cytotoxicity of MF-6, TS, and TS-L6 toward HER2-expressing cells BT474, NCI-N87, SK-BR-3, and CT26.WT-HER2 was evaluated. Tumor cells were cultured with serially diluted drugs for 6 days. The cell growth inhibition ratio was calculated as the reduced cell viability signal in the drug-treated group compared with no drug treatment. The compound concentration and cell growth inhibition ratio were fitted with a four-parameter method to generate a dose–response curve.
f, Bystander killing effect of TS-L6 was evaluated using HER2-positive cell line NCI-N87 and HER2-negative cell line MDA-MB-468. When 5 μg/mL TS was added in the co-culture of the two cell lines, MDA-MB-468 was not affected (80.6 ± 9.7% of no drug group, P>0.05), but the growth of NCI-N87 was inhibited (54.9 ± 0.7% of no drug group, P<0.001). When 5 μg/mL TS-L6 was added, both NCI-N87 (10.1 ± 0.5% of no drug group, P<0.001) and MDA-MB-468 (5.0 ± 0.5% of no drug group, P<0.001) were inhibited.
As measured by a topoisomerase I-based DNA relaxation assay, MF-6 was a more potent inhibitor of topoisomerase I compared with DXd and SN38. The ratio of supercoiled plasmid increased as the concentration of compounds increased, revealing the inhibition of relaxed or nicked plasmid formation (Figure 1c), and the inhibition ratio was calculated according to the intensity of the supercoiled plasmid band in gel electrophoresis. After log transformation, the compound concentration within a certain range was fit linearly with the topoisomerase I activity inhibition ratio (Figure S1). The half maximal inhibitory concentration (IC50) of MF-6, DXd, and SN38 was calculated to be 7.7, 24.7, and 207.3 μmol/L according to the linear fitting equation.
Cytotoxicity assessment of small molecular compounds and ADC
The cytotoxicity of MF-6, DXd, and SN38 was assessed with four human cancer cells, BT474, SK-BR-3, NCI-N87, and BxPC-3. The IC50 of MF-6, DXd and SN38 toward those four cells was 6.84/15.62/36.26 nmol/L for BT474, 0.66/7.81/4.50 nmol/L for NCI-N87, 1.38/6.33/5.57 nmol/L for SK-BR-3 and 0.72/15.46/5.25 nmol/L for BxPC-3. MF-6 was more cytotoxic than DXd and SN38 toward all four cell lines (Figure 1d, Table 1). The cytotoxicity of MF-6, TS, and TS-L6 was assessed with three human cancer cell lines and a mouse cell line CT26.WT-HER2 (Figure 1e, Table 1), whose HER2 cell surface expression level was measured by flow cytometry with trastuzumab as detecting antibody (Figure S2). The IC50 of TS and TS-L6 toward those three cells was 0.54/0.50 nmol/L for BT474, 0.23/0.24 nmol/L for NCI-N87 and 0.33/0.37 nmol/L for SK-BR-3. Table 1 summarizes the cytotoxicity of compounds and ADC toward tumor cells. In three HER2-expressing human cell lines, trastuzumab inhibited cell growth because HER2 was a functional cell growth receptor that was blocked by trastuzumab.17 Because HER2 was not functional on CT26.WT-HER2 cell lines, TS-L6 and TS did not inhibit growth. TS-L6 exhibited similar cytotoxicity compared with TS, which indicated that the cytotoxicity of TS-L6 was due primarily to TS and not the drug.
Table 1.
Cytotoxicity (IC50) of compounds and ADC toward cancer cell lines.
| Cell line | IC50 (nmol/L)/Maximal inhibition ratio |
Cell line | HER2 level§ | IC50 (nmol/L)/Maximal inhibition ratio |
||||
|---|---|---|---|---|---|---|---|---|
| MF-6 | DXd | SN38 | MF-6 | TS | TS-L6 | |||
| BT474 | 6.84/72.1% | 15.62/71.7% | 36.26/72.4% | BT474 | 186.1 | 1.02#/80.7% | 0.54/81.6% | 0.50/67.0% |
| NCI-N87 | 0.66/91.7% | 7.81/88/3% | 4.50/87.8% | NCI-N87 | 524.5 | 0.17/98.5% | 0.23/46.9% | 0.24/66.6% |
| SK-BR-3 | 1.38/89.0% | 6.33/85.7% | 5.57/87.6% | SK-BR-3 | 171.6 | 0.59/98.4% | 0.33/51.7% | 0.37/54.3% |
| BxPC-3 | 0.72/96.5% | 15.46/98.9% | 5.25/98.3% | CT26.WT-HER2 | 60.6 | 3.25/95.5% | N/A* | N/A |
§ :The relative HER2 expression level was calculated as the ratio of mean fluorescence intensity of positive group with primary HER2 antibody added to the negative control group with fluorescence-labeled secondary antibody only.
# :The differences between two results of MF-6 on BT474, NCI-N87 and SK-BR-3 were due to the different cytotoxicity evaluation condition in two studies, such as cell number and incubation time.
* :No significant cell growth inhibition.
Bystander killing effect of TS-L6
In the bystander killing assay, to measure the ratio of HER2-positive NCI-N87 and HER2-negative MDA-MB-468 cells, we used a commercial human HER2 antibody (not trastuzumab) in flow cytometry to avoid competitive antigen binding with trastuzumab. When 5 μg/mL TS was added in the co-culture of the two cell lines, MDA-MB-468 was not affected (80.6 ± 9.7% of no drug group, P>0.05), but the growth of NCI-N87 was inhibited (54.9 ± 0.7% of no drug group, P<0.001) due to HER2 signal pathway inhibition. When 5 μg/mL TS-L6 was added, both NCI-N87 (10.1 ± 0.5% of no drug group, P<0.001) and MDA-MB-468 (5.0 ± 0.5% of no drug group, P<0.001) were inhibited (Figure 1f). Although MDA-MB-468 was HER2-negative, it was inhibited by the released MF-6 in HER-2 positive NCI-N87, which indicated the bystander killing effect of TS-L6.
In vitro drug release
The bridging VA (Valine-Alanine) linker in TS-L6 was digested by both cathepsin B and cathepsin L, which released 45% and 25% MF-6 at the optimal pH condition after 96 h. Conversely, two unusual cathepsins, C and S, did not digest TS-L6 at any pH (data not shown). The GGFG (Glycine-Glycine-Phenylalanine-Glycine) linker was more sensitive to cathepsin L, which released nearly all the DXd from its ADC after 72 h, whereas cathepsin B barely catalyzed DXd release. The traditional vcMMAE (Valine-Citrulline MMAE) linker-drug was more sensitive to both cathepsin B and L compared with VA and GGFG linker; after only 0.5 h, nearly all the MMAE was released (Figure 2a). In a cellular model, when NCI-N87 tumor cell was treated with TS-L6 or TS-GGFG-DXd, ADCs were internalized and drugs were released in lysosome. The released MF-6 in tumor cells was lower compared with released DXd at all the times. Seventy-two hours after incubation, the released MF-6 and DXd was 13.7 ± 0.5 and 57.5 ± 5.6 fmol/million cells (P<0.001). Moreover, the ratio of released MF-6 to total MF-6 carried by internalized TS-L6 (1.04 ± 0.04%, 72 hours after incubation) was also lower than DXd (2.11 ± 0.20%, P<0.001) (Figure 2b).
Figure 2.
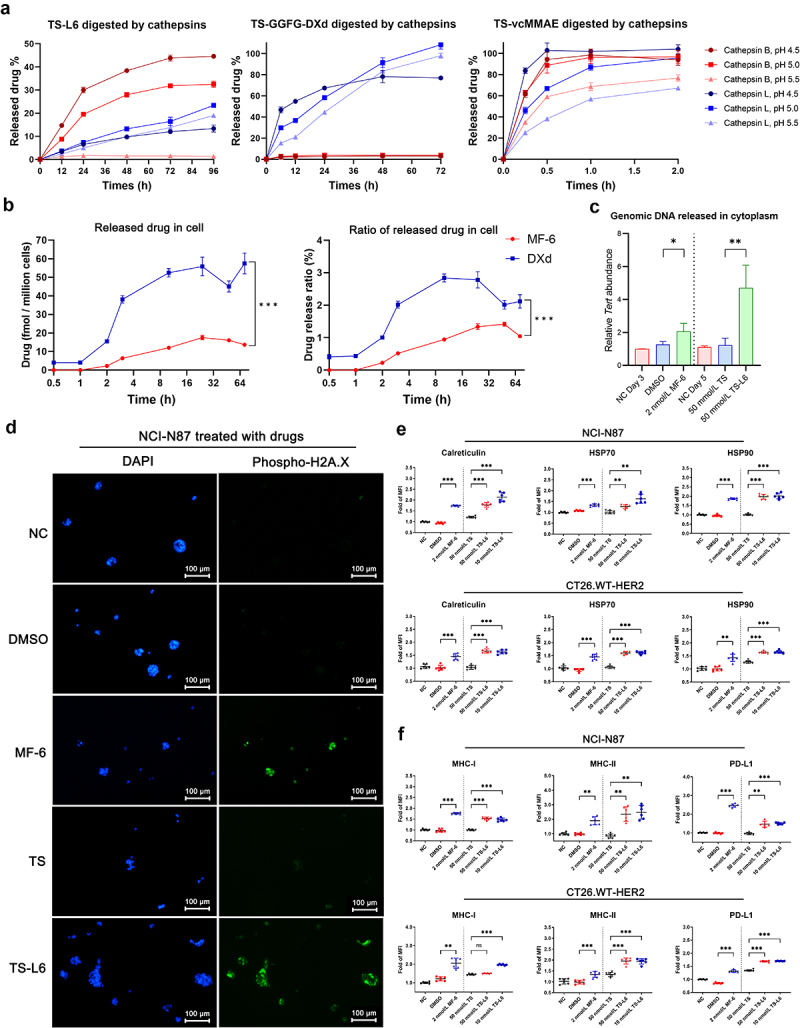
Genomic DNA damage and DAMPs upregulation in tumor cells with drug treatment.
a, The drug release rate of TS-L6, TS-GGFG-DXd, and TS-vcMMAE was evaluated using cathepsin B and L at pH 4.5, 5.0, and 5.5. The cathepsin B and cathepsin L released 45% and 25% MF-6 from TS-L6 at the optimal pH condition after 96 h. The GGFG linker was more sensitive to cathepsin L, which released approximately all the DXd after 72 h; cathepsin B barely catalyzed DXd release. The traditional VC linker was more sensitive to both cathepsin B and L compared with VA and GGFG linker; after only 0.5 h, approximately all the MMAE was released. The release rates were TS-L6<TS-GGFG-DXd<TS-vcMMAE. Three independent tests were performed, and the data are shown as mean and SD.
b, The drug release of TS-L6 and TS-GGFG-DXd in tumor cells was evaluated on NCI-N87. After incubation for different times, the released drugs in cells were isolated for quantitative analysis. The released MF-6 in tumor cells was lower compared with released DXd at all the times (left), and the ratio of released MF-6 to total MF-6 carried by internalized TS-L6 was also lower compared with DXd (right). Three independent tests were performed, and the data are shown as mean and SD.
c, Genomic DNA of NCI-N87, indicated by the Tert gene, was released into the cytoplasm after MF-6 and TS-L6 treatment. When treated with 2 nmol/L MF-6 for 3 days, the amount of genomic DNA of NCI-N87 was significantly increased compared with DMSO (2.07 ± 0.48 vs. 1.27 ± 0.18, P<0.05). Similarly, when treated with 50 nmol/L TS-L6 for 5 days, the amount of genomic DNA was significantly increased compared with 50 nmol/L TS (4.71 ± 1.38 vs. 1.24 ± 0.42, P<0.01). Three independent tests were performed, and the data are shown as mean and SD.
d, Phosphorylated H2A.X (γ-H2A.X) level of NCI-N87 treated with MF-6 and TS-L6, which was a biomarker of genomic DNA damage, was assessed by immunofluorescence. The γ-H2A.X (green, stained with phosphorylated H2A.X antibody) was located in the nucleus (blue, stained with DAPI) and its nuclear abundance increased after MF-6 and TS-L6 treatment, thereby indicating genomic DNA damage.
e, When NCI-N87 and CT26.WT-HER2 were treated with 2 nmol/L MF-6 for 3 days or 50 nmol/L TS-L6 for 5 days, the cell surface DAMPs calreticulin, HSP70, and HSP90 significantly increased, compared with DMSO or 50 nmol/L TS treatment. For NCI-N87 cells, MF-6 treatment significantly increased calreticulin (1.74 ± 0.03 vs. 0.94 ± 0.03 folds, P<0.001), HSP70 (1.34 ± 0.06 vs. 1.08 ± 0.02 folds, P<0.001), and HSP90 (1.86 ± 0.05 vs. 0.96 ± 0.05 folds, P<0.001) on cell surface compared with DMSO. Similarly, compared with the 50 nmol/L TS treatment, 50 nmol/L TS-L6 treatment led to increase in calreticulin (1.78 ± 0.11 vs. 1.21 ± 0.04 folds, P<0.001), HSP70 (1.27 ± 0.09 vs. 1.01 ± 0.08 folds, P<0.01), and HSP90 (1.97 ± 0.13 vs. 1.01 ± 0.05 folds, P<0.001). Mouse cell line CT26.WT-HER2 also exhibited upregulated DAMPs after 2 nmol/L MF-6 or 50 nmol/L TS-L6 treatment. Six independent tests were performed, and the data are shown as mean and SD.
f, When NCI-N87 and CT26.WT-HER2 were treated with 2 nmol/L MF-6 for 3 days or 50 nmol/L TS-L6 for 5 days, the MHC-I, MHC-II, and PD-L1 on the tumor cell surface significantly increased, compared with DMSO or 50 nmol/L TS treatment. For NCI-N87 cells, 2 nmol/L MF-6 treatment significantly increased MHC-I (1.77 ± 0.04 vs. 0.97 ± 0.07 folds, P<0.001), MHC-II (1.91 ± 0.28 vs. 0.98 ± 0.07 folds, P<0.01) and PD-L1 (2.46 ± 0.09 vs. 0.99 ± 0.03 folds, P<0.001) on cell surface compared with DMSO. Similarly, compared with the 50 nmol/L TS treatment, 50 nmol/L TS-L6 treatment led to increase in MHC-I (1.53 ± 0.07 vs. 1.00 ± 0.03 folds, P<0.001), MHC-II (2.35 ± 0.50 vs. 0.85 ± 0.14 folds, P<0.01) and PD-L1 (1.47 ± 0.16 vs. 0.96 ± 0.05 folds, P<0.001). Mouse tumor cell CT26.WT-HRR2 also exhibited upregulated MHC-I, MHC-II, and PD-L1 after MF-6 or TS-L6 treatment. Six independent tests were performed, and the data are shown as mean and SD.
ns, *, ** and *** represent no significance, P<0.05, 0.01, and 0.001, respectively.
MF-6 and TS-L6 induced cancer cell genomic DNA damage
After NCI-N87 cells were treated with 2 nmol/L MF-6 for 3 days or treated with 50 nmol/L TS or 50 nmol/L TS-L6 for 5 days, the genomic DNA released into the cytoplasm was evaluated by real-time PCR measurement of the amount of genomic Tert DNA sequence relative to 16S rRNA sequence. The MF-6 and TS-L6 treatments significantly increased genomic DNA released into the cytoplasm, compared with dimethyl sulfoxide (DMSO) (2.07 ± 0.48 vs. 1.27 ± 0.18, P<0.05) or TS (4.71 ± 1.38 vs. 1.24 ± 0.42, P<0.01) treatment, respectively (Figure 2c). In addition, the immunofluorescence of phosphorylated histone H2A.X, a marker of genomic DNA damage,18 showed that MF-6 or TS-L6 treatment significantly raised the phosphorylated histone H2A.X level compared with DMSO or TS treatment of NCI-N87 (Figure 2d), BT474 and CT26.WT-HER2 (Figure S3).
Upregulated damage-associated molecular patterns and antigen presentation after drug treatment
We used HER2-expressing cell lines, human tumor cell NCI-N87, BT474 and mouse tumor cell CT26.WT-HER2 to evaluate the ability of MF-6 and TS-L6 to induce DAMPs, including calreticulin, HSP70, and HSP90. Compared with DMSO, 2 nmol/L MF-6 treatment significantly increased calreticulin (1.74 ± 0.03 vs. 0.94 ± 0.03 folds, P<0.001), HSP70 (1.34 ± 0.06 vs. 1.08 ± 0.02 folds, P<0.001), and HSP90 (1.86 ± 0.05 vs. 0.96 ± 0.05 folds, P<0.001) on tumor surface. Similarly, compared with the 50 nmol/L TS treatment, 50 nmol/L TS-L6 treatment led to an increase in calreticulin (1.78 ± 0.11 vs. 1.21 ± 0.04 folds, P<0.001), HSP70 (1.27 ± 0.09 vs. 1.01 ± 0.08 folds, P<0.01), and HSP90 (1.97 ± 0.13 vs. 1.01 ± 0.05 folds, P<0.001). Another human tumor cell BT474 (Figure S4) and mouse tumor cell CT26.WT-HRR2 (Figure 2e) also exhibited upregulated DAMPs after MF-6 or TS-L6 treatment.
Because tumor antigen expression on the tumor cell surface is essential for APCs recognition, we investigated the influence of MF-6 and TS-L6 on the expression of components of the tumor antigen presentation machinery. Compared with DMSO, 2 nmol/L MF-6 treatment significantly increased MHC-I (1.77 ± 0.04 vs. 0.97 ± 0.07 fold, P<0.001) and MHC-II (1.91 ± 0.28 vs. 0.98 ± 0.07 fold, P<0.01) on NCI-N87 cell surface. Compared with the 50 nmol/L TS treatment, 50 nmol/L TS-L6 treatment led to an increase in MHC-I (1.53 ± 0.07 vs. 1.00 ± 0.03 fold, P<0.001) and MHC-II (2.35 ± 0.50 vs. 0.85 ± 0.14 fold, P<0.01) (Figure 2f). Similarly, another human tumor cell BT474 (Figure S4) and mouse tumor cell CT26.WT-HER2 (Figure 2f) also exhibited upregulated MHC-I and II after MF-6 or TS-L6 treatment. Specifically, the expression of genes encoding β₂ microglobulin (B2m), an essential component of MHC-I, were upregulated after drug treatment of NCI-N87. As well, the genes that direct peptide cleavage (Erap1), peptide transport (Tap1 and Tap2), and transporter-MHC interactions (Tapbp)18 were upregulated (Figure S5) as measured by RT-PCR. The primers used in real time-PCR to detect those genes were listed in Table S1. Interestingly, PD-L1 on NCI-N87, BT474 and CT26.WT-HER2 cell surface was also increased after MF-6 (for NCI-N87, 2.46 ± 0.09 vs. 0.99 ± 0.03-fold, P<0.001 compared with DMSO) and TS-L6 treatment (for NCI-N87, 1.47 ± 0.16 vs. 0.96 ± 0.05-fold, P<0.001 compared with TS) (Figure 2f, Figure S4), which indicated potential immune escape. MF-6 upregulated DAMPs and enhanced antigen presentation in NCI-N87 at a lower concentration compared with DXd (Figure 3b). Generally, 1–2 nmol/L MF-6 upregulated DAMPs and MHC-I, MHC-II, and PD-L1 on the cell surface, whereas 4 nmol/L DXd was insufficient to produce the same effect.
Figure 3.
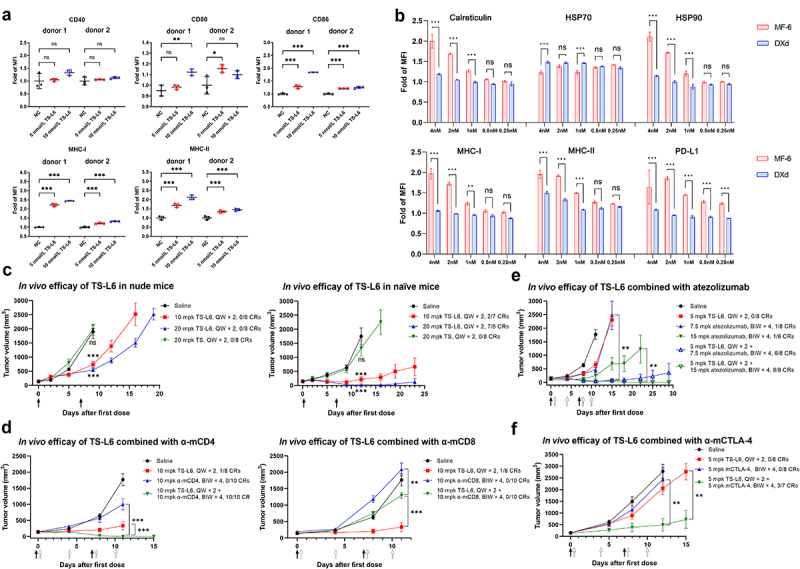
Dendritic cell activation and the T cells’ contribution to TS-L6 efficacy.
a, Dendritic cells from two healthy donors were activated by NCI-N87, which had been pre-treated with TS-L6. NCI-N87 was pre-treated with 5 nmol/L or 10 nmol/L TS-L6 for 2 days, and then co-cultured with dendritic cells for another 2 days to activate dendritic cells. The dendritic cell activating markers CD80, CD86, MHC-I, and MHC-II increased after dendritic cells were incubated with tumor cells committed to ICD induced by TS-L6. Three independent tests were performed, and the data are shown as mean and SD.
b, MF-6 exhibited greater potency in upregulating DAMPs, MHC-I, MHC-II, and PD-L1 on the tumor cell surface compared with DXd. After being treated with serial dilutions of MF-6 or DXd, the molecules, including calreticulin, HSP70, HSP90, MHC-I, MHC-II and PD-L1 on NCI-N87 surface increased when MF-6 reached 1-2 nmol/L, but those molecules had not increased when DXd reached up to 4 nmol/L. Three independent tests were performed, and the data are shown as mean and SD.
c, In vivo efficacy of TS-L6 was assessed with nude mice (left) and naïve BALB/c mice (right) bearing CT26.WT-HER2 tumors. In both nude and naïve BALB/c mice, administration of 20 mg/kg (mpk) TS once weekly × 2 (QW × 2) did not show any efficacy. Both 10 mg/kg and 20 mg/kg TS-L6 administrated once weekly × 2 inhibited tumor growth in nude and naïve mice, but TS-L6 was more efficacious in naïve mice. In nude mice, 10 mg/kg (TGI = 65.9 ± 17.0%, P<0.001) and 20 mg/kg TS-L6 (TGI = 76.8 ± 10.9%, P<0.001) showed tumor inhibition but no CRs were reached. In naïve mice, the tumor growth in the 10 mg/kg (TGI = 95.8 ± 14.4%, P<0.001) and 20 mg/kg TS-L6 (TGI = 107.9 ± 4.2%, P<0.001) groups was significantly inhibited with 29% (2/7) and 75% (6/8) CRs, respectively. The tumor volumes of drug-treated groups were statistically compared with the volumes of the saline group on the day when the saline group mice were euthanized due to large tumors. Black arrows represent drugs administrated. The tumor volume data are shown as mean and SEM.
d, TS-L6in vivo efficacy increased when combined with anti-mCD4 (left) and decreased with mCD8 (right) antibody. When cytotoxic CD8+ T cells were depleted using an anti-mCD8 monoclonal antibody, the efficacy of the TS-L6 was abrogated. Compared with TS-L6 treatment, the combination of TS-L6 and anti-mCD8 antibody significantly reduced antitumor efficacy (TGI = 88.5 ± 24.9% vs 28.7 ± 17.4%, P<0.001). When we ablated CD4+ T cells using a depleting anti-mCD4 monoclonal antibody, the TGI was 47.6% at day 11 in anti-mCD4 antibody group, but all eight mice reached CRs in the TS-L6 combined with anti-mCD4 antibody group. The tumor volumes of the combination drug-treated groups were statistically compared with the volumes of the single drug-treated group at the time when the single drug-treated mice were euthanized due to large tumors. Black and white arrows represent ADC and anti-mCD4/CD8 antibody administrated, respectively. The tumor volume data are shown as mean and SEM.
e,fIn vivo efficacy of TS-L6 combined with atezolizumab (E) and anti-mCTLA-4 antibody (F) indicated an increased combination efficacy with immune checkpoint inhibitors. Even at a relatively low dose of 5 mg/kg for TS-L6, both 7.5 mg/kg and 15 mg/kg atezolizumab significantly enhanced ADC efficacy to 75% (6/8) and 100% (8/8) CRs 29 days post-administration, respectively. When we combined TS-L6 with anti-mCTLA-4 antibody, the efficacy of TS-L6 was enhanced with 84.3 ± 29.6% TGI (tumor volume P<0.01 compared with TS-L6 treatment TGI 27.5 ± 28.8%) 12 days post-administration and 43% (3/7) CRs after 15 days, whereas the anti-mCTLA-4 antibody alone showed limited efficacy with only 13.1 ± 53.7% TGI (tumor volume P>0.05 compared with saline treatment) after 12 days. Black and white arrows represent ADC and immune checkpoint inhibitors administrated, respectively. The tumor volume data are shown as mean and SEM.
ns, *, ** and *** represent no significance, P<0.05, 0.01, and 0.001, respectively.
Activation of dendritic cells co-cultured with tumor cells committed to immunogenic cell death
After DAMPs on the surface of tumor cell NCI-N87 were induced by TS-L6 or TS-GGFG-DXd treatment, human dendritic cells from two healthy donors were added and incubated for another 2 days. The dendritic cell activation markers CD40, CD86, CD80, MHC-I, and MHC-II were measured by flow cytometry. In dendritic cells from the healthy donors, all the dendritic cell-activating markers except CD40 were significantly upregulated after co-culturing with tumor cells (Figure 3a).
T cells contributed to the antitumor activity of TS-L6
To assess the importance of an intact immune system for TS-L6 efficacy, we examined immunodeficient nude mice that lack T cells and immunocompetent naïve BALB/c mice. In nude mice, after two doses, 20 mg/kg TS did not show any efficacy (tumor growth inhibition, TGI = −4.0 ± 31.8%, tumor volume P>0.05), whereas 10 mg/kg (TGI = 65.9 ± 17.0%, tumor volume P<0.001) and 20 mg/kg TS-L6 (TGI = 76.8 ± 10.9%, tumor volume P<0.001) showed tumor inhibition but no complete response (CRs, defined as tumor disappeared) were reached. In naïve mice, 20 mg/kg TS showed limited efficacy (TGI = 26.9 ± 51.7%, tumor volume P>0.05), but the tumor growth in the 10 mg/kg (TGI = 95.8 ± 14.4%, tumor volume P<0.001) and 20 mg/kg TS-L6 (TGI = 107.9 ± 4.2%, tumor volume P<0.001) groups was significantly inhibited with 29% (2/7) and 75% (6/8) CRs, respectively (Figure 3c).
To demonstrate that T cells contributed to antitumor activity, we used an anti-mCD8 monoclonal antibody to deplete cytotoxic CD8+ T cells in immunocompetent mice. The depletion of CD8+ T cells abrogated the efficacy of the TS-L6 (Figure 3d); compared with TS-L6 treatment, the combination of TS-L6 and anti-mCD8 antibody significantly reduced antitumor efficacy (TGI = 88.5 ± 24.9% vs 28.7 ± 17.4%, tumor volume P<0.001), which demonstrated that CD8+ T cells were required for full antitumor activity of the TS-L6. Interestingly, when we ablated CD4+ T cells using a depleting anti-mCD4 monoclonal antibody, the TGI was 47.6% at day 11 in anti-mCD4 antibody group, but all eight mice reached CRs in the TS-L6 combined with anti-mCD4 antibody group (Figure 3d). The surprising efficacy of combining TS-L6 with anti-mCD4 antibody might have been due to the depletion of Treg cells, which are critical immune-suppressing cells. Thus, mouse CD25 (also named IL-2 receptor alpha) highly expressed on Treg cells was selected as a Treg depletion target.19 TS-L6 at 5 mg/kg once weekly (QW) for × 2 combined with 10 mg/kg anti-mCD25 monoclonal antibody twice weekly (BIW) × 4 were administrated to CT26.WT-HER2 tumor-bearing mice. However, no significant tumor growth inhibition was observed in the combination group compared with the single drug group (Figure S6).
Enhanced antitumor activity by TS-L6 combined with immuno-oncology drugs
The foregoing finding that T cells contributed to ADC activity led to the hypothesis that the antitumor activity of an ADC could be enhanced by T cell-modulating immunotherapy. To address this hypothesis, we evaluated the efficacy of TS-L6 combined with a mouse PD-L1 blocking agent, atezolizumab, which has an affinity for human PD-L1 similar to mouse, and a mouse CTLA-4 blocking agent, anti-mCTLA-4 monoclonal antibody. Even at a relatively low dose of 5 mg/kg for TS-L6, both 7.5 mg/kg and 15 mg/kg atezolizumab significantly enhanced ADC efficacy to 75% (6/8) and 100% (8/8) CRs 29 days post-administration, respectively (Figure 3e). When we combined TS-L6 with anti-mCTLA-4 antibody, the efficacy of TS-L6 was enhanced with 84.3 ± 29.6% TGI (tumor volume P<0.01 compared with TS-L6 treatment TGI 27.5 ± 28.8%) 12 days post-administration and 43% (3/7) CRs after 15 days, whereas the anti-mCTLA-4 antibody alone showed limited efficacy with only 13.1 ± 53.7% TGI (tumor volume P>0.05 compared with saline treatment) after 12 days (Figure 3f).
Immuno-memory of cured mice
On the basis of these results, we performed a tumor re-challenge study with completely cured mice to investigate whether treatment of tumor-bearing mice with TS-L6 resulted in immune-memory because adaptive immunity had an important function in ADC efficacy (Figure 4a). CT26.WT-HER2 tumor-bearing mice (n = 115) were administered two doses of 20 mg/kg TS-L6 with one-week interval when average initial tumor volume reached 100–150 mm3. About 2 months after the first dose, 68% (78/115) of the mice reached CRs, whereupon we randomized the cured mice to conduct a re-challenge test; naïve mice of the same age were used as control. When mice were re-challenged with CT26.WT-HER2 tumor cells, after 21 days the cured mice reached 100% (8/8) tumor rejection compared with 0/8 tumor rejection by the control mice (Figure 4c). When re-challenged with parental CT26.WT tumor cells, the cured mice also showed significant tumor growth inhibition and 38% (3/8) tumor rejection was reached 21 days after re-challenge (Figure 4c). However, when re-challenged with EMT6, an irrelevant mouse breast cancer cell, both the cured mice and naïve mice did not show any tumor rejection or growth inhibition (Figure 4c).
Figure 4.
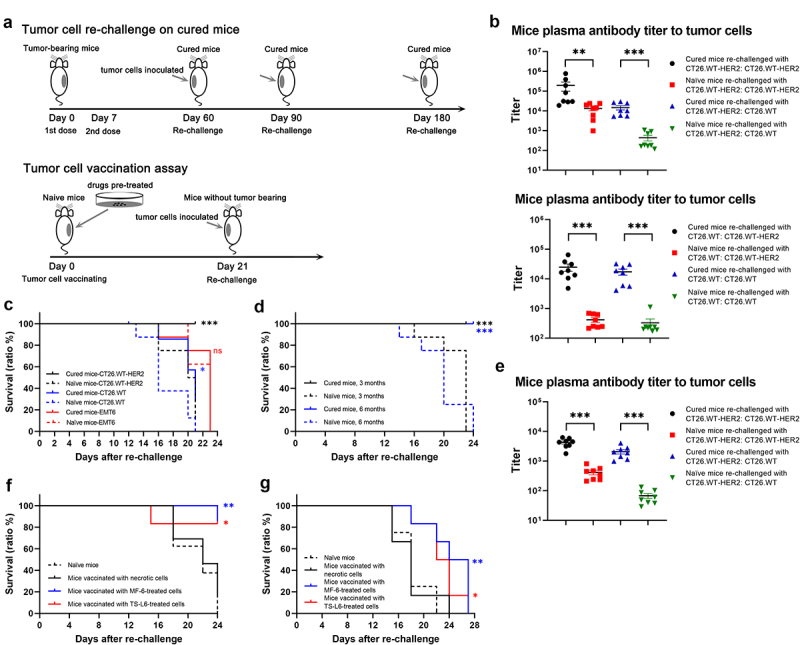
Tumor cells re-challenge of cured mice and tumor cell vaccination.
a, Schematic of tumor re-challenge (upper) and tumor cell vaccination assay (bottom). In the tumor re-challenge study, CT26.WT-HER2-tumor bearing mice were completely cured with TS-L6 (20 mg/kg, once weekly × 2), followed by tumor re-challenge at days 60, 90 and 180 post-initial dosing. In the tumor vaccination assay, mice were vaccinated with 3 million tumor cells pre-treated with MF-6 or TS-L6, followed by tumor re-challengeat day 21.
b, When the tumor re-challenge at 2 months post-initial dosing was completed, the plasma antibody titers to CT26.WT-HER2 and CT26.WT cells were measured by a cell-based ELISA. The cured mice re-challenged with either CT26.WT-HER2 (upper) or CT26.WT (bottom) showed higher antibody titers to CT26.WT-HER2 or CT26.WT cells compared with naïve mice. Eight mice in each group were used, and the data are shown as mean and SD. The titer data were statistically analyzed after logarithm transformation.
c, Tumor re-challenge was conducted with mice cured by single-drug TS-6 (20 mg/kg, once weekly × 2) 2 months after initial dosing. All cured mice (8/8) rejected CT26.WT-HER2 tumor cells (black lines), whereas 3 of 8 of the cured mice rejected parental CT26.WT tumor cell re-challenge (blue lines). Neither the cured mice nor the naïve mice showed any tumor rejection when challenged with EMT6 (red lines). A log-rank test was used to compare the probability of survival, and the same method was for survival curve comparison.
d, All mice (8/8) cured by single-drug TS-L6 rejected CT26.WT-HER2 tumor cell challenge for months (black line) and 6 months (blue line) after initial dosing.
e, When the tumor re-challenged at 6 months post-initial dosing was completed, the plasma antibody titers to CT26.WT-HER2 and CT26.WT were measured with a cell-based ELISA. The cured mice re-challenged with CT26.WT-HER2 showed higher antibody titer to CT26.WT-HER2 or CT26.WT cells compared with naïve mice. Eight mice/group were used, and the data are shown as mean and SD. The data were statistically analyzed after logarithm transformation.
f, g, Mice vaccinated with CT26.WT-HER2 cells pre-treated with 10 nmol/L MF-6 for 24 h (blue line) or 100 nmol/L TS-L6 for 72 h (red line) exhibited a higher tumor rejection ratio and higher survival probability than mice vaccinated with necrotic cells (black line) when re-challenged with CT26.WT-HER2 (f) or parental CT26.WT (g).
ns, *, ** and *** represented no significance, P<0.05, 0.01, and 0.001, respectively.
When the re-challenge tests were completed, we used a cell-based ELISA assay to measure the antibody titers to CT26.WT-HER2 and CT26.WT in mouse plasma. The plasma from cured mice after CT26.WT-HER2 cells re-challenging exhibited higher antibody titers to both CT26.WT-HER2 cells (titer range 18,921 to 781,250 with median 57,381 vs. range 981 to 23,948 with median 14,290, P<0.01) and CT26.WT cells (titer range 5,258 to 29,356 with median 10,378 vs. range 118 to 1,074 with median 184, P<0.001) compared with naïve control mice (Figure 4b, Table S2). Similarly, the plasma from cured mice after CT26.WT cells re-challenging exhibited higher antibody titers to both CT26.WT-HER2 cells (titer range 4,805 to 64,812 with median 17,774 vs. range 214 to 690 with median 328, P<0.001) and CT26.WT cells (titer range 4,095 to 32,332 with median 17,539 vs. range 170 to 1,116 with median 230, P<0.001) compared with naïve control mice (Figure 4b, Table S2). The CT26.WT-HER2 tumor cell re-challenge was also conducted on mice cured by combination of TS-L6 with either atezolizumab or anti-mCD4 antibody. All the mice cured by 5 mg/kg TS-L6 combined with 7.5 mg/kg atezolizumab, 15 mg/kg atezolizumab, or 10 mg/kg TS-L6 combined with 10 mg/kg anti-mCD4 antibody reached 100% tumor rejection (6/6, 8/8, and 8/8) in the re-challenge test (Figure S7).
Durability of immuno-memory response in cured mice
To investigate the durability of immune-memory response, we re-challenged the cured mice with CT26.WT-HER2 tumor cells 3 and 6 months after the first ADC dosing. All the cured mice (8/8) rejected tumor after re-challenge, at both 3 and 6 months, which indicated a durable immune-memory (Figure 4d). When the re-challenge test was completed with cured mice 6 months after the first dosing, we measured the antibody titers in plasma. The plasma from cured mice exhibited higher titers to both CT26.WT-HER2 cells (titer range 1,790 to 6,250 with median 4,424 vs. range 209 to 809 with median 395, P<0.001) and CT26.WT cells (titer range 982 to 4,018 with median 1,950 vs. range 29 to 132 with median 55, P<0.001) compared with naïve control mice (Figure 4e, Table S2).
MF-6 and TS-L6-treated cells vaccinated against tumor challenge
The tumor cell re-challenge test results hinted to us that tumor cells pre-treated with MF-6 or TS-L6 might be used as a tumor vaccine (Figure 4a). After CT26.WT-HER2 cells were treated with either 10 nmol/L MF-6 for 24 h or 100 nmol/L TS-L6 or 72 h, the cells were committed to ICD, but still alive. CT26.WT-HER2 cells treated by three freeze-thaw cycles were used as a negative necrotic cell control. We inoculated three million treated cells into the right flank of immunocompetent BALB/c mice. Three weeks later, the mice without tumors were inoculated again with three million CT26.WT-HER2 or CT26.WT cells into the left flank, and naïve BALB/c mice of the same age were used as control. The mice vaccinated with MF-6-treated TS-L6-treated cells rejected 86% (6/7) and 83% (5/6) CT26.WT-HER2 cells, whereas the naïve mice and mice vaccinated with necrotic cells rejected 0% (0/8) and 15% (2/13) cells (Figure 4f). The MF-6 and TS-L6-treated cells also provided vaccination against the parental CT26.WT cells, although vaccination against CT26.WT was weaker compared with CT26.WT-HER2 (Figure 4g). Compared with mice vaccinated with necrotic cells, the mice vaccinated with cells pre-treated with 10 nmol/L MF-6 or 100 nmol/L TS-L6 had significantly prolonged survival when re-challenged with tumor cells.
Increased immune cell infiltration in tumor after TS-L6 treatment
To measure immune cell infiltration and activation in tumor tissue after drug treatment, tumor tissues were isolated from mice 6 days after the first dose of 20 mg/kg TS or TS-L6. The tissues were immediately fixed in neutral formalin overnight and embedded in paraffin, then cut into 3–5 μm slides for immunohistochemistry (IHC). As an activation marker of dendritic cells, the expression level of CD86 in the TS-L6 treatment group was greater than the TS group, which indicated activation of dendritic cells in the tumor (Figure 5a). Compared with the blank control or TS group, TS-L6 treatment increased CD3+ T cell and cytotoxic CD8+ T cell tumor infiltration six days post-dose (Figure 5b). In addition, expression of both CD163 and CD206, markers of type M2 macrophage,20 was decreased in the TS-L6 treatment group, which indicated transformation of immuno-suppressive type M2 macrophages into immuno-active type M1 macrophages (Figure 5c). To quantify the results of IHC, we randomly selected five fields in the microscopy and counted the positive cells in total 200 cells. The quantification and statistics of the IHC summarized in Figure 5d show that TS-L6 treatment significantly increased CD86+, CD3+ and CD8+ cells and decreased CD163+ and CD206+ cells in tumors, compared with TS treatment.
Figure 5.
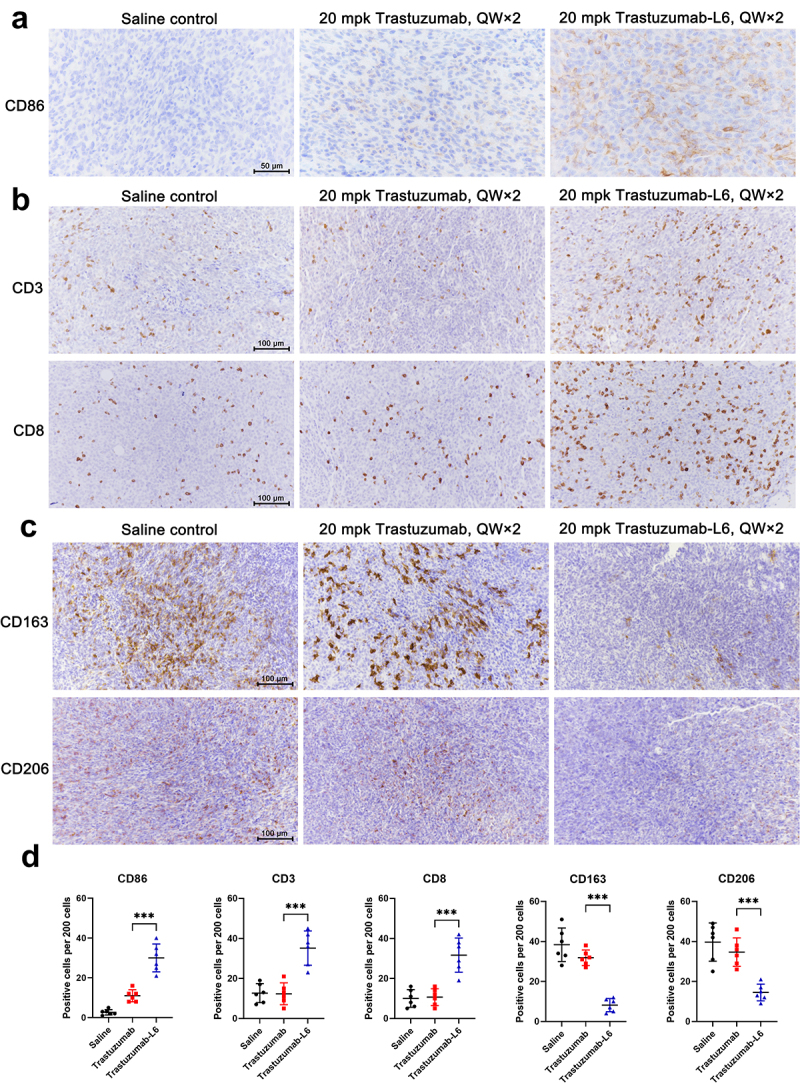
Immune cell infiltration in tumor tissue after TS-L6 treatment.
Six days after 20 mg/kg TS-L6 administration, tumor tissues were isolated, fixed, and embedded for IHC. a, A tumor from the TS-L6 treatment group exhibited more CD86+ cells compared with saline control and the TS treatment group. b, The CD3+ T cells (upper) and CD8+ T cells (bottom) infiltration was enhanced after TS-L6 treatment. c, Innate immune cells, mainly macrophages, showed decreased CD163 and (upper) CD206 (bottom) expression in TS-L6 treated tumors. D, The amounts of CD86, CD3, CD8, CD163, and CD206 positive cells were collected and the statistical differences between the TS and TS-L6-treated groups were analyzed using Student’s t test. Five independent tests were performed, and the data are shown as mean and SD.
ns, *, ** and *** represented no significance, P<0.05, 0.01, and 0.001, respectively.
Discussion
Most antitumor therapies, especially radiotherapy and chemotherapy, are considered to be directly tumoricidal, killing tumor cells or inducing cell cycle arrest. Because chemotherapy targets not only tumor cells but also rapidly proliferating host immune cells, investigators have assumed that cytotoxic chemotherapeutics induce immunosuppressive rather than immunostimulatory effects. As a result, the importance of host immune responses to cytotoxic drugs has been neglected.21–25 Similarly, the importance of an intact host immune system has not been sufficiently considered in developing ADC products. For example, ADC preclinical in vivo efficacy studies are usually conducted on immunodeficient mice, such as athymic nude mice to investigate the direct killing effect of the ADC on human tumor cells or patient-derived tumors. In such experiments, the importance of the immune system, especially T cells and B cells, has been ignored because their functions or numbers are impaired in the models. Several research groups challenged this conventional notion, and have found that dying tumor cells, whether induced by ADCs or chemotherapeutics, can be immunogenic, a phenomenon termed immunogenic cell death. The DAMPs released by tumor cells committed to ICD can be acquired by APCs, especially dendritic cells and macrophages, and finally active cytotoxicity T cells, connecting innate and adaptive immunity.
In this study, we designed a more potent topoisomerase I inhibitor MF-6 than DXd in cytotoxicity and inducing ICD, and developed a HER2-targeted ADC, TS-L6, that contained MF-6, and a cleavable Val-Ala bridging linker which released toxin more slowly than the Gly-Gly-Phe-Gly linker coupled to DXd. Although TS-L6 exhibited limited or even no cytotoxicity toward tumor cells, it did lead to damage of genomic DNA, and induction of tumor cell ICD. The tumor cells committed to ICD by TS-L6 could subsequently activate dendritic cells in vitro, which indicated activation of innate immunity.
On the basis of the foregoing evidence, we used a syngeneic tumor cell model with mouse cell line CT26.WT-HER2 to investigate the in vivo efficacy and antitumor mechanism of TS-L6. Compared with immunodeficient nude mice, immunocompetent mice showed better responses to TS-L6 signal drug treatment. Considering that T cell function was absent in the nude mice, we assumed that T cells were essential for TS-L6 efficacy. This assumption was verified by experiments that showed depletion of cytotoxic CD8+ T cells abrogated the efficacy of the TS-L6. Surprisingly, depletion of CD4+ T cells enhanced TS-L6 efficacy. This surprising efficacy of combining TS-L6 with an anti-mCD4 antibody might have been due to the elimination of Treg cells, which are critical immune-suppressing cells. However, when we used anti-mCD25 to deplete mouse CD25, another Treg-specific molecule, we did not observe any significant enhanced efficacy. Couper et al. reported that CD25 was expressed on cytotoxic CD8+ T cells in addition to Treg cells.26 As a result, when CD25+ cells were depleted, both cytotoxic T cells and Treg cells were affected, which was the reason why efficacy was not enhanced when we used anti-mCD25 antibody. This suggests that investigating the combination of TS-L6 and another antibody that specifically targets Treg cells as a therapeutic strategy may be worthwhile.
To prove that adaptive antitumor immunity was acquired from ADC treatment, the mice completely cured by TS-L6 still rejected tumor cells re-challenged even 6 months after initial TS-L6 administration, which was about one-fourth of a mouse lifetime.27 Thus, the cured mice acquired durable antitumor immunity to reject tumor relapse. When re-challenged with parent CT26.WT tumor cells, only 3 of 8 mice rejected tumor. Because the CT26.WT and CT26.WT-HER2 cells originated from two different vendors, and tumor cells are highly prone to acquire mutations, the different tumor rejection ratio for the two cell lines may have been due to different tumor-associated antigens. The adaptive antitumor immunity of the cured mice, especially the humoral immunity, was also demonstrated by the increased antitumor cell antibodies in the mouse blood. The adaptive antitumor immunity could also be induced via tumor vaccine. CT26.WT-HER2 cells, committed to ICD by MF-6 or TS-L6 treatment but still alive, were used to vaccinate mice. The mice vaccinated with tumor cells rejected tumor re-challenge; thus, MF-6 or TS-L6 treatment was essential to enhance dendritic cells and subsequent T cell activation and antigen presentation.
Recently, cancer immunotherapy, especially with PD1/PD-L1 and CTLA-4 blocking antibodies, has become the major theme of cancer treatment for several cancer types.28 Mechanistically, PD1/PD-L1 antibodies block the interaction between PD-L1/L2 and the PD1 receptor, thereby removing the immunosuppressive signal on T cells and restoring the antitumor function of exhausted T cells. Despite the success of this treatment strategy, the response rate of PD1/PD-L1 blocking antibodies is relatively low, from 20% to 40%.29 The tumor response to PD1/PD-L1 blocking agents is highly dependent on tumor immunogenicity, such as tumor mutation burden, tumor neoantigen abundance, PD-L1 expression level, and an immune-active tumor microenvironment. We have now shown that TS-L6 induced tumor cell ICD and tumor-associated antigen presentation, promoted dendritic cell activation and maturation, elicited immune cell tumor infiltration, and finally induced adaptive antitumor immunity. We also found that the type M2 macrophage, a critical immune-suppressing cell, was reduced after TS-L6 treatment, which indicated the reversion of the tumor microenvironment from immune-suppressing to immune-activating. All this evidence suggested that the immunomodulator activity of TS-L6 converted the immune “cold” tumor into “hot” tumor.
TS-L6 seemed to be naturally suitable to combine with immune checkpoint inhibitors to treat cancer. Even at a low dose of 5 mg/kg, the combination of TS-L6 plus atezolizumab or anti-mCTLA-4 exhibited high antitumor activity. The addition of TS-L6 to immune checkpoint inhibitors treatment might enhance the antitumor efficacy and expand the benefit of immunotherapy to tumor types that do not respond to single immunotherapy. Furthermore, most ADCs are used as an end-line therapy to treat patients who received several rounds of radiotherapy and/or chemotherapy and whose immune systems were thereby seriously damaged. Because an intact immune system was important in the activities of ADCs and immune checkpoint inhibitors, we suggest that it is worth considering using ADC monotherapy or a combination with immune checkpoint inhibitors as the primary treatment when a patient still possesses an intact immune system.
ENHERTU® (trastuzumab deruxtecan, DS-8201a, T-DXd), a milestone product in treating breast cancer, is composed of trastuzumab, the DNA topoisomerase I inhibitor DXd, and a Gly-Gly-Phe-Gly tetrapeptide linker. Compared with DXd, MF-6 exhibited greater activity in inhibiting topoisomerase I and in killing tumor cells and inducing ICD. Although MF-6 might have greater antitumor activity as an ADC, we were concerned that MF-6 might cause serious systemic toxicity and the therapy window would thereby be narrow. Thus, we designed a site-specific dipeptide bridging linker Val-Ala that had a slower drug release rate compared with the Gly-Gly-Phe-Gly linker; slow release should reduce potential systemic toxicity. All mice that received up to 20 mg/kg TS-L6 increased in body weight, and there were no deaths or abnormal responses; thus, TS-L6 was safe in the animal model. Although growth of CT26.WT-HER2 was not inhibited by TS-L6, the DAMPs, MHC-I and MHC-II were upregulated, indicative of ICD of tumor cells. The mice bearing CT26.WT-HER2 tumors responded well to TS-L6 treatment, especially the immunocompetent mice. Because the direct cell killing activity of TS-L6 was limited, the in vivo antitumor efficacy of TS-L6 was due to its ICD-inducing activity, which was demonstrated by increased T cell infiltration, especially the cytotoxic CD8+ T cells and enhanced dendritic cell activation in the tumor tissues. Usually, the more rapidly a drug is released due to linker design, the greater the expected efficacy because only the cytotoxicity of ADCs is considered and immunomodulator activity is overlooked. However, excess toxin released in a short amount of time might exceed the ability to metabolize and excrete the drug, causing drug accumulation and adverse events. We demonstrated that an ADC that had a slow drug release rate still exhibited satisfactory antitumor activity via its immunomodulator activity, with potentially lower systemic toxicity. The relatively slow drug release rate might also contribute to durable efficacy because the toxin was stored as an intact ADC for a long time.
In conclusion, we designed and investigated TS-L6, a HER2-targeting ADC having a site-specific bridging linker and a DNA topoisomerase I inhibitor that was more potent than DXd. The main mode of action of TS-L6 was demonstrated to be immunomodulation, via induction of tumor cell ICD and antigen presentation, promotion of dendritic cell activation and maturation, and recruitment of T cells into tumor, not direct tumor cell killing. Using a slowly releasing linker, the TS-L6 exhibited high safety and surprisingly good antitumor activity. We demonstrated that even an ADC with no significant cytotoxicity could still provide long-lasting antitumor efficacy via immunostimulatory effect, which theoretically would widen therapy window and reduce adverse effects in clinical studies. The investigation of TS-L6 provided a new idea in designing ADCs: the immunostimulatory activity of toxin should be sufficiently exploited. Moreover, the combination of TS-L6 with anti-PD-L1 or anti-CTLA-4 antibody might overcome the low response of single immune checkpoint inhibitors. Furthermore, MF-6-based ADC technology is theoretically widely capable of being used with antibodies that target other tumor molecules. ADCs with the same drug and linker but different targets are expected to have similar immunostimulatory activities. The results of this study provide preliminary evidence for the antitumor mechanism of TS-L6. The efficacy and safety of TS-L6 and potential benefits in combination with immune checkpoint inhibitors still require assessment in clinical studies.
Materials and methods
Cell lines, antibodies, and animals
The following cells lines were purchased from the American Type Culture Collection: human breast cancer cell lines BT474 and SK-BR-3, human gastric cancer cell line NCI-N87, human pancreatic cancer cell line BxPC-3, mouse colon cancer cell line CT26.WT, and mouse breast cancer cell line EMT6. Cells were cultured in appropriate media containing 10% fetal bovine serum (FBS). Human breast cancer cell line MDA-MB-468 was purchased from the National Collection of Authenticated Cell Cultures of Chinese Academy of Sciences and cultured in medium containing 10% FBS. CT26.WT-HER2, a cell line stably transfected with the full-length human HER2 gene, was purchased from BYinno Biotechnology Co., Ltd. and cultured in Dulbecco’s modified eagle medium containing 10% FBS and 20 μg/mL puromycin (Gibco, Cat# 15070–063). The dendritic cells were purchased from Milestone (Shanghai) Biological Science & Technology Co. Ltd. and the informed consent for the scientific research in this study was obtained from the dendritic cells’ donors.
Trastuzumab (TS) was produced in CHO-K1 cells that expressed a recombinant monoclonal antibody produced by Mabwell Biotechnology. For in vivo experiments, anti-human PD-L1 monoclonal antibody atezolizumab, which was cross-reactive with mouse PD-L1, was purchased from Roche Pharmaceutical. Anti-mouse CD4 antibody (α-mCD4, clone# GK1.5), anti-mouse CD8 antibody (α-mCD8, clone# 53–6.7), anti-mouse CD25 antibody (α-mCD25, clone# PC-61.5.3), and anti-mouse CTLA-4 antibody (α-mCTLA-4, clone# 9D9) were purchased from Bio X Cell.
All mouse studies were performed in an approved veterinary research facility, Jiangsu Hanjiang Biotechnology Co., Ltd., and approved by the local Animal Use and Care Committee. Female 6- to 8-week-old BALB/c and nude mice were used in this study; 3–6 mice were housed together in sterilized cages and maintained under pathogen-free conditions. The mice were euthanized with CO2 when endpoints were reached, including tumor volume exceeding 3000 mm3, 10% body weight reduction, end of studies, or other signs that indicated euthanasia for ethical reasons.
Antibody–drug conjugate production
Trastuzumab-L6 was produced in three steps. First, trastuzumab was reduced with tris (2-carboxyethyl) phosphine hydrochloride to break the interchain disulfide bonds. Then, MF-L6, consisting of linker and drug, was added and incubated with reduced trastuzumab for 1 h. Lastly, the free small molecule and organic solvent were removed by ultrafiltration, and the ADC was purified by hydrophobic interaction chromatography to obtain a homogeneous ADC with a drug–antibody ratio of 4.0. The linker-drug unit was located at the positions of interchain disulfide bonds to form a “bridge”; thus, the theoretical drug–antibody ratio was 4.0.
DNA topoisomerase I inhibition activity assessment
The inhibition of topoisomerase I by MF-6 and two other camptothecin-derived compounds, SN38 and DXd, was assessed by measuring the inhibition level of enzyme.30 Two hundred and fifty nanograms of supercoiled plasmid pHOT-1, 1.5 μL (1.5 Unit) human topoisomerase I, 2 μL 10 × reaction buffer (Human Topoisomerase I assay Kit, TopoGEN, Cat# TG1015-3A), and serially diluted compounds were mixed; deionized water was added to a final volume of 20 μL. The final concentrations of MF-6 and DXd were 0.4 to 100 μmol/L, SN38 was 0.032 to 500 μmol/L, and an equal volume of DMSO was used as blank control. The reaction mixture was incubated at 37°C for 30 min, followed by adding 4 μL 10 × gel loading stop buffer containing 0.125% bromophenol blue, 25% glycerol, and 5% Sarkosyl. Then, 10 μL of the reaction mixture was loaded in an 0.8% agarose gel without nucleic acid dye, and electrophoresis was conducted at 75 V voltage for 2 h. After electrophoresis, the gel was stained with 1 × TAE containing 1/10000 Ultra GelRed (Vazyme, Cat#GR501) to stain DNA for 30 min and photographed under UV light. The intensity of supercoiled plasmid band in each group was measured using automatic software, and the inhibition ratio was calculated as follows:
Cytotoxicity assay
The cytotoxicity of MF-6, SN38, and DXd was assessed on four human cancer cell lines, BT474, SK-BR-3, NCI-N87, and BxPC-3. Cells were cultured as monolayers in 5% CO2 at 37°C, harvested by trypsinization, and seeded in 96-well plates at 5,000 to 10,000 cells per well. Serially diluted drugs were added, and DMSO was used as negative control. After 4 days incubation, CellTiter 96® AQueous One Solution Cell Proliferation Assay MTS reagent (Promega, Cat#G3581) was added to determine cell viability; the optical density at 490 nm was measured after 1.5 h with a microplate reader (Molecular Devices, Spectrum Max M5). The IC50 for each drug was calculated according to the 4-parameter fitting results from SoftMax Pro version 7.0.2 GxP.
Human cancer cell lines BT474, SK-BR-3, NCI-N87 and mouse cancer cell line CT26.WT-HER2 were used to assess cytotoxicity of TS-L6, TS, and MF-6. After cells were seeded in 96-well plate, serially diluted drugs were added and incubated for up to 6 days, followed by adding CellCounting-Lite 3D Luminescent Cell Viability Assay reagent (Vazyme, Cat#DD1102–02). After 25 min incubation, the luminescence was measured in a microplate reader (Molecular Devices, Spectrum Max M5), and the IC50 for each drug was calculated as described above.
Bystander killing effect
To evaluate the bystander killing effect of TS-L6, HER2-positive NCI-N87 and HER2-negative MDA-MB-468 cell lines were seeded together in a 6-well plate as 5 × 105 cells and 1 × 105 cells, respectively. The supernatant was removed after overnight incubation, and 5 μg/mL TS or TS-L6 was added; no added drug was the control. After 4 days of coculture, the supernatant was removed, and total viable cells were detached by trypsinization. The total cell number in each well was determined using a cell counter. The ratios of NCI-N87 and MDA-MB-468 cells relative to the total viable cells were determined by staining with a mouse monoclonal anti-HER2 antibody (Abcam, Cat# ab264541) on ice for 1 h. After washing, the cells were stained with goat anti-mouse IgG H&L-Alexa Fluor® 488 (Abcam, Cat#ab150113) on ice for 1 h. After washing, fluorescent signals of 2 × 104 stained cells were measured using a flow cytometer (Beckman Coulter, CytoFLEX), and the ratio of HER2-positive and HER2-negative cells was calculated. Finally, the number of NCI-N87 and MDA-MB-468 cells in each well was calculated, and the bystander killing effect was evaluated based on the ratio of MDA-MB-468 in drug-treated group compared with control.
Detection of genomic DNA in cytosolic extracts
MF-6 could damage tumor cell genomic DNA, followed by release of genomic DNA into the cytoplasm and upregulation of DNA damage marker phosphorylated histone H2A.X. About one million NCI-N87 cells were seeded in a 6-well plate and treated with 2 nmol/L MF-6 or DMSO for 3 days or treated with 50 nmol/L TS or TS-L6 for 5 days. After treatment, cells were harvested and washed 3 times with cold phosphate-buffered saline (PBS), and finally resuspended in 0.5 mL buffer containing 150 mmol/L NaCl, 50 mmol/L HEPES pH 7.4, and 20 μg/mL digitonin. The homogenates were incubated end over end for 10 min to enable selective plasma membrane permeabilization. The homogenates were then centrifuged at 1000 g for 3 min to pellet intact cells. The cytosolic supernatants were transferred to fresh tubes and centrifuged at 17,000 g for 10 min to pellet cellular debris, yielding cytosolic preparations free of nuclear, mitochondrial, and endoplasmic reticulum contamination.31 The DNA in the cytosolic preparations was isolated using FastPure Gel DNA Extraction Mini Kit (Vazyme, Cat#DC301–01) and used as real-time PCR template. Quantitative real-time PCR was performed using nucleus DNA primer Tert, which codes telomerase reverse transcriptase, with 16S rRNA as housekeeping gene. The forward and reverse primers for Tert were 5’-AGCAACTTCTTCGGGTGTG-3’ and 5’-ATGTGAGTGTTGGGGAAGG−3’. The forward and reverse primers for 16S rRNA were 5’-GCTAAACCTAGCCCCAAACC−3’ and 5’- TTCTTGGGTGGGTGTGGGTAT−3’.
Immunofluorescence and immunohistochemistry
In an immunofluorescence assay, NCI-N87, BT474 or CT26.WT-HER2 tumor cells were fixed in 4% paraformaldehyde in tris-buffered saline (TBS) for 20 min at ambient temperature, washed twice with TBS, and permeabilized with 0.5% Triton X-100 in TBS for 10 min. After washing, the cells were blocked with 2% bovine serum albumin (BSA) and 2% FBS in TBS for 1 h. The cells were then incubated with phosphorylated histone H2A.X (Ser139) antibody (CST, Cat#9718, 1:200 dilution) at 4°C overnight. After washing, the cells were incubated with fluorescein isothiocyanate (FITC)-conjugated goat-anti-rabbit IgG (H+L) secondary antibody (Jackson ImmunoResearch, Cat#111-095-144, 1:200 dilution) and 1 μg/mL of 4’,6-diamidino-2-phenylindole (DAPI) in 2% BSA and 2% FBS in TBS for 1 h at ambient temperature. The cells were washed 3 times, and the slides were photographed using fluorescent microscopy (Nikon, TS2R-FL).
Formalin-fixed, paraffin-embedded tumor tissues were cut into 3- to 5-μm-thick sections and mounted on positively charged slides for immunohistochemistry. Slides were deparaffinized in xylene and rehydrated in graded ethanol. Antigen retrieval was conducted in acidic antigen retrieval buffer (10 mmol/L citrate, pH 6.0) at 97°C for 20 min for CD3/CD206 and in basic antigen retrieval buffer (10 mmol/L Tris and 1 mmol/L EDTA, pH 9.0) for CD8/CD86/CD163. Then, slides were covered with 3% hydrogen peroxide to inactivate endogenous peroxidase and blocked with 20% goat serum. The primary antibodies were anti- mouse CD3 (Novus, Cat#NB600, 1:40 dilution), anti-mouse CD8 (Abcam, Cat#ab217344, 1:1000 dilution), anti-mouse CD86 (Novus, Cat#NBP2–25208, 1:200 dilution), anti-mouse CD163 (Abcam, Cat#ab182422, 1:400 dilution), and anti-mouse mannose receptor (CD206) (Abcam, Cat#ab64693, 1:10,000 dilution). Incubation with primary antibodies was at 4°C overnight, except for CD206, which was incubated at ambient temperature for 30 min. After washing with PBS, the slides were incubated with horseradish peroxidase (HRP)-labeled goat-anti mouse secondary antibody (Jackson ImmunoResearch, Cat#115-035-003, 1:1000 dilution) or HRP-labeled goat-anti rabbit secondary antibody (Thermo, Cat#31460, 1:1000 dilution) at ambient temperature for 1 h. After washing, DAB chromogenic reagent (Thermo, Cat#8801–4965–72) was added and reacted for proper time at ambient temperature in the dark. The chromogenic reaction was stopped by washing the reagent with deionized water, and slides were counterstained with hematoxylin, followed by adding neutral resin. Images were acquired by microscopy (Nikon, TS2R-FL) with a 20 or 40 × objective lens and a digital scanning system.
Drug release with cathepsin B and L digestion
The linkers of TS-L6, TS-GGFG-DXd, and TS-vcMMAE, valine-alanine (VA linker), Glycine-Glycine-Phenylalanine-Glycine (GGFG linker), and valine-citrulline (VC linker), respectively, were digested by cathepsin B and L, the most prevalent lysosomal cathepsins. Drug release rates were measured at pH 4.5, 5.0 and 5.5. ADCs were diluted to 0.9 mg/mL (6 μmol/L) with appropriate digestion buffer. The cathepsin B (Sigma, Cat#C8571), which was activated with cathepsin B activation buffer (30 mmol/L dithiothreitol, 15 mmol/L EDTA, pH 5.5) prior to use, and cathepsin L (Sigma, Cat#C6854) were diluted to 240 nmol/L with corresponding digestion buffer. The diluted ADCs were mixed with equal volumes of diluted cathepsins and incubated at 37°C. Acetonitrile or acetonitrile containing 33% DMSO was added to terminate the reaction and precipitate the protein, and the supernatant was isolated by centrifugation. The released drugs in the supernatant were analyzed with an ultra-performance liquid chromatography system (Waters, H-class) using a 2.1 × 50 mm chromatographic reverse-phase column (Waters, Cat# 186002350); the mobile phase A was aqueous 0.1% phosphoric acid, and mobile phase B was acetonitrile phosphate solution.
Drug release in tumor cells
Five million NCI-N87 cells were seeded in a 10 cm-cell culture dish, and TS-L6 or TS-GGFG-DXd was added to a final concentration of 1 μg/mL. After drug treatment for 0.5, 1, 2, 3, 10, 24, 48, and 72 h in 5% CO2 at 37°C, the supernatant was removed and cells were harvested by trypsinization and washed 3 times with cold PBS. The cells were lysed with Cell Lysis Buffer II (Invitrogen, Cat#FNN0021) containing Protease Inhibitor Cocktail (MedChemExpress, Cat#HY-K0010) and supernatant was collected by centrifugation. The cell lysis was divided into two equal aliquots for released drug and ADC concentration testing, respectively. A quantitative high-performance liquid chromatography tandem mass spectrometry method (HPLC-MS) was applied to measure the concentration of released MF-6 or DXd. The cell lysis was added with acetonitrile containing 0.01% formic acid (v/v) to precipitate protein and the supernatant isolated by centrifugation was injected into HPLC (Shimadzu, LC-30AD) tandem MS (AB SCIEX Triple Quad 6500) for quantitative analysis. An ELISA assay was applied to measure the TS-L6 and TS-GGFG-DXd concentration. Recombinant human HER2 extra-cellular domain (Acro Biosystems, Cat#HE2-H5212) was coated on ELISA plate to capture antibodies with HRP-labeled goat anti-human IgG (H&L) (Abcam, Cat#ab209702) as detecting reagent, and tetramethylbenzidine (TMB) was used as chromogenic reagent.
Tumor cells treated with drug and co-cultured with dendritic cells
BT474, NCI-N87, and CT26.WT-HER2 cells were seeded in a 6-well plate, treated with 2 nmol/L MF-6 for 3 days with DMSO as negative control, or treated with 10 nmol/L or 50 nmol/L TS-L6, 50 nmol/L TS for 5 days with medium as negative control. After drug treatment, the amount of calreticulin, HSP90, HSP70, MHC-I, MHC-II, and PD-L1 on the cell surface was assessed by flow cytometry. In the tumor cell and dendritic cell co-culture assay, NCI-N87 cells were seeded in a 6-well plate as 15,000 per well, followed by 5 nmol/L or 10 nmol/L TS-L6 incubation for 2 days. After the supernatant was removed, 1 million dendritic cells were added and incubated for another 2 days. Finally, the suspended dendritic cells were collected for analysis and adherent tumor cells were discarded. After being blocked with human Fc block reagent (Becton Dickinson, Cat#564220) and stained with antibodies against human CD40, CD80, CD86, MHC-I and MHC-II, the dendritic cells were quantitated by flow cytometry.
Flow cytometry
Tumor cells BT474, NCI-N87, and CT26.WT-HER2 treated with drugs were stained with antibodies for flow cytometry analysis. Anti-Calreticulin (Abcam, Cat#ab92516, 1:100 dilution), anti-HSP70 (Abcam, Cat#ab181606, 1:250 dilution), anti-HSP90 (Abcam, Cat#ab59459, 1:200 dilution), and FITC-labeled atezolizumab were used to stain both human and mouse cells; anti-human MHC-I (R&D, Cat#NBP1-43122, 1:500 dilution) and anti-human MHC-II antibody (Abcam, Cat#ab55152, 1:200 dilution) were used to stain BT474 and NCI-N87, and FITC-labeled anti-mouse MHC-I antibody (Abcam, Cat#ab95572, 1:500 dilution) and APC labeled anti-mouse MHC-II antibody (Abcam, Cat#ab93559, 1:500 dilution) were used to stain CT26.WT-HER2. FITC-labeled goat anti-rabbit IgG (H+L) antibody (Jackson ImmunoResearch, Cat#111-095-144, 1:200 dilution) or FITC-labeled goat anti-mouse IgG (H+L) antibody (Proteintech, Cat#SA0003–1, 1:200 dilution) was used as secondary antibody if necessary before cells were examined by flow cytometry.
Dendritic cells, after being co-cultured with tumor cells and blocked with human Fc block reagent, were stained with FITC-labeled anti-human CD80 (Biolegend, Cat#305206, 1:20 dilution), FITC-labeled anti-human CD40 (Biolegend, Cat#334306, 1:20 dilution), Alexa Fluor® 488-labeled anti-human CD86 (Abcam, Cat#ab290990, 1:500 dilution), PE-labeled anti-human HLA Class I (MHC-I) (Abcam, Cat#ab58998, 1:500 dilution), and FITC-labeled anti-human HLA-DR (MHC-II) antibody (Biolegend, Cat#307604, 1:20 dilution), followed by flow cytometry analysis.
In vivo efficacy measurement
Mouse CT26.WT-HER2 tumor cells were cultured as monolayers, harvested by trypsinization, and implanted subcutaneously into either immunocompetent BALB/c or athymic nude mice. Three million tumor cells were implanted in the right flank of 6- to 8-week-old female mice for tumor formation. Tumor volume and body weight measurements were collected twice weekly, and tumor volume was calculated using the equation (Length × Width2/2. When the average volume of tumors reached approximately 100–150 mm3, the mice were divided by the randomized block method into saline control and treatment groups based on tumor volume, and treatment was initiated (day 0). TS-L6 (5, 10, and 20 mg/kg), TS (20 mg/kg), atezolizumab (7.5 and 15 mg/kg), anti-mCD4 antibody (10 mg/kg), anti-mCD8 antibody (10 mg/kg), anti-mCD25 antibody (10 mg/kg), and anti-mCTLA-4 antibody (5 mg/kg) were administered intravenously at a volume of 10 mL/kg. As control, saline was administered at the same volume. TS-L6 and TS were administered once weekly (QW) on day 0 and 7, whereas atezolizumab and anti-mCD4/mCD8/mCD25/mCTLA-4 antibodies were administered twice weekly (BIW) on day 0, 4, 7, and 10. For drugs combinations, TS-L6 was combined with atezolizumab, anti-mCD4 antibody, anti-mCD8 antibody, anti-mCD25 antibody, and ant-mCTLA-4 antibody, respectively.
Complete response (CR) was defined as disappeared tumor. Tumor growth inhibition (TGI) was calculated as follows:
For tumor re-challenge, when immunocompetent mice bearing CT26.WT-HER2 tumors were completely cured by TS-L6 or TS-L6 combined with another antibody, the cured mice were subcutaneously injected in the left flank with 3 million CT26.WT-HER2, CT26.WT or EMT6 cells. Naïve immunocompetent mice of the same age were used as control.
Tumor vaccination assay with drug pre-treated cells
For the tumor cell vaccination study, CT26.WT-HER2 cells were treated with either 10 nmol/L MF-6 for 24 h or 100 nmol/L TS-L6 for 72 h, so that the cells were committed to ICD, but were still alive. CT26.WT-HER2 cells treated three times by freeze-thaw cycles (necrotic cells) were used as negative control. Three million treated cells were inoculated into the right flank of immunocompetent BALB/c mice. Three weeks later, mice without tumors were inoculated again with three million CT26.WT-HER2 or CT26.WT cells into the left flank with naïve mice as blank control; mice vaccinated with necrotic cells were the negative control. Mice were fed for another 3 weeks and tumor volume was measured twice a week.
Cell-based enzyme-linked immunosorbent assay
The anti-CT26.WT-HER2 and anti-CT26.WT antibody titers were measured with plasmas from mice subjected to tumor re-challenge. Cells were seeded in a 96-well plate at 20,000 cells per well for overnight incubation. After discarding the supernatant, the cells were washed three times with PBS and fixed with 4% paraformaldehyde at ambient temperature for 10 min, followed by washing and permeabilization with 0.2% Triton X-100 for 10 min. Cellular peroxidase was inactivated with 3% hydrogen peroxide for 15 min, followed by blocking with 5% skim milk for 2 h. The plasmas were serially diluted with 1% skim milk in PBST and added as 100 μL per well. After 2 h incubation at ambient temperature, the cells were washed and incubated with HRP-labeled goat-anti mouse secondary antibody (Jackson ImmunoResearch, Cat#115-035-003, 1:10000 dilution) at ambient temperature for 1 h. After washing, TMB chromogenic reagent was added for 10 min at ambient temperature, terminated by 1 mol/L phosphoric acid. The optical density at 450 nm was measured with a microplate reader (Molecular Devices, Spectrum Max M5). The antibody titer in plasma was defined as the maximal dilution that showed a positive signal, with 2.1 times the negative plasma signal as the threshold value.
Statistical analysis
All data were analyzed statistically with GraphPad Prism 9.4.1 software (GraphPad Software, LLC). Flow cytometric data, antitumor effects, and antibody titer in mouse plasma were analyzed using one-way ANOVA or Student’s t test. For survival curve analyses in the tumor re-challenge and vaccination studies, Kaplan–Meier analysis was performed followed by log-rank test to compare the probability of survival. A P value<0.05 was considered to be statistically significant. Data were depicted as mean with standard deviation (SD) or standard error of mean (SEM).
List of Abbreviations
- ADC
Antibody-drug conjugate
- APCs
Antigen-presenting cells
- BIW
Twice weekly
- CRs
Complete responses
- CTLA-4
Cytotoxic T lymphocyte-associated antigen-4
- DAMPs
Damage-associated molecular patterns
- DAR
Drug-antibody ratio
- DMSO
Dimethyl sulfoxide
- ELISA
Enzyme-linked immunosorbent assay
- HER2
Human epidermal growth factor receptor 2
- HMGB1
High-mobility group box 1
- HPLC-MS
High-performance liquid chromatography tandem mass spectrometry
- HRP
Horseradish peroxidase
- HSP70
Heat-shock protein 70
- HSP90
Heat-shock protein 90
- IC50
Half maximal inhibitory concentration
- ICD
immunogenic cell death
- IHC
Immunohistochemistry
- MHC-I
Histocompatibility complex-class I
- MHC-II
Histocompatibility complex-class II
- MMAE
Monomethyl auristatin E
- Mpk
mg/kg
- PD1
Programmed death 1
- PD-L1
Programmed death-ligand 1
- QW
Once weekly
- SD
Standard deviation
- SEM
Standard error of mean
- TGI
Tumor growth inhibition
- Treg
T regulatory cell
- TS-L6
Trastuzumab-L6
Supplementary Material
Acknowledgments
This study was supported by the Shanghai Science and Technology Support Program (20S11904800) and the Shanghai Natural Science Foundation (20ZR1459500). The authors thank AiMi Academic Services (www.aimieditor.com) for English language editing and review services.
Funding Statement
The work was supported by the The Shanghai Natural Science Foundation [20ZR1459500]; The Shanghai Science and Technology Support Program [20S11904800].
Disclosure statement
No potential conflict of interest was reported by the authors.
Authors’ Contributions
Maomao An: Conceptualization, designing study plan, reviewing draft. Xiaoding Tan: Conceptualization, designing study plan, project management, writing – original draft and editing. Wei Zhou: Designing small molecules, project management, preparing small molecules and antibody-drug conjugates. Peng Fang: Conceptualization, conducting studies, project management, writing – original draft and editing. Kaiying Li and Meng You: Cellular studies, statistical analysis, Yuxia Cao, Xiaohong Zhu, Wendi Li, Lei Shi and Haiying Wen: Cellular studies, ELISA and flow cytometry. Xin Wei: Molecular biological studies; Lu Wang: Immunofluorescence and immunohistochemistry studies. Xiaowei Sun: HPLC and HPLC tandem MS analysis. Dongan Yu: Animal studies. Datao Liu: Resources, project management. Hui Xu, Huikai Zhu and Zhenzhen Wang: Preparing small molecules and antibody-drug conjugates. Hui Shen: Project management, editing drafts.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2023.2220466
References
- 1.Criscitiello C, Morganti S, Curigliano G.. Antibody–drug conjugates in solid tumors: a look into novel targets. J Hematol Oncol. 2021;14(1):20. doi: 10.1186/s13045-021-01035-z. PMID: 33509252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rios-Doria J, Harper J, Rothstein R, Wetzel L, Chesebrough J, Marrero A, Chen C, Strout P, Mulgrew K, McGlinchey K, et al. Antibody–drug conjugates bearing pyrrolobenzodiazepine or tubulysin payloads are immunomodulatory and synergize with multiple Immunotherapies. Cancer Research. 2017;77(10):2686–16. doi: 10.1158/0008-5472.CAN-16-2854. PMID: 28283653. [DOI] [PubMed] [Google Scholar]
- 3.Gerber HP, Sapra P, Loganzo F, May C.. Combining antibody-drug conjugates and immune-mediated cancer therapy: what to expect? Biochem Pharmacol. 2016;102:1–6. doi: 10.1016/j.bcp.2015.12.008. PMID: 26686577. [DOI] [PubMed] [Google Scholar]
- 4.Iwata TN, Ishii C, Ishida S, Ogitani Y, Wada T, Agatsuma T. A HER2-targeting antibody–drug conjugate, trastuzumab deruxtecan (DS-8201a), enhances antitumor immunity in a mouse model. Mol Cancer Ther. 2018;17(7):1494–503. doi: 10.1158/1535-7163.MCT-17-0749. PMID: 29703841. [DOI] [PubMed] [Google Scholar]
- 5.Montes de Oca R, Alavi AS, Vitali N, Bhattacharya S, Blackwell C, Patel K, Seestaller-Wehr L, Kaczynski H, Shi H, Dobrzynski E, et al. Belantamab mafodotin (GSK2857916) drives immunogenic cell death and immune-mediated antitumor responses in vivo. Mol Cancer Ther. 2021;20(10):1941–55. doi: 10.1158/1535-7163.MCT-21-0035. PMID: 34253590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yaghoubi S, Karimi MH, Lotfinia M, Gharibi T, Mahi-Birjand M, Kavi E, Hosseini F, Sineh Sepehr K, Khatami M, Bagheri N, et al. Potential drugs used in the antibody–drug conjugate (ADC) architecture for cancer therapy. J Cell Physiol. 2020;235(1):31–64. doi: 10.1002/jcp.28967. PMID: 31215038. [DOI] [PubMed] [Google Scholar]
- 7.Galluzzi L, Vitale I, Warren S, Adjemian S, Agostinis P, Martinez AB, Chan TA, Coukos G, Demaria S, Deutsch E, et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J ImmunoTher Cancer. 2020;8(1):e000337. doi: 10.1136/jitc-2019-000337. PMID: 32209603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aaes TL, Vandenabeele P. The intrinsic immunogenic properties of cancer cell lines, immunogenic cell death, and how these influence host antitumor immune responses. Cell Death Differ. 2021;28:843–60. doi: 10.1038/s41418-020-00658-y. PMID: 33214663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Almanza A, Carlesso A, Chintha C, Creedican S, Doultsinos D, Leuzzi B, Luis A, McCarthy N, Montibeller L, More S, et al. Endoplasmic reticulum stress signalling - from basic mechanisms to clinical applications. FEBS J. 2019;286(2):241–78. doi: 10.1111/febs.14608. PMID: 30027602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen X, Cubillos-Ruiz JR. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat Rev Cancer. 2021;21:71–88. doi: 10.1038/s41568-020-00312-2. PMID: 33214692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12:860–75. doi: 10.1038/nrc3380. PMID: 23151605. [DOI] [PubMed] [Google Scholar]
- 12.Muller P, Kreuzaler M, Khan T, Thommen DS, Martin K, Glatz K, Savic S, Harbeck N, Nitz U, Gluz O, et al. Trastuzumab emtansine (T-DM1) renders HER2+ breast cancer highly susceptible to CTLA-4/PD-1 blockade. Sci Translational Med. 2015;7(315):315ra188. doi: 10.1126/scitranslmed.aac4925. PMID: 26606967. [DOI] [PubMed] [Google Scholar]
- 13.Nakada T, Sugihara K, Jikoh T, Abe Y, Agatsuma T. The latest research and development into the antibody-drug conjugate, [fam-] trastuzumab deruxtecan (DS-8201a), for HER2 cancer therapy. Chem Pharm Bull (Tokyo). 2019;67:173–85. doi: 10.1248/cpb.c18-00744. PMID: 30827997. [DOI] [PubMed] [Google Scholar]
- 14.Drake PM, Rabuka D. Recent developments in ADC technology: preclinical studies signal future clinical trends. BioDrugs. 2017;31:521–31. doi: 10.1007/s40259-017-0254-1. PMID: 29119409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mahalingaiah PK, Ciurlionis R, Durbin KR, Yeager RL, Philip BK, Bawa B, Mantena SR, Enright BP, Liguori MJ, Van Vleet TR. Potential mechanisms of target-independent uptake and toxicity of antibody-drug conjugates. Pharmacology & Therapeutics. 2019;200:110–25. doi: 10.1016/j.pharmthera.2019.04.008. PMID: 31028836. [DOI] [PubMed] [Google Scholar]
- 16.Ferraro E, Drago JZ, Modi S. Implementing antibody-drug conjugates (ADCs) in HER2-positive breast cancer: state of the art and future directions. Breast Cancer Res. 2021;23:84. doi: 10.1186/s13058-021-01459-y. PMID: 34380530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hudis CA. Trastuzumab — mechanism of action and use in clinical practice. N Engl J Med. 2007;357:39–51. doi: 10.1056/NEJMra043186. PMID: 17611206. [DOI] [PubMed] [Google Scholar]
- 18.Wang Z, Chen J, Hu J, Zhang H, Xu F, He W, Wang X, Li M, Lu W, Zeng G, et al. cGAS/STING axis mediates a topoisomerase II inhibitor–induced tumor immunogenicity. Journal Of Clinical Investigation. 2019;129(11):4850–62. doi: 10.1172/JCI127471. PMID: 31408442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: can Treg cells be a new therapeutic target? Cancer Sci. 2019;110:2080–89. doi: 10.1111/cas.14069. PMID: 31102428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–55. doi: 10.1016/S1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 21.Zitvogel L, Apetoh L, Ghiringhelli F, Andre F, Tesniere A, Kroemer G. The anticancer immune response: indispensable for therapeutic success? J Clin Invest. 2008;118:1991–2001. doi: 10.1172/JCI35180. PMID: 18523649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307. doi: 10.1038/nri1806. PMID: 16557261. [DOI] [PubMed] [Google Scholar]
- 23.Wu J, Waxman DJ. Immunogenic chemotherapy: dose and schedule dependence and combination with immunotherapy. Cancer Lett. 2018;419:210–21. doi: 10.1016/j.canlet.2018.01.050. PMID: 29414305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garg AD, More S, Rufo N, Mece O, Sassano ML, Agostinis P, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: immunogenic cell death induction by anticancer chemotherapeutics. Oncoimmunology. 2017;6:e1386829. doi: 10.1080/2162402X.2017.1386829. PMID: 29209573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kersten K, Salvagno C, de Visser KE. Exploiting the immunomodulatory properties of chemotherapeutic drugs to improve the success of cancer immunotherapy. Front Immunol. 2015;6:516. doi: 10.3389/fimmu.2015.00516. PMID: 26500653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Couper KN, Lanthier PA, Perona-Wright G, Kummer LW, Chen W, Smiley ST, Mohrs M, Johnson LL. Anti-CD25 antibody-mediated depletion of effector T cell populations enhances susceptibility of mice to acute but not chronic Toxoplasma gondii infection. J Immunol. 2009;182:3985–94. doi: 10.4049/jimmunol.0803053. PMID: 19299696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Willinghamm MD, Brodt MD, Lee KL, Stephens AL, Ye J, Silva MJ. Age-related changes in bone structure and strength in female and male BALB/c mice. Calcif Tissue Int. 2010;86:470–83. doi: 10.1007/s00223-010-9359-y. PMID: 20405109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Miguel M, Calvo E. Clinical challenges of immune checkpoint inhibitors. Cancer Cell. 2020;38:326–33. doi: 10.1016/j.ccell.2020.07.004. PMID: 32750319. [DOI] [PubMed] [Google Scholar]
- 29.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–55. doi: 10.1126/science.aar4060. PMID: 29567705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kawato Y, Aonuma M, Hirota Y, Kuga H, Sato K. Intracellular roles of SN-38, a metabolite of the camptothecin derivative CPT-11, in the antitumor effect of CPT-11. Cancer Res. 1991;51:4187–91. [PubMed] [Google Scholar]
- 31.West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520(7548):553–57. doi: 10.1038/nature14156. PMID: 25642965. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.