


Abstract
Defects in the human gene XPV result in the variant form of the genetic disease xeroderma pigmentosum (XP-V). XPV encodes DNA polymerase η, a novel DNA polymerase that belongs to the UmuC/DinB/Rad30 superfamily. This polymerase catalyzes the efficient and accurate translesion synthesis of DNA past cis-syn cyclobutane di-thymine lesions. In this report we present the cDNA sequence and expression profiles of the mouse XPV gene and demonstrate its ability to complement defective DNA synthesis in XP-V cells. The mouse XPV protein shares 80.3% amino acid identity and 86.9% similarity with the human XPV protein. The recombinant mouse XPV protein corrected the inability of XP-V cell extracts to carry out DNA replication, by bypassing thymine dimers on template DNA. Transfection of the mouse or human XPV cDNA into human XP-V cells corrected UV sensitivity. Northern blot analysis revealed that the mouse XPV gene is expressed ubiquitously, but at a higher level in testis, liver, skin and thymus compared to other tissues. Although the mouse XPV gene was not induced by UV irradiation, its expression was elevated ~4-fold during cell proliferation. These results suggest that DNA polymerase η plays a role in DNA replication, though the enzyme is not essential for viability.
INTRODUCTION
DNA is continuously damaged by endogenous as well as exogenous agents in living organisms. To maintain the integrity of the genetic material, cells possess multiple pathways to repair various types of DNA damage, such as the nucleotide excision and base excision repair pathways (1). However, some damage is poorly repaired. For example, cyclobutane pyrimidine dimers (CPD) are one of the major DNA lesions induced by UV irradiation and >50% of them are not repaired within 24 h in mammalian cells (1). Unrepaired damage can interfere with basic cellular processes such as DNA replication and transcription and can lead to mutations and cell death. Inhibition of DNA replication by DNA lesions can be overcome by a post-replication repair pathway. This pathway has ‘damage tolerance’ mechanisms of repair, including translesion synthesis, sister chromatid exchange and template switching (1).
Xeroderma pigmentosum (XP) is a human autosomal recessive disease clinically associated with extreme sensitivity to sunlight and a high incidence of skin cancer (2). XP has been classified into eight complementation groups, XP-A–XP-G and the XP variant (XP-V). Unlike the classical XP cells (XP-A–XP-G), XP-V cells show no defect in nucleotide excision repair; however, they are defective in replication after UV irradiation and demonstrate UV hypermutability (3,4). Some groups have reported that extracts from XP-V cells lacked bypass replication when UV-induced CPDs were present (5–7). From these results, the XPV gene product is suspected to be part of the post-replication repair pathway and to act as a translesion synthesis factor. The UV hypermutability of XP-V cells could be due to abnormal replication after UV irradiation. We recently identified the XPV gene product from HeLa cell extracts and found that it acts as a novel DNA polymerase which can bypass CPD damage (8,9). The XPV protein incorporated two dAMPs against the di-thymine dimer (8), suggesting that XPV is a relatively accurate translesion polymerase. Johnson et al. (1999) reported that the yeast, Saccharomyces cerevisiae, RAD30 gene encodes a novel DNA polymerase (polymerase η) which can replicate past CPD damage by incorporating two dAMPs (10). As XPV shares amino acid homology with Rad30 and has similar biochemical activity, we concluded that XPV is the corresponding human DNA polymerase η.
The Rad30 and XPV proteins have no obvious motifs of known DNA polymerases but share homology with the Escherichia coli DinB and UmuC and yeast Rev1 proteins, which are involved in translesion synthesis following damage-induced mutagenesis. Recent biochemical studies revealed that all of these proteins possess DNA polymerase activity (11–14). These novel polymerases belong to the UmuC/DinB/Rad30 superfamily. This family is classified into two sub-families, of relatively accurate and mutagenic polymerases, by the fidelity of the newly synthesized DNA that bypasses the lesion. As mentioned above, DNA polymerase η inserts only adenine bases opposite the thymine residues of CPD, which suggests that polymerase η belongs to the relatively accurate polymerase family. In contrast, E.coli Pol IV (DinB), Pol V (the complex of UmuC and UmuD′; UmuD′2C) and yeast Rev1 belong to the mutagenic polymerases. In yeast, not only Rev1 but the Rev3 and Rev7 proteins are also key elements of the mutagenic pathway. A complex of Rev3 and Rev7, designated DNA polymerase ζ, can replicate past a CPD lesion (15). In addition, the human homologs of the dinB, REV1 and REV3 genes have been cloned, suggesting that both relatively accurate and mutagenic translesion pathways are present in mammals (16–20). In XP-V patients unrepaired lesions are more likely to be bypassed by the mutagenic pathway, because of inactivation of the relatively accurate pathway. As a result, XP-V patients suffer from a high incidence of skin cancer. However, regulation of the two sub-pathways in mammalian cells is still unclear and the mouse model of translesion synthesis may be an effective tool to understand the roles of these pathways in vivo.
In this study we have isolated a mouse homolog of XPV and examined its expression profile during cell proliferation and after UV irradiation. Moreover, we show that both human and mouse XPV can complement the defect in human XP-V cells in vivo and in vitro.
MATERIALS AND METHODS
Screening of a mouse cDNA library
A mouse FM3A cDNA library in λgt10 was screened with the coding region of human XPV (hXPV) cDNA as probe. One positive clone was obtained from 6 × 105 plaques. A 2 kb insert was amplified by PCR using λgt10 forward and reverse primers with ExTaq (TaKaRa). The PCR product was subcloned into pGEM-T Easy vector (Promega) and sequenced with an automated DNA sequence analyzer (Prism310; Applied Biosystems). This clone contained a partial mouse XPV (mXPV) cDNA, corresponding to the 3′-end of the coding region and 3′-untranslated region (UTR) (DDBJ/EMBL/GenBank accession no. AB037184). We designed two primers for amplification of the entire coding region of mXPV: primer 1 (5′-GAGTTCCAGGACAGCCAGAG-3′) and primer 2 (5′-GAAGCAAGTGAGCTTGCCAG-3′). The entire coding region of mXPV and the 5′-UTR were amplified by PCR from a 10.5 day 129Sv mouse cDNA library in λgt11 with ExTaq. The first PCR was performed using a λgt11 forward primer and primer 1 as reverse primer. The second PCR was performed with the first PCR product as template and a λgt11 forward primer and primer 2 as reverse primer. A 2.5 kb PCR product was identified by Southern blotting and subcloned into vector pGEM-T Easy, generating pGEM/mXPV. The subcloned PCR fragment contained the open reading frame of mXPV and the 5′-UTR (DDBJ/EMBL/GenBank accession no. AB027128).
Cell culture and cell-free extracts
WI38-VA13, Swiss 3T3 and partially and transiently transformed XP7TA (XP-V) (8) cells were cultured at 37°C with 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) (Nissui) supplemented with 10% fetal calf serum (FCS). HeLa cells were cultured at 37°C with 5% CO2 in DMEM supplemented with 5% calf serum. An insect cell line, Spodoptera frugiperda Sf9, was cultured at 27°C in TMN-FH medium supplemented with 10% heat-inactivated (56°C, 30 min) FCS. TMN-FH medium was prepared from Grace’s insect cell culture medium (Gibco BRL), TC yeastolate and TC lactalbumin hydrolysate (Difco).
Cell extracts for cell-free DNA replication assays were prepared as described (21).
Plasmid constructs
To produce mXPV protein tagged with hexa-histidine at its C-terminal end, the mXPV cDNA was cloned into the pFASTBAC1 vector (Gibco BRL) as follows. First, the translation initiation and termination sites of mXPV in vector pGEM-T Easy were converted to an NdeI site and an XhoI site, respectively, with oligonucleotides 5′-GTCGCCTTGAAACATATGGCTCCTGGG-3′ (for the NdeI site) and 5′-CACTGACACATCTCGAGTGCCTGGGGCATC-3′ (for the XhoI site) and a site-directed mutagenesis system, Mutan-K (TaKaRa). The resulting plasmid was digested with NdeI and XhoI and the 2.1 kb mXPV cDNA fragment was inserted into the NdeI and XhoI sites of pET-24a (Novagen), generating pET-24a/mXPVHis6. This plasmid was digested with Bpu1102I and the recessed 3′-end was filled with T4 DNA polymerase (TaKaRa), followed by XbaI digestion. The resulting mXPVHis6 fragment was subcloned into pFASTBAC1, generating pFASTBAC1/mXPVHis6. Baculovirus expressing mXPVHis6 protein was prepared with the BAC to BAC Baculovirus Expression System (Gibco BRL) according to the method described by the manufacturer.
pRc/CMV (Invitrogen) was used as an expression vector in mammal cells. hXPV and mXPV cDNA fragments prepared by NotI digestion of pBS/XPV (9) and pGEM/mXPV were subcloned into the NotI site of the pRc/CMV vector, generating pRc/CMV/hXPV and pRc/CMV/mXPV, respectively. pRc/CMV/hXPV contains the 3′-UTR with three mRNA degradation signals, while pRc/CMV/mXPV does not.
Purification of recombinant XPV protein from insect cells
Mouse XPV protein tagged with hexa-histidine was expressed in Sf9 cells infected for 72 h with recombinant virus. Cells were collected and washed twice with ice-cold phosphate-buffered saline (PBS). Pellets were suspended with six packed cell volumes of ice-cold buffer A [20 mM sodium phosphate, pH 7.2, 0.25 mM phenylmethylsulfonyl fluoride, 10 mM 2-mercaptoethanol, 10% glycerol, proteinase inhibitors (0.4 µg/ml antipain, 0.4 µg/ml aprotinin, 0.2 µg/ml leupeptin and 0.16 µg/ml pepstatin A) and 0.1 mM EGTA] containing 0.3 M NaCl and 1% Nonidet P-40. After incubation on ice for 20 min with occasional agitation, the suspension was centrifuged at 12 000 g for 1 h. The supernatant was adjusted to 20 mM imidazole and passed through a 5 ml Hitrap DEAE column (Amersham Pharmacia) equilibrated with buffer A containing 20 mM imidazole and 0.3 M NaCl. The flow-through was directly loaded onto a Ni–NTA agarose (Qiagen) column equilibrated with the same buffer. Mouse XPV-His6 was eluted from the column with a linear gradient of imidazole from 20 to 250 mM in buffer A. The fractions containing mXPV protein were dialyzed against buffer A containing 0.1 M NaCl and then loaded onto a MonoS PC1.6/5 column (Amersham Pharmacia) equilibrated with the same buffer. The proteins were eluted with a linear gradient of NaCl from 0.1 to 0.5 M in buffer A. The peak fractions were stored at –80°C.
hXPV protein was expressed and purified as described previously (9).
DNA polymerase α was purified from mouse FM3A cells as described previously (21).
DNA polymerase and damage bypass DNA polymerase assays
DNA polymerase and damage bypass DNA polymerase assays were conducted as reported previously (8). Purified recombinant protein was incubated with singly primed phagemid DNA and [α-32P]dCTP (74 kBq) at 37°C for 30 min in 10 µl standard reactions containing 40 mM Tris–HCl, pH 8.0, 60 mM KCl, 5 mM MgCl2, 100 µM each four dNTPs, 10 mM dithiothreitol, 250 µg/ml bovine serum albumin and 2% glycerol. DNA polymerase activity was measured as dCMP incorporation into acid-insoluble materials.
The 5′-[32P]primer–template DNA was prepared by mixing the 13mer primer labeled at its 5′-end with a 30mer template containing the lesion [5′-CTCGTCAGCATCTTCATCATACAGTCAGTG-3′ with the CPD or (6–4) photoproduct at positions 17 and 18 from the 3′-end (shown in bold) or without damage; 22,23]. Purified protein was incubated with 4 nM 5′-[32P]primer–template DNA at 37°C for 15 min in 10 µl standard reactions as described above. Reactions were terminated by addition of formamide and boiling. The products were subjected to 20% polyacrylamide, 7 M urea gel electrophoresis and autoradiographed.
DNA transfection and UV survival
pRc/CMV, pRc/CMV/hXPV or pRc/CMV/mXPV plasmid DNA was transfected into XP7TA cells using Lipofectamine 2000 reagent (Gibco BRL) following the instructions of the manufacturer. After G418 selection (200 µg/ml) for 3 weeks, cells transfected with pRC/CMV/hXPV or pRC/CMV/mXPV were exposed to 2 J/m2 UV light and then cultured for 4 days in the presence of 1 mM caffeine. As expected, a large population of these transformants with hXPV or mXPV cDNA survived after this selection, while the transformants containing the vector with no insert did not. G418 plus UV-selected mass populations of XP7TA + pRc/CMV/hXPV and XP7TA + pRc/CMV/mXPV, G418-selected XP7TA + pRc/CMV and non-selected mass populations of WI38-VA13 and XP7TA cells were characterized further by UV survival experiments. For this purpose, we measured S phase-dependent [3H]thymidine incorporation as a measure of proliferating cells. Cells were plated at densities varying from 1 × 104 to 2 × 105 per well in 6-well dishes. One day after plating, cells were exposed to 0, 1, 2, 4 or 8 J/m2 UV light and then incubated with culture medium containing 1 mM caffeine. Four days later, before reaching confluency, the cells were incubated with culture medium containing [3H]thymidine (1 µCi/ml) for 1 h. The cells were then rinsed twice with PBS and lysed in 10 mM Tris, 1 mM EDTA, 0.1% SDS. Nucleic acids were precipitated in 5% trichloroacetic acid and blotted onto GF/C glassfiber filters (Whatmann). The radioactivity incorporated into acid-insoluble materials was scintillation counted.
Analysis of mXPV gene expression
For tissue northern blot analyses, mouse MTN blot membrane (Clontech) and Mouse Multiple Choice Northern Blot no. 5 (OriGene), containing 2 µg of poly(A)+ RNA per track, were hybridized with random primed probes generated from the coding region of mXPV cDNA or β-actin.
To examine the expression profile of mXPV during cell proliferation, Swiss 3T3 cells were synchronized in the G0/G1 phase by serum starvation for 48 h in the presence of 0.5% FCS and released by addition of growth medium containing 10% FCS. After serum stimulation, the cells were harvested at the times indicated. Total RNA was extracted with Isogen (Nippon Gene) and poly(A)+ RNA was isolated with Oligotex-dT30 ‘Super’ (TaKaRa) as described by the manufacturer. Poly(A)+ RNA (2 µg) samples were fractionated by electrophoreses in 1.2% agarose gels containing 6% formaldehyde and transferred to nylon membranes (Hybond-N; Amersham Pharmacia). Hybridization was performed overnight at 42°C in hybridization/prehybridization solution 2 (Clontech) containing the probe fragment labeled with [α-32P]dCTP by random labeling (Amersham Pharmacia). The membranes were washed twice at room temperature with 2× SSC and 0.05% SDS and then once at 42°C with 0.1× SSC and 0.1% SDS. Autoradiography was performed at –80°C with Hyperfilm MP (Amersham Pharmacia). Quantitation of the hybridized signals was carried out with a Fuji BAS-2500 bioimaging analyzer.
To examine the induction of mXPV by UV irradiation, Swiss 3T3 cells were irradiated with 10 J/m2 UV light. The mRNA was prepared at the indicated times after UV irradiation. RNA preparation and northern blot analysis were carried out as described above.
RESULTS
Cloning of mXPV cDNA
To isolate the mXPV cDNA, an FM3A cDNA library was screened with an hXPV cDNA as probe and one positive clone containing a 2.0 kb insert was obtained. Sequence analysis revealed that the clone was a mouse homolog of XPV (mXPV) and contained the 3′-portion of the coding sequence and the 3′-UTR, including a polyadenylation signal (GenBank accession no. AB037184). Based on the sequence, we designed primers for PCR to obtain a cDNA containing the entire coding sequence for mXPV. Using these primers, the mXPV cDNA was obtained from a 129Sv cDNA library as described in Materials and Methods (GenBank accession no. AB027128). The mXPV cDNA contained 2085 bp of open reading frame, encoding a putative protein of 694 amino acid residues with a calculated molecular weight of 76.1 kDa. The sequence alignment of the hXPV and mXPV proteins is shown in Figure 1. Comparison of the amino acid sequences of the two proteins revealed an overall 80.3% amino acid identity and 86.9% similarity. We found previously that the N-terminal half of the hXPV protein has seven amino acid sequences conserved among damage bypass replication proteins (9). These regions were also well conserved in the mXPV protein. In contrast, the C-terminal portion, especially amino acids beyond residue 500, were less conserved between the human and mouse proteins, although the last 80 amino acids, including a putative nuclear localization signal, were conserved in both proteins. Thus, the N-terminal amino acids appear to be important for the translesion activity of polymerase η.
Figure 1.

Alignment of deduced amino acid sequences of hXPV and mXPV proteins. Identical and similar residues are boxed in dark and light gray, respectively. The conserved regions in human and other related translesion polymerases (Rad30, DinB, UmuC and Rev1) are boxed from A to G (9). Two putative nuclear localization signals are underlined (9).
CPD bypass DNA polymerase activity of recombinant mXPV protein
To examine the properties of the mXPV protein, we expressed the recombinant protein tagged with hexa-histidine at its C-terminal end using a baculovirus protein expression system. As shown in Figure 2A, the mXPV and hXPV proteins were purified to near homogeneity after a three-step purification. While the mXPV protein is 20 amino acids smaller than hXPV, the recombinant mXPV migrated slightly slower than the recombinant human protein on SDS–PAGE, suggesting that the mouse protein has unique structural sequences, perhaps in the less conserved C-terminal region.
Figure 2.

CPD bypass activity of recombinant mXPV protein. (A) Recombinant hXPV and mXPV proteins tagged with hexa-histidine at their C-terminal end were expressed in the baculovirus system and purified by sequential chromatography on Hitrap DEAE, Ni–NTA agarose and MonoS. The purified fractions (0.27 ng) of both proteins were subjected to SDS–PAGE and silver stained. M, 10 kDa protein ladder marker (Gibco BRL). (B) DNA polymerase activity of recombinant proteins. Recombinant hXPV (open circles) or mXPV (open squares) proteins were incubated at 37°C with reaction mixtures containing single primed DNA templates and [α-32P]dCTP. After 30 min incubation, acid-insoluble material was measured by scintillation counting. (C) The damage bypass DNA polymerase activity. The recombinant hXPV (0.05 ng in lanes 2, 8 and 14; 0.5 ng in lanes 3, 9 and 15) or mXPV (0.075 ng in lanes 4, 10 and 16; 0.75 ng in lanes 5, 11 and 17) protein or mouse DNA polymerase α (8.2 ng) was incubated at 37°C for 15 min with the 5′-32P-labeled 13mer primer annealed with the 30mer DNA fragment containing a lesion [CPD, lanes 7–12; (6–4) photoproduct, lanes 13–18] or not containing a lesion (lanes 1–6). After incubation, the products were subjected to PAGE and detected by autoradiography. The position of the thymine dimer on the template is indicated by the bridge.
The DNA polymerase activities of purified recombinant proteins were measured by incorporation of [α-32P]dCMP into singly primed phagemid templates (Fig. 2B). mXPV showed DNA polymerase activity, although the activity was slightly lower than that of hXPV when equal amounts of the proteins were examined. The amounts of mouse and human proteins with the same level of DNA polymerase activity were added to reactions in the following experiments.
Damage bypass DNA polymerase activity of mXPV was assayed with a 30mer template DNA containing a lesion annealed with a labeled 13mer primer. Both hXPV and mXPV bypassed CPD damage and synthesized DNA up to 30 nt in length (Fig. 2C, lanes 8–11), whereas DNA polymerase α stopped DNA synthesis 1 nt before the CPD (Fig. 2C, lane 12). mXPV and hXPV incorporated dAMPs to bypass the CPD lesion (data not shown). On the (6–4) photoproduct-containing template neither hXPV nor mXPV protein bypassed the lesion (Fig. 2C, lanes 14–17). From these results we conclude that the mXPV gene we identified here encodes a mouse DNA polymerase η.
We also confirmed that the mouse polymerase η complemented the defective translesion synthesis in XP-V cell extracts in vitro as well as the human enzyme (data not shown).
Correction of UV sensitivity of XP-V cells by transfection of XPV cDNA
XP-V cells are known to be slightly more sensitive to UV light than normal cells. However, the sensitivity of XP-V cells to UV light is enhanced in the presence of caffeine (24). So we examined whether the sensitivity of XP-V cells to UV light was corrected by transfection of hXPV or mXPV cDNA in the presence of caffeine. The hXPV and mXPV cDNAs were subcloned into the pRC/CMV vector, in which the genes are expressed under the regulation of a CMV promoter, and transfected into XP7TA cells. Stably transfected mass populations were obtained as described in Materials and Methods. These transformants, untransfected XP7TA and translesion-proficient WI38-VA13 cells were examined for UV sensitivity (Fig. 3). WI38-VA13 cells demonstrated normal resistance to UV irradiation in the presence or absence of 1 mM caffeine (data not shown). On the other hand, XP7TA (XP-V) cells and the transformants containing the vector with no insert were extremely sensitive to UV irradiation in the presence of caffeine. The sensitivity of the transformants containing hXPV or mXPV cDNA to UV irradiation was corrected and the cells demonstrated normal resistance to UV irradiation, although the correction by human cDNA was not complete. This partial complementation by hXPV cDNA might be caused by instability of the mRNA or protein. It is possible that the mRNA of hXPV used in this experiment might be more unstable than that of mXPV, because hXPV cDNA in this construct contained three motifs for mRNA selective degradation (ATTTA) in its 3′-UTR, whereas no degradation signal was included in the construct for mXPV. Although these points must be clarified in the future, we conclude here that mXPV as well as hXPV can complement the defects in XP-V cells both in vivo and in vitro.
Figure 3.

Complementation of UV sensitivities of XP-V cells by hXPV and mXPV cDNA. After UV irradiation, cells were cultured in the presence of 1 mM caffeine for 4 days. The number of proliferating cells was measured by [3H]thymidine incorporation. Open circle, WI38-VA13; closed circle, XP7TA; open square, XP7TA + hXPV cDNA; closed square, XP7TA + mXPV cDNA; open triangle, XP7TA + vector (pRC-CMV).
Expression of the mXPV gene in mouse tissues
Expression of mXPV in various mouse tissues was examined by northern blot analysis (Fig. 4). The mXPV mRNA was detected as an ~3.2 kb transcript, which is in agreement with the size of the cDNA, in all tissues examined except testis, where a slightly smaller transcript was observed. When the expressed level of mXPV was normalized against that of β-actin in each tissue, expression of the mXPV gene was higher in testis, liver, skin and thymus than other tissues, especially in non-proliferating tissues such as brain, heart and skeletal muscle.
Figure 4.
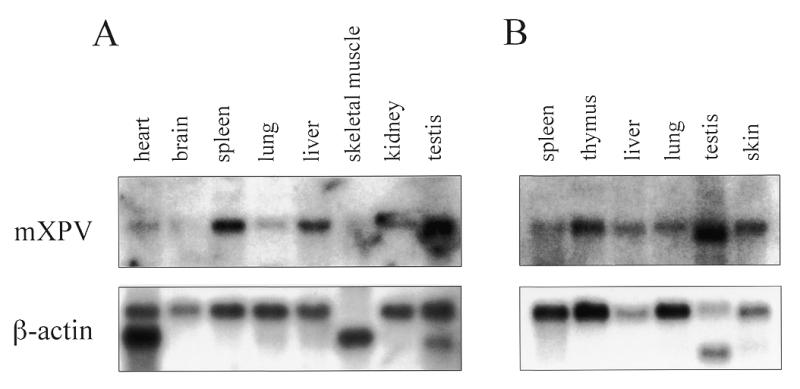
Expression of mXPV mRNA in adult mouse tissues. The mXPV cDNA probe was hybridized with 2 µg of poly(A)+ RNA from each tissue as indicated (upper). A β-actin cDNA was used as a control (lower). (A) Clontech. (B) OriGene.
Induction of mXPV transcripts in the cells stimulated to proliferate
The steady-state level of mRNA for DNA polymerase α, which is involved in DNA replication, is known to increase markedly when quiescent cells are stimulated to proliferate by serum addition (25,26). In contrast, DNA polymerase β, which is involved in the base excision repair pathway, is expressed constantly throughout cell proliferation (27) but is induced by DNA-damaging agents (28). To determine the expression profile of mXPV (polymerase η) during cell proliferation, we arrested Swiss 3T3 cells in the G0 phase by serum deprivation and then induced their proliferation by the addition of serum. DNA synthesis, measured by incorporation of [3H]thymidine into cells, increased from 12 h after serum stimulation and reached its maximum at 20 h (Fig. 5B). As reported previously (26), the expression level of the 68 kDa subunit of DNA polymerase α was suppressed by serum starvation (compare log phase with 0 h in Fig. 5A) and increased dramatically prior to DNA synthesis following the addition of serum (compare Fig. 5A with B). The mRNA level of mXPV was also suppressed by serum starvation, increased gradually after serum stimulation and rose to a maximum (~4-fold) at 20 h (Fig. 5A and B). Expression of EF-1a was not affected by deprivation or addition of serum (Fig. 5A). These results suggest that mXPV might play a role in the proliferation of cells, although the stimulation profile and induction rate of mXPV were different from those of DNA polymerase α.
Figure 5.


Expression profiles of mXPV in Swiss 3T3 cells. (A and B) Serum-starved mouse Swiss 3T3 cells were stimulated with 10% serum and mRNA was isolated at the indicated times and subjected to northern blot analysis. (A) mRNA levels of mXPV, DNA polymerase α 68 kDa subunits (Polα 68K) and EF-1α in each fraction. EF-1α was used as an internal control. An asynchronous log phase (log) control is also shown. (B) Autoradiograms were quantitated and relative expression was normalized to EF-1α at each time point. The values were plotted relative to the values for 0 h. Closed circle, mXPV; open square, Polα 68K. DNA synthesis in Swiss 3T3 cells, measured by incorporation of [3H]thymidine into the acid-insoluble fractions, is plotted as a function of time (h) after the addition of serum to quiescent cells (open triangle). (C) Swiss 3T3 cells in log phase were exposed to UV light (10 J/m2) and poly(A)+ RNA was isolated at the indicated times after UV irradiation and subjected to northern blot analysis with the probes indicated. (D) Autoradiograms were quantified and relative expression was normalized to the level of EF-1α at each time point. The values were plotted relative to the values for 0 h. Closed circle, mXPV; open square, Polα 68K; closed triangle, p21.
In Swiss 3T3 cells two transcripts of ~4.5 and ~3.2 kb were detected, whereas only the smaller one was detected in mRNA from various mouse tissues (Figs 4 and 5A). These transcripts might arise as a result of alternative splicing or alternative use of polyadenylation signals, but we have not confirmed these possibilities. It is possible that this might reflect the differences between somatic cells and cultured cell lines, because we detected only the smaller transcript in two independent northern blots of different tissues (Fig. 4), whereas both transcripts were detected in northern blots of RNA extracted from FM3A and 129Sv cells as well as Swiss 3T3 cells (data not shown).
mXPV was not induced by UV irradiation
Yeast RAD30, which encodes yeast polymerase η, is induced by DNA damage (29,30). To investigate whether XPV (mammalian polymerase η) is also induced by DNA damage, Swiss 3T3 cells in log phase were irradiated with 10 J/m2 UV light and mRNA was isolated for northern blot analysis at the indicated times after UV irradiation. The levels of p21 and EF-1α mRNA were measured as controls, as the former is known to be induced by DNA damage and the latter is not. As expected, expression of p21 increased from 1 h after UV irradiation and rose to a maximum at 4 h, while the expression of EF-1α was not influenced by UV irradiation (Fig. 5C). In contrast, expression of mXPV decreased at 2 h after UV irradiation but recovered gradually and returned to the level of unirradiated cells at 24 h (Fig. 5D). Expression of the 68 kDa subunit of DNA polymerase α also decreased after UV irradiation, reached its lowest level at 4 h and then recovered gradually. There are two possible explanations for this reduction in mRNA after UV irradiation. First, the gene is down-regulated by UV irradiation, including indirect effects via cell cycle arrest. Second, the transcripts of the genes are unstable. Inhibition of de novo syntheses of the transcripts by UV irradiation or rapid degradation of the transcripts would result in a reduction in the amount of the transcripts in cells. As mXPV has a potential degradation signal for mRNA (ATTTA) in its 3′-UTR (data not shown), the latter case is likely to occur, although we could not exclude the former possibility. In any case, we concluded from these results that the expression of mXPV was not induced by UV irradiation, in contrast to that of yeast RAD30.
DISCUSSION
In recent years novel DNA polymerases, which can bypass DNA lesions, have been identified in organisms ranging from bacteria to humans and categorized into the UmuC/DinB/Rad30 superfamily (31–34). XPV, human DNA polymerase η, belongs to this family and can replicate past UV-induced CPD lesions in a relatively accurate manner. In this paper we have isolated and characterized a cDNA of the mouse homolog of XPV (mXPV).
mXPV and hXPV correct the defects of XP-V cells in vivo and in vitro
Recombinant mXPV protein demonstrated CPD bypass polymerase activity, indicating that mXPV is the mouse DNA polymerase η. hXPV protein also demonstrated this activity. mXPV protein could correct the translesion defect of XP-V cell extracts in our cell-free system (data not shown). Moreover, transfection of mXPV or hXPV cDNA into XP-V cells resulted in complementation of the defect and loss of UV sensitivity. To our knowledge, this is the first report to show that the XPV gene can correct the UV sensitivity of XP-V cells.
During DNA replication, when the replication fork is blocked by a lesion the replicative polymerases must be exchanged with a translesion DNA polymerase, i.e. polymerase η. Recently we found that the human polymerase η replicates undamaged DNA with very low fidelity (35), suggesting that the function of the polymerase is restricted to synthesis of a few nucleotides past the lesion. Thus, it would be expected that there are mechanisms for efficient and regulated polymerase switching, such as a specific protein–protein interaction between polymerase η and the replication machinery. Our finding that the mouse protein substituted for the human protein indicates that the conserved N-termini of XPV proteins are responsible for these functions. Studies of the molecular mechanism of translesion DNA synthesis and identification of the functional domains in XPV proteins are required to clarify these mechanisms.
Expression profile of the mXPV gene
The northern blot analysis revealed that expression of mXPV was not induced by UV irradiation (Fig. 5C and D). In yeast, in contrast, RAD30 was shown to be induced ~4-fold by UV irradiation (29,30). Thus, the transcriptional regulation of polymerase η is different in mammals compared to yeast.
Although mXPV was not induced by UV irradiation, expression of mXPV was elevated ~4-fold during cell proliferation (Fig. 5A and B). mXPV expression increased with the onset of DNA synthesis, whereas the level of mRNA for the DNA polymerase α 68 kDa subunit rose to a maximum before DNA synthesis. Not only DNA polymerase α but also chromosomal replicative DNA polymerases δ and ɛ are known to be induced before DNA synthesis (36,37). Although the induction profile of mXPV transcription was different from those of replicative DNA polymerases, this result indicates that expression of mXPV is dependent on cell proliferation. In addition, tissue northern blot analysis showed that levels of mRNA for mXPV were elevated in testis, thymus, liver and skin compared with the levels in other tissues (Fig. 4). These tissues contain a significant proportion of proliferating cells, suggesting that mXPV plays a role in DNA replication, although further analyses are required to reach a firm conclusion. In yeast and humans, however, mutations in the polymerase η gene are not lethal to cells, indicating that polymerase η is not essential for the replication of chromosomal DNA. Recently we found that the human polymerase η can bypass spontaneously induced oxidative damage, e.g. abasic sites in addition to CPD lesions (38). Although polymerase η is not involved in the basal DNA replication process, it is possible that polymerase η is required for efficient progression of DNA replication in cells.
Translesion synthesis in rodent cells
UV-induced DNA damage is known to be repaired by the nucleotide excision repair (NER) pathway. In rodent cells pyrimidine dimers in genomic DNA are known to be poorly repaired by NER compared to that in human cells (39). We have shown here that the mXPV gene carried out a function similar to the human gene in rodent cells, strongly suggesting that relatively accurate translesion synthesis is functioning in rodent cells. Additionally, the mouse homolog of REV3 has been isolated, suggesting that mutagenic translesion synthesis could also function in rodent cells (40). However, the relationship between the repair pathway and translesion synthesis and their contributions to cell survival or carcinogenesis remain unclear. In rodents, UV-sensitive mutant cells, referred to as ERCC (excision repair cross complementing) cells, have been isolated as 11 complementation groups. Cell lines representing eight of these groups have mutations in the genes responsible for XP and another UV-sensitive hereditary disorder Cockayne’s syndrome (41). Although the mutations in the remaining three complementation groups (ERCC7, 9 and 10) have not been identified, our preliminary experiments demonstrate that expression of the mXPV gene in these cells is normal (data not shown). Since mXPV mutant cells have not been cloned yet, XPV-deficient mice may provide a good model to analyze these mechanisms, in addition to the function of XPV in vivo.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Dr Marito Araki, Rika Kusumoto, Mayumi Yuasa, Tomokazu Nogimori and other members of Dr Hanaoka’s laboratory at Osaka University for helpful discussions. We thank Dr Hisato Kondoh (IMCB, Osaka University) and Dr Masako Izumi (RIKEN) for providing the 129Sv cDNA library and the EF-1α probe, respectively. This work was supported by grants from the Ministry of Education, Science, Sports and Culture of Japan, from CREST and from the Biodesign Research Program of RIKEN.
DDBJ/EMBL/GenBank accession nos AB037184, AB027128
REFERENCES
- 1.Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis. ASM Press, Washington, DC.
- 2.Bootsma D., Kraemer,K.H., Cleaver,J. and Hoeijmakers,J.H.J. (1998) In Vogelstein,B. and Kinzler,K.W. (eds), The Genetic Basis of Human Cancer. McGraw-Hill, New York, NY, pp. 245–274.
- 3.Lehmann A.R., Kirk-Bell,S., Arlett,C.F., Paterson,M.C., Lohman,P.H., de Weerd-Kastelein,E.A. and Bootsma,D. (1975) Proc. Natl Acad. Sci. USA, 72, 219–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maher V.M., Ouellette,L.M., Curren,R.D. and McCormick,J.J. (1976) Nature, 261, 593–595. [DOI] [PubMed] [Google Scholar]
- 5.Cordeiro-Stone M., Zaritskaya,L.S., Price,L.K. and Kaufmann,W.K. (1997) J. Biol. Chem., 272, 13945–13954. [DOI] [PubMed] [Google Scholar]
- 6.Svoboda D.L., Briley,L.P. and Vos,J.M. (1998) Cancer Res., 58, 2445–2448. [PubMed] [Google Scholar]
- 7.Ensch-Simon I., Burgers,P.M. and Taylor,J.S. (1998) Biochemistry, 37, 8218–8226. [DOI] [PubMed] [Google Scholar]
- 8.Masutani C., Araki,M., Yamada,A., Kusumoto,R., Nogimori,T., Maekawa,T., Iwai,S. and Hanaoka,F. (1999) EMBO J., 18, 3491–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Masutani C., Kusumoto,R., Yamada,A., Dohmae,N., Yokoi,M., Yuasa,M., Araki,M., Iwai,S., Takio,K. and Hanaoka,F. (1999) Nature, 399, 700–704. [DOI] [PubMed] [Google Scholar]
- 10.Johnson R.E., Prakash,S. and Prakash,L. (1999) Science, 283, 1001–1004. [DOI] [PubMed] [Google Scholar]
- 11.Wagner J., Gruz,P., Kim,S.R., Yamada,M., Matsui,K., Fuchs,R.P. and Nohmi,T. (1999) Mol. Cell, 4, 281–286. [DOI] [PubMed] [Google Scholar]
- 12.Tang M., Shen,X., Frank,E.G., O’Donnell,M., Woodgate,R. and Goodman,M.F. (1999) Proc. Natl Acad. Sci. USA, 96, 8919–8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reuven N.B., Arad,G., Maor-Shoshani,A. and Livneh,Z. (1999) J. Biol. Chem., 274, 31763–31766. [DOI] [PubMed] [Google Scholar]
- 14.Nelson J.R., Lawrence,C.W. and Hinkle,D.C. (1996) Nature, 382, 729–731. [DOI] [PubMed] [Google Scholar]
- 15.Nelson J.R., Lawrence,C.W. and Hinkle,D.C. (1996) Science, 272, 1646–1649. [DOI] [PubMed] [Google Scholar]
- 16.Gerlach V.L., Aravind,L., Gotway,G., Schultz,R.A., Koonin,E.V. and Friedberg,E.C. (1999) Proc. Natl Acad. Sci. USA, 96, 11922–11927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ogi T., Kato,T.,Jr, Kato,T. and Ohmori,H. (1999) Genes Cells, 4, 607–618. [DOI] [PubMed] [Google Scholar]
- 18.Lin W., Xin,H., Zhang,Y., Wu,X., Yuan,F. and Wang,Z. (1999) Nucleic Acids Res., 27, 4468–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gibbs P.E., McGregor,W.G., Maher,V.M., Nisson,P. and Lawrence,C.W. (1998) Proc. Natl Acad. Sci. USA, 95, 6876–6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin W., Wu,X. and Wang,Z. (1999) Mutat. Res., 433, 89–98. [DOI] [PubMed] [Google Scholar]
- 21.Eki T., Enomoto,T., Masutani,C., Miyajima,A., Takada,R., Murakami,Y., Ohno,T., Hanaoka,F. and Ui,M. (1991) J. Virol., 65, 4874–4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murata T., Iwai,S. and Ohtsuka,E. (1990) Nucleic Acids Res., 18, 7279–7286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iwai S., Shimizu,M., Kamiya,H. and Ohtsuka,E. (1996) J. Am. Chem. Soc., 118, 7642–7643. [Google Scholar]
- 24.Arlett C.F., Harcourt,S.A. and Broughton,B.C. (1975) Mutat. Res., 33, 341–346. [DOI] [PubMed] [Google Scholar]
- 25.Wahl A.F., Geis,A.M., Spain,B.H., Wong,S.W., Korn,D. and Wang,T.S. (1988) Mol. Cell. Biol., 8, 5016–5025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miyazawa H., Izumi,M., Tada,S., Takada,R., Masutani,M., Ui,M. and Hanaoka,F. (1993) J. Biol. Chem., 268, 8111–8122. [PubMed] [Google Scholar]
- 27.Zmudzka B.Z., Fornace,A., Collins,J. and Wilson,S.H. (1988) Nucleic Acids Res., 16, 9587–9596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fornace A.J. Jr, Zmudzka,B., Hollander,M.C. and Wilson,S.H. (1989) Mol. Cell. Biol., 9, 851–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McDonald J.P., Levine,A.S. and Woodgate,R. (1997) Genetics, 147, 1557–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roush A.A., Suarez,M., Friedberg,E.C., Radman,M. and Siede,W. (1998) Mol. Gen. Genet., 257, 686–692. [DOI] [PubMed] [Google Scholar]
- 31.Friedberg E.C. and Gerlach,V.L. (1999) Cell, 98, 413–416. [DOI] [PubMed] [Google Scholar]
- 32.Woodgate R. (1999) Genes Dev., 13, 2191–2195. [DOI] [PubMed] [Google Scholar]
- 33.Cordonnier A.M. and Fuchs,R.P. (1999) Mutat. Res., 435, 111–119. [DOI] [PubMed] [Google Scholar]
- 34.Johnson R.E., Washington,M.T., Prakash,S. and Prakash,L. (1999) Proc. Natl Acad. Sci. USA, 96, 12224–12226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsuda T., Bebenek,K., Masutani,C., Hanaoka,F. and Kunkel,T.A. (2000) Nature, 404, 1011–1013. [DOI] [PubMed] [Google Scholar]
- 36.Yang C.L., Chang,L.S., Zhang,P., Hao,H., Zhu,L., Toomey,N.L. and Lee,M.Y. (1992) Nucleic Acids Res., 20, 735–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tuusa J., Uitto,L. and Syväja,J.E. (1995) Nucleic Acids Res., 23, 2178–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Masutani C., Kusumoto,R., Iwai,S. and Hanaoka,F. (2000) EMBO J., in press. [Google Scholar]
- 39.Mellon I., Spivak,G. and Hanawalt,P.C. (1987) Cell, 51, 241–249. [DOI] [PubMed] [Google Scholar]
- 40.Van Sloun P.P.H., Romeijn,R.J. and Eeken,J.C.J. (1999) Mutat. Res., 433, 109–116. [DOI] [PubMed] [Google Scholar]
- 41.Wood R.D. (1996) Annu. Rev. Biochem., 65, 135–167. [DOI] [PubMed] [Google Scholar]