

ABSTRACT
Upon reformatting of an antibody to single-chain variable fragment format, a region in the former variable/constant domain interface of the heavy chain becomes accessible for preexisting (PE) anti-drug antibody (ADA) binding. The region exposed because of this reformatting contains a previously hidden hydrophobic patch. In this study, mutations are introduced in this region to reduce PE ADA reactivity and concomitantly reduce the hydrophobic patch. To enhance our understanding of the importance of individual residues in this region with respect to PE ADA reactivity, a total of 50 molecules for each of two antibodies against different tumor-associated antigens were designed, produced, and characterized by an arsenal of biophysical methods. The aim was to identify suitable mutations that reduce, or completely eliminate, PE ADA reactivity to variable fragments, without compromising biophysical and pharmacodynamic properties. Computational methods were used to pinpoint key residues to mutate and to evaluate designed molecules in silico, in order to reduce the number of molecules to produce and characterize experimentally. Mutation of two threonine residues, Thr101 and Thr146 in the variable heavy domain, proved to be critical to eliminate PE ADA reactivity. This may have important implications in optimizing early drug development for antibody fragment-based therapeutics.
KEYWORDS: Anti-drug antibodies, antibody, antibody fragment, biotherapeutics, immunogenicity, multispecific, scFv
Introduction
Antibody therapeutics are a rapidly developing class of pharmaceuticals with a multitude of approvals per year.1 Thus far, approved antibodies have almost exclusively been in immunoglobulin G (IgG) format.2 More recently, antibody fragment formats, such as single-chain variable fragment (scFv) and variable domain of the heavy chain of heavy chain-only antibodies (VHH) have emerged and are often assembled into bi- and multi-specifics.3 When derived from a non-human species, such antibodies are routinely humanized to mitigate the risk of immunogenicity.
Despite advances in antibody discovery techniques, both humanized and fully human antibodies may still elicit an immune response leading to the generation of anti-drug antibodies (ADAs)4 that can have an effect on pharmacokinetics, pharmacodynamics, efficacy, and/or safety.5 For these reasons, immunogenicity assessments are commonly carried out during drug development, and detailed guidelines6 on how to perform these, as well as how to include the information in drug labeling,7 have been published. However, the assessment of these so-called treatment-emergent (TE) ADAs can be challenging if preexisting (PE) ADAs are present, since the latter can lead to an elevated cut-point in the immunogenicity assay and subsequently an inaccurate assessment of the true treatment-induced immunogenicity due to false negatives.8,9 PE ADAs are reactive with the biologics and present in subjects prior to the first administration.10 The most commonly reported source of PE ADAs are nonspecific immunoglobulins. One type of PE ADAs bind to neoepitopes in antibody fragments, such as VHHs, scFvs and antigen-binding fragments (Fab), that are not exposed in full-length antibodies. One example of this is anti-hinge antibodies that, after proteolytic cleavage of the Fab domain, bind to the newly exposed hinge region.11 Furthermore, PE ADAs that bind to the scFv part of an IgG-scFv12 and to the first approved scFv, brolucizumab, have been reported.13 Moreover, according to Bivi et al.,12 PE ADA reactivity correlates with the overall risk of TE ADAs.
PE ADAs binding to the framework sequences of VHHs14,15 have also been described. For the latter, significant reduction of PE ADA reactivity was achieved by addition of a single alanine residue at the C-terminus.15,16 An alanine is typically present at this position in natural human IgG and is the first residue of the CH1 domain. PE ADAs bind to the same region in scFvs and VHHs, namely the variable heavy (VH) region that becomes exposed upon reformatting to scFv or VHH format. This region is located in the interface to the constant domain in IgG (Figure 1A) and Fab (Figure 1B) format and becomes accessible to PE ADAs when the associated constant domain is not present in the scFv format (Figure 1C), and it appears to be a hot spot for PE ADA reactivity. Nieba and coworkers were early investigators of this region and coined the phrase “former V/C domain interface” to refer to it.17 It has been shown in a more recent study that PE ADAs do not appear to bind to the corresponding exposed region in the former V/C domain interface of the variable light (VL) chain.14
Figure 1.
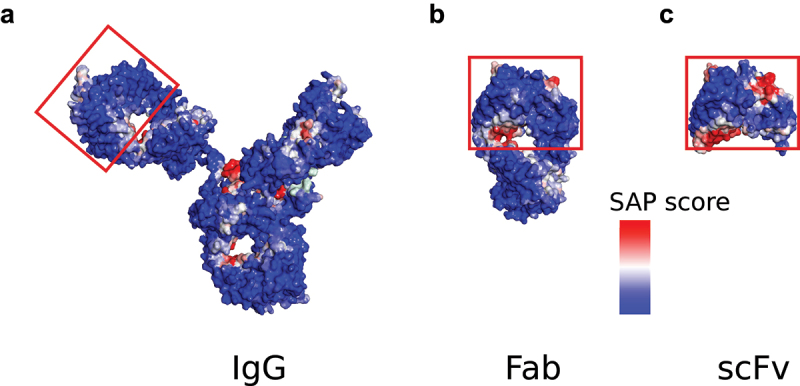
The SAP scores at R = 10 Å mapped for various formats. (a) IgG (1hzh.Pdb, (b) Fab (1fvd.Pdb) and (c) scFv (1fvc.Pdb). The region to which PE ADAs bind is located in the lower left corner of the red boxes in (c). In (a) and (b) the region is not accessible to PE ADAs since it is buried by the constant domain in these cases.
In this study, we investigated in detail the PE ADA reactivity to the exposed, hydrophobic region in the former (heavy) V/C domain interface of two scFvs with different binding specificities against tumor-associated antigens (TAA). These two scFvs are hereafter referred to as the αTAA1 scFv and the αTAA2 scFv. Using a collection of biophysical methods,18–20 we identified double mutations on residues 101 and 146 that completely eliminate PE ADA reactivity while maintaining favorable developability and antigen-binding properties. The scFvs in this study (all of which are of Vk1-VH3 type) contain a so-called λ-cap™, i.e., the framework region 4 of the light chain (FW4L) has been replaced by a λ-type framework region 4 germline sequence. It has previously been shown that scFvs containing a λ-cap have higher stability and lower aggregation propensity and thereby lower risk of immunogenicity.21
Results
Identification of newly exposed residues of scFvs in the former V/C domain interface
Prior to the investigation of the exposed region in the former V/C domain interface, we calculated SAP scores in the IgG (Figure 1A), Fab (Figure 1B) and scFv format (Figure 1C), as well as the relative change in fractional SAS between the Fab and scFv formats (data not shown). Five framework residues (Leu12, Leu144, Thr146, Ser148, Ser149) in the former V/C domain interface were shown to be more accessible to solvent in the scFv format. However, since PE ADAs are significantly larger than solvent molecules, residues for which there is no difference in fractional SAS can also be more accessible to PE ADAs in the absence of a constant domain because not all residues with PE ADA accessibility are necessarily subject to solvent accessibility changes. Solvent accessibility thus only estimates a subset of residues of PE ADA accessibility. As can be seen in Figure 3A, both parental αTAA scFvs have predicted hydrophobic patches in the former V/C domain interfaces as well as in the complementary-determining region (CDRs). The SAP scores for individual residues constituting the hydrophobic patch for four proprietary inhouse crystal structures are shown in Supplementary Table 1. The focus of this study is the exposed region in framework regions of VH. This region is well separated from the CDRs, and modifications here should therefore only have minor effects on functional properties such as binding affinity. The number of positive sera in the PE ADA reactivity experiments (20 donor sera in total) was 9 for the αTAA1 scFv and 8 for the αTAA2 scFv (Figure 2), which is similar to what has been reported elsewhere.14,15 In the following sections, all conclusions and statements apply to both αTAA scFvs unless otherwise explicitly stated. Differences in PE ADA reactivities between αTAA1 scFvs and αTAA2 scFvs that are sometimes observed are likely caused by differences in CDR sets.
Figure 3.
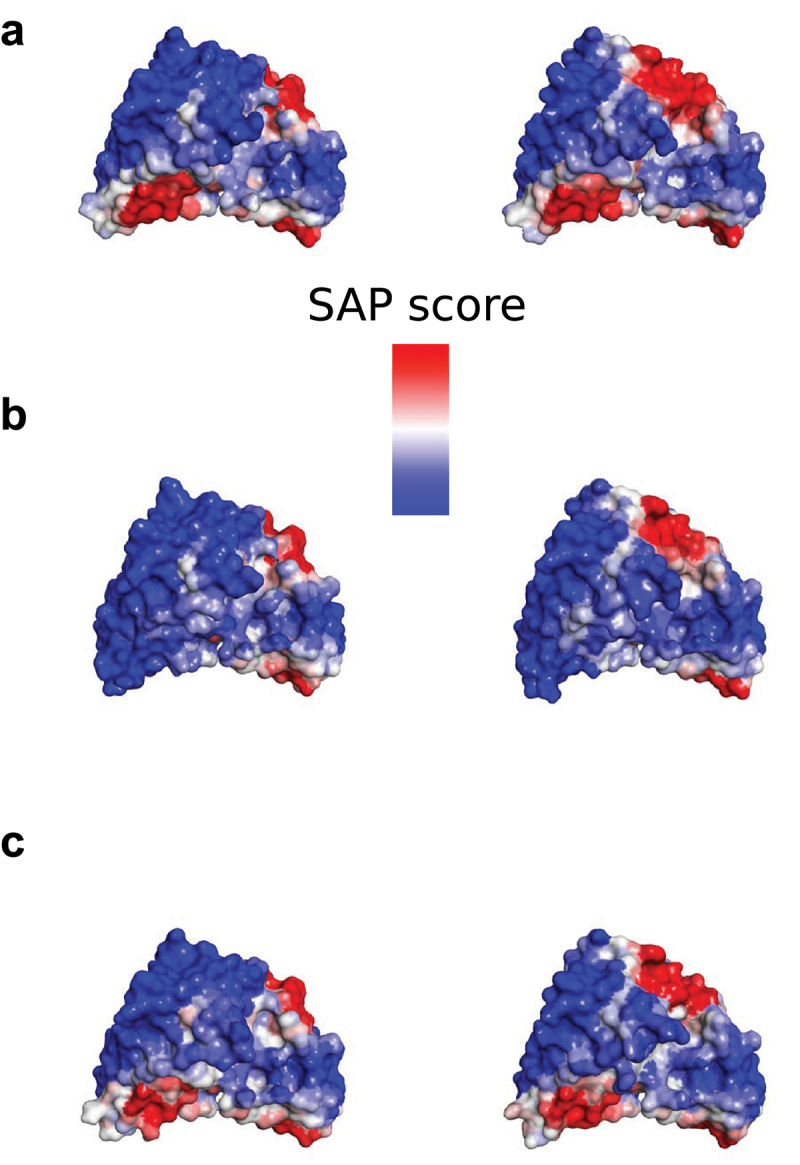
The SAP scores at R = 10 Å mapped for αTAA1 scFv (left) and αTAA2 scFv (right). (a) parental, (b) L12 R-V103T-L144Q and (c) T101S-T146K. The molecules are oriented so that the predicted hydrophobic patch in the former (heavy) V/C domain interface appears to the lower left and CDRs (partly obscured) in the upper regions of each image.
Figure 2.
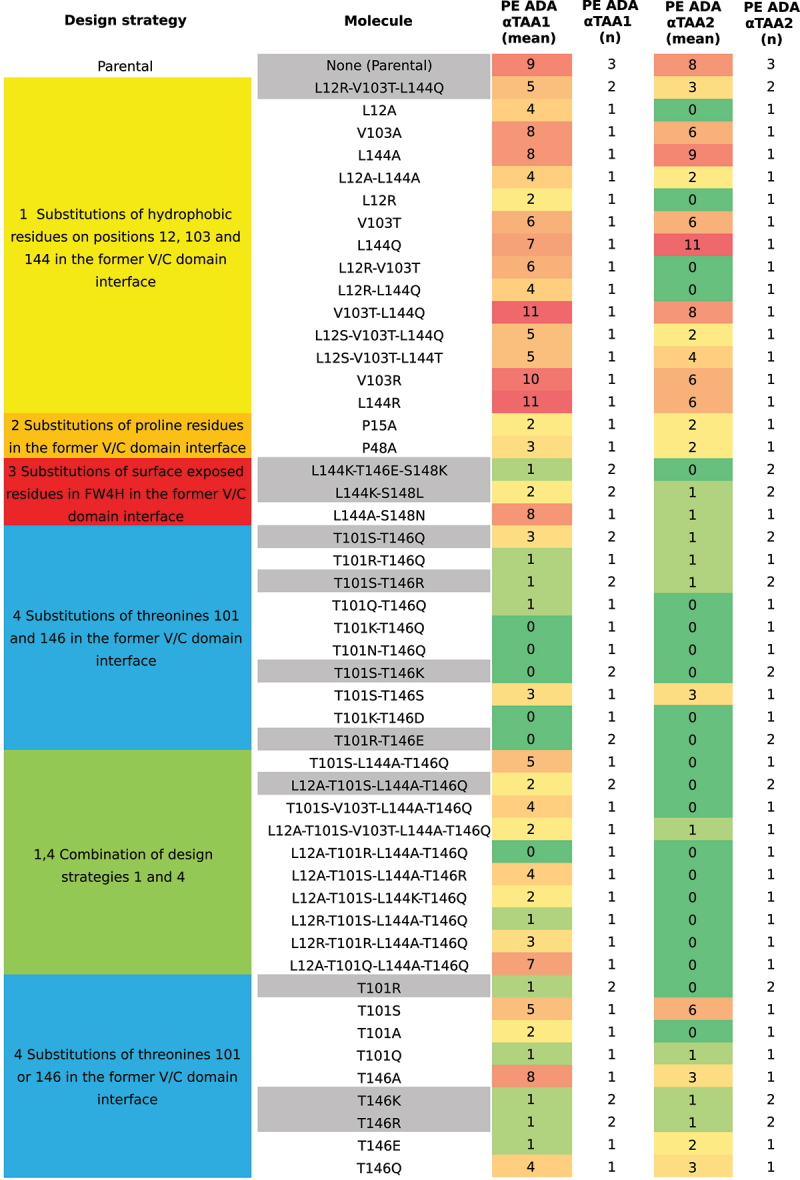
Design strategies, molecules, and PE ADA reactivities for the αTAA1 and αTAA2 scFvs investigated. Molecules are referred to by the mutations applied separated by hyphens.
Overview of the different design strategies to investigate PE ADA reactivity
An array of different molecules of the two αTAA scFvs have been designed, produced, and investigated (Figure 2). The following sections will describe these molecules and their PE ADA reactivities. The starting point for our study was the investigation of the importance of residues Leu12, Val103 and Leu144 for PE ADA reactivity since they were reported earlier to create the hydrophobic patch in the former V/C domain interface. Replacement of hydrophobic residues in the patch by more hydrophilic residues has been reported to increase production yield and decrease aggregation while not detrimentally affecting solubility and stability.17 There are also existing patents claiming that mutation of these residues into serines and/or threonines can lead to increased solubility22 and reduced immunogenicity.23 The mutations in our reference molecule (L12R-V103T-L144Q; 5 and 3 positive sera) are very effective in reducing the extent of the exposed hydrophobic patch (Figure 3B). However, these three mutations are not able to completely eliminate PE ADA reactivity. Therefore, the residues Leu12, Val103 and Leu144, and residues in close spatial proximity (Figure 4), were investigated in more detail, to potentially design molecules with completely eliminated PE ADA reactivity while maintaining developability and antigen-binding characteristics.
Figure 4.
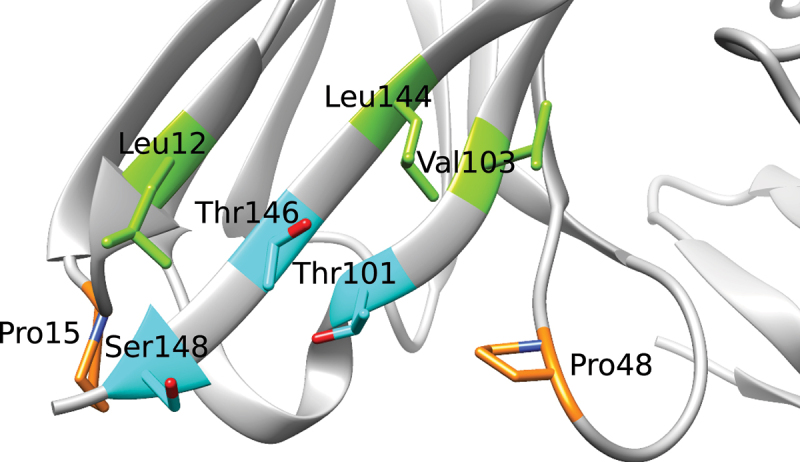
Close-up of the exposed region in the former (heavy) V/C domain interface with sidechains of mutated residues indicated using AHo numbering. The corresponding Chothia/Kabat/IMGT numbering are: Leu11/11/12, Pro14/14/15, Pro41/41/47, Thr87/87/99, Val89/89/101, Leu108/108/123, Thr110/110/125, and Ser112/112/127. Figure 2
In addition to Leu12, Val103 and Leu144, the effects of mutating the nearby prolines (Pro15 and Pro48),16 and the three exposed beta-strand residues (Leu144, Thr146, Ser148 [Figure 4 and Supplementary Figure 1]) at the C-terminus in framework region 4 of the heavy chain (FW4H) have been investigated. Furthermore, residues Thr101 and Thr146 have been investigated in detail. Residues Thr101 and Thr146 are surrounded by Leu12, the two prolines, the FW4H region residues described above, where Thr146 is positioned in the center of the exposed patch. The sidechains of both threonines are solvent exposed and could potentially influence PE ADA reactivity, despite not being hydrophobic amino acids. The two threonines were primarily mutated to amino acids with charged or polar side chains such as lysine, arginine, aspartic acid, glutamic acid, serine, asparagine, and glutamine. For many of the positions described above, a mutation to alanine was also tested as a strategy to reduce the hydrophobicity and/or bulkiness of the sidechain. The designed molecules should neither lead to new predicted stretches of T cell epitopes,24–27 nor critical post-translational motifs (PTM), i.e., N-glycosylation, aspartic acid isomerization (DG), deamidation (NG) or oxidation (M). Additionally, the mutations should not be predicted to destabilize the structure, nor should they lower the isoelectric point (pI). A total of 50 molecules (including parental and reference) were designed, produced, and analyzed for each αTAA scFv. The entire set of investigated molecules is shown in Figure 2, and the following sections describe the design strategies in more detail and the PE ADA reactivity results obtained for the designed molecules.
Substitutions of hydrophobic residues at positions 12, 103 and 144 (strategy 1)
At first, the effects of alanine substitutions at positions 12, 103 and 144 (Figure 4) were investigated, since replacing leucine and valine with a smaller hydrophobic amino acid such as alanine should reduce the hydrophobic patch and therefore agree with our initial hypothesis of achieving reduced PE ADA reactivity by reducing the hydrophobic patch. Of the three residues 12, 103 and 144, replacing Leu12 (L12A; 4 and 0 positive sera) has the largest effect with respect to reducing PE ADA reactivity, whereas alanine substitutions on 103 (V103A; 8 and 6 positive sera) and 144 (L144A; 8 and 9 positive sera) do not result in significantly reduced PE ADA reactivity. This finding suggests that, contrary to our initial hypothesis, a pure decrease in the size of the hydrophobic patch does not reduce PE ADA reactivity, but also that the effect is residue specific. The double alanine mutations at positions 12 and 144 (L12A-L144A; 4 and 2 positive sera) also lead to a reduction of PE ADA reactivity, which is identical or less pronounced than observed for L12A alone, depending on the αTAA scFv. Other double alanine mutation molecules have not been tested.
The results observed for the alanine substitution are confirmed by mutations to amino acids with charged or polar side chains. A single mutation to arginine (L12R; 2 and 0 positive sera) leads to a higher reduction of PE ADA reactivity than L12A, whereas V103T (6 and 6 positive sera), V103R (10 and 6 positive sera), L144Q (7 and 11 positive sera) and T144R (11 and 6 positive sera) do not lead to significant reductions. When analyzing double mutation molecules where residues 12, 103 and 144 have been replaced by charged or polar residues (L12R-V103T [6 and 0 positive sera], L12R-L144Q [4 and 0 positive sera], and V103T-L144Q [11 and 8 positive sera]), we found that no combination has a greater effect on PE ADA reactivity than the single mutation molecule L12R. Molecules with a double mutation that includes L12R perform slightly worse than the single mutation molecule L12R, and the combination which does not involve position 12 (i.e., V103T-L144Q) does not reduce the PE ADA reactivity at all. When analyzing triple mutation molecules for the same positions and with the same kind of replacements (charged or polar residues), the combinations with L12S, i.e., L12S-V103T-L144Q (5 and 2 positive sera) are similarly effective in reducing PE ADA reactivity as the reference molecule L12R-V103T-L144Q and the triple mutation molecule known from patents, L12S-V103T-L144T (5 and 4 positive sera). Furthermore, none of the triple mutation molecules are superior to L12R in reducing PE ADA reactivity.
In conclusion, the analysis of positions 12, 103 and 144 reveals a crucial role of Leu12 in the PE ADA reactivity. A single mutation to alanine or arginine leads to a substantial decrease in PE ADA reactivity. This is confirmed by double and triple mutation molecules involving position 12. Importantly, a single substitution at position 12 is responsible for the entire effect and additional substitutions at positions 103 and 144 do not further decrease the PE ADA reactivity. It appears that Leu12 is part of an epitope that PE ADAs bind to. Interestingly, Lin and coworkers15 claimed that Leu12 is not a major contributor to the identified PE ADA epitope since replacements of Leu12 by glutamic acid, glutamine or serine only led to minor reductions in PE ADA reactivity in their study. Although the present study does not contain any of these mutations, it does show a dramatic reduction of PE ADA reactivity caused by the L12R mutation.
Substitutions of proline residues (strategy 2)
Substitution of proline residues to alanine has been described previously, as a means to reduce PE ADA reactivity.16 Mutation of either of the two prolines (Figure 4) to alanine (P15A [2 and 2 positive sera], or P48A [3 and 2 positive sera]) is successful in reducing PE ADA reactivity (Figure 2).
Substitutions of exposed residues in FW4H (strategy 3)
In addition to the already described residues mentioned above, residues 146 and 148 are part of the same β-strand as Leu144, the central β-strand of the hydrophobic patch, and their side chains are solvent exposed (Figure 4). Due to the strong structural similarities of kappa light chains, lambda light chains and shark VHH domains (Supplementary Figure 1), we substituted corresponding residues from these domains at positions 144, 146 and 148. This created molecules L144K-T146E-S148K (kappa light chain inspired), L144K-(T146T)-S148L (lambda light chain inspired) and L144A-(T146T)-S148N (shark VHH inspired). The kappa light chain inspired mutations (L144K-T146E-S148K; 1 and 0 positive sera) and the lambda light chain inspired mutations (L144K-S148L; 2 and 1 positive sera) were successful in reducing PE ADA reactivity. Both molecules introduce a net positive charge in FW4H. The shark VHH inspired mutations (L144K-S148L; 8 and 1 positive sera), which introduce no charged residues, were only successful for the αTAA2 scFv. Taking into consideration that L144R does not lead to a reduction of PE ADA reactivity, the results suggest that Thr146 and/or Ser148 are part of the identified PE ADA epitope. For all other design strategies, mutation of Ser148 was avoided, since it is located sequentially close to the linker in multi-specific antibody formats such as a Multispecific Antibody-based Therapeutics by Cognate Heterodimerization (MATCH) molecule.21
Substitutions of threonines 101 and 146 (strategy 4)
Residues Thr101 and Thr146 are surrounded by Leu12 and the two prolines (Pro15, Pro48) and are positioned centrally in the exposed region (Figure 4). For this reason and following the observation above that Thr146 may be part of a common PE ADA epitope, Thr101 and Thr146 were investigated in further detail. The reason for the focus on double mutation molecules is the assumption that complete elimination of PE ADA reactivity through single mutations will only be possible through radical (non-conservative) modifications of the identified PE ADA epitope. These radical single mutations are likely to compromise the biophysical properties, whereas double mutations allow more subtle changes over a larger fraction of the epitope.
Based on in silico mutation calculations using Rosetta, 10 of 49 possible double mutation molecules were investigated for which the two threonines are replaced by any of the seven charged or polar residues, namely arginine, lysine, glutamic acid, aspartic acid, glutamine, asparagine, or serine. Of the 49 possible molecules, T101S-T146Q was predicted to be the most stable double mutation. For this reason, T101S-T146Q was used as a starting point to create 10 additional multi-mutation molecules. Four molecules were designed to explore potential additive effect(s) when introducing alanine, charged or polar residues at one or more of the positions 12, 103 and 144. Changes were made to one (T101S-L144A-T146Q), two (L12A-T101S-L144A-T146Q, T101S-V103T-L144A-T146Q) or three (L12A-T101S-V103T-L144A-T146Q) of the residues 12, 103 and 144 in the starting point (T101S-T146Q). Six additional multi-mutation molecules were constructed for which one or two positively charged residues and/or larger polar residues were introduced in L12A-T101S-L144A-T146Q. In total, nine single mutations at positions 101 or 146 were included to accumulate more information about the effect of various single mutations on PE ADA reactivity.
For alanine substitutions of the threonine, a significant decrease in PE ADA reactivity is observed in case of residue 101 (T101A; 2 and 0 positive sera), whereas at position 146, no effect is seen for the αTAA1 scFv and a partial effect is seen for the αTAA2 scFv (T146A; 8 and 3 positive sera). For glutamine substitutions, the situation is similar. At position 101, a significant reduction is observed (T101Q; 1 and 1 positive sera), whereas at position 146 the corresponding substitution showed only a partial effect (T146Q; 4 and 3 positive sera). Interestingly, a mutation to serine at position 101 shows a very small effect (T101S; 5 and 6 positive sera), presumably due to a similar polar side-chain size as threonine. Along this line, a mutation to an amino acid with a large and positively charged side chain, such as arginine, leads to an almost complete elimination of PE ADA reactivity (T101R; 1 and 0 positive sera). A mutation to an amino acid with a large and positively charged side chain is similarly effective at position 146 (T146R [1 and 1 positive sera], and T146K [1 and 1 positive sera]). Introducing an amino acid with a large and negatively charged side chain is also very effective at position 146 (T146E; 1 and 2 positive sera). The data clearly suggest that both Thr146 and Thr101 are directly involved in binding PE ADAs and therefore are part of the identified PE ADA epitope. As mentioned previously, Thr146 is more accessible to solvent in the scFv format than in the Fab format. For Thr101, on the other hand, there is no difference in solvent accessibility between Fab and scFv formats, but it evidently becomes more accessible to PE ADAs in the scFv format due to the absence of the constant domain. This illustrates that solvent accessibility is not a reliable estimator of PE ADA accessibility.
The findings for single substitutions translate very well to double mutations at positions 101 and 146. A complete elimination of PE ADA reactivity is observed for molecules harboring residues with charged side chains at both positions, i.e., T101K-T146D (0 and 0 positive sera) and T101R-T146E (0 and 0 positive sera). An amino acid with a positively charged side chain at position 101 or 146 in combination with a more conservative mutation at the other position is sufficient to completely eliminate PE ADA reactivity when using lysine (T101K-T146Q (0 and 0 positive sera) and T101S-T146K (0 and 0 positive sera)) and to almost completely eliminate PE ADA reactivity when using arginine (T101R-T146Q [1 and 1 positive sera], and T101S-T146R [1 and 1 positive sera]). Two molecules with conservative, non-charged substitutions show a complete (T101N-T146Q; 0 and 0 positive sera) or a nearly complete (T101Q-T146Q; 1 and 0 positive sera) elimination of PE ADA reactivity. Although T101K and T101N were not tested individually, other data in this study suggest an additive effect of mutations on PE ADA reactivity, which is not unexpected if both residues are part of a common PE ADA epitope. Using conservative mutations at both positions (T101S-T146S [3 and 3 positive sera] or T101S-T146Q [3 and 1 positive sera]) yields a substantial reduction of PE ADA reactivity. This is to some extent expected because of the similarities with respect to side-chain properties. The combination of substitutions at positions 101 and 146 and at positions 12, 103, or 144 worked particularly well for αTAA2 scFvs, and PE ADA reactivity is completely eliminated for all but one (L12A-T101S-V103T-L144A-T146Q; 2 and 1 positive sera) of the 10 multi-mutation molecules. The largest reductions of PE ADA reactivity for both αTAA scFvs are observed for L12A-T101S-L144A-T146Q (2 and 0 positive sera), L12A-T101R-L144A-T146Q (0 and 0 positive sera), L12A-T101S-L144K-T146Q (2 and 0 positive sera) and L12R-T101S-L144A-T146Q (1 and 0 positive sera). The remaining five multi-mutation molecules (T101S-L144A-T146Q, T101S-V103T-L144A-T146Q, L12A-T101S-L144A-T146R, L12R-T101R-L144A-T146Q and L12A-T101Q-L144A-T146Q) did show an intermediate effect (≥3 and 0 positive sera). For these molecules, it is unclear why no additive effect could be observed for αTAA1 scFvs.
PE ADA reactivities for individual donor sera
Non-preselected sera were used for the study to mimic a patient-like population. These sera exhibited different PE ADA reactivity against the tested molecules. In addition to the similarities between αTAA1 and αTAA2 scFv molecules shown in Figure 2, the two αTAA scFvs also exhibit considerable similarities with respect to donor serum response, as can be seen in Figure 5. Overall, the two different αTAA scFvs exhibit a similar pattern of PE ADA response, although αTAA1 scFvs exhibit slightly higher magnitude reductions than αTAA2 scFvs. Nonetheless, we found common substitutions that completely eliminate PE ADA reactivity for both αTAA scFvs. In the cases where more substantial differences between αTAA1 and αTAA2 scFvs are observed (also seen in Figure 2), these are very likely caused by differences in CDR sets.
Figure 5.

Heatmaps of the PE ADA reactivities from sera of 20 human donors to the parental and modified αTAA1 scFv molecules (below diagonals), and parental and modified αTAA2 molecules (above diagonals). The values in the heatmaps range from 0% (green) up to 100% (red), with all values ≤30% set to 0%. Corresponding heatmaps with numerical values for all cells are provided in Supplementary Figures 9 (αTAA1) and 10 (αTAA2). The color-coding of design strategies (leftmost column) is identical to that in Figure 2.
Interestingly, donor serum 20 and partially donor serum 7 showed the presence of PE ADA reactivity for the αTAA2 multi-mutation molecules, despite the absence of PE ADA reactivity for the parental molecule (Figure 5), suggesting that simultaneous modifications at too many residues may create a new PE ADA epitope.
Biophysical and functional measurements
In addition to the PE ADA reactivity results described above, binding affinity by surface plasmon resonance (SPR), thermal stability by nano differential scanning fluorimetry (nDSF), and hydrophobicity by hydrophobic interaction chromatography- high-performance liquid chromatography (HIC-HPLC) were assessed for all produced molecules to screen their biophysical properties (Supplementary Table 2).
The affinities of the αTAA scFvs to their respective target were screened by SPR. The parental αTAA1 scFv binds with an affinity of 48.8 pM (±23.8 pM) and the parental αTAA2 scFv binds with an affinity of 1.8 nM (±0.4 nM). The αTAA1 scFvs show binding affinities in the range 14–91 pM, and the αTAA2 scFvs show binding affinities in the range 0.7–3.3 nM. With our screening SPR setup analyzing three concentrations of the scFv (analyte), the observed affinity differences are not considered significant. In addition, there is no correlation between the introduced mutation and change in affinity for either αTAA scFv. Since the introduced modifications are distantly located from the CDRs, these results are not unexpected (Supplementary Figure 2).
Similarly, for either αTAA scFv, HIC-HPLC results indicate that mutations lead to very small variations in apparent hydrophobicity, despite hydrophobic amino acids being replaced by amino acids with charged or polar side chains in a majority of substitutions (Supplementary Figure 3).
For some mutations, a minor increase in hydrophobicity (<10% decrease in (NH4)2SO4 concentration) is observed for both αTAA scFvs. This concerns all molecules with a polar modification at position 101 and 146 (T101Q, T101Q-T146Q, T101N-T146Q and T101S-T146S) except T101S-T146Q. In addition, T101Q and T101R-T146E also lead to increased hydrophobicity.
The thermal stability of all molecules was assessed by nDSF. The αTAA1 scFvs exhibit a three-state unfolding (Supplementary Figures 4A and 4C) whereas the αTAA2 scFvs do not (Supplementary Figures 4B and 4D). However, for αTAA1 scFvs only the second transition at 80.7°C (referred to as Tm2) can be resolved for the parental molecule. However, a decreased Tm1 is observed for several molecules, permitting the first transition to be resolved, and revealing a lower stability of the VH domain compared to the VL domain. The first transition of the parental molecule was determined by non-linear regression using a three-state model at 74.0°C (referred to as Tm1). The αTAA2 scFvs show a clear two-state unfolding with a Tm at 69.0°C. Overall, the mutations affect the thermal unfolding similarly for both αTAA scFvs (Pearson correlation coefficient: 0.94) and lead to decreased thermal stabilities. However, in most cases, the effect is minor, i.e., a decrease of ≤2°C (Supplementary Figure 5).
A significant decrease in thermal stability was observed for all scFvs harboring an amino acid with a positively charged side chain at position 101 (e.g., T101R or T101K). This led on average to a decrease of 8.1°C, 3.7°C for Tm1, Tm2 of αTAA1 scFvs, and of 4.9°C for Tm of αTAA2 scFvs. The effect is less pronounced for other modifications at this site (e.g., T101S; Supplementary Figure 5). Rosetta calculations on our in-house structures predict T101R to be a clearly destabilizing mutation. The residue preceding Thr101, namely Asp100, interacts with Arg77 (Figure 6A) to form a highly conserved buried salt bridge in the center of the so-called charge cluster in the lower core region of VL and VH.28 It appears that the addition of an arginine or lysine at position 101 disrupts charge interactions formed by the residues surrounding Arg77 and Asp100, leading to compromised thermal stability for T101R (Figure 6B). The destabilizing effect can be partially reduced by introducing, at position 146, an amino acid with a negatively charged side chain (T101R-T146E or T101K-T146D), which results in a decrease of 5.2°C, 1.6°C for Tm1, Tm2 of αTAA1 scFv, and of 2.1°C for Tm of αTAA2 scFv. This effect may be due to a new salt bridge being formed between residue 101 and 146, leading to an increase in thermal stability.
Figure 6.
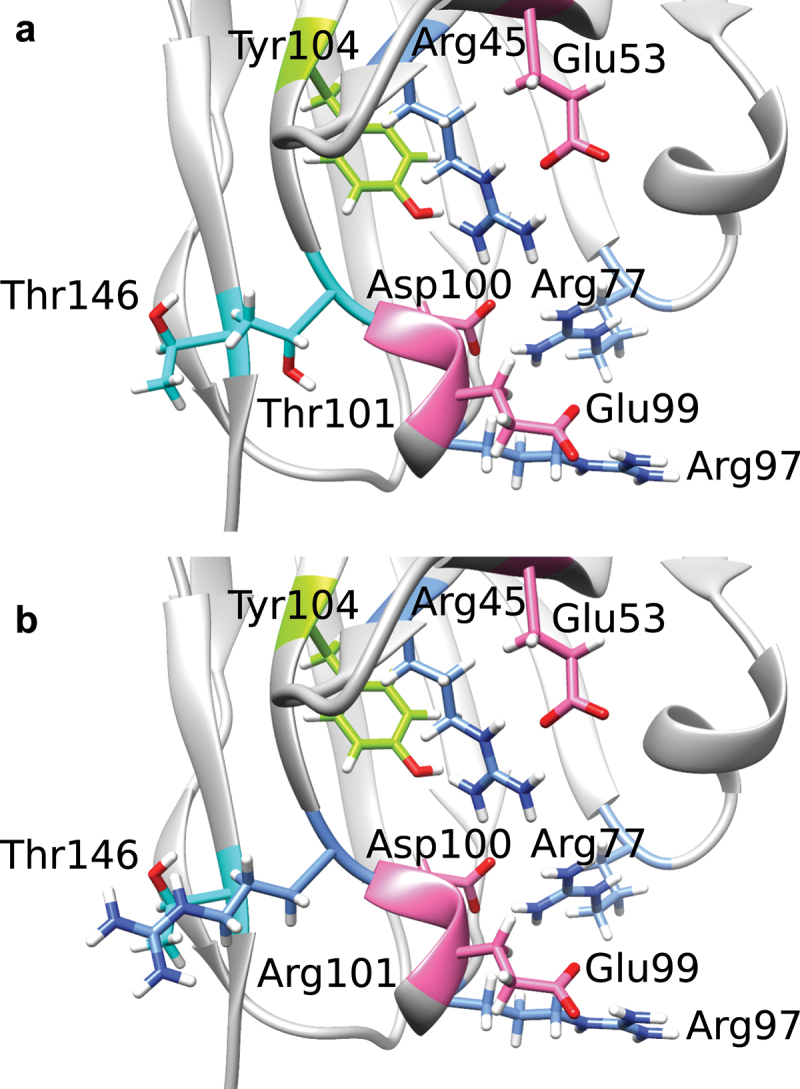
Residues surrounding the buried Arg77-Asp100 salt bridge forming the charge cluster in the lower core as well as Thr101 and Thr146 in the repacked Rosetta structure (a), based on crystal structure of αTAA1, and the resulting Rosetta structure after T101R has been applied (b).
Another molecule that exhibits reduced thermal stability is L12A-T101Q-L144A-T146Q (−6.7°C, −2.8°C for Tm1, Tm2 of αTAA1 scFv, and −3.8°C for Tm of αTAA2 scFv), which in this case appears to be an additive effect of the individual mutations (Supplementary Figure 5). As observed for the molecules just described, Tm1 of αTAA1 scFvs is the most sensitive to modifications. A medium effect on thermal stability is observed for molecules containing T101N or T101Q in combination with T146Q. Interestingly, T101S or T101S-T146Q does not show a substantial change in thermal stability. Overall, the data reveals that Thr101 is an important residue for thermal stability and its modification must be well chosen to not significantly reduce thermal stability.
Selection of a subset of molecules for an extended characterization
To further characterize the molecules that showed significantly reduced PE ADA reactivity, 12 molecules for each αTAA were chosen (hereafter referred to as selected molecules and indicated with a gray background below the keyword ‘Molecule’ in Figure 2). The parental molecule and L12R-V103T-L144Q (reference molecule) were selected to be available for comparisons. The light chain inspired molecules, L144K-T146E-S148K (kappa), and L144K-S148L (lambda) were selected based on their reduced PE ADA reactivity. Since molecules with double mutations at positions 101 and 146 have shown reduced PE ADA reactivity up to complete elimination, four such molecules were selected. T101S-T146Q was selected as it was predicted to be the most stable double mutation molecule. T101S-T146K and T101R-T146E were selected due to complete elimination of PE ADA reactivity and T101S-T146R was selected since it only exhibited one positive sera for each αTAA. Being among the most potent single mutation molecules in reducing PE ADA reactivity, the molecule T101R was selected despite its reduced thermal stability, since further characterization of this molecule should reveal whether a decreased thermal stability also leads to increased monomer loss after storage. T146K and T146R exhibited similarly low PE ADA reactivities and were therefore also selected. Finally, the best molecule that harbors mutations at positions 12, 103 or 144 as well as at positions 101 and 146, namely L12A-T101S-L144A-T146Q, was selected.
Short-term stability study
The subset of 12 selected molecules for each αTAA were subjected to an extended characterization, including a short-term stability study. Modified scFvs were compared to their parental scFv in their colloidal, conformational, and storage stability by means of PEG precipitation assay, chemical unfolding, thermal onset of aggregation and stability after 4 weeks of storage at 4°C and 40°C at 10 mg/ml in 20 mM histidine, pH 6. Within the stability study, samples were analyzed for content loss, monomer loss, formation of charge variants and fragmentation. For the extended characterization, selected molecules were purified, and the identity confirmed by MS.
For the parental αTAA1 scFv, no changes in protein concentration were observed after 4 weeks of storage at 4°C or 40°C (10 ± 1 mg/ml). For the parental αTAA2 scFv, an increase after 4 weeks of storage was observed (+1.8 mg/ml at 40°C, and +1.4 mg/ml at 4°C), most probably due to evaporation of water (Figures 7A and 7C). After 4 weeks of storage at 40°C, a higher loss of monomeric content was observed for the parental αTAA1 scFv (−4.4% of monomer content) than for the parental αTAA2 scFv (−1.3% of monomer content, Figure 7D). After 4 weeks of storage at 4°C, no significant loss of monomeric content was observed for either αTAA1 scFv (−0.4% of monomer content) or αTAA2 scFv (−0.1% of monomer content, Figure 7B).
Figure 7.
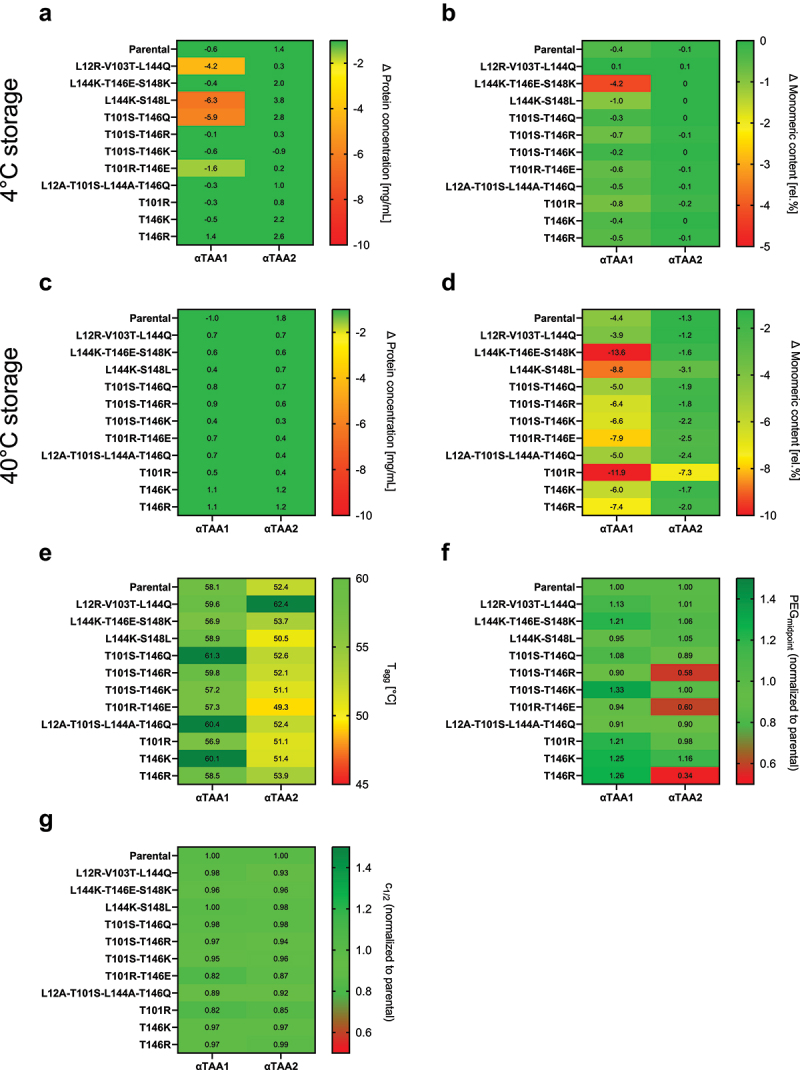
Data for extended characterization of the selected molecules. The obtained data is shown as heatmaps. Each analysis was color-coded to visualize liabilities of the tested molecules. Each cell is labeled with the corresponding value. A change in protein concentration after 4 weeks of storage at 4°C (a) and 40°C (c) of more than 2 mg/mL is regarded critical and colored yellow and red with increasing protein loss. A change in monomeric loss after 4 weeks of storage at 4°C (b) of more than 2% and at 40°C (d) of more than 8%, is regarded as a risk for developability and colored yellow and red with increasing monomer loss (d). The Tagg at a temperature below 50°C is regarded critical and colored in yellow and red with decreasing temperature (e). For the PEG precipitation assay (f) and the chemical unfolding (g), the obtained values of the PEGmidpoint and c1/2 were normalized to the respective value of the parental molecule. The absolute values can be found in the supplementary information. A reduction of the relative solubility by more than 20% is regarded as a significant decrease in colloidal stability. In case of the chemical unfolding, a reduction of c1/2 of more than 10% is regarded as a significant change in stability.
After 4 weeks of storage at 4°C, several αTAA1 scFv molecules showed a substantial decrease in protein concentration, namely the L12R-V103T-L144Q reference molecule (−4.2 mg/ml), L144K-S148L (−6.3 mg/ml) and T101S-T146Q (−5.9 mg/ml), while T101R-T146E showed a less pronounced decrease in concentration (−1.6 mg/ml) (Figure 7A and Supplementary Table 3). Interestingly, neither the molecule L12A-T101S-L144A-T146Q nor the corresponding αTAA2 scFv molecule showed signs of reduced solubility. After 4 weeks of storage at 40°C, none of the selected molecules showed any change in protein concentration (Figure 7C and Supplementary Table 3). The monomer stability at 4°C was significantly affected for the αTAA1 molecule L144K-T146E-S148K (−4.2% [Figure 7B and Supplementary Table 4]).
After 4 weeks of storage at 40°C, all molecules except L12R-V103T-L144Q exhibited a higher loss of monomeric content than the parental molecule (Figure 7D and Supplementary Table 4). For both αTAA, a substantial loss of monomeric content was observed for molecule T101R (−11.9% [αTAA1] and −7.3% [αTAA2]). Analogously to Tm values, this increased rate of aggregation can be partially reduced by an amino acid with a negatively charged side chain at position 146 (T101R-T146E: −7.9% [αTAA1], and −2.5% [αTAA2]). Another molecule exhibiting increased aggregation for both scFvs was L144K-S148L (−8.8% [αTAA1]; −3.1% [αTAA2]). The triple mutation molecule, L144K-T146E-S148K, exhibits the highest loss of monomer content (13.6%) for αTAA1 scFv, whereas for αTAA2 scFv the loss is only 1.6% in monomer content, which is similar to the parental molecule. Molecules T146K, T146R, T101S-T146R, and T101S-T146K exhibited only a minor increase of monomer loss after 4 weeks of storage at 40°C compared to their parental scFv (6.4–7.4% [αTAA1], 1.7–2.2% [αTAA2]).
The short-term stability data shows that modifications at the V/C domain interface can have a significant effect on stability, both at 4°C and at 40°C. In line with the thermal stability data above, the mutation of Thr101 to arginine leads to a substantially decreased monomer stability under stress conditions. Part of the stability can be regained by mutating Thr146 to glutamic acid. At 4°C, we observed that mutating Leu144 to glutamine, glutamic acid, or lysine, and/or mutating Ser148 to a leucine, possibly in combination with other mutations, can reduce solubility or monomer stability of αTAA1 scFvs in 20 mM histidine, pH 6 (Figures 7A and 7B). The data for 40°C shows that a modification at Leu144 and Ser148 leads to decreased stability, since an increased monomer loss has been observed for both αTAA scFvs (Figure 7D).
Formation of charge variants and fragmentation
The formation of charge variants was investigated after 4 weeks of storage at 40°C, by cation-exchange (CEX)-HPLC analysis. The starting target purity was 96.8% for the parental αTAA1 scFv and 82.3% for the parental αTAA2 scFv (Supplementary Table 5). After 4 weeks of storage at 40°C, the main peak area decreased by 0.3% for αTAA1 scFv molecules and 0.4% for the αTAA2 scFv molecules, indicating excellent chemical stability. Similar target purities as well as changes in purity over time were observed for most scFvs (Supplementary Table 5). The largest decrease in purity was observed for αTAA2 scFvs, namely L12A-T101S-L144A-T146Q (−5.7%), T101S-T146Q (−3.3%) and T101R-T146E (−2.9%). The selected molecules were further analyzed for fragmentation using reduced capillary gel electrophoresis (cGE). No significant increase in low molecular weight species (LMWS) could be observed after 4 weeks of storage at 40°C (Supplementary Table 6, LMWS formation ≤2%). The only molecule showing an increased amount of LMWS formation during storage was L144K-T146E-S148K (−3.7%). In summary, chemical stability was only affected to a minor extent.
Thermal onset of aggregation
The thermal propensity of aggregation was assessed for the selected molecules of each αTAA using the onset temperature of aggregation. Higher values of the onset temperature of aggregation (Tagg) were obtained for the αTAA1 parental molecule than for the αTAA2 parental molecule (58.1°C vs 52.9°C). The onset of aggregation is in general not affected by the introduced mutations (difference from parental ≤2°C, Figure 7E and Supplementary Figure 6). Interestingly, a significantly altered thermal onset of aggregation was observed for only the αTAA2 scFv molecule L12R-V103T-L144Q (Tagg of 62.4°C (+10°C)), but not for the corresponding αTAA1 scFv.
Solubility assessment by PEG precipitation assay
To investigate the effect of the described mutations on solubility, a PEG precipitation assay was performed to analyze the relative solubility. The parental αTAA1 scFv molecule showed a higher PEGmidpoint value than the parental αTAA2 scFv molecule (15% vs 21.3%, Figure 7F and Supplementary Figure 7). The solubility was similar for most molecules compared to the parental molecules. However, position 146 appears to be critical for solubility in the αTAA2 scFv. The highest solubility was observed for T146K (PEGmidpoint 24.7% PEG8000 (w/v)), whereas T146E (in T101R-T146E; PEGmidpoint 14.1% PEG8000 (w/v)) and T146R as well as T101S-T146R (PEGmidpoint 7.3% and 12.4% PEG8000 (w/v), respectively) led to the lowest solubilities. For αTAA1, on the other hand, mutations at this position generally lead to an improvement of solubility that is maximized for T101S-T146K. The data thus indicate that in individual cases solubility can be affected very differently depending on the CDRs.
Chemical unfolding
Chemical unfolding was analyzed using guanidinium HCl. Both αTAA scFvs show a two-state unfolding (c1/2 values of 3.9 M [αTAA1], 3.2 M [αTAA2], Figure 7G and Supplementary Figure 8). In this assay, T101R again has a destabilizing effect, both as a single mutation and in combination with another mutation (e.g., T101R-T146E; c1/2 values of 3.2 M [αTAA1], 2.8 M [αTAA2]). In addition, L12A-T101S-L144A-T146Q shows a decrease in stability (c1/2 values of 3.4 M [αTAA1], 3.0 M [αTAA2]) for both αTAA scFvs. All other selected molecules show comparable results, i.e., no change in stability is observed.
Discussion
In this work, a previously hidden region that becomes accessible for PE ADA binding in the scFv format was investigated. Two very different scFvs, with different target specificities, were used to investigate PE ADA reactivity. Four strategies were used to investigate positions 12, 15, 48, 101, 103, 144, 146 and 148. We could identify residues that are part of the binding interface for PE ADAs. From the analysis of positions 12, 103 and 144, it becomes obvious that position 12 is the only residue of the three that has a direct effect in reducing PE ADA reactivity. Positions 15 and 48 also have a direct effect toward reducing PE ADA reactivity, but mutations at positions 101 and 146 are the most effective for reducing PE ADA reactivity. Finally, mutations at position 148 also appear to reduce PE ADA reactivity. Taken together, this indicates that the PE ADA binding interface comprises Leu12, Pro15, Pro48, Thr101, Thr146 and Ser148, whereas, contrary to earlier reports,23 Val103 and Leu144 are less likely to be involved.
Based on the analysis of the individual amino acids, alanine and arginine are suitable substitutions at position 12 (L12A, L12R) for reducing PE ADA reactivity. For positions 15 and 48, only alanine substitutions have been tested (P15A, P48A), but the essential effect of these mutations may be increased loop flexibility. The threonine at position 101 can be replaced by alanine, serine, glutamine, and arginine/lysine (T101A, T101S, T101Q, T101R/K) with different effects on PE ADA reactivity, but higher reduction in PE ADA reactivity is seen for amino acids with larger side chains. High reduction in PE ADA reactivity is also seen for amino acids with larger side chains at position 146. For position 148, single mutants have not been tested, but the other results obtained suggest that mutations to larger amino acids are beneficial also here.
Since mutations to reduce PE ADA reactivity should not interfere with the developability potential of the molecules, all molecules were investigated for thermal stability, hydrophobicity, and affinity. Extended biophysical characterization was performed for a subset of selected molecules. As already mentioned, the parental scFvs were chosen based on differences in the CDR sets, the presence or absence of a VH-VL disulfide bridge as well as different grafting strategies. As a result, these scFvs also exhibit different biophysical properties and stabilities. The αTAA1 scFvs show a higher hydrophobicity by HIC-HPLC and slightly lower solubility in the PEG screen. On the other hand, αTAA1 scFvs exhibit higher unfolding midpoints, higher onset of aggregation and higher midpoint of unfolding using denaturants. This emphasizes that the observed effects in the reduction of PE ADA arise purely from the interaction with donor sera antibodies and are not due to generic instabilities of the molecules.
Five double mutation molecules were identified that completely eliminate PE ADA reactivity (T101K-T146Q, T101N-T146Q, T101S-T146K, T101K-T146D and T101R-T146E). Among these, T101S-T146K and T101R-T146E were selected for the extended biophysical characterization. With all experimental data taken together, T101S-T146K (0 and 0 positive sera) is the best mutation since PE ADA reactivity is completely eliminated while favorable developability and antigen-binding properties are maintained (Figure 8). By comparison, T101R-T146E has a slightly reduced thermal stability, earlier unfolding by denaturant, and exhibits reduced solubility after storage at 4°C or in the PEG screen. T101S-T146R (1 and 1 positive sera) also exhibited mostly very good biophysical properties as observed for T101S-T146K, but shows reduced solubility in the PEG screen. This effect is probably caused by the arginine at position T146, as it is also observed for the single mutation T146R. The remaining double mutation molecule subjected to the extended biophysical characterization, T101S-T146Q, shows substantial loss in monomer content after storage at 4°C.
Figure 8.
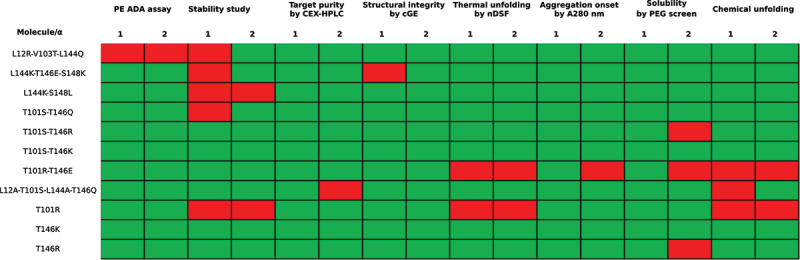
Evaluation of mutations for integration into human scFv acceptor framework. For each assessment, an acceptance criterium was chosen. These were set as follows. PE ADA assay: Sum of positive sera for both αTAA scFvs ≤3. Stability study (after 4 weeks at 4°C and 40°C): Loss in protein concentration ≤2 mg/ml, loss of monomeric content ≤2× loss observed for parental molecule; Target purity: Loss in purity ≤5% after 4 weeks at 40°C. Thermal unfolding Tm: ≥ −2°C compared to parental molecule. Onset temperature of aggregation, Tagg: ≥ −2°C compared to parental molecule. Solubility in PEG screen: PEGmidpoint ≥0.8* PEGmidpoint of parental molecule. Chemical unfolding: c1/2 ≥ 0.9*c1/2 of parental molecule. Box is marked green when the criterium is met and red when the criterium is not satisfied.
The SAP scores mapped for T101S-T146K (0 and 0 positive sera) molecules are shown in Figure 3C. Comparisons with L12R-V103T-L144Q (5 and 3 positive sera) shown in Figure 3B, and the fact that PE ADA reactivities differ significantly indicate that reduction of the hydrophobic patch does not in itself lead to effective reduction of PE ADA reactivity. T101S alone is not very successful in reducing PE ADA reactivity (5 and 6 positive sera), but T146K (1 and 1 positive sera) is almost as good as T101S-T146K in reducing PE ADA reactivity.
Unexpectedly, the very similar molecule T146R is an unfavorable single mutation molecule since it exhibits lower solubility in the PEG screen. This indicates that it may be difficult (if at all possible) to find a single mutation that is able to completely eliminate PE ADA reactivity without compromising biophysical properties.
In the extended biophysical characterization, molecules arising from different strategies than substitution at positions 101 and 146 were also tested in order to see their effects on stability, even though they do not show complete PE ADA elimination. For example, the reference molecule L12R-V103T-L144Q showed precipitation after storage at 4°C in 20 mM histidine, pH 6. The insertion of lambda or kappa light chain sequences triggered increased oligomerization after storage at 40°C, and for αTAA1 scFvs also after storage at 4°C storage. Combination of strategies one and four in L12A-T101S-L144A-T146Q showed a slight increase in charge variants after 40°C storage, as well as unfolding at lower denaturant concentrations.
As mentioned above, substitutions at positions 101 and 146 can lead to complete elimination of detectable PE ADA reactivity. This suggests that, at least for the two chosen CDR sets, the exposed region in the former V/C domain interface is an exclusive binding epitope for PE ADA. It is possible that an increase in the number of investigated sera leads to the identification of new antibodies or that the observations are specific for the VH3 framework. However, other frameworks share sequence similarities at the mentioned positions. In particular, T101S-T146K, with its simplicity (only two mutations) and beneficial properties, is a double mutation that may be broadly applicable to antibody variable fragments. Since, as demonstrated, the application of T101S-T146K mutations was beneficial for two scFvs with different binding and biophysical properties, it stands to reason that their application is likely to also be beneficial in other scFvs.
Materials and methods
Proteins
The two αTAA scFvs used in this study were selected because they have different binding specificities. The use of more than one scFv created diversity in the CDR with the purpose of reducing bias from CDR specific effects, e.g., with respect to PE ADA reactivity and stability. The identity between the two αTAA scFvs is 80.9% over the whole region and 44.6% over the CDRs. Non-modified αTAA scFvs are referred to as parental molecules throughout the study and all numbering of residues and sequence positions are according to the Honegger (AHo) numbering scheme.29 Modified αTAA scFvs are referred by the mutations applied to obtain them, separated by hyphens (e.g., L12R-V103T-L144Q, which is the reference molecule of this study). The two CDR sets have rabbit origin and were reformatted into a human Vk1-VH3 consensus framework stabilized by a so-called lambda (λ)-cap™ of the FW4L.21 In addition, for the αTAA1 scFv there are four rabbit back-mutations in the interface region of the framework and an additional disulfide bridge, whereas for αTAA2 scFv, being a standard “CDR graft”, there are neither any additional rabbit back-mutations in the framework nor any additional non-canonical disulfide bridges. These two αTAA scFvs were considered suitable for this extensive experimental study because they can be purified to a monomeric content of >95% by a generic process, but still differ in their biophysical parameters. In total, 50 molecules were designed, produced, and characterized (Figure 2) for each αTAA scFv.
Protein expression and purification
Expression plasmids of the scFvs were purchased at GeneUniversal using the pcDNA 3.1 backbone (ThermoFisher Scientific). The expression was performed in Chinese hamster ovary (CHO) cells using the ExpiCHO™ Expression System (ThermoFisher Scientific). Expression was conducted according to manufacturer instructions. Proteins were purified from clarified harvest using affinity chromatography (Protein A and/or Protein L) using PrismA and/or Capto-L resin (Cytiva). When necessary, scFvs were polished by size-exclusion chromatography (SEC) using a Superdex 75(Cytiva) to a final monomeric content >95%.
PE ADA reactivity
PE ADAs were detected by analysis of their interactions with coated scFv. Specificity of the interaction was assessed by the competition of the coated scFv with the soluble scFv. For coating, the scFvs were diluted in phosphate-buffered saline (PBS), pH 7.4 (Lonza, #BE17-517Q) to a concentration of 100 ng/ml and 25 μl per well were applied onto a 96-well half-area plate (Greiner Bio-One). After incubation for 2 h at room temperature, the wells were blocked for 1 h at room temperature with 150 µL of blocking buffer (PBS +0.1% Tween 20 [AppliChem, #A4974] + 1% BSA [Sigma-Aldrich, #A3294]). The PE ADA reactivity was tested with 20 individual normal human sera from 10 females and 10 males (Dunn Labortechnik). For the direct interaction, the human sera were diluted 20-fold using low cross buffer (Candor, #100 500) and 25 μl applied onto the coated wells at room temperature for 15 min. For the competition with soluble scFv, the human sera were diluted 10-fold with low cross buffer, mixed 1:1 with the corresponding scFv (diluted to 67 nM in low cross buffer) and incubated for 1 h at room temperature prior to applying to the immobilized scFv as stated above. The minimal required dilution of the assay was at 1:20. For detection, an anti-human Fc-IgG-HRP (Jackson ImmunoResearch, 309-035-008) diluted in low cross buffer to 100 ng/ml was applied and incubated for 1 h at room temperature. 3,3’, 5,5”-tetramethylbenzidine (SeraCare, #5120–0083) was added and the reaction stopped with 1 M H2SO4 (AppliChem, #212176) after 15 min at room temperature. Absorbance was measured at 450 nm using a Tecan plate reader. Between each step, plates were washed three times (PBS, 0.005% Tween 20, pH 7.4). Plates are incubated on a “belly dancer shaker” at 40 rpm.
Thermal stability by nano differential scanning fluorimetry
Thermal unfolding of the molecules was assessed by nDSF using a Prometheus device (Nanotemper Technologies). All proteins were present at 1 ± 0.1 mg/ml in 20 mM Histidine, pH 6 (Carl Roth, #3852). Melting curves were generated from 20°C to 95°C with a 1°C/min increase. Data was analyzed by the PR.ThermControl v2.3.1 and PR.Stability v1.1 software. Most molecules with αTAA1 specificity showed two thermal midpoints. However, in most cases, the two unfolding events could not be resolved and only the main unfolding event was assessed using the first derivative of the fluorescence ratio (350 nm/330 nm). When possible, both midpoints were assessed.
Binding affinity by surface plasmon resonance
For the determination of the binding kinetics and affinity of the molecules, human TAA1-Fc (Sino Biological) and human TAA2 (Acro Biosystems) were immobilized at the surface density of 100–500 Response Units (RU) on a new CMD200M SPR sensor prism chip (Xantec) in an SPR 24 system (Sierra Sensor – Bruker) with standard mine coupling. The samples were then injected at concentrations of 10 nM, 2 nM and 0.4 nM over the ligand-immobilized and reference spots. The wild type molecules were used as a control, while an empty spot was used as reference. After each cycle with analyte, the surface was regenerated with a 3 M MgCl2 solution, thereby allowing the next analyte cycle to have only free ligand available. Dissociation constants (KD) were globally calculated by the Bruker SPR software based on the binding association (kon) and dissociation (koff) rate, by fitting a 1:1 Langmuir model to the curves obtained by the cycles with the three different concentrations of the analyte.
Hydrophobicity by HIC-HPLC
ScFvs were tested for their hydrophobicity using HIC-HPLC applying a Pro-Pac HIC-10 column (5 µm, 300 Å, 4.6 × 100 mm, ThermoFisher Scientific, #063655). 20 μg of protein are injected and eluted from the column using a gradient from 1.5 M to 0 M (NH4)2SO4 (AppliChem, #A1032) in 20 mM Histidine, pH 5.5 (Carl Roth, #3852 & #9455) in 12 min with a flow of 1 ml/min. The retention time is subsequently correlated to the respective (NH4)2SO4 concentration. For the determination of the (NH4)2SO4 concentration, a delay volume of 2.81 ml was applied to account for column volume and dead volume of the system.
Short-term stability study
The scFvs were dialyzed into 20 mM histidine, pH 6 (Carl Roth, #3852 & #9455) and concentrated using Amicon Centrifugal filters (10 kDa MWCO, Merck Millipore) to 10 mg/ml. Concentrated samples were analyzed by analytic SEC, analytical CEX and cGE. Samples were stored at refrigerated conditions (2–8°C, no humidity control) and at 40°C (75% rel. humidity) in Eppendorf Safe Lock Tubes. Samples were analyzed after 2 and 4 weeks for their protein content and monomeric content. Samples stored at 40°C for 4 weeks were also analyzed for charge molecule distribution using CEX-HPLC and fragmentation using cGE.
Concentration and monomeric content determination by size-exclusion HPLC
Concentration and monomeric content determination of the scFvs after purification and during the stability study was assessed using analytical size-exclusion HPLC (SE-HPLC). Samples were applied at their present concentration. A 5 μg of scFv were injected onto a Protein KW402.5-4F column (Shodex, #F6989201) using a Hitachi Chromaster HPLC system at 25°C and a flow rate of 0.35 ml/min. The mobile phase consisted of 50 mM sodium acetate, 250 mM sodium chloride, pH 6.0. The elution profile was monitored at 280 nm.
Charge variants by CEX-HPLC
Charge variants were assessed during the stability study using analytical cation exchange chromatography. A 30 μg of scFv were injected onto a MabPac SCX-10 (ThermoFisher Scientific, #075602) using a Hitachi Chromaster HPLC system. Main peak and charge molecules were eluted applying a pH gradient from pH 4 to pH 11 within 15 min and a flowrate of 0.85 ml/min at 25°C. Composition of the mobile phase has previously been reported and consist of 15.6 mM CAPS (Sigma-Aldrich, #C2632), 9.4 mM CHES (AppliChem, #A1065), 4.6 mM TAPS (Sigma-Aldrich, #M8389), 9.9 mM HEPPSO (Molekula, #44974537), 8.7 mM MOPSO (Sigma-Aldrich, #M8389), 11 mM MES (AppliChem, #A0689), 13 mM acetic acid (AppliChem, #361008), and 9.9 mM formic acid (Honeywell, #09676). pH was adjusted to 4 and 11 using NaOH.30 Species eluting prior to or after the main peak were grouped as acidic and basic variants, respectively.
Structural integrity by cGE
cGE was assessed using the Protein Express Chip on the LabChip GX Touch HT system (PerkinElmer, #760499) according to the manufacturer’s protocol. The scFvs were diluted to 1 mg/ml with 20 mM histidine, pH 6 (Carl Roth, #3852 & #9455), denatured at 70°C for 10 min and analyzed under non-reducing and reducing conditions. Data was integrated using the LabChip GX Reviewer Software (Perkin Elmer).
Thermal onset of aggregation
The thermal onset of aggregation was determined using the backscattering optics of a Prometheus device (Nanotemper technologies). Samples, present in 20 mM histidine, pH 6.0 (Carl Roth, #3852 & #9455) at 10 mg/ml, were heated to 95°C with 1°C/min. Samples were measured in duplets. The onset was determined automatically with the ThermControl software (Nanotemper Technologies).
Solubility assessment by polyethylene glycol (PEG) precipitation assay
Selected molecules were tested for their relative solubility using PEG-induced precipitation. ScFvs were incubated at 1 mg/ml in 20 mM histidine, pH 6.0 (Carl Roth, #3852 & #9455) with increasing PEG8000 concentrations from 0% (w/v) to 33.3% (w/v) in 12 non-linear steps. After incubation overnight, the supernatant was filtered (1.2 μm Supor filter, Pall), and remaining protein content was determined. The protein concentration was plotted against the logarithm of the PEG8000 concentration, and the data fitted using a 4-parameter logistic sigmoidal fit. The PEG8000 concentration at which 50% protein precipitation was observed has been reported.
Chemical unfolding by guanidinium-HCl
For chemical unfolding experiments, scFvs were incubated in 24 solutions ranging from 1.5 M to 5 M guanidinium-HCl in 20 mM histidine pH 6.0 (Carl Roth, #3852 & #9455). Final protein concentration was 0.5 mg/ml. Samples were allowed to reach equilibrium overnight at room temperature and were measured for their intrinsic fluorescence at 330 nm and 350 nm using a Prometheus device (Nanotemper Technologies). The change in fluorescence ratio (350 nm/330 nm) was used to determine both c1/2 and ΔGunfolding using the PR.Stability v1.1 software (Nanotemper Technologies).
Mass spectrometry
The mass of the scFvs analyzed in the extended biophysical characterization was verified by electrospray ionization-mass spectrometry (ESI-MS). Samples were 5-fold diluted with 1% trifluoroacetic acid and transferred to an autosampler vial for LC/MS. 2 μl of sample were injected into an ACQUITY UPLC@ BioResolve-RP-mAb 2.7 μm 2.1 × 150 mm, 450 Å column (Waters, USA) and desalted using a gradient from 15% to 85% buffer B (0.1% formic acid, 25% propan-2-ol in acetonitrile) at a flow rate 200 μl/min at 50°C. The analysis was performed on a Synapt G2 mass spectrometer directly coupled to the UPLC station. Mass spectra were acquired in the positive-ion mode by scanning the m/z range from 400 to 5000 da with a scan duration of 1 s and an interscan delay of 0.1 s. The spray voltage was set to 3 kV, the cone voltage to 50 V, and the source temperature to 100°C. The data were recorded with the MassLynx 4.2 Software (Waters, UK). Where possible, the recorded m/z data of single peaks were deconvoluted into mass spectra by applying the maximum entropy algorithm MaxEnt1 (MaxLynx) with a resolution of the output mass 0.5 Da/channel and Uniform Gaussian Damage Model at the half height of 0.7 Da.
Computational procedures
The spatial aggregation propensity (SAP) score, which is a measure of the surface-exposed hydrophobicity, was calculated using the methods of Chennamsetty31 as implemented in Discovery Studio 2020/2022.32,33 Per-residue aggregation propensity scores were calculated as the average of per-atom aggregation propensity scores over all atoms of each corresponding residue. Per-atom aggregation propensity scores, in turn, were obtained from solvent accessible surface (SAS) values by evaluating the product of the fractional SAS (the ratio of the actual side chain SAS to the SAS of side-chain atoms in a fully exposed residue of the same type) and the residue hydrophobicity,34 for all residues within the cutoff radius of each atom (5 and 10 Å cutoff radii were used in the calculations), with SAS values calculated for the side chain of each residue using CHARMm.35
Four proprietary inhouse crystal structures were prepared for Rosetta ΔΔG calculations by removing non-protein compounds including waters and ions, and renumbering residues to start at one and proceed using consecutive numbers. After this, input structures were subjected to energy minimization using harmonic distance constraints on Cα atoms within 9 Å of one another. The ΔΔG prediction calculations themselves were performed by the ddg_monomer application provided in Rosetta (version 2020.08.61164) using the high-resolution protocol,36 and calculating 50 structures for each mutation. For each mutation or combination of simultaneous mutations, the ddg_monomer application estimates ΔΔG as the difference between the mean of the three top scoring parental structures and the mean of the three top scoring mutant structures.
MHC class II binding prediction was carried out using tools developed at the Immune Epitope Database (IEDB), assembled into a computational pipeline using in-house scripts that is fully compliant with IEDB guidelines for identification of promiscuous peptides.24–27
Homology models were generated using the Antibody Modelling Cascade protocol of Discovery Studio 2020/2022.32,33 Templates were assembled from a set of non-redundant antibody structures from the RCSB PDB database37 augmented with eight proprietary inhouse structures, by applying the Discovery Studio Antibody Modelling Cascade protocol using the top 5 framework construction method with a 15% sequence similarity cutoff, and with the number of framework models set to 10, number of CDR models set to 10, and maximum templates per loop set to 5.38
Graphical representations were generated with UCSF Chimera.39,40
Supplementary Material
Acknowledgments
The authors want to thank Wei Xu and Xingguang Cai for critically reviewing the manuscript. Boris Abramovic is thanked for his support in the laboratory.
Funding Statement
The author(s) reported that there is no funding associated with the work featured in this article.
Author contributions
Conceptualization – MUJ, CW, CH, SW; Investigation – MUJ, CW, DAR, DM, DD, DF, SC, JT, NG, FMS, SW; Original draft preparation – MUJ, CW; Figures and visualizations – MUJ, CW; Review and editing – MUJ, CW, FMS, JT, DU, SW.
Abbreviations
ADA: anti-drug antibody; AHo: Honegger; CDR: complementary-determining region; CEX-HPLC: cation-exchange HPLC; cGE: capillary gel electrophoresis; CHO: Chinese hamster ovary; nDSF: nano differential scanning fluorimetry; ESI-MS: electrospray ionization-mass spectrometry; Fab: fragment antigen-binding; FW4H: framework region 4 of the heavy chain; FW4L: framework region 4 of the light chain; HIC: hydrophobic interaction chromatography; HPLC: high-performance liquid chromatography; IEDB: Immune Epitope Database; IgG: immunoglobulin G; LMWS: low molecular weight species; MATCH: Multispecific Antibody-based Therapeutics by Cognate Heterodimerization; PBS: phosphate-buffered saline; PD: pharmacodynamics; PE: preexisting; PEG: polyethylene glycol; pI: isoelectric point; PTM: post-translational motif; SAP: spatial aggregation propensity; SAS: solvent accessible surface; scFv: single-chain variable fragment; SEC: size-exclusion chromatography; SE-HPLC: size-exclusion HPLC; SPR: surface plasmon resonance; TAA: tumor-associated antigen; Tagg: onset temperature of aggregation; TE: treatment-emergent; V/C: variable/constant; VH: variable heavy; VL: variable light; VHH: variable domain of heavy chain of heavy chain-only antibody
Disclosure statement
This research was funded by Numab Therapeutics AG. The authors were all employed by Numab Therapeutics AG at the time when the study was conducted. Current affiliation of Dietrich Alexander Reichardt: F. Hoffman-La Roche AG, Basel, Switzerland. Current affiliation of Dana Mahler and Diana Diem: GlycoEra AG, Wädenswil, Switzerland
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2023.2215887.
References
- 1.Kaplon H, Chenoweth A, Crescioli S, Reichert JM.. Antibodies to watch in 2022. MAbs. 2022;14:2014296. PMID: 35030985. doi: 10.1080/19420862.2021.2014296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carter PJ, Rajpal A. Designing antibodies as therapeutics. Cell. 2022;185:2789–16. PMID: 35868279. doi: 10.1016/j.cell.2022.05.029. [DOI] [PubMed] [Google Scholar]
- 3.Lou H, Cao X. Antibody variable region engineering for improving cancer immunotherapy. Cancer Commun (Lond). 2022;42(9):804–27. PMID: 35822503. doi: 10.1002/cac2.12330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harding FA, Stickler MM, Razo J, DuBridge RB. The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. MAbs. 2010;2:256–65. PMID: 20400861. doi: 10.4161/mabs.2.3.11641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xue L, Fiscella M, Rajadhyaksha M, Goyal J, Holland C, Gorovits B, Morimoto A. Pre-existing biotherapeutic-reactive antibodies: survey results within the American Association of Pharmaceutical Scientists. Aaps J. 2013;15(3):852–55. PMID: 23620231. doi: 10.1208/s12248-013-9492-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guidance for industry: immunogenicity assessment for therapeutic protein products. 2014. Date.
- 7.Immunogenicity information in human prescription therapeutic protein and select drug product labeling —Guidance for industry. 2022. Date.
- 8.Kumar SC, DelCarpini JA, Qu Q, Kane M, Gorovits B. Mitigation of pre-existing antibodies to a biotherapeutic in non-clinical species when establishing anti-drug antibody assay cutpoint. Aaps J. 2017;19(1):313–19. PMID: 27873117. doi: 10.1208/s12248-016-0011-2. [DOI] [PubMed] [Google Scholar]
- 9.Schneider AK, Vainshtein I, Roskos LK, Chavez C, Sun B, Liang M. An immunoinhibition approach to overcome the impact of pre-existing antibodies on cut point establishment for immunogenicity assessment of moxetumomab pasudotox. J Immunol Methods. 2016;435:68–76. PMID: 27220271. doi: 10.1016/j.jim.2016.05.007. [DOI] [PubMed] [Google Scholar]
- 10.Shankar G, Arkin S, Cocea L, Devanarayan V, Kirshner S, Kromminga A, Quarmby V, Richards S, Schneider CK, Subramanyam M, et al. Assessment and reporting of the clinical immunogenicity of therapeutic proteins and peptides—Harmonized terminology and tactical recommendations. Aaps J. 2014;16(4):658–73. PMID: 24764037. doi: 10.1208/s12248-014-9599-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Falkenburg WJ, van Schaardenburg D, Ooijevaar-de Heer P, Tsang ASMW, Bultink IE, Voskuyl AE, Bentlage AE, Vidarsson G, Wolbink G, Rispens T. Anti-hinge antibodies recognize IgG Subclass– and protease-restricted neoepitopes. J Immunol. 2017;198(1):82–93. PMID: 27864476. doi: 10.4049/jimmunol.1601096. [DOI] [PubMed] [Google Scholar]
- 12.Bivi N, Moore T, Rodgers G, Denning H, Shockley T, Swearingen CA, Gelfanova V, Calderon B, Peterson DA, Hodsdon ME, et al. Investigation of pre-existing reactivity to biotherapeutics can uncover potential immunogenic epitopes and predict immunogenicity risk. MAbs. 2019;11(5):861–69. PMID: 31099718. doi: 10.1080/19420862.2019.1612699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.BEOVU® (brolucizumab-dbll) prescribing information . Available Date: https://www.novartis.com/us-en/sites/novartis_us/files/beovu.pdf (accessed December 24).
- 14.Holland MC, Wurthner JU, Morley PJ, Birchler MA, Lambert J, Albayaty M, Serone AP, Wilson R, Chen Y, Forrest RM, et al. Autoantibodies to Variable Heavy (VH) chain ig sequences in humans impact the safety and clinical pharmacology of a VH domain antibody antagonist of TNF-α receptor 1. J Clin Immunol. 2013;33(7):1192–203. PMID: 23832582. doi: 10.1007/s10875-013-9915-0. [DOI] [PubMed] [Google Scholar]
- 15.Lin J, Lee SL, Russell AM, Huang RF, Batt MA, Chang SS, Ferrante A, Verdino P, Henry KA. A structure-based engineering approach to abrogate pre-existing antibody binding to biotherapeutics. PLos One. 2021;16(7):e0254944. PMID: 34297759. doi: 10.1371/journal.pone.0254944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cordy JC, Morley PJ, Wright TJ, Birchler MA, Lewis AP, Emmins R, Chen YZ, Powley WM, Bareille PJ, Wilson R, et al. Specificity of human anti-variable heavy (VH) chain autoantibodies and impact on the design and clinical testing of a VH domain antibody antagonist of tumour necrosis factor-α receptor 1. Clin Exp Immunol. 2015;182(2):139–48. PMID: 26178412. doi: 10.1111/cei.12680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nieba L, Honegger A, Krebber C, Pluckthun A. Disrupting the hydrophobic patches at the antibody variable/constant domain interface: improved in vivo folding and physical characterization of an engineered scFv fragment. Protein Eng. 1997;10(4):435–44. PMID: 9194169. doi: 10.1093/protein/10.4.435. [DOI] [PubMed] [Google Scholar]
- 18.Gibson TJ, McCarty K, McFadyen IJ, Cash E, Dalmonte P, Hinds KD, Dinerman AA, Alvarez JC, Volkin DB. Application of a high-throughput screening procedure with PEG-induced precipitation to compare relative protein solubility during formulation development with IgG1 monoclonal antibodies. J Pharm Sci. 2011;100(3):1009–21. PMID: 21280052. doi: 10.1002/jps.22350. [DOI] [PubMed] [Google Scholar]
- 19.Haverick M, Mengisen S, Shameem M, Ambrogelly A. Separation of mAbs molecular variants by analytical hydrophobic interaction chromatography HPLC: overview and applications. MAbs. 2014;6:852–58. PMID: 24751784. doi: 10.4161/mabs.28693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Temel DB, Landsman P, Brader ML. Orthogonal methods for characterizing the unfolding of therapeutic monoclonal antibodies: differential scanning calorimetry, isothermal chemical denaturation, and intrinsic fluorescence with concomitant static light scattering. Methods Enzymol. 2016;567:359–89. PMID: 26794361. doi: 10.1016/bs.mie.2015.08.029. [DOI] [PubMed] [Google Scholar]
- 21.Egan TJ, Diem D, Weldon R, Neumann T, Meyer S, Urech DM. Novel multispecific heterodimeric antibody format allowing modular assembly of variable domain fragments. MAbs. 2017;9:68–84. PMID: 27786600. doi: 10.1080/19420862.2016.1248012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borras L, Urech D. 2009. WO 2009/155725.
- 23.Borras L, Gunde T, Urech D. 2011. WO 2011/075861.
- 24.Andreatta M, Karosiene E, Rasmussen M, Stryhn A, Buus S, Nielsen M. Accurate pan-specific prediction of peptide-MHC class II binding affinity with improved binding core identification. Immunogenet. 2015;67:641–50. PMID: 26416257. doi: 10.1007/s00251-015-0873-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andreatta M, Trolle T, Yan Z, Greenbaum JA, Peters B, Nielsen M, Berger B. An automated benchmarking platform for MHC class II binding prediction methods. Bioinf. 2018;34(9):1522–28. PMID: 29281002. doi: 10.1093/bioinformatics/btx820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greenbaum J, Sidney J, Chung J, Brander C, Peters B, Sette A. Functional classification of class II human leukocyte antigen (HLA) molecules reveals seven different supertypes and a surprising degree of repertoire sharing across supertypes. Immunogenet. 2011;63:325–35. PMID: 21305276. doi: 10.1007/s00251-011-0513-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oseroff C, Sidney J, Kotturi MF, Kolla R, Alam R, Broide DH, Wasserman SI, Weiskopf D, McKinney DM, Chung JL, et al. Molecular determinants of T cell epitope recognition to the common Timothy grass allergen. J Immunol. 2010;185(2):943–55. PMID: 20554959. doi: 10.4049/jimmunol.1000405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ewert S, Huber T, Honegger A, Pluckthun A. Biophysical properties of human antibody variable domains. J Mol Biol. 2003;325(3):531–53. PMID: 12498801. doi: 10.1016/s0022-2836(02)01237-8. [DOI] [PubMed] [Google Scholar]
- 29.Honegger A, Pluckthun A. Yet another numbering scheme for immunoglobulin variable domains: an automatic modeling and analysis tool. J Mol Biol. 2001;309(3):657–70. PMID: 11397087. doi: 10.1006/jmbi.2001.4662. [DOI] [PubMed] [Google Scholar]
- 30.Kroner F, Hubbuch J. Systematic generation of buffer systems for pH gradient ion exchange chromatography and their application. J Chromatogr A. 2013;1285:78–87. PMID: 23489486. doi: 10.1016/j.chroma.2013.02.017. [DOI] [PubMed] [Google Scholar]
- 31.Chennamsetty N, Voynov V, Kayser V, Helk B, Trout BL. Design of therapeutic proteins with enhanced stability. Proceedings of the National Academy of Sciences of the United States of America. 2009; 106:11937–42. doi: 10.1073/pnas.0904191106. PMID: 19571001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.BIOVIA, Dassault Systèmes, Discovery Studio, v20.1.0.19295, San Diego: Dassault Systèmes, 2019. [Google Scholar]
- 33.BIOVIA, Dassault Systèmes, Discovery Studio, v22.1.021297, San Diego: Dassault Systèmes, 2021. [Google Scholar]
- 34.Black SD, Mould DR. Development of hydrophobicity parameters to analyze proteins which bear post- or cotranslational modifications. Anal Biochem. 1991;193(1):72–82. PMID: 2042744. doi: 10.1016/0003-2697(91)90045-u. [DOI] [PubMed] [Google Scholar]
- 35.Brooks BR, Brooks CL, Mackerell AD Jr., Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, et al. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30(10):1545–614. PMID: 19444816. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kellogg EH, Leaver-Fay A, Baker D. Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins Struct Funct Bioinf. 2011;79(3):830–38. PMID: 21287615. doi: 10.1002/prot.22921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28(1):235–42. PMID: 10592235. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kemmish H, Fasnacht M, Yan L, Gill AC. Fully automated antibody structure prediction using BIOVIA tools: validation study. PLos One. 2017;12(5):e0177923. PMID: 28542300. doi: 10.1371/journal.pone.0177923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–12. PMID: 15264254. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 40.Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, Ferrin TE. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 2021;30(1):70–82. PMID: 32881101. doi: 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.