

ABSTRACT
Objective
To explore the clinical characteristics of mitochondrial encephalomyopathy, lactic acidosis, and stroke‐like episodes (MELAS) caused by mitochondrial DNA‐encoded complex I subunit (mt‐ND) variants.
Methods
In this retrospective study, the clinical, myopathological and brain MRI features of patients with MELAS caused by mt‐ND variants (MELAS‐mtND) were collected and compared with those of MELAS patients carrying the m.3243A > G variant (MELAS‐A3243G).
Result
A total of 18 MELAS‐mtND patients (female: 7; median age: 24.5 years) represented 15.9% (n = 113) of all patients with MELAS caused by mtDNA variants in our neuromuscular center from January 2012 to June 2022. In this MELAS‐mtND cohort, the two most common variants were m.10191 T > C (4/18, 22.2%) and m.13513 G > A (3/18, 16.7%). The most frequent symptoms were seizures (14/18, 77.8%) and muscle weakness (11/18, 61.1%). Compared with 87 MELAS‐A3243G patients, MELAS‐mtND patients were significantly more likely to have a variant that was absent in blood cells (40% vs. 1.4%). Furthermore, MELAS‐mtND patients had a significantly lower MDC score (7.8 ± 2.7 vs. 9.8 ± 1.9); less hearing loss (27.8% vs. 54.0%), diabetes (11.1% vs. 37.9%), and migraine (33.3% vs. 62.1%); less short stature (males ≤ 165 cm; females ≤ 155 cm; 23.1% vs. 60.8%) and higher body mass index (20.4 ± 2.5 vs. 17.8 ± 2.7). MELAS‐mtND patients had significantly more normal muscle pathology (31.3% vs. 4.1%) and fewer RRFs/RBFs (62.5% vs. 91.9%), COX‐deficient fibers/blue fibers (25.0% vs. 85.1%) and SSVs (50.0% vs. 81.1%). Moreover, brain MRI evaluated at the first stroke‐like episode showed significantly more small cortical lesions in MELAS‐mtND patients (66.7% vs. 12.2%).
Interpretation
Our results suggested that MELAS‐mtND patients have distinct clinical, myopathological and brain MRI features compared with MELAS‐A3243G patients.
Introduction
Mitochondrial encephalomyopathy, lactic acidosis, and stroke‐like episodes (MELAS) is one of the most frequent maternally inherited mitochondrial disease (MD) and seriously impacts the health of patients with no effective treatment. 1 The pathogenic variant m.3243A > G, first described by Goto et al., 2 has been demonstrated to account for approximately 80.0% of MELAS cases. 3 However, the clinical features of MELAS caused by other pathogenic variants are often overlooked.
The first description of MELAS caused by a mitochondrial DNA‐encoded complex I subunit (mt‐ND) variant (MELAS‐mtND) (m.13513G > A, mt‐ND5) was reported by Santorelli et al. in 1997. 4 Henceforth, a growing number of studies revealed that mt‐ND is the second most common genetic subtype of MELAS after the MT‐TL1 gene (encoding mt‐tRNALeu (UUR)). 5 , 6 , 7 However, most of the previous studies were restricted to sporadic case reports 8 , 9 , 10 or focused on isolated complex I (CI) deficiency. 11 , 12 , 13 MELAS‐mtND has long been overlooked. To our knowledge, even in the largest mt‐ND variant cohort, only three MELAS‐mtND patients were enrolled. 14 Our previous studies found that MELAS‐mtND patients exhibited a variant that was absent in blood cells 8 and presented a migratory tiny lesion on brain MRI 15 which is not common in MELAS caused by m.3243A > G variant (MELAS‐A3243G). The rarity of mt‐ND variants has limited previous studies, but the advancements in the technology and the decreasing cost of next‐generation sequencing (NGS) techniques now enable us to know more about the details of MELAS‐mtND.
MDs including MELAS are difficult to diagnose due to extraordinary variable phenotypes and genotypes and the lack of specific laboratory indicators. 16 Over the past decades, consensus diagnostic criteria for MDs have been proposed and refined for adults and children. 17 , 18 The mitochondrial disease criteria (MDC) which evaluate the clinical symptoms, metabolic abnormalities, neuroimaging and muscle pathology of MDs is useful in the diagnosis of MDs. 19 , 20 However, the diagnostic value of the MDC in patients with MELAS caused by different mtDNA variants is rarely reported.
Here, we present a cohort of 18 MELAS‐mtND patients focusing on the clinical, neuroimaging, and pathological findings, molecular genetics, and the MDC score and compared them with those of MELAS‐A3243G patients.
Materials and Methods
Identification of patients and clinical data
We conducted a retrospective study of MELAS patients in the local database of mitochondrial diseases from January 2012 to June 2022. The inclusion criteria were as follows: (1) clinically diagnosed with MELAS according to standard clinical criteria, meeting at least two category A criteria (headaches with vomiting, seizures, hemiplegia, cortical blindness, and acute focal lesions in neuroimaging) and one category B criteria (high plasma or cerebrospinal fluid (CSF) lactate, mitochondrial abnormalities in muscle biopsy), 21 and (2) carrying mt‐ND variants or the m.3243A > G variant. For the novel mt‐ND variant, the possibility of mutation in nDNA was precluded. A total of 18 MELAS‐mtND and 87 MELAS‐A3243G patients were identified. Clinical data were collected by experienced neurologists from all MELAS‐mtND and MELAS‐A3243G patients including age, sex, family history, CNS symptoms (seizures, migraine, cortical blindness, and ataxia), muscular symptoms (muscle weakness, exercise intolerance and ophthalmoplegia), multisystem involvement (cardiac abnormalities, hearing loss and diabetes), brain MRI, and lactate levels. The heteroplasmy levels were measured in at least one of four tissues (skeletal muscle, blood cells, urothelium, or oral mucosa) by NGS. Height and body mass index (BMI) were collected from 13 MELAS‐mtND patients and 51 MELAS‐A3243G patients (age > 16 years). Additionally, brain MRI from the first acute onset of stroke‐like episodes to the last follow‐up was obtained from 18 MELAS‐mtND patients and 49 MELAS‐A3243G patients. The MDC score, which was used to assess clinical features, was calculated in 16 MELAS‐mtND patients and 74 MELAS‐A3243G patients who underwent muscle biopsy. All these data were compared between the MELAS‐mtND and MELAS‐A3243G groups. Informed consent was obtained from all patients enrolled in this study. This study was approved by the Ethics Committee of Qilu Hospital.
Muscle histochemistry
Sixteen MELAS‐mtND patients and 74 MELAS‐A3243G patients underwent muscle biopsies. Routine histological and immunohistochemical examinations were conducted according to standard protocols. 22 The sections were stained with hematoxylin and eosin (HE), modified Gomoritrichrome (MGT), NADH‐tetrazolium reductase (NADP‐TR), succinate dehydrogenase (SDH), cytochrome C oxidase (COX), succinate reductase/cytochrome C oxidase (S/C), oil red O (ORO), and adenosine triphosphatase (ATPase pH 9.4, 4.6 and 4.3). 23 Muscle pathological features were compared between MELAS‐mtND and MELAS‐A3243G groups.
Statistical analysis
The data were analyzed by GraphPad Prism Software (GraphPad Prism 8, Inc). Normality of distribution was determined using the D'Agostino and Pearson test. Two side unpaired t tests were used to compare normally distributed data, including body mass index (BMI) and MDC score. One‐way ANOVA was used in multiple comparisons, such as different patterns of MDC score. Mann–Whitney U test was used to compare nonnormal distributed data (onset age). Chi‐square test or Fisher's exact test analyses were conducted to compare categorical variables, including clinical features such as short stature, muscular presentation, multisystem involvement, CNS involvement, muscle pathology, and neuroimaging between the MELAS‐mtND group and MELAS‐A3243G group. Significance levels were defined as p value < 0.05 (*p < 0.05, **p < 0.01, ***p < 0.001).
Results
Genetic findings
A total of 18 MELAS‐mtND patients was included in this study. They represent 15.9% (18/113) of all patients with genetically confirmed MELAS patients caused by mtDNA variants in our mitochondrial database from January 2012 to June 2022. The two most common variants in the MELAS‐mtND group were m.10191 T > C (4/18) in mt‐ND3 and m.13513G > A (3/18) in mt‐ND5 (Fig. 1). The other mt‐ND variants detected were m.3901G > A (mt‐ND1) (n = 1), m.3946G > A (mt‐ND1) (n = 1), m.10158 T > C (mt‐ND3) (n = 1), m.11138A > G (mt‐ND4) (n = 1), m.11777C > A (mt‐ND4) (n = 1), m.11406 T > A (mt‐ND4) (n = 1), m.12706 T > C (mt‐ND5) (n = 1), m.13042G > A (mt‐ND5) (n = 1), m.13046 T > C (mt‐ND5) (n = 1), m.14487 T > C (mt‐ND6) (n = 1), and m.14597A > G (mt‐ND6) (n = 1). Among these variants, two were novel variants (m.3901G > A and m.11138A > G), four were reported pathogenic variants (m.3946G > A, m.13046 T > C, m.11406 T > A and m.14597A > G) and the remainder were confirmed pathogenic variants according to the MITOMAP database (MITOMAP: A Human Mitochondrial Genome Database. http://www.mitomap.org, 2019). The variant loads in muscle samples were higher than those in blood cells and oral mucosa (Figs 1 and 2A). There was also higher proportion of variant levels in the urothelium than in the blood cells (Figs 1 and 2A). Interestingly, 6/15 (40.0%) of MELAS‐mtND patients showed no detectable variants in their blood cells, while only 1/72 (1.4%) in MELAS‐A3243G group (p < 0.001). The mt‐ND variants that were absent in blood were m.3901G > A (n = 1), m.13042G > A (n = 1), m.13513G > A (n = 1), m.11406 T > A (n = 1), m.12706 T > C (n = 1), and m.14487 T > C (n = 1).
Figure 1.
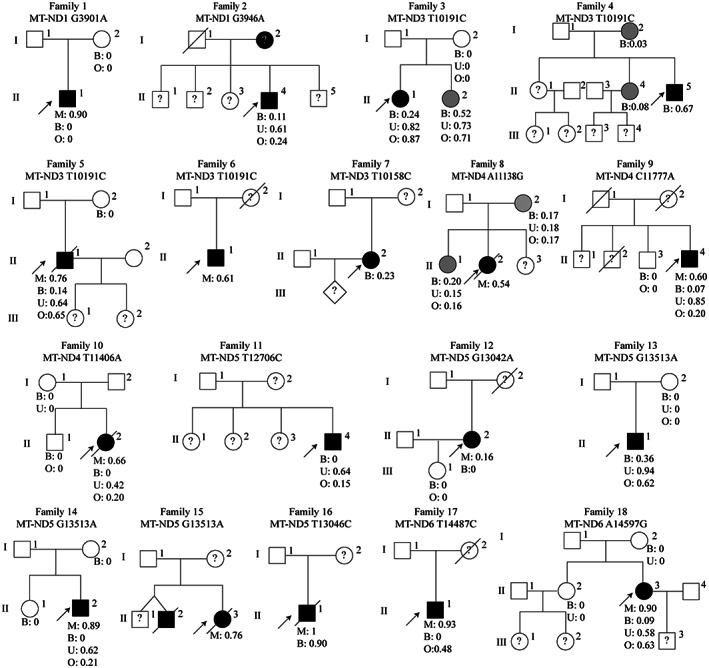
Pedigree diagram for 18 unrelated Chinese families with mt‐ND variants. The arrow indicates the proband. Squares symbolize males, and circles symbolize females. Black symbols and gray symbols indicate symptomatic patients and asymptomatic carriers, respectively. Question marks indicate family members who refused to undergo genetic analysis. B, blood cells; M, muscle sample; O, oral mucosa; U, urothelium.
Figure 2.
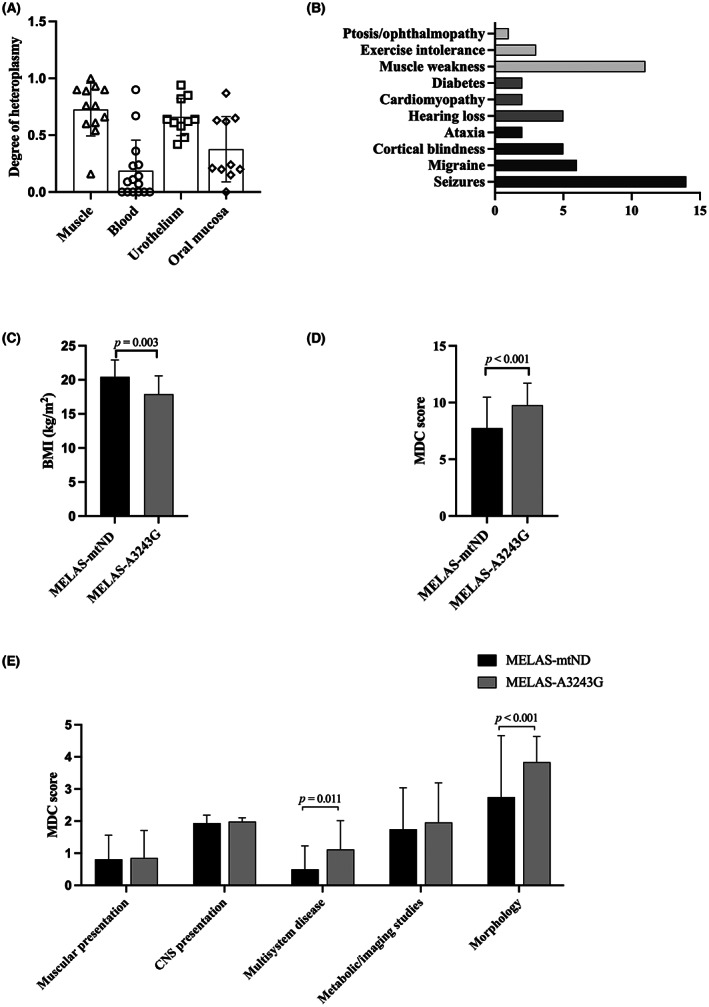
Genetic heteroplasmy and clinical findings in MELAS‐mtND patients. (A) Genetic heteroplasmy in different tissues of MELAS‐mtND patients. (B) Frequency of symptoms of 18 MELAS‐mtND patients. The gray column represents muscular involvement, the dark gray column represents the multisystem involvement, and the black column represents central nerve system involvement. (C) Comparison of BMI between MELAS‐mtND and MELAS‐A3243G patients. (D and E) Comparison of the MDC score between MELAS‐mtND and MELAS‐A3243G patients.
Clinical features and laboratory findings of the mt‐ND variant cohort
This MELAS‐mtND cohort consisted of 11 males and 7 females. Five (27.8%) individuals were maternally inherited. The median onset age was 24.5 years old (IQR: 16–45 years). A total of five patients (27.8%) presented with clinical syndromes ≥ 40 years of age, and seven patients (38.9%) presented with clinical syndromes ≤ 20 years of age. During follow‐up, five (27.8%) MELAS‐mtND patients died of generalized seizures. Short stature in this study was defined as male ≤ 165 cm and female ≤ 155 cm 24 at age > 16 years old. Three (3/13, 23.1%) MELAS‐mtND patients exhibited short stature. The most common symptoms associated with MELAS‐mtND were encephalopathy, including seizures (14/18, 77.8%), migraine (6/18, 33.3%), cortical blindness (5/18, 27.8%), and ataxia (2/18, 11.1%). Seven patients (38.9%) showed multisystem involvement, including hearing loss (5/18, 27.8%), cardiac abnormalities (2/18, 11.1%), and diabetes (2/18, 11.1%). Eleven patients manifested muscle involvement as muscle weakness (11/18, 61.1%), exercise intolerance (3/18, 16.7%), and ptosis/ophthalmopathy (1/18, 5.6%) (Table 1 and Fig. 2B). Two patients were excluded from MDC scoring because they had no prior biopsy. Regarding the MDC, 62.5% (10/16) of MELAS‐mtND patients had a score predicting definite mitochondrial disorder, 25.0% (4/16) had a probable mitochondrial disorder, and 12.5% (2/16) had a possible mitochondrial disorder.
Table 1.
Comparison of demographic, clinical, and muscle pathology findings between MELAS‐mtND and MELAS‐A3243G patients.
| MELAS‐mtND | MELAS‐A3243G | p Value | |
|---|---|---|---|
| n | 18 | 87 | |
| Sex (female:male) | 7:11 | 37:50 | 0.776 |
| Onset age (years) (median [range]) | 24.5 [4–58] | 29 [2–76] | 0.569 |
| Short stature | 3/13, 23.1% | 31/51, 60.8% | 0.027 |
| BMI | 20.4 ± 2.5, n = 13 | 17. 8 ± 2.7, n = 51 | 0.003 |
| Muscular presentation | |||
| Muscle weakness | 11/18, 61.1% | 43/87, 49.4% | 0.367 |
| Exercise intolerance | 3/18, 16.7% | 26/87, 29.9% | 0.386 |
| Ophthalmoplegia | 1/18, 5.6% | 9/87, 10.3% | >0.999 |
| Multisystem involvement | |||
| Cardiomyopathy | 2/18, 11.1% | 17/87, 19.5% | 0.517 |
| Diabetes | 2/18, 11.1% | 33/87, 37.9% | 0.030 |
| Hearing loss | 5/18, 27.8% | 47/87, 54.0% | 0.043 |
| CNS involvement | |||
| Migraine | 6/18, 33.3% | 54/87, 62.1% | 0.025 |
| Seizure | 14/18, 77.8% | 62/87, 71.3% | 0.774 |
| Cortical blindness | 5/18, 27.8% | 36/87, 41.4% | 0.282 |
| Ataxia | 2/18, 11.1% | 12/87, 13.8% | >0.999 |
| Metabolic | |||
| Lactate elevated | 10/18, 55.6% | 45/87, 51.7% | 0.767 |
| Muscle pathology | |||
| Normal | 5/16, 31.3% | 3/74, 4.1% | 0.004 |
| RRFs/RBFs | 10/16, 62.5% | 68/74, 91.9% | 0.002 |
| COX‐deficient fibers/blue fibers | 4/16, 25.0% | 63/74, 85.1% | <0.001 |
| SSVs | 8/16, 50.0% | 60/74, 81.1% | 0.009 |
BMI, body mass index; CNS, central nerve system; RBFs, ragged blue fibers; RRFs, ragged red fibers; SSVs, strongly SDH‐reactive vessels.
Comparing the clinical data of 18 MELAS‐mtND patients with 87 MELAS‐A3243G patients (Table 1), the most striking findings were that MELAS‐mtND patients exhibited higher BMI (20.4 ± 2.5 vs. 17.8 ± 2.7, p = 0.003) (Fig. 2C), less short stature (23.1% vs. 60.8%, p = 0.027), less hearing loss (27.8% vs. 54.0%, p = 0.043), less diabetes (11.1% vs. 37.9%, p = 0.030), and less migraine (33.3% vs. 62.1%, p = 0.025). Comparing the MDC score between these two groups, a lower MDC score was observed in MELAS‐mtND patients (7.8 ± 2.7 vs. 9.8 ± 1.9, p < 0.001) (Fig. 2D), which is consistent with the less definite mitochondrial disorder (62.5% vs. 90.5%, p = 0.004) in MELAS‐mtND group. The differences between these two groups were mainly observed in the multisystem involvement (p = 0.011) and muscle pathology (p < 0.001) (Fig. 2E).
Brain MRI features
Stroke‐like lesions (SLLs) at the acute onset of stroke‐like episodes to the last follow‐up were available from 18 MELAS‐mtND patients and 49 MELAS‐A3243G patients. The phenotypes of the initial SLLs were classified as classic or nonclassic according to Yu Hongo's report. 25 In summary, classic SLLs were defined as lobular edematous lesions extending along the cerebral cortex and incongruent to the vascular territory. Nonclassic SLLs were defined as those other than classic SLLs, including single/multiple/disseminated small cortical lesions and, cerebellar lesions, with or without basal ganglion and brain stem lesions (Fig. 3).
Figure 3.
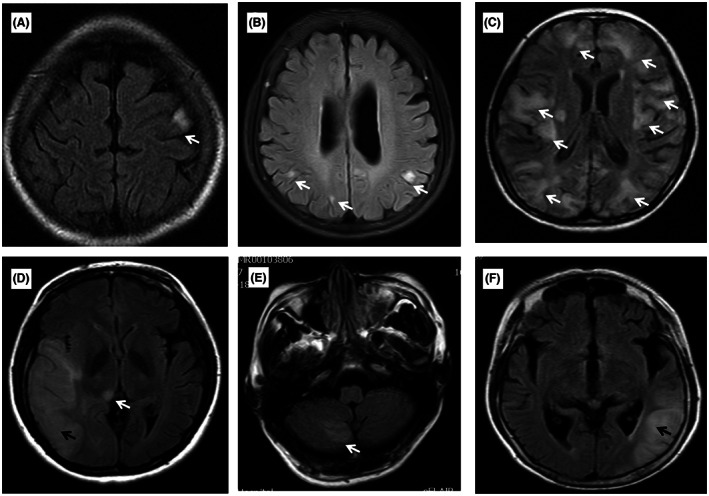
Classic and nonclassic SLLs of MELAS‐mtND patients. The white arrow indicates nonclassic SLLs. The black arrow indicates classic SLLs. (A) nonclassic SLLs with single small cortical lesion; (B) nonclassic SLLs with multiple isolated small cortical lesions; (C) nonclassic SLLs with disseminated cortical lesions; (D) classic SLLs with nonclassic basal ganglion lesion; (E) nonclassic SLLs with cerebellar lesions; (F) classic SLLs with lesions in the temporal lobe.
Of the 18 initial SLLs of MELAS‐mtND patients, 12 (66.7%) were nonclassic, including single small cortical lesion (n = 7), isolated multiple small cortical lesions (n = 3), and multiple small cortical lesions with basal ganglion lesions (n = 2), while the remaining six were classic. Of the 12 nonclassic SLLs, one developed to extensively disseminated, seven developed into classic SLLs at the last follow‐up (range 6–36 months), and the remaining four consistently presented single/multiple SLLs during the 2–6 years follow‐up. Compared to MELAS‐A3243G patients, MELAS‐mtND patients exhibited more nonclassic SLLs (66.7% vs. 12.2%, p < 0.001). Furthermore, SLLs in MELAS‐mtND group were found most commonly in the parietal lobe (7/18, 38.9%), followed by 22.2% (4/18) in the temporal lobe, 27.8% (5/18) in the occipital lobe, 11.1% (2/18) in the frontal lobe and 5.6% (1/18) in the insula. Compared to MELAS‐A3243G patients, SLLs in the temporal (22.2% vs. 65.3%, p = 0.002) and occipital lobes (27.8% vs. 55.1%, p = 0.047) were less frequent in the MELAS‐mtND group (Table 2).
Table 2.
Comparison of the initial SLLs between MELAS‐mtND and MELAS‐A3243G patients.
| SLLs | MELAS‐mtND | MELAS‐A3243G | p Value |
|---|---|---|---|
| n | 18 | 49 | |
| Nonclassic SLLs | 12/18, 66.7% | 6/49, 12.2% | <0.001 |
| Frontal lobe | 2/18, 11.1% | 5/49, 10.2% | >0.999 |
| Parietal lobe | 7/18, 38.9% | 19/49, 38.8% | 0.993 |
| Temporal lobe | 4/18, 22.2% | 32/49, 65.3% | 0.002 |
| Occipital lobe | 5/18, 27.8% | 27/49, 55.1% | 0.047 |
| Insular | 1/18, 5.6% | 3/49, 6.1% | >0.999 |
| Cerebellum | 1/18, 5.6% | 2/49, 4.1% | >0.999 |
| Basal ganglion | 2/18, 11.1% | 1/49, 2.0% | 0.174 |
| Brain stem | 0/18, 0 | 0/49, 0 | >0.999 |
SLLs, stroke‐like lesions.
Muscle histopathological features
Sixteen patients underwent muscle biopsy in our MELAS‐mtND cohort. Five of them (31.3%) presented normal muscle biopsy (no RRFs [ragged red fibers], RBFs [ragged blue fibers], COX‐deficient fibers, blue fibers, and SSVs [strongly SDH‐reactive blood vessels]). One patient showed only SSVs and COX‐deficient fibers, while the remaining 10 patients (10/16, 62.5%) displayed mitochondrial abnormalities as RRFs/RBFs with COX‐deficient fibers/blue fibers (3/16, 18.85%) or SSVs (7/16, 43.8%). Of the 10 patients (62.5%) with RRFs/RBFs, only four (40%) exhibited typical RRFs/RBFs (<1%), and the remaining six patients were atypical. Compared with muscle pathology of MELAS‐A3243G group, the MELAS‐mtND group showed a high proportion of normal muscle pathology (31.3% vs. 4.1%, p = 0.004). Furthermore, muscle pathology changes in MELAS‐mtND were significantly slighter, including RRFs/RBFs (62.5% vs. 91.9%, p = 0.002), COX‐deficient fibers/blue fibers (25.0% vs. 85.1%, p < 0.001), and SSVs (50.0% vs. 81.1%, p = 0.009). (Table 1 and Fig. 4).
Figure 4.
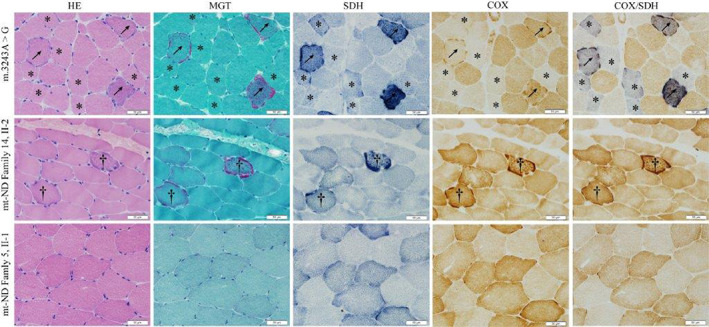
Comparison of muscle pathology between MELAS‐mtND and MELAS‐A3243G patients. Muscle pathology of one MELAS‐A3243G patient and two MELAS‐mtND patients (Family 5, II‐1 and Family 14, II‐2). Arrows represent RRFs with blue fibers. †Represent RRFs without blue fibers. *Represent COX decreased fibers presented with blue fibers but without RRFs.
Discussion
In this study, we conducted a retrospective study to summarize the characteristics of 18 patients with MELAS caused by mt‐ND variants by comparing them with 87 patients with MELAS caused by the m.3243A > G variant. This cohort, to our knowledge, is the largest cohort of MELAS‐mtND patients to date. 14 , 26 , 27 The present study shows that MELAS‐mtND patients present with lower mutation heteroplasmy in blood cells, lower proportion of migraine, lower multisystem involvement (hearing loss and diabetes), and lower presentation of RRFs and COX‐deficient fibers/blue fibers, but higher stature, higher BMI and higher probability of small cortical SLLs in brain MRI compared with MELAS‐A3243G patients. Furthermore, MELAS‐mtND patients had lower MDC scores than MELAS‐A3243G patients, and the difference was mainly in muscle presentation and multisystem involvement.
One of the remarkable features of MELAS‐mtND patients is that the mutation load in the blood cells was too low to be detected by NGS in almost half of the MELAS‐mtND cases. If only blood samples had been used for genetic testing, we would have failed to identify six MELAS‐mtND patients. Notably, although absent in the blood cells, those mutation loads were high in the muscle tissue. Thus, using correct tissue for genetic testing is essential in patients with mt‐ND variants. Although sporadic cases have reported the absence of mt‐ND variants in blood cells, 4 , 6 , 28 , 29 very few studies have investigated the mt‐ND variants heteroplasmy in MELAS patients, with only one study reporting that 1/7 patients with MELAS caused by the m.13513G > A variant had no detectable variant in blood cells. 30 In our cohort, of the three patients with MELAS caused by the m.13513G > A variant, only one showed a variant absent in blood cells. On the contrary, variants absent in blood cells were also observed in mtDNA encoded complex IV subunits (mt‐COX) 31 and mtDNA encoded complex III subunits (mt‐CYTB) 32 gene variants. In contrast to those mutations in mitochondrial protein coding genes (mt‐ND, mt‐COX, and mt‐CYB), 31 , 32 variants absent in blood were rare in patients with m.3243A > G. 33 , 34 , 35 Additionally, MELAS‐mtND diagnosis can be more difficult, because some older asymptomatic individuals can carry low levels of heteroplasmy. 36
In this study, MELAS‐mtND patients presented with greater stature, higher BMI, lower proportions of migraine, hearing loss, and diabetes. These results were consistent with MDC scores. Thus, the MDC may help to orient further genetic screening. Previous studies have documented that adult MD patients with the m.3243A > G variant have a shorter stature and lower BMI than MD patients with a single large‐scale deletion, 37 and short stature patients were more likely to have diabetes and cardiovascular involvement. 38 However, the differences between the MELAS‐mtND and MELAS‐A3243G groups were not been previously described. Previous studies showed that mitochondrial protein coding gene variants were associated with higher BMI in adults with unknown mechanisms. 39 Considering that multisystem involvement was less common in the MELAS‐mtND group, heteroplasmy levels of mt‐ND variants may be lower in the organs associated with growth and development. Additionally, distinct myopathology may also contribute to the differences in height and BMI as muscle function was found to be associated with bone development. 37 , 40
This study also clearly demonstrated a diversity of SLLs in patients with mt‐ND variants. Classic SLLs are the main presentation of adult‐onset MELAS. 31 Interestingly, in our series of patients with mt‐ND variants, the phenotypic diversity of SLLs was more complex, and single and/or multiple small cortical SLLs were more frequent than that in m.3243A > G patients. We have reported a patient with m.10191 T > C who recurrently presented tiny lesions at the left rolandic cortex over two years follow‐up. 15 In addition, 58.3% (7/12) of patients with nonclassic SLLs developed classic SLLs over the 3‐year follow‐up. One of our cases, who harboring m.14487 T > C variant, exhibited a small cortex lesion in the occipital lobe and developed classic SLLs 2 years later. 41 In addition, these single, multiple, or disseminated lesions were all confined to the cerebral cortex.
In a prospective cohort study of the brain MRI features of Japanese MELAS patients (13 MELAS‐A3243G), 25 34.1% of SLLs were nonclassic; however, most of these cases were pediatric‐onset. In our cohort, of the six pediatric‐onset patients (mt‐ND n = 4; m.3243A > G n = 2), only two mt‐ND variant patients showed nonclassic SLLs. In another retrospective study with 30 patients with CI deficiency caused by mt‐ND or nDNA, 42 100.0% showed bilateral brainstem lesions (30/30), 26.7% (8/30) presented with supratentorial SLLs, while the isolated SLLs were never observed. The obvious contrast between these studies with ours might due to the difference in phenotype and age of onset, with most of their patients presenting with leigh syndrome and 86.7% (26/30) were pediatric‐onset while only 22.2% (4/18) had pediatric onset in our cohort. Thus, MELAS‐mtND patients may frequently show single/multiple/disseminated small cortical lesions, basal ganglia lesions, and/or brain stem lesions. Nonclassic SLLs may have easily led to underdiagnosis or misdiagnosis in some cases in our patients, with a time to diagnosis of more than 6 years. Thus, it is important for clinicians to perform a differential diagnosis of mitochondrial disease, especially mt‐ND variants, when nonclassic SLLs appeared.
Although, the absence of RRFs and/or COX‐deficient fibers in the muscle pathology of patients with mt‐ND pathogenic variants was commonly reported in Leigh syndrome, 43 , 44 its occurrence probability in adult MELAS patients is rarely reported. Our cohort showed MELAS‐mtND patients showed a lower presentation of RRFs and COX‐deficient fibers/blue fibers in contract with MELAS‐A3243G patients. Consistently, a study showed that 38.5% (5/13) of patients carrying mt‐ND without morphologic muscle changes while 6.5% (6/93) in MELAS‐A3243G. 45 Both RRFs and COX‐deficient fibers are pathological hallmarks of mitochondrial disease, in which RRFs represent the proliferation of abnormal mitochondria in muscle fibers. There are several factors that may influence the formation of RRFs and COX‐deficient fibers, including the genotype and genetic heteroplasmy among muscle fibers and the course of the disease. mt‐ND variants mainly affect the function of CI, which may affect the activity of complex IV less than m.3243A > G variant does. In addition, considering that all the patients with normal muscle pathology had adult onset in our cohort, it is unlikely that pathological changes were too early to form; hence, there could be a different underlying mechanism. Recently, the mitochondrial integrated stress response (ISR) 46 and mTORC1 47 were reported to involve in the formation of RRFs and COX‐deficient fibers. Furthermore, study showed that impairment of CI activity in myotube did not activate the ISR. 48 Hence, ISR and the mTORC1 pathway may be associated with the different pathological features between MELAS‐mtND and MELAS‐A3243G.
This study was limited by the inherent to single‐center observational design and the small sample size. mtDNA mutations are known to exhibit variability in phenotype–genotype expression, and the severity of MELAS is highly dependent on the degree of heteroplasmy. Therefore, more cohort studies are needed to draw conclusions about the relative severity of mutations in mt‐ND without considering the level of heteroplasmy.
Nonetheless, this retrospective study supports the view that mt‐ND variants cause a common pattern of clinical, muscle pathology and brain MRI findings. We suggest considering mt‐ND variants when a patient presents with small cortical SLLs and/or basal ganglion lesions with normal stature and BMI, no family history and normal muscle pathology. Finally, further genetic analysis of the muscle tissue, other than blood cells, would facilitate faster gene identification.
Author Contributions
Kunqian Ji, Chuanzhu Yan and Wei Wang designed the studies. Wei Wang and Kunqian Ji wrote the paper with the contribution of Yuying Zhao. Kunqian Ji, Chuanzhu Yan, Yuying Zhao, Fuchen Liu, and Wei Li reviewed and revised the manuscript. Wei Wang, Xuebi Xu, Yuan Sun, Yan Lin, Ying Zhao, Zhihong Xu, Jiayin Wang, Hong Ren, Bin Wang collected the data. Wei Wang and Xiaotian Ma analyzed the data. Dandan Zhao and Dongdong Wang performed the muscle histopathological studies. All authors read and approved the final manuscript.
Funding Information
This work was supported by the National Key Research and Development Program of China (2021YFC2700904), National Natural Science Foundation of China (No. 82171394), and the Taishan Scholars Program of Shandong Province and the Qingdao Key Health Discipline Development Fund.
Conflict of Interest
All authors report no competing interests.
Disclosures
There are no financial conflicts of interest to disclose.
Acknowledgements
The authors would like to thank the MELAS patients for their participation in this study.
Funding Statement
This work was funded by National Key Research and Development Program of China grant 2021YFC2700904; National Natural Science Foundation of China grant 82171394; Qingdao Key Health Discipline Development Fund; Taishan Scholars Program of Shandong Province.
Contributor Information
Chuanzhu Yan, Email: czyan@sdu.edu.cn, Email: chuanzhuyan@163.com.
Kunqian Ji, jikunqian@email.sdu.edu.cn, Email: jikunqian@qq.com.
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors (Kunqian Ji and Chuanzhu Yan) and first authors (Wei Wang and Yuying Zhao), upon reasonable request.
References
- 1. La Morgia C, Maresca A, Caporali L, Valentino ML, Carelli V. Mitochondrial diseases in adults. J Intern Med. 2020;287(6):592‐608. [DOI] [PubMed] [Google Scholar]
- 2. Goto Y, Nonaka I, Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;348(6302):651‐653. [DOI] [PubMed] [Google Scholar]
- 3. El‐Hattab AW, Adesina AM, Jones J, Scaglia F. MELAS syndrome: clinical manifestations, pathogenesis, and treatment options. Mol Genet Metab. 2015. Sep‐Oct;116(1–2):4‐12. [DOI] [PubMed] [Google Scholar]
- 4. Santorelli FM, Tanji K, Kulikova R, et al. Identification of a novel mutation in the mtDNA ND5 gene associated with MELAS. Biochem Biophys Res Commun. 1997;238(2):326‐328. [DOI] [PubMed] [Google Scholar]
- 5. Valentino ML, Barboni P, Rengo C, et al. The 13042G→A/ND5 mutation in mtDNA is pathogenic and can be associated also with a prevalent ocular phenotype. J Med Genet. 2006;43(7):e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Naini AB, Lu J, Kaufmann P, et al. Novel mitochondrial DNA ND5 mutation in a patient with clinical features of MELAS and MERRF. Arch Neurol. 2005;62(3):473‐476. [DOI] [PubMed] [Google Scholar]
- 7. Liolitsa D, Rahman S, Benton S, Carr LJ, Hanna MG. Is the mitochondrial complex I ND5 gene a hot‐spot for MELAS causing mutations? Ann Neurol. 2003;53(1):128‐132. [DOI] [PubMed] [Google Scholar]
- 8. Lin Y, Xu X, Zhao D, et al. A novel m.11406 T > A mutation in mitochondrial ND4 gene causes MELAS syndrome. Mitochondrion. 2020;54:57‐64. [DOI] [PubMed] [Google Scholar]
- 9. Lin J, Zhao CB, Lu JH, et al. Novel mutations m.3959G>A and m.3995A>G in mitochondrial gene MT‐ND1 associated with MELAS. Mitochondrial DNA. 2014. Feb;25(1):56‐62. [DOI] [PubMed] [Google Scholar]
- 10. Lertrit P, Noer AS, Jean‐Francois MJ, et al. A new disease‐related mutation for mitochondrial encephalopathy lactic acidosis and strokelike episodes (MELAS) syndrome affects the ND4 subunit of the respiratory complex I. Am J Hum Genet. 1992;51(3):457‐468. [PMC free article] [PubMed] [Google Scholar]
- 11. Kirby DMCM, Cleary MA, Dahl HH, Dennett X, Thorburn DR. Respiratory chain complex I deficiency an underdiagnosed energy generation disorder. Neurology. 1999;52(6):1255‐1264. [DOI] [PubMed] [Google Scholar]
- 12. Distelmaier F, Koopman WJH, van den Heuvel LP, et al. Mitochondrial complex I deficiency: from organelle dysfunction to clinical disease. Brain. 2009;132(Pt 4):833‐842. [DOI] [PubMed] [Google Scholar]
- 13. Fiedorczuk K, Sazanov LA. Mammalian mitochondrial complex I structure and disease‐causing mutations. Trends Cell Biol. 2018;28(10):835‐867. [DOI] [PubMed] [Google Scholar]
- 14. Swalwell H, Kirby DM, Blakely EL, et al. Respiratory chain complex I deficiency caused by mitochondrial DNA mutations. Eur J Hum Genet. 2011;19(7):769‐775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ji K, Ren H, Zhao X, Yan C. Migratory Rolandic encephalopathy caused by the mitochondrial ND3 variant. Neurology. 2022. Jan 11;98(2):80‐81. [DOI] [PubMed] [Google Scholar]
- 16. Li Y, Lin J. Current insight into MELAS: clinical perspectives and multimodal MRI. J Magn Reson Imaging. 2018;47(2):583‐584. [DOI] [PubMed] [Google Scholar]
- 17. Walker UA, Collins S, Byrne E. Respiratory chain encephalomyopathies: a diagnostic classification. Eur Neurol. 1996;36(5):260‐267. [DOI] [PubMed] [Google Scholar]
- 18. Bernier FP, Boneh A, Dennett X, Chow CW, Cleary MA, Thorburn DR. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology. 2002;59(9):1406‐1411. [DOI] [PubMed] [Google Scholar]
- 19. Morava E, van den Heuvel L, Hol F, et al. Mitochondrial disease criteria: diagnostic applications in children. Neurology. 2006;67(10):1823‐1826. [DOI] [PubMed] [Google Scholar]
- 20. Wolf NI, Smeitink JAM. Mitochondrial disorders: a proposal for consensus diagnostic criteria in infants and children. Neurology. 2002;59(9):1402‐1405. [DOI] [PubMed] [Google Scholar]
- 21. Yatsuga S, Povalko N, Nishioka J, et al. MELAS: a nationwide prospective cohort study of 96 patients in Japan. Biochim Biophys Acta. 2012;1820(5):619‐624. [DOI] [PubMed] [Google Scholar]
- 22. Pearl GS, Ghatak NR. Muscle biopsy. Arch Pathol Lab Med. 1995;119(4):303‐306. [PubMed] [Google Scholar]
- 23. Old SL, Johnson MA. Methods of microphotometric assay of succinate dehydrogenase and cytochrome c oxidase activities for use on human skeletal muscle. Histochem J. 1989;21(9–10):545‐555. [DOI] [PubMed] [Google Scholar]
- 24. Tatlisumak T, Putaala J, Innilä M, et al. Frequency of MELAS main mutation in a phenotype‐targeted young ischemic stroke patient population. J Neurol. 2016;263(2):257‐262. [DOI] [PubMed] [Google Scholar]
- 25. Hongo Y, Kaneko J, Suga H, et al. A cluster of disseminated small cortical lesions in MELAS: its distinctive clinical and neuroimaging features. J Neurol. 2019;266(6):1459‐1472. [DOI] [PubMed] [Google Scholar]
- 26. Zhang Z, Zhao D, Zhang X, et al. Survival analysis of a cohort of Chinese patients with mitochondrial encephalomyopathy with lactic acidosis and stroke‐like episodes (MELAS) based on clinical features. J Neurol Sci. 2018;385:151‐155. [DOI] [PubMed] [Google Scholar]
- 27. Zhao D, Hong D, Zhang W, et al. Mutations in mitochondrially encoded complex I enzyme as the second common cause in a cohort of Chinese patients with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke‐like episodes. J Hum Genet. 2011;56(11):759‐764. [DOI] [PubMed] [Google Scholar]
- 28. Ravn K, Wibrand F, Hansen FJ, Horn N, Rosenberg T, Schwartz M. An mtDNA mutation, 14453G→A, in the NADH dehydrogenase subunit 6 associated with severe MELAS syndrome. Eur J Hum Genet. 2001;9(10):805‐809. [DOI] [PubMed] [Google Scholar]
- 29. Lebon S, Chol M, Benit P, et al. Recurrent de novo mitochondrial DNA mutations in respiratory chain deficiency. J Med Genet. 2003;40(12):896‐899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shanske S, Coku J, Lu J, et al. The G13513A mutation in the ND5 gene of mitochondrial DNA as a common cause of MELAS or Leigh syndrome: evidence from 12 cases. Arch Neurol. 2008;65(3):368‐372. [DOI] [PubMed] [Google Scholar]
- 31. Wang W, Sun Y, Lin Y, et al. A novel nonsense variant in MT‐CO3 causes MELAS syndrome. Neuromuscul Disord. 2021. Jun;31(6):558‐565. [DOI] [PubMed] [Google Scholar]
- 32. Keightley JA, Anitori R, Burton MD, Quan F, Buist NR, Kennaway NG. Mitochondrial encephalomyopathy and complex III deficiency associated with a stop‐codon mutation in the cytochrome b gene. Am J Hum Genet. 2000;67(6):1400‐1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Grady JP, Pickett SJ, Ng YS, et al. mtDNA heteroplasmy level and copy number indicate disease burden in m.3243A>G mitochondrial disease. EMBO Mol Med. 2018;10(6):e8262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xia C‐Y, Liu Y, Liu H, Zhang Y‐C, Ma Y‐N, Qi Y. Clinical and molecular characteristics in 100 Chinese pediatric patients with m.3243A>G mutation in mitochondrial DNA. Chin Med J (Engl). 2016;129(16):1945‐1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Elson JL, Swalwell H, Blakely EL, McFarland R, Taylor RW, Turnbull DM. Pathogenic mitochondrial tRNA mutations – which mutations are inherited and why? Hum Mutat. 2009;30(11):E984‐E992. [DOI] [PubMed] [Google Scholar]
- 36. Fayet G, Jansson M, Sternberg D, et al. Ageing muscle: clonal expansions of mitochondrial DNA point mutations and deletions cause focal impairment of mitochondrial function. Neuromuscul Disord. 2002;12:484‐493. [DOI] [PubMed] [Google Scholar]
- 37. Boal RL, Ng YS, Pickett SJ, et al. Height as a clinical biomarker of disease burden in adult mitochondrial disease. J Clin Endocrinol Metab. 2019;104(6):2057‐2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hou Y, Xie Z, Zhao X, Yuan Y, Dou P, Wang Z. Appendicular skeletal muscle mass: a more sensitive biomarker of disease severity than BMI in adults with mitochondrial diseases. PloS ONE. 2019;14(7):e0219628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Flaquer A, Baumbach C, Kriebel J, et al. Mitochondrial genetic variants identified to be associated with BMI in adults. PloS ONE. 2014;9(8):e105116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Matsumoto M, Awano H, Lee T, Takeshima Y, Matsuo M, Iijima K. Patients with Duchenne muscular dystrophy are significantly shorter than those with Becker muscular dystrophy, with the higher incidence of short stature in Dp71 mutated subgroup. Neuromuscul Disord. 2017;27(11):1023‐1028. [DOI] [PubMed] [Google Scholar]
- 41. Xu XB, Ji KQ, Lyu JW, et al. Late‐onset mitochondrial disease in a patient with MELAS and mitochondrial DNA T14487C mutation. Chin Med J (Engl). 2019;132(6):716‐718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lebre AS, Rio M, Faivre d'Arcier L, et al. A common pattern of brain MRI imaging in mitochondrial diseases with complex I deficiency. J Med Genet. 2011;48:16‐23. [DOI] [PubMed] [Google Scholar]
- 43. Alston CL, Morak M, Reid C, et al. A novel mitochondrial MTND5 frameshift mutation causing isolated complex I deficiency, renal failure and myopathy. Neuromuscul Disord. 2010;20(2):131‐135. [DOI] [PubMed] [Google Scholar]
- 44. Oldfors A, Sewry CA, Dubowitz V. Muscle Biopsy: A Practical Approach. Elsevier; 2013. [Google Scholar]
- 45. Lu Y, Deng J, Zhao Y, et al. Patients with MELAS with negative myopathology for characteristic ragged‐red fibers. J Neurol Sci. 2020;408:116499. [DOI] [PubMed] [Google Scholar]
- 46. Forsström S, Jackson CB, Carroll CJ, et al. Fibroblast growth factor 21 drives dynamics of local and systemic stress responses in mitochondrial myopathy with mtDNA deletions. Cell Metab. 2019. Dec 3;30(6):1040‐54.e7. [DOI] [PubMed] [Google Scholar]
- 47. Lyu JW, Xu XB, Ji KQ, et al. Activated mTOR signaling pathway in myofibers with inherited metabolic defect might be an evidence for mTOR inhibition therapies. Chin Med J (Engl). 2019;132(7):805‐810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mick E, Titov DV, Skinner OS, Sharma R, Jourdain AA, Mootha VK. Distinct mitochondrial defects trigger the integrated stress response depending on the metabolic state of the cell. Elife. 2020;9:e49178. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors (Kunqian Ji and Chuanzhu Yan) and first authors (Wei Wang and Yuying Zhao), upon reasonable request.