

Abstract
Human telomerase reverse transcriptase (hTERT) is a catalytic subunit of human telomerase and is a critical determinant of the enzymatic activity of telomerase. Expression of hTERT is known to be regulated mainly at the transcriptional level. In the present study, using transient expression assays, we identified a 400 bp silencer of the hTERT promoter between –776 and –378 upstream of the proximal core promoter. The inhibitory effects of this silencer were enhanced with cellular differentiation. A computer-assisted homology search identified multiple binding motifs for myeloid-specific zinc finger protein 2 (MZF-2) within this region. Mutation introduced in these sites resulted in significant activation of hTERT transcription. Gel shift assays demonstrated that MZF-2 proteins specifically bound to these sites. Overexpression of MZF-2 in cells led to down-regulation of hTERT transcription as well as telomerase activity. These findings suggest that the 400 bp region upstream of the hTERT core promoter that we identified functions as a negative regulatory region and that MZF-2 may be an effector of negative regulation of hTERT.
INTRODUCTION
Telomeres are the distal ends of chromosome DNA and are composed of tandem repeats of the sequence TTAGGG (1). Possible functions of telomeres include prevention of DNA degradation, end-to-end fusion and chromosome loss, which contribute to the maintenance of chromosomal integrity (2). Telomerase is a specialized ribonucleoprotein complex that elongates telomere sequences (1). Since expression of this enzyme is repressed in normal somatic cells, telomeres are progressively shortened with cell division due to the inability of DNA polymerase to completely replicate the ends of chromosomes (3). Critically short telomeres resulting from a substantial number of cell divisions induce cellular senescence. Telomerase activation is therefore required for cells to overcome replicative senescence and to obtain immortality. Use of a recently developed, highly sensitive detection system for telomerase activity, the TRAP assay, has revealed that telomerase activity can be detected in the vast majority of human tumors, but in only a few types of normal somatic cells (4,5), suggesting that telomerase activation is a critical step in carcinogenesis (6,7).
Human telomerase reverse transcriptase (hTERT) is the catalytic subunit protein that is responsible for the enzymatic activity of telomerase (8–12). High levels of expression of hTERT have been observed in telomerase-positive tumor tissues and immortal cell lines, but not in normal tissues and telomerase-negative cells (11,12). There is a significant correlation between level of hTERT expression and telomerase activity in a variety of types of cancers (13–17). Introduction of hTERT cDNA into telomerase-negative normal cells leads to activation of telomerase in these cells (18,19) and normal cell clones with high levels of hTERT expression attain immortal capacity (20). These findings suggest that hTERT is a rate limiting factor for telomerase activity and that regulation of hTERT may critically determine the status of telomerase activity in cells.
Recent studies, including our own, have identified promoter sequences of hTERT, enabling us to examine transcriptional regulation of hTERT (21,22). These studies demonstrated that expression of hTERT is regulated mainly at the transcriptional level and that the core promoter of the hTERT gene is essential for transcriptional activation in cancer cells and immortalized cells. This core promoter encompasses the proximal 181 bp region upstream of the transcription start site. c-Myc and Sp1 specifically bind to the consensus motifs within this region and play essential roles in transactivation of hTERT (23–25). Up-regulation of these factors during the course of carcinogenesis is therefore thought to be responsible for telomerase activation.
In contrast, negative regulation of hTERT has not yet been well studied. Examination of the hTERT promoter is required for an understanding of the mechanisms of negative regulation of telomerase. In the present study we have identified and characterized the negative regulatory region of the hTERT promoter and found that multiple consensus sequences for myeloid zinc finger protein 2 (MZF-2) play significant roles in negative regulation of this promoter.
MATERIALS AND METHODS
Cell culture
C33A, SiHa, ME180 and HeLa cell lines derived from cervical cancers, as well as TSU-PR1, PC3 and DU145 cell lines derived from prostate cancers, were obtained from the American Type Culture Collection. Leukemia cell line K562 was obtained from the Human Science Research Rescue Bank. The former cells were grown in Dulbecco’s modified Eagle’s medium and the latter in RPMI 1640, both of which were supplemented with 10% fetal calf serum, in the presence of 5% CO2. To induce cellular differentiation, K562 cells were incubated with phorbol 12-myristate 13-acetate (PMA) at a final concentration of 80 nM for 48–72 h.
Plasmid construction
The structures of hTERT promoter–luciferase constructs were described in a previous study (21). These reporter plasmids were constructed with the enhancer/promoter-less pGL3-basic luciferase reporter plasmid (Promega, Madison, WI). The MZF-2 expression vector (pEF-BOS/MYC-MZF2), kindly provided by Dr S. Nagata (Osaka University, Osaka, Japan), contained the MZF-2 cDNA driven by the elongation factor promoter and c-myc epitope peptide sequences in the N-terminal end of human MZF-2 cDNA (26). PCR-based site-directed mutagenesis was performed to construct the mutant reporter plasmid pGL3-776-MZF2MT (27) in which four consensus binding motifs for MZF-2 in the potential silencer of the hTERT promoter were altered by substitution mutation.
Luciferase assay
Transient transfection of luciferase reporter plasmids into adherent cell lines C33A, SiHa, HeLa and ME100 and non-adherent K562 cells was performed using LipofectAMINE (Life Technologies, Gaithersburg, MD) and Tfx-20 (Promega), respectively, according to the protocols recommended by the manufacturers. Luciferase assays were performed using the Dual-Luciferase Reporter Assay System (Promega), in which Renilla luciferase plasmids were co-transfected as control plasmids to standardize transcription efficiency. All experiments were performed at least three times with each plasmid and the results are average values of relative luciferase activity.
Stretch PCR assay
For quantitative analyses of telomerase activity, stretch PCR assays were performed using the Telochaser system according to the manufacturer’s protocol (Toyobo, Tokyo, Japan). The PCR products were electrophoresed on a 7% polyacrylamide gel and visualized with SYBR Green I nucleic acid gel stain (FMC BioProducts, Rockland, ME). To monitor the efficiency of PCR amplification, 10 ng of internal controls from λ phage DNA sequences (Toyobo) together with 50 pmol of specific primers (Toyobo) were added to the PCR mixture per reaction. Relative telomerase activity was determined by measuring band intensities of telomerase ladders and comparing them with those of internal controls. Band intensity was measured using NIH Image analysis software.
RT–PCR assay
Preparation of total RNA from cell lines was performed by the AGPC method described previously (28). First strand cDNAs were synthesized with AMV reverse transcriptase (Pharmacia) with 1 µg total RNA from the cell lines. The cDNAs synthesized from each RNA sample were assayed in 50 µl of reaction mixture containing 2 mM Tris–HCl, pH 8.0, 10 mM KCl, 0.01 mM EDTA, 0.1 mM DTT, 5% glycerol, 1 mM MgCl2, 0.05% Tween-20, 0.05% Nonidet P-40, 5 U Taq DNA polymerase, 200 µM each dATP, dCTP, dTTP and dGTP, 5 µCi [32P]dCTP and 0.4 µM sense and antisense primers. The reaction mixture was heated at 90°C for 1 min and then 26 cycles of PCR were performed, including denaturation at 94°C for 1 min, annealing at 65°C for 1 min and extension at 72°C for 1.5 min. The PCR products were electrophoresed on an 8% polyacrylamide gel and dried gels were analyzed by autoradiography. For PCR reactions, sense and antisense oligonucleotide primers for MZF-2 (5′-CCGGAGATGGGT- CACAGTCC-3′ and 5′-TTGCTGAACACCTTGCCAC-3′) were used. The efficiency of cDNA synthesis from each sample was estimated by PCR with GAPDH-specific primers as previously described (27).
Gel shift assay
Nuclear extracts from Cos7 and C33A cells were prepared as previously described (26). For overexpression of MZF-2 in Cos7 cells, 10 µg MZF-2 expression vector were transfected into 105 Cos7 cells by the DEAE–dextran method (28) in 60 mm culture dishes. After 48 h transfection, cells were harvested and nuclear extracts were prepared. As a negative control, blank vectors were transfected in the same fashion. In gel shift assays 5–10 µg nuclear extracts were incubated with 0.5 µg poly(dI·dC) in the presence or absence of unlabeled competitors on ice for 20 min in a 25 µl reaction volume containing 10% glycerol, 25 mM HEPES, pH 7.9, 50 mM KCl, 0.5 mM PMSF, 1 mM dithiothreitol and 4 mg/ml bovine serum albumin. Following incubation, 10 000 c.p.m. of 32P-end-labeled oligonucleotide probes containing the MZF-2 consensus motifs at –687 (5′-GTTTGCTCATGGTGGGGACCCCTCGCCG-3′) or mutant oligonucleotides (5′-GTTTGCTCATGGTAAAGACCCCTCGCCG-3′) were added and the reaction was incubated at 4°C for an additional 20 min. Following electrophoresis on a 4% polyacrylamide gel, the gel was dried and subjected to autoradiography. For supersift assays, anti-c-Myc epitope tag antibody (9E10; SantaCruz) was pre-incubated with nuclear extracts for 1 h at 4°C, followed by a binding reaction.
RESULTS
Identification and characterization of silencer elements in hTERT promoter
To identify the regulatory region responsible for the transcriptional regulation of hTERT, a series of luciferase reporter plasmids containing various lengths of hTERT promoter were prepared and tested by luciferase assay using a variety of cell types. As shown in Figure 1, the 1375 bp full-length promoter and 5′-truncated promoters exhibited significant transcriptional activity in all cell types examined, though levels of activity varied among cell types. The 5′ deletions of the promoter up to –776 significantly reduced transcriptional activity. However, further deletion from –776 to –378 resulted in a significant increase in transcriptional activity. In particular, deletion from –578 to –378 resulted in the largest increase in transcriptional activity. The proximal 373 and 181 bp regions exhibited high levels of transcriptional activity comparable with that of the 1375 bp promoter. However, 5′ deletion of the proximal 181 bp region resulted in a stepwise reduction in transcriptional activity. These findings suggest that the proximal 181 bp region functions as a minimal core promoter, in agreement with recent observations (21,22), and that a negative regulatory element is present in the region between –776 and –378.
Figure 1.

Transcriptional activity of the hTERT promoter in various cell lines. (A) A schematic representation of the reporter plasmids. The 1395 bp DNA and 5′-truncated fragments of hTERT promoter upstream of the ATG initiation codon were inserted into the luciferase (LUC) reporter vector pGL3-Basic in the sense orientation. The 181 bp core promoter is shown as a black box. Numbers refer to the number of bases upstream (–) or downstream (+) of the transcription start site. (B) Luciferase reporter plasmids with full-length and 5′-truncated promoters of hTERT were transfected into various cell lines and luciferase assays were performed. N, pGL3-Basic, which lacks the enhancer/promoter, was used as a negative control; P, pGL3-Control containing the SV40 enhancer/promoter used as a positive control. The transcriptional activities in each reporter plasmid are indicated as relative luciferase activities. The values for the positive control are indicated as 100%.
To examine the roles played by this potential silencer during cellular differentiation, myeloid cell line K562 cells were treated with the differentiation-inducing agent PMA (29) and luciferase assays were performed using a series of deletion mutants of reporter plasmids. As shown in Figure 2, the transcriptional activity in each reporter plasmid was decreased in the presence of PMA, suggesting that cellular differentiation represses hTERT transcription, consistent with the previous finding that telomerase activity is inhibited by cellular differentiation through down-regulation of hTERT (11). Interestingly, transcriptional repression by PMA was dramatically enhanced in pGL3-578, which contains the proximal 200 bp region of the silencer, but not in pGL3-378 or pGL3-181, which lack the silencer. These findings suggest that the potential silencer, especially its proximal 200 bp region, augments the effects of differentiation-inducing reagents on transcriptional repression.
Figure 2.

Effects of cellular differentiation on transcriptional activity of the hTERT promoter. K562 cells were transfected with various luciferase reporter plasmids containing full-length or 5′-truncations of the 1395 bp hTERT promoter in the absence or presence of PMA. The transcriptional activities in the presence of PMA are indicated as luciferase activities relative to those in the absence of PMA. Bars indicate standard deviations (SD).
Multiple sites for MZF-2 consensus motifs are responsible for transcriptional repression
A computer-assisted homology search identified putative binding sites for several transcription factors, such as NF-κB, MyoD and GATA, within the silencer (Fig. 3). In addition, we found four binding motifs for MZF-2 in this region. The presence of multiple sites for MZF prompted us to examine the roles played by these sites in transcriptional regulation. Reporter plasmids in which all four binding sites for MZF-2 were mutated (pGL3-776-MZF2MT) were tested by luciferase assay using C33A and SiHa cells. As shown in Figure 4, mutation of these sites resulted in a 2- to 3-fold activation of transcription, suggesting that the four MZF binding sites play roles in transcriptional repression.
Figure 3.

Putative binding sites for transcription factors in the negative regulatory region of hTERT promoter. Mutated sequences (MT) in each putative MZF-2 motif are shown below the wild-type sequences (WT).
Figure 4.

The effects of mutation in the putative MZF-2 binding motifs on the transcriptional activity of hTERT. Wild-type pGL3-776 or mutated reporter plasmid (pGL3-776-MZF2MT) in which four putative binding motifs for MZF-2 were abrogated by substitution mutation (see Fig. 4) were transfected into C33A and SiHa cells and luciferase assays were performed. The transcriptional activities in pGL3-776-MZF2MT are indicated as luciferase activities relative to control activity in pGL3-776, which is shown as 100%. Bars indicate standard deviations (SD).
MZF-2 protein binds to the hTERT promoter
To examine whether MZF-2 can bind to the putative binding sites in the hTERT promoter, a gel shift assay was performed using each putative site as a probe. As shown in Figure 5A, a specific band was observed with probes spanning a putative site at –687 using nuclear extracts from Cos7 cells transfected by MZF-2 expression vectors, while no specific band was observed with use of extracts from control Cos7 cells into which blank vectors had been transfected. This band was supershifted by addition of an antibody to the c-myc epitope tag. No specific band was observed with mutated probes in which the MZF-2 consensus motif had been altered. Similar band patterns were observed using probes spanning three other putative sites for MZF-2 (data not shown). The specific band was observed in nuclear extracts from some cancer lines, such as C33A (Fig. 5B) and SiHa (data not shown), which had not been transfected with MZF-2 expression vectors, indicating constitutive expression of MZF-2 in these cells.
Figure 5.
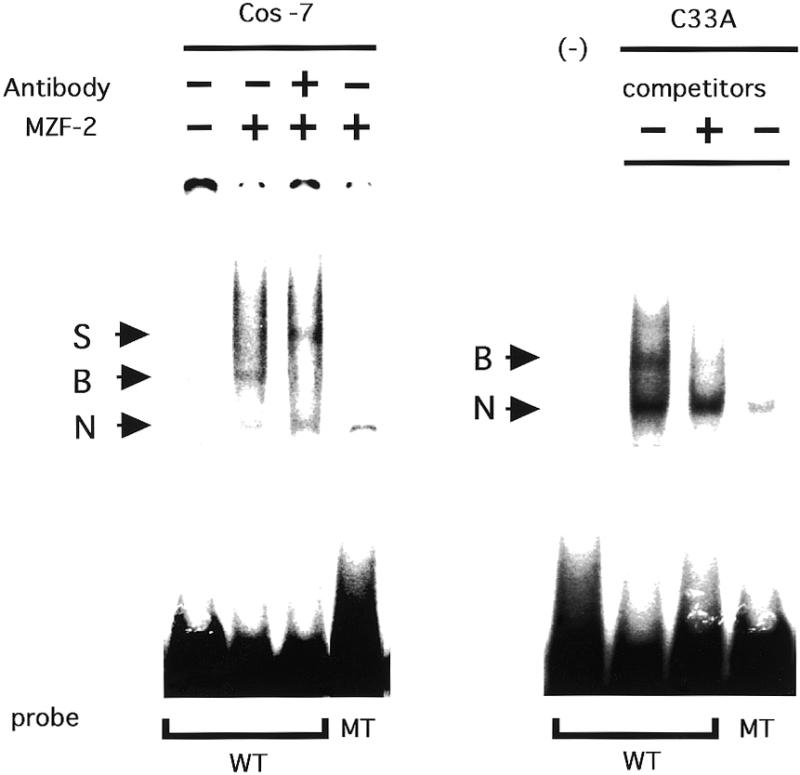
MZF-2 can specifically bind to the putative binding motifs in the hTERT promoter. (A) Nuclear extracts were prepared from Cos7 cells transfected with MZF-2 expression vectors or blank vectors and were incubated with the 32P-labeled probes spanning the wild-type (WT) or mutated (MT) sequences of the putative MZF-2 binding motif at –687. Anti-c-myc epitope tag antibody (myc Ab) was used for supershift assays to determine the specific band formed with MZF-2. (B) Nuclear extracts were prepared from C33A cells and were incubated with wild-type (WT) or mutant (MT) probes for MZF-2 binding motifs at –687 in the absence or presence of cold homologous competitors. B, specific binding complexes; S, supershifted bands; N, non-specific bands.
Overexpression of MZF-2 represses transcription of hTERT and telomerase activity
To examine the effects of MZF-2 on hTERT transcription, a MZF-2 expression vector was co-transfected with the pGL3-776 reporter plasmid, which contains the silencer, into SiHa and C33A cells and luciferase assays were performed (Fig. 6A). Overexpression of MZF-2 in these cells resulted in a significant reduction in transcriptional activity, while no repression was observed in control cells transfected with vectors without the MZF-2 gene. We further examined the effects of MZF-2 overexpression on telomerase activity. C33A cells were transfected with MZF-2 expression vectors and telomerase activity was determined by quantitative telomerase assay. Telomerase activity in MZF2-transfected cells was significantly lower than that in control cells, which had been transfected with vectors without the MZF-2 gene (Fig. 6B). These results suggest that MZF-2 may be an effector of negative regulation of hTERT transcription.
Figure 6.
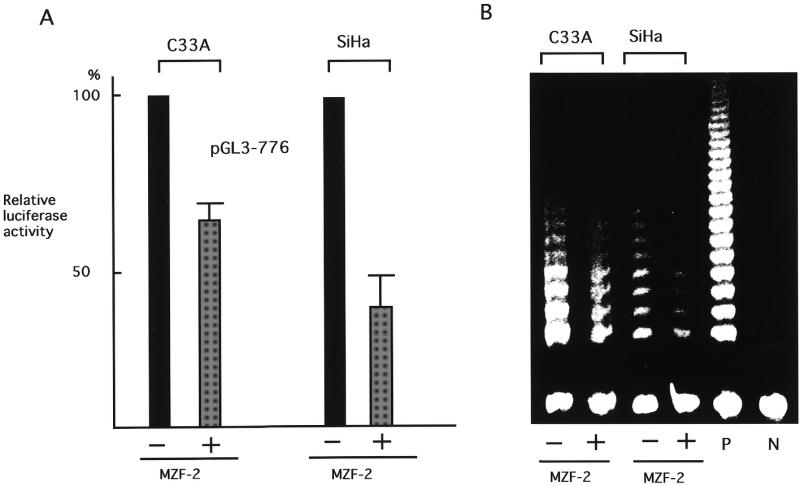
The effects of overexpression of MZF-2 on the transcriptional activity of hTERT and telomerase activity. (A) MZF-2 expression vectors or blank vectors were co-transfected into C33A and SiHa cells with pGL3-776 reporter plasmids and luciferase assays were performed. The transcriptional activities in pGL3-776 co-transfected with MZF-2 expression vectors are indicated as luciferase activities relative to control activity in pGL3-776 co-transfected with blank vector, which is shown as 100%. Bars indicate standard deviations (SD). (B) Telomerase activities in C33A cells transfected with MZF-2 expression vectors or blank vectors were examined by quantitative stretch PCR assay. P, positive control samples using extracts from telomerase-positive C33A cell lines; N, negative control samples using lysis buffer only.
Expression of MZF-2 in various established cell lines
The expression of MZF-2 was examined by RT–PCR in cell lines established from tumors which were telomerase-positive. Interestingly, specific MZF-2 bands were observed in all cell types examined (data not shown). These findings suggest that MZF-2 is constitutively expressed in a wide variety of cell types. Since the inhibitory effects of the silencer on the hTERT promoter were enhanced by cellular differentiation, we examined whether MZF-2 is induced by treatment of K562 cells with PMA. However, expression of MZF-2 was not induced to a significant extent by this treatment (data not shown).
DISCUSSION
In the present study we have demonstrated that the 400 bp region between –776 and –378 in the hTERT promoter functions as a transcriptional silencer. This is probably the first study demonstrating a negative regulatory element for the hTERT promoter. The inhibitory effects of this element were observed in a variety of cell types, suggesting that this element may function as a constitutive silencer.
Telomerase is down-regulated by cellular differentiation, which is accompanied by inhibition of hTERT mRNA expression. The present study in fact showed that PMA treatment of myeloid cells resulted in a significant down-regulation of promoter activity. Interestingly, the inhibitory effect of PMA on the activity of the hTERT promoter was significantly enhanced with cellular differentiation in reporter plasmids that contained this silencer element, but not in those lacking it. This element may thus play roles in differentiation-dependent transcriptional repression of hTERT. In particular, the proximal 200 bp region between –578 and –378 appears to play a central role in this repression. It would be of interest to identify the transcription factors involved in this regulation, which may be induced by cellular differentiation. We confirmed that MZF-2 is not induced by cellular differentiation, suggesting that factors other than MZF-2 may interact with the silencer in differentiation-dependent transcriptional repression of hTERT. Identification of these unknown regulatory factors may significantly contribute to our understanding of the molecular mechanisms through which cellular differentiation inhibits telomerase.
In the present study our interest was focused on multiple consensus motifs for the zinc finger transcription factor MZF-2 present in the silencer. Abrogation of these sites by substitution mutations led to significant transactivation of hTERT. Gel shift analyses identified specific binding of MZF2 to these motifs. Overexpression of MZF-2 in cells resulted in a marked reduction in transcriptional activity. These findings suggest that MZF-2 may mediate transcriptional down-regulation of hTERT. MZF-2 is a myeloid-specific transcription factor involved in the development of neutrophils (26). The initial study demonstrated that this factor is expressed preferentially in myeloid cells. Although the expression of MZF-2 in normal cell lines and normal tissues has not been examined, our study clearly demonstrated that MZF-2 is expressed not only in myeloid cells but in established cell lines of various origins, suggesting that it may contribute to regulation of hTERT in a wide variety of cell types. The finding that telomerase-positive cell lines express MZF-2 shows that MZF-2 is not a rate limiting determinant of telomerase activity in these cell lines. However, MZF-2 might function as a significant inhibitor of telomerase under some cellular conditions in which it is efficiently induced. We are currently attempting to determine these conditions.
The molecular mechanisms through which MZF-2 inhibits the promoter activity of hTERT remain unclear. Recent progress in understanding the mechanisms of transcriptional repression by specific transcription factors has revealed that these factors interact with co-repressors, such as N-CoR and mSin3 (30). It will therefore be necessary to examine whether MZF-2 interacts with these co-repressors in vitro and in vivo. A previous study suggested that MZF-2 may be functionally inactive in the absence of unknown myeloid-specific co-activators which bind to the activation domain of MZF-2 through protein–protein interaction and facilitate the transactivating function of MZF-2 (30). Therefore, in cells lacking expression of such co-activators, MZF-2 may bind to the promoters of target genes as a functionally inactive form. Inactive MZF-2 may function as a transcriptional repressor by competing with other transcription factors recognizing the same or overlapping sites, leading to a decrease in transcriptional efficiency. Further extensive studies will be required to identify the molecular mechanisms through which MZF-2 functions as a telomerase inhibitor. Such studies may enable development of novel anticancer strategies involving inhibition of telomerase activity in cancer cells.
Acknowledgments
ACKNOWLEDGEMENT
We thank Dr S. Nagata (Department of Genetics, Osaka University Medical School, Japan) for kindly providing MZF-2 expression vectors.
REFERENCES
- 1.Greider C.W. (1996) Annu. Rev. Biochem., 65, 337–365. [DOI] [PubMed] [Google Scholar]
- 2.Greider C.W. (1991) Cell, 67, 645–647. [DOI] [PubMed] [Google Scholar]
- 3.Watson J.D. (1972) Nature, 239, 197–201. [DOI] [PubMed] [Google Scholar]
- 4.Kim N.W., Piatyszek,M.A., Prowse,K.R., Harley,C.B., West,M.D., Ho,P.L.C., Oviello,G.M., Wright,W.E., Weinrich,S.L. and Shay,J.W. (1994) Science, 266, 2011–2015. [DOI] [PubMed] [Google Scholar]
- 5.Shay J.W. and Bacchetti,S. (1997) Eur. J. Cancer, 33, 787–791. [DOI] [PubMed] [Google Scholar]
- 6.Counter C.M., Avilion,A.A., LeFeuvre,C.E., Stewart,N.G., Greider,C.W. and Harley,C.B. (1992) EMBO J., 11, 1921–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Counter C.M., Hirte,H.W., Bacchetti,S. and Harley,C.B. (1994) Proc. Natl Acad. Sci. USA, 91, 2900–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feng J., Funk,W.D., Wang,S.S., Weinrich,A.A.A., Chiu,C.P., Adams,R.R., Chang,E., Allsopp,R.C., Yu,J., Le,S., West,M.D., Harley,C.B., Andrew,W.H., Greider,C.W. and Villeponteau,B. (1995) Science, 269, 1236–1241. [DOI] [PubMed] [Google Scholar]
- 9.Harrington L., McPhail,T., Mar,V., Zhou,W., Oulton,R., Bass,M.B., Arruda,I. and Robinson,M.O. (1997) Science, 275, 973–977. [DOI] [PubMed] [Google Scholar]
- 10.Nakayama J., Saito,M., Nakamura,H., Matsuura,A. and Ishikawa,F. (1997) Cell, 88, 875–884. [DOI] [PubMed] [Google Scholar]
- 11.Meyerson M., Counter,C.M., Eaton,E.N., Ellisen,L.W., Steiner,P., Caddle,S.D., Ziaugra,L., Beijersbergen,R.L., Davidoff,M.J., Liu,Q., Bacchetti,S., Haber,D.A. and Weinberg,R.A. (1997) Cell, 90, 785–795. [DOI] [PubMed] [Google Scholar]
- 12.Nakamura T.M., Morin,G.B., Chapman,K.B., Weinrich,S.L., Andrews,W.H., Lingner,J., Harley,C.B. and Cech,T.R. (1997) Science, 277, 955–959. [DOI] [PubMed] [Google Scholar]
- 13.Ito H., Kyo,S., Kanaya,T., Takakura,M., Inoue,M. and Namiki,M. (1998) Clin. Cancer Res., 4, 1603–1608. [PubMed] [Google Scholar]
- 14.Kanaya T., Kyo,S., Takakura,M., Ito,H., Namiki,M. and Inoue,M. (1998) Int. J. Cancer, 78, 539–543. [DOI] [PubMed] [Google Scholar]
- 15.Takakura M., Kyo,S., Kanaya,T., Tanaka,M. and Inoue,M. (1998) Cancer Res., 58, 1558–1561. [PubMed] [Google Scholar]
- 16.Kyo S., Kanaya,T., Takakura,M., Tanaka,M. and Inoue,M. (1999) Int. J. Cancer, 80, 60–63. [DOI] [PubMed] [Google Scholar]
- 17.Kyo S., Kanaya,T., Takakura,M., Tanaka,M., Yamashita,A., Inoue,H. and Inoue,M. (1999) Int. J. Cancer, 80, 804–809. [DOI] [PubMed] [Google Scholar]
- 18.Weinrich S.L., Pruzan,R., Ma,L., Ouellette,M., Tesmer,V.M., Holt,S.E., Bodnar,A.G., Lichtsteiner,S., Kim,N.W., Trager,J.B., Taylor,R.D., Carlos,R., Andrews,W.H., Wright,W.E., Shay,J.W., Harley,C.B. and Morin,G.B. (1997) Nature Genet., 17, 498–502. [DOI] [PubMed] [Google Scholar]
- 19.Nakayama J., Tahara,H., Tahara,E., Saito,M., Ito,K., Nakamura,H., Nakanishi,T., Tahara,E., Ide,T. and Ishikawa,F. (1998) Nature Genet., 18, 65–68. [DOI] [PubMed] [Google Scholar]
- 20.Bodnar A.G., Ouellette,M., Frolkis,M., Holt,S.E., Chiu,C.P., Morin,G.B., Harley,C.B., Shay,J.W., Lichtsteiner,S. and Wright,W.E. (1998) Science, 279, 349–352. [DOI] [PubMed] [Google Scholar]
- 21.Takakura M., Kyo,S., Kanaya,T., Hirano,H., Takeda,J., Yutsudo,M. and Inoue,M. (1999) Cancer Res., 59, 551–559. [PubMed] [Google Scholar]
- 22.Cong Y.S., Wen,J. and Bacchetti,S. (1999) Hum. Mol. Genet., 8, 137–142. [DOI] [PubMed] [Google Scholar]
- 23.Wu K.J., Grandori,C., Amacker,M., Simon-Vermot,N., Polack,A., Lingner,J. and Dalla-Favera,R. (1999) Nature Genet., 21, 220–224. [DOI] [PubMed] [Google Scholar]
- 24.Greenberg R.A., O’Hagan,R.C., Deng,H., Xiao,Q., Hann,S.R., Adams,R.R., Lichtsteiner,S., Chin,L., Morin,G.B. and DePinho,R.A. (1999) Oncogene, 118, 1219–1226. [DOI] [PubMed] [Google Scholar]
- 25.Kyo S., Takakura,M., Taira,T., Kanaya,T., Itoh,H., Yutsudo,M., Ariga,H. and Inoue,M. (2000) Nucleic Acids Res., 28, 669–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murai K., Murakami,H. and Nagata,S. (1998) Proc. Natl Acad. Sci. USA, 95, 3461–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Higuchi R. (1990) In Innis,M.A., Gelfand,D.H., Sninsky,J.J. and White,T.J. (eds), PCR Protocols. Academic Press, San Diego, CA, pp. 177–183.
- 28.Chomczynski P. and Sacchi,N. (1987) Anal. Biochem., 162, 156–159. [DOI] [PubMed] [Google Scholar]
- 29.Irving J., Eang,J., Wistrom,C., Pikaert.M. and Villoponteau.,B. (1992) Exp. Cell Res., 202, 161–166. [DOI] [PubMed] [Google Scholar]
- 30.Horlein A.J., Naar,A.M., Heinzel,T., Torchia,J., Gloss,B., Kurokawa,R., Ryan,A., Kamei,Y., Soderstrom,M. and Glass,C.K. (1995) Nature, 377, 397–404. [DOI] [PubMed] [Google Scholar]