

Abstract
LRRK2 variants are implicated in both familial and sporadic PD. LRRK2‐PD has a generally benign clinical presentation and variable pathology, with inconsistent presence of Lewy bodies and marked Alzheimer's disease pathology. The mechanisms underlying LRRK2‐PD are still unclear, but inflammation, vesicle trafficking, lysosomal homeostasis, and ciliogenesis have been suggested, among others. As novel therapies targeting LRRK2 are under development, understanding the role and function of LRRK2 in PD is becoming increasingly important. Here, we outline the epidemiological, pathophysiological, and clinical features of LRRK2‐PD, and discuss the arising therapeutic approaches targeting LRRK2 and possible future directions for research.
Introduction
Parkinson's disease (PD) is the fastest‐growing neurological condition worldwide, with 12–17 million PD patients projected by 2040. 1 Despite these alarming data, no disease‐modifying therapy is currently available for this devastating disease. Most clinical trials on potential disease‐modifying drugs so far have treated PD as a single entity, which may have contributed to their failure. With our evolving understanding of the genetic and pathophysiological basis of PD, new approaches targeting specific genetic subtypes of PD are emerging. 2 , 3
One of the most common genetic risk factors in PD is variants in leucine‐rich repeat protein kinase‐2 (encoded by LRRK2). 4 In 1978, autosomal dominant PD patients were observed over five generations in a Japanese family, 5 and in 2002 linkage analyses in this family identified the disease‐associated locus, PARK8. 6 A mutation in the LRRK2 gene, p.I2020T, was identified in this locus, and other variants associated with PD have been identified in the same gene in 2004. 7 , 8 LRRK2 variants account for ~4% of familial PD up to 36% in some ancestries and ~1% up to 39% of sporadic PD. 9 , 10 One of the most frequent LRRK2 mutations in PD is p.G2019S, 9 albeit uncommon in certain populations. In Asians, for example, p.G2019S is rare whereas p.G2385R and p.R1628P are most frequently associated with PD. 11 A common LRRK2 haplotype, p.N551K‐p.R1398H‐p.K1423K, is associated with decreased risk for PD across different populations. 4 , 12 , 13 , 14
In this review, we describe the current knowledge about genetic variants, structure, suggested mechanisms, pathology, and phenotype. We finally discuss the therapeutic approaches under development targeting LRRK2 and possible future directions for research.
LRRK2 Protein Structure, LRRK2 Variants, and Ethnic Distribution
The LRRK2 protein is a multi‐domain enzyme including catalytic kinase, armadillo, ankyrin, leucine‐rich repeats, C‐terminal WD40, and GTPase domains (Fig. 1). 15 , 16 The latter consists of a Ras‐like GTPase called ROC (Ras of complex) and a dimerization domain called COR (C‐terminal of ROC). 17 , 18
Figure 1.
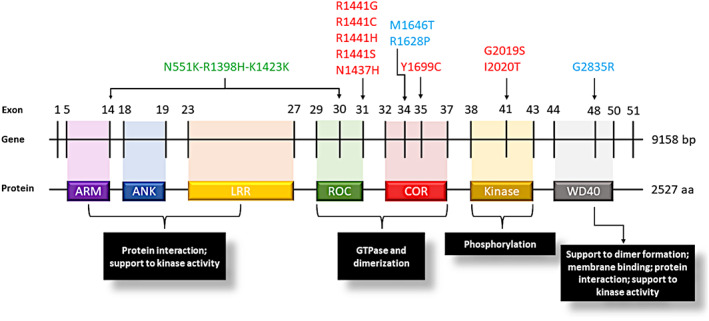
Schematic representation of LRRK2 gene and LRRK2 protein with its functional domains. LRRK2 pathogenic variants are indicated in red, risk variants in light blue, the protective haplotype in green. aa, amino acids; ANK, ankyrin repeat region; ARM, armadillo repeat region; Bp, base pairs; COR, C‐terminal‐of‐Roc domain; Kinase, protein tyrosine kinase‐like domain; LRR, leucine‐rich repeats; ROC, ras‐of‐complex GTPase domain; WD40, WD40 repeat region.
p.G2019S and p.I2020T are located within the kinase domain, and they seem to directly increase LRRK2 kinase activity. p.G2019S is a dominantly inherited variant with reduced penetrance, very common among Ashkenazi Jewish and North African Berbers, reported respectively in up to 28% and 39% of PD patients in these groups. 9 However, it is rarely observed in other groups such as East Asians. 11 Other confirmed pathogenic, dominant variants in PD are p.N1437H and p.R1441G/C/H/S, located in the ROC domain, and p.Y1699C in the COR domain. 19 , 20 These variants seem to also increase LRRK2 kinase activity, albeit indirectly by compromising the GTPase function. 18 , 21
Additional LRRK2 variants have been reported as risk factors for PD. For example, p.R1628P and p.G2385R, located respectively in the COR and WD40 LRRK2 domains, are the most frequent risk variants in the East Asian PD population, 22 , 23 increasing PD risk by about twofold, 4 , 24 Another population‐specific variant is p.M1646T, a common variant associated with a mild increase in PD risk in Europeans, but not in Asians or Arab‐Berbers, 4 , 14 , 25 This variant is located in the COR domain of LRRK2, and it was reported to be associated with an increased GCase activity, 4 , 25 Finally, an intergenic variant, rs76904798, located at the 5′ end of the LRRK2 region, is associated with increased expression of LRRK2 and with a higher hazard ratio in PD patients for progression to stage three of the Hoehn and Yahr scale (H&Y). 26 , 27 , 28
In contrast, LRRK2 p.N551K‐p.R1398H‐p.K1423K is a common haplotype associated with a reduced risk for developing PD. 4 , 12 , 13 , 14 It has been observed across populations (Europeans, Asians, and Berbers) and is associated with reduced LRRK2 kinase activity, 13 opposite to the LRRK2 deleterious variants. Located in the ROC domain, p.R1398H appears the most likely variant driving this association. 4 , 12
LRRK2 penetrance and putative genetic modifiers
LRRK2 penetrance in PD is incomplete and age‐dependent, ranging from 17% up to 85%, with no sex differences. 9 , 11 Variants in MAPT 29 , 30 have been reported to increase PD risk in LRRK2 variant carriers, but this effect has not been confirmed elsewhere. 31 In a genome‐wide association study, the SNP rs77395454 in CORO1C was associated with PD penetrance among LRRK2 carriers, and showed an interaction at the protein level with LRRK2 kinase using co‐immunoprecipitation. 30 Variants in the GAK 30 , 32 and PARK16 30 , 33 loci have also been suggested to modify both LRRK2‐PD penetrance and age at onset (AAO), whereas DNM3 variants have been associated with decreased LRRK2‐PD AAO. 31 However, further studies are required to confirm and better define the role of such variants in LRRK2‐PD penetrance and/or AAO. DNM3 variants, for instance, were not confirmed as LRRK2‐PD AAO modifiers in other studies and showed possible ethnic‐specific effects. 34 , 35
Polygenic risk score analyses comprising variants associated with PD36 also showed an association with an increased PD penetrance in LRRK2 variant carriers, implying that the cumulative effect of such common variants might also act as a genetic modifier. 30 , 37 Overall, the role of the putative genetic modifiers in LRRK2‐PD requires further investigation to confirm their role in PD penetrance.
Pathology
The typical PD pathology is characterized by the loss of nigrostriatal dopaminergic neurons and the accumulation of phosphorylated α‐synuclein (α‐syn), the major component of Lewy Bodies (LBs). 38 Unlike idiopathic PD (iPD), LRRK2‐PD does not show the typical LB pathology in about half of the cases studied, 39 , 40 while frequently displaying an Alzheimer's disease (AD)‐like pathology with senile plaques 2 and/or phosphorylated tau‐composed neurofibrillary tangles. 39 , 40
As detailed in Table 1, where we summarize the pathological findings of 69 autopsies from multiple studies in LRRK2‐PD patients, only 43/69 (62%) of these patients had LB pathology. This frequency differs between carriers of the p.G2019S variant and other LRRK2 variants, with 76% of the former showing LB pathology, in contrast with only 41% of the latter. Tau pathology is reported in a larger proportion of LRRK2‐PD, in 48/68 (71%) of the patients. Similar to LB pathology, tau pathology is represented differently between carriers of p.G2019S and other LRRK2 variants, appearing in 90% and 38% of the autopsies, respectively. A study using antibodies specific for AD‐type tau (co‐presence of both 3 and 4 microtubule‐binding repeat isoforms) demonstrated that tau pathology was found in 100% (11/11) of LRRK2‐PD patients (LB pathology was found in 64% of them) and that AD‐type tau is the prominent type of tau present in these patients. Abundant Aβ pathology, consistent with AD, was also found in most of the cases. 40 More rarely, LRRK2‐PD exhibits ubiquitin‐positive inclusions or the typical frontotemporal dementia pathology with TAR DNA‐binding protein 43 (TDP‐43) deposits, 41 or pure nigrostriatal neurodegeneration reported in p.I2020T carriers, who show a pure degeneration in about 50% of the cases. 42
Table 1.
LRRK2‐PD pathology reported by human brain autopsies in separate studies.
| Report | Autopsies (n) | Variants (n) | LB pathology (n+/n) | Tau pathology (n+/n) | Other inclusions (n+/n) |
|---|---|---|---|---|---|
| [10] | 3 | p.G2019S | 3/3 | 1/3 | – |
| 139, 140, 141 | 33 | p.G2019S | 2/3 | 3/3 | – |
| 142 | 1 | p.G2019S | 0/1 | 1/1 | PSP‐like (1/1) |
| 143 | 8 | p.G2019S | 8/8 | 6/8 | – |
| 144 | 3 | p.G2019S | 2/3 | 3 /3 | – |
| 145 | 4 | p.G2019S | 4/4 | 4/4 | Olfactory bulb LBs (4/4) |
| 146 | 3 | p.G2019S | 3/3 | 3/3 | – |
| [39] | 4 |
p.G2019S (3) p.R1441G (1) |
2/4 (2/3 p.G2019S, 0/1 p.R1441G) | 3/4 (3/3 p.G2019S, 0/1 p.R1441G) | – |
| 147 | 1 | p.G2019S | 1/1 | 1/1 | – |
| 42,8,148 | 6 | p.R1441C (4), p.Y1699C (2) | 2/6 (2/4, p.R1441C, 0/2 p.Y1699C) | 2/6 (1/4 p.R1441C, 1/2 p.Y1699C) | PSP‐like (1/6, R1441C); TDP‐43 (1/6, R1441C) |
| [53] | 1 | p.Y1699C | 1/1 | 1/1 | Olfactory bulb LBs |
| 149 | 1 | p.I1371V | 1/1 | 1/1 | – |
| 150 | 2 | p.R793M (1), p.L1165P (1) | 2/2 | 2 /2 | TDP‐43 in TC (2/2) |
| 139 | 1 | p.R1441R | 1/1 | n.d. | – |
| 151, 152 | 8 | p.I2020T | 1/8 | 0/8 | Glial cytoplasmic inclusions (1/8) |
| 153 | 1 | p.R1441G | 0/1 | 1/1 | Aß in SN |
| 154 | 1 | p.N1437H | 1/1 | 1/1 | Ubiquitin inclusions |
| 155 | 3 | p.R1441H | 0/3 | 0/3 | – |
| [40] | 11 | p.G2019S (9), p.L1165P (1), p.R793M (1) | 7/11 (5/9 p.G2019S, 1/1 p.L1165P, 1/1 p.R793M) | 11/11 | – |
| 156 | 4 | p.G2019S | 2/4 | 4/4 | Ubiquitin inclusions (1/4) |
| TOTAL | 69 | – | 43/69 (62%) | 48/68 (71%) | – |
| TOTAL – p.G2019S | 42 | – | 32/42 (76%) | 38/42 (90%) | – |
| TOTAL – other LRRK2 variants | 27 | – | 11/27 (41%) | 10/26 (38%) | – |
Aß, amyloid beta; LBs, Lewy bodies; n, number of subjects; n+, number of subjects with the pathology; PSP, progressive supranuclear palsy; SN, substantia nigra; TC, temporal cortex; TDP‐43, TAR DNA‐binding protein 43.
Clinical presentation
Although at the individual‐level LRRK2‐PD resembles iPD in clinical manifestations and response to therapy, 9 as a group, LRRK2‐PD has some notable differences, including a more benign phenotype with less frequent non‐motor symptoms. LRRK2‐PD patients show a slower progression and milder cognitive impairment compared to iPD patients. They perform better in attention, executive functions and language tests, tend to develop cognitive deficits only in more advanced stages of PD, and dementia in general appears less frequently than in iPD. 9 , 43 Additionally, hyposmia and autonomic dysfunction are less prevalent in LRRK2‐PD patients. Abnormal olfaction is present in 36%–49% of LRRK2‐PD patients, compared to 75%–81% of iPD patients. 9 , 44 , 45 LRRK2‐PD also shows a reduced frequency of orthostatic hypotension 46 and gastrointestinal dysfunction, 47 as well as greater cardiac [123I]metaiodobenzylguanidine uptake on scintigraphy, 48 compared to iPD. Another study, however, showed a similar prevalence of autonomic dysfunctions in LRRK2‐PD and iPD. 49 While rapid eye movement sleep behavior disorder (RBD), a common prodromal symptom of synucleinopathies, is present in about 25%–58% of iPD patients, in LRRK2‐PD it is displayed in only 0%–15% of the cases. 50 , 51 , 52 Whether the frequency of psychiatric symptoms, including anxiety and depression, is different in LRRK2‐PD compared to iPD, is controversial. Some studies showed that LRRK2‐PD patients have an increased risk of psychiatric symptoms, compared to iPD patients, 53 , 54 while others showed reduced risk. 55
Although LRRK2‐PD may display a milder phenotype compared to iPD, some characteristics could be more severe. LRRK2‐PD patients are more prone to manifest postural instability and gait difficulty. 43 In addition, the average AAO of LRRK2‐PD is slightly lower than iPD with a higher proportion of individuals developing PD earlier than 40 years of age. 9 , 56 Other differences of LRRK2‐PD compared to iPD include a larger involvement of lower limbs in the motor dysfunction 57 and an absence of the male predominance observed in iPD. 58 , 59 Finally, LRRK2‐PD shows more frequently atypical phenotypes, such as tauopathy‐like symptoms, progressive aphasia and choreoathetosis. 45
Only few attempts were made to compare the clinical phenotype between different LRRK2 variants, typically with inconsistent results. A meta‐analysis attempted to fill this gap and suggested some potential differences between carriers of the p.G2019S and p.G2385R variants. For example, p.G2019S, but not p.G2385R, was associated with dyskinesia, and p.G2385R was associated with less severe H&Y stages. 60 A systematic review showed that motor fluctuations were more frequently associated with the p.R1441C/G/H/S mutations compared to p.G2019S. Similarly, tremor and postural instability were more frequent in carriers of p.R1441G compared to p.G2019S. No other differences in motor symptoms, AAO or levodopa response emerged between LRRK2 mutations, 61 demonstrating that, overall, it might be challenging for clinicians to distinguish the LRRK2 mutations in individual patients based on their PD presentation.
Inflammation
LRRK2 is particularly expressed in immune cells, including lymphocytes B, macrophages, neutrophils, whereas it is substantially less expressed in the brain, except for medium‐sized spiny neurons of the nucleus striatum and microglia. 62 , 63 Microglia arguably play a determinant role in LRRK2‐PD neurodegeneration. In addition to their trophic function, microglial cells act as scavengers, internalizing and clearing extracellular debris, including pathological α‐syn. Furthermore, they are responsible for inducing neuroinflammation by the recruitment of peripheral immune cells and release of cytokines. 64 , 65 α‐syn is one of the triggers of such release and LRRK2 inhibition was shown to significantly mitigate this effect in human microglia cell lines. 66 In the human frontal cortex and substantia nigra, LRRK2 expression may be modulated by microglia‐specific open chromatin regions. 67
Numerous additional lines of evidence, many of which have been extensively reviewed elsewhere, 68 , 69 suggested a link between LRRK2‐mediated inflammation and PD development. B/T cells and CD16+ monocytes of PD patients express increased levels of LRRK2, compared to healthy controls, 19 increased levels of cytokines positively correlate with LRRK2 expression in PD patient monocytes. 70 In microglia differentiated from patient‐derived iPSC, p.G2019S was shown to influence microglial activation and cytokine production in response to interferon‐γ. 65
Another hint for the relationship between PD, LRRK2 and inflammatory processes is provided by the inflammatory bowel diseases (IBDs), chronic autoimmune diseases affecting the digestive tract, including Crohn's disease (CD) and ulcerative colitis (UC). IBD patients show a 20–90% increased risk for developing PD. 71 The LRRK2 p.N2081D variant has been associated with an increased risk for CD and a mild increase in PD risk, and with elevated kinase activity. 72 The LRRK2 p.N551K‐p.R1398H‐p.K1423K protective haplotype, in contrast, is associated with reduced kinase activity and reduced risk for both diseases. 4 , 72 LRRK2 variants are also associated with leprosy, a dermato‐neurological infectious disease produced by Mycobacterium leprae, and with type‐1 reaction (T1R), one of the main complications of this disease, which causes an autoimmune response against the peripheral nerves. 73 These associations with immune and infectious diseases strengthen the notion that LRRK2 has an important role in the immune system and inflammation which may contribute to PD pathogenesis in LRRK2 variant carriers.
The association between LRRK2 variants and inflammation in PD has been challenged by a recent study, 74 showing how LRRK2‐PD patients did not differ from healthy controls in the count of any leukocyte subpopulation or neutrophil‐to‐lymphocyte ratio, a biomarker of systemic inflammation. These results suggest that LRRK2 might mediate immune processes through different pathways, with a more prominent role of inflammatory mediators and a dysregulation of specific immune cell subpopulations like microglia, while having a marginal effect on the number of peripheral leukocytes. 74
Other cellular mechanisms
Multiple LRRK2‐mediated mechanisms have been proposed in PD pathophysiology, including inflammation, lysosomal, autophagy, and endolysosomal trafficking dysfunction, apoptosis, ciliogenesis, and numerous others 18 , 75 (Fig. 2). In recent years its role in and around the lysosome has gained traction as a potentially important mechanism in PD, and in this section, we mainly focus on this role, as other mechanisms have been extensively reviewed elsewhere 18 , 75 and we discussed inflammation separately above. Thus far, RAB8 and RAB10 are the most validated LRRK2 substrates, while RAB29 and VPS35 are potential upstream activators. 76 , 77 Variants in RAB29 and VPS35, but not in RAB8/10, have also been associated with PD risk. 36 , 78
Figure 2.
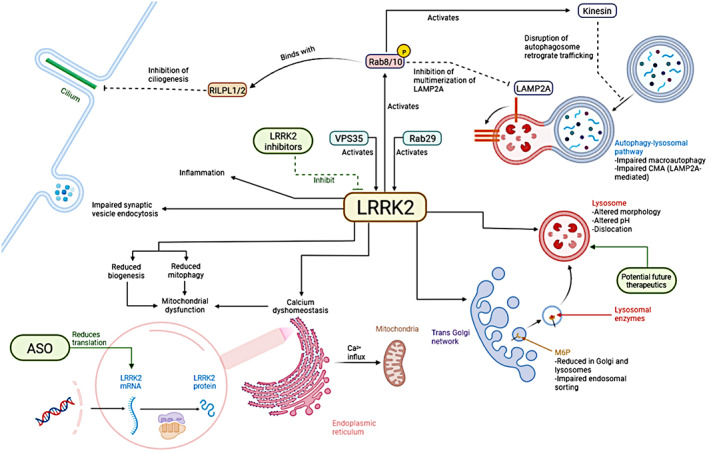
Principal mechanisms where LRRK2 has been implicated in Parkinson's disease and therapeutic targets. ASO, antisense oligonucleotides; CMA, chaperon‐mediated autophagy; M6P, mannose‐6‐phosphate, deputed to transfer of lysosomal enzymes from the Golgi to the lysosome. Created with Biorender.
Under stress conditions, RAB29 recruits LRRK2 to the lysosomal membranes, where it phosphorylates Rab8/10 to maintain lysosomal homeostasis. 79 LRRK2 mutations have been associated with alteration in lysosomal morphology, distribution, pH, and function. 33 , 80 , 81 , 82 Wildtype, but not mutated VPS35 protein, rescues endolysosomal alterations driven by LRRK2 mutations in primary rodent neurons. 33 LRRK2 also plays a key role in vesicular endocytosis and trafficking. Induced pluripotent stem cells (iPSC)‐derived dopaminergic neurons from PD patients carrying the p.R1441G mutation showed impaired synaptic vesicle endocytosis (SVE), with a reduction of SVE proteins and functional synaptic vesicles, along with the accumulation of enlarged vesicles. 83 Multiple proteins phosphorylated by LRRK2 have been proposed to be involved in these processes, including endophilin A, auxilin, and synaptojanin 1. 84 In addition, mannose‐6‐phosphate receptor (M6PR), implicated in endosomal sorting, including the transportation of lysosomal hydrolases to late endosomes, was shown to be deficient at the lysosomes and Golgi of primary cortical neurons with p.G2019S mutation, suggesting an impairment in endolysosomal function. 33 , 84 , 85
LRRK2 variants have also been implicated in calcium dyshomeostasis, triggering endoplasmic reticulum (ER) stress. 86 , 87 This effect was not observed in LRRK2‐p.G2019S neurons, but when these were cocultured with LRRK2‐p.G2019S astrocytes, α‐syn treatment was more harmful to neurons, suggesting a cell‐specific effect on astrocytes. LRRK2 variants also affect lysosomal homeostasis, autophagy, and intra−/extracellular clearance of α‐syn in astrocytes. 87
LRRK2 is further implicated in the autophagy‐lysosomal pathway (ALP), playing a role both in macroautophagy and chaperon‐mediated autophagy. 88 , 89 , 90 , 91 , 92 LRRK2 mutations are associated with the accumulation of ALP substrates (including α‐synuclein) and autophagic vacuoles. 93 , 94 They are also associated with Rab10‐mediated enhancement of kinesin activity, which interferes with the normal retrograde trafficking of autophagosomes and the degradation of their content within the lysosomes. 95 , 96 Mitophagy was also demonstrated to be affected by LRRK2 mutations, like p.G2019S and R1441C, both associated with decreased mitophagy in multiple human cell lines including fibroblasts, DA neurons, and microglial cells. 97 In mouse brains with LRRK2 mutation, increased LRRK2 phosphorylation of Rab8/10 enhances the activity of Rab interacting lysosomal‐like proteins 1 and 2 (RILPL1/2), 98 , 99 which alter centrosome cohesion and ciliogenesis. 100 This mechanism was suggested to impair the signaling of neuroprotective factors sent by cholinergic neurons in the striatum to the dopaminergic neurons in the substantia nigra, with a possible increased vulnerability to neurodegeneration. 98 , 101 LRRK2 does not only affect mitophagy but may also be associated with mitochondrial dysfunction. 102 , 103 In carriers of LRRK2 p.G2019S, mtDNA damage was found specifically in midbrain dopaminergic neurons compared to cortical neurons. 102 One explanation proposed for the mitochondrial dysfunction is the LRRK2 variant‐associated calcium dyshomeostasis, which would in turn increase the calcium influx into the mitochondria from the ER. 103
These data suggest that LRRK2 involvement in PD may be mediated through mechanisms including endolysosomal trafficking, autophagy dysfunction, mitochondrial dysfunction, calcium dyshomeostasis, and inflammation. These processes, like lysosomal dysfunction and inflammation, might also be interconnected with each other in PD pathophysiology. 104 , 105 It is possible that other mechanisms are at play, and further studies of LRRK2 are warranted to characterize them.
LRRK2 and GBA1
GBA1 variants are common genetic risk factors for PD, accounting for 5%–20% of PD patients in different populations. 106 GBA1 encodes β‐glucocerebrosidase (GCase), a lysosomal enzyme that hydrolyzes glucocerebroside and glucosylsphingosine. 107 PD patients carrying GBA1 variants display reduced Gcase activity, and there is a large variance in Gcase activity among non‐carriers of GBA1 variants, 108 , 109 suggesting the existence of other modifiers of Gcase activity. The TMEM175 T393M and LRRK2 p.G2019S variants are such potential modifiers, as both may affect Gcase activity. 110
It is still unclear whether LRRK2 risk variants are associated with increased or reduced Gcase activity. One study on iPSC‐derived dopaminergic neurons showed that LRRK2 variants were associated with decreased Gcase activity, with a mechanism that would involve Rab10 phosphorylation. 111 However, in two studies performed in peripheral blood, LRRK2 variant carriers exhibited increased Gcase activity. 25 , 109 These discrepant results can be explained by the different tissues that have been analyzed, that is, LRRK2 variants might be associated with decreased Gcase activity in dopaminergic neurons and increased Gcase activity in blood cells. 25 A cell‐type‐specific effect of LRRK2 was further supported by a recent study that showed decreased Gcase activity in p.G2019S knock‐in mouse brains and p.G2019S iPSCs‐derived neurons but increased in patient‐derived blood cells and fibroblasts. 112 Another hypothesis that has been suggested is that iPSCs cannot fully reproduce the aging processes happening in the human tissues, hence not fully representing Gcase activity in PD patients as they age. 25 Finally, the different methods to estimate GCase activity and divergent specificities of the substrates used in the studies can be another confounder. 25 , 113
From a clinical perspective, it seems that the pathophysiology of GBA1‐PD, characterized by a decreased Gcase activity, is different from LRRK2‐PD. First, the clinical presentation of GBA1‐PD is more severe than LRRK2‐PD, with earlier onset, faster progression, and more frequent non‐motor symptoms, including neuropsychiatric symptoms, RBD, autonomic dysfunction, and others (Table 2). 59 , 114 , 115 , 116 , 117 Three independent studies 118 , 119 , 120 also demonstrated that PD patients carrying both GBA1 and LRRK2 mutations manifest a milder phenotype compared to those carrying only GBA1 variants, in contrast to what we would expect if LRRK2 mutations further disrupted GCase activity. Moreover, the neuropathology of GBA1‐PD resembles the typical iPD pathology with LB deposition, whereas LRRK2‐PD often lacks this feature (Table 1). 39 , 40 , 121 Finally, while LRRK2 variants are associated just with PD among the synucleinopathies, 122 GBA1 variants are also associated with dementia with Lewy bodies, 116 , 117 a synucleinopathy that shares several features with PD, including LB deposition, but with cognitive decline preceding the motor symptoms. 123 All these differences between GBA1 and LRRK2 suggest that the development of LRRK2‐PD is not connected with the GCase impairment observed in GBA1‐PD, but other pathways might rather be involved. Furthermore, an association between LRRK2 variants and an increased GCase activity might explain, at least in part, the milder phenotype observed in LRRK2‐PD. It is also possible that in a subset of these patients LRRK2 variants, and not GBA1, represent the main driver of the disease. Functional studies on LRRK2 kinase mechanisms of action will be necessary to address these hypotheses and clarify how LRRK2‐PD pathophysiology differs from GBA1‐PD.
Table 2.
Prevalence, penetrance, pathology, and clinical features of LRRK2‐PD versus GBA1‐PD.
| LRRK2 | GBA1 | References | |
|---|---|---|---|
| Prevalence in PD | 0–39% | 5–20% | [9, 106, 157, 158] |
| Male/female representation | Comparable | Males overrepresented | [58, 59, 159] |
| Penetrance in PD | 17–85% | 10–30% | [9, 11, 160] |
| Pathology | Less frequent LBs; More frequent AD‐like pathology. TDP‐43 deposits, ubiquitin‐positive inclusions and pure nigral degeneration have also been reported | Typical PD pathology, with LB deposition | [2, 39, 40, 41, 42] |
| Clinical presentation | |||
| Motor features | Comparable to iPD | Comparable to iPD, but faster motor progression, more frequent dysphagia, dyskinesia and motor fluctuations | [9, 43, 116, 117, 161] |
| Cognitive decline | Less severe, later onset, dementia less frequent | More severe, earlier onset, faster progression, dementia more frequent | [9, 43, 116, 161] |
| Psychiatric symptoms | Less frequent | More frequent | [53, 54, 55, 161] |
| Autonomic symptoms | Less frequent | More frequent | [46, 48, 49, 55, 161] |
| Hyposmia | Less frequent | More frequent | [9, 45, 55, 162] |
| RBD | Less frequent | More frequent | [59, 162, 163] |
| Longitudinal features | |||
| Onset | Comparable to iPD or slightly earlier | Earlier | [9, 56, 159] |
| Overall progression | Slower | Faster | [9, 164] |
AD, Alzheimer's disease; iPD, idiopathic Parkinson's Disease; LBs, Lewy bodies; PD, Parkinson's disease; PIGD, postural instability and gait disorders; Prevalence in PD, prevalence of LRRK2/GBA variant carriers in PD patients; RBD, REM sleep behavior disorder; TDP‐43, TAR DNA‐binding protein 43.
Therapy
LRRK2‐targeted treatments
The discovery that LRRK2 deleterious variants lead to increased kinase activity 9 , 18 laid the foundations for LRRK2 kinase inhibitors as a potential treatment for LRRK2‐PD (Table 3). The main LRRK2 inhibitors currently developed are DNL151 and DNL201, which already passed phase I and Ib clinical trials with most of the participants developing no or mild adverse effects at clinically relevant doses (https://www.denalitherapeutics.com, 2021). 124 An indirect mechanism proposed to control LRRK2 activity is the inhibition of its GTPase activity, which demonstrated positive outcomes both in vitro and in vivo, with reduced LRRK2 autophosphorylation and neuroinflammation, as well as suppression of neurodegeneration. Since LRRK2 is one of the only four ROC GTPases in humans, GTPase inhibitors represent promising medications in PD therapy due to their potential advantage of a greater specificity. 125 However, evidence for the involvement of LRRK2 GTPase in PD development is still not completely clear and its activity appears more difficult to modulate. 51 , 126 Another potential resource for LRRK2‐targeted therapy is represented by antisense oligonucleotides (ASO), RNA molecules that decrease the expression or alter the splicing of LRRK2. When injected into the brain ventricles of a PD murine model with and without p.G2019S, reduced LRRK2 levels, α‐syn inclusions, and nigral dopaminergic loss were observed. 127 In LRRK2‐PD human fibroblast‐deriving iPSCs, ASO restored endoplasmic reticulum Ca2+ homeostasis and mitophagy rate. 128 Compounds targeting specific LRRK2 mutations are also recently under development. 129
Table 3.
Clinical trials for therapeutics targeting LRRK2.
| Clinical trial | Drug name | Drug type | Phase | Funding body |
|---|---|---|---|---|
| NCT03710707 | DNL201 | Kinase inhibitor | Phase Ib (completed) | Denali Therapeutics Inc. (South San Francisco, CA, USA) |
| NCT04056689 | BIIB122/DNL151 | Kinase inhibitor | Phase Ib (completed) | Denali Therapeutics Inc. (South San Francisco, CA, USA) and Biogen (Cambridge, MA, USA) |
| NCT05348785 | BIIB122/DNL151 | Kinase inhibitor | Phase IIb (ongoing) | Denali Therapeutics Inc. (South San Francisco, CA, USA) and Biogen (Cambridge, MA, USA) |
| NCT05418673 | BIIB122/DNL151 | Kinase inhibitor | Phase III (ongoing) | Denali Therapeutics Inc. (South San Francisco, CA, USA) and Biogen (Cambridge, MA, USA) |
| NCT03976349 | BIIB094 | ASO | Phase I (ongoing) | Biogen (Cambridge, MA, USA) |
ASO, antisense oligonucleotides.
Safety concerns and limitations
Some safety concerns need to be addressed for LRRK2 therapy. First, along with immune cells, LRRK2 is also highly expressed in the kidneys and lungs. In mice, histopathological abnormalities have been reported in kidneys after LRRK2 inhibition 130 and, in primates, LRRK2 inhibition produced reversible lamellar bodies in the lungs, but they did not lead to any clinical manifestation. 131 In the DNL151/201 phase 1/1b clinical trials no significant alteration in the pulmonary or renal function emerged (https://www.denalitherapeutics.com, 2021). 124 In addition, loss of function LRRK2 variants reduce LRRK2 protein levels but are not associated with any phenotypic or pathological alteration. 132 However, it should be noted that, similar to other PD therapies, LRRK2‐targeted therapy will plausibly be chronic, and these experiments are not sufficiently representative of typical long‐term treatments. 51 This is especially true for LRRK2‐PD, which progresses slowly and would require unrealistically long clinical trials and large cohorts to observe the long‐term effects (beneficial and/or harmful) of the therapy. Another safety concern is suggested by the role of LRRK2 in the defense from opportunistic infections. 133 In particular, LRRK2 loss of function is associated with increased vulnerability to some infections, particularly of intracellular pathogens. 134 , 135 The impact of a decreased LRRK2 activity on the immune system will therefore be a major aspect to consider in LRRK2‐targeted therapy. Another issue to consider is the interaction of LRRK2 with GCase activity, both in the early and advanced stages of clinical trials. 2 , 18 As mentioned previously, the relationship between LRRK2 and GCase is controversial. If increased LRRK2 kinase activity is associated with decreased GCase activity, 111 then LRRK2 therapy might produce positive effects in PD patients with reduced GCase activity, including carriers of GBA1 variants. However, if LRRK2 kinase is associated with increased GCase activity, as suggested by two studies in humans, 25 , 109 then LRRK2 therapy might have the collateral effect of reducing GCase activity, which demands caution, especially in cases when it is already impaired.
Biomarkers for LRRK2 therapy
Appropriate biomarkers to measure LRRK2 activity also need to be identified. On top of measuring the effectiveness of the LRRK2 therapy in clinical trials, they could also be employed to stratify patients based on their response and define the most appropriate dose for each subgroup. Proposed biomarkers, already used in DNL151/201 clinical trials, 124 include phosphorylation of LRRK2 substrates, such as Rab10, or LRRK2 auto‐phosphorylation activity, both associated with LRRK2 kinase function and proven to be appropriate biomarkers to measure target engagement. 51 Another possible biomarker, potentially easier to measure on a clinical setting, is the urinary bis(monoacylglycerol)phosphate (BMP), a phospholipid localized on the late endosome and lysosomal membranes, which regulates the activity of the lysosomal hydrolases and is significantly increased in carriers of the LRRK2 p.G2019S variant. 124 , 136
Challenges in LRRK2 trial design and recruitment
When planning clinical trials for LRRK2, we will face several major challenges, especially around recruitment and trial design. Specifically, LRRK2 mutations are rare, and as indicated above, carriers with LRRK2 mutations might progress slower than others. This means that in order to see an effect on the chosen endpoint, a larger and longer trial might be required. While LRRK2 mutations are the most common cause of autosomal dominant PD, they are still overall rare and represent a small portion of PD patients. Therefore, performing large and long trials might be challenging. This can be remedied, at least in part, by large international collaborations around these trials, some of which are already ongoing 137 (https://www.parkinson.org/advancing‐research/our‐research/pdgeneration).
Future perspectives
LRRK2 is a widely studied gene in PD and considerable advances have been made in our knowledge about the mechanisms that may lead to LRRK2‐PD, but this deeper knowledge also raised new questions. Future studies will need to clarify, for example, how LRRK2 regulates the endolysosomal pathway and what is the role of primary cilia in PD pathogenesis. Furthermore, inflammation could be a determinant driver in PD development so it will be crucial to elucidate the relationship of LRRK2 with the inflammation in PD and the mechanisms that alter such relationship.
Since increased LRRK2 activity has been observed also in some iPD patients, 138 understanding the role played by LRRK2 in PD development could also further our understanding of iPD pathophysiology in some patients. It is possible that LRRK2‐targeting treatments might be beneficial for this subgroup of iPD patients, and future research should test this possibility. Understanding the differences between LRRK2‐PD from iPD might also provide key clues to comprehending PD pathogenesis and progression. For example, answering why LBs are often not observed in LRRK2‐PD patients might shed light on other drivers of PD development.
Over the last years, considerable progress has been made on LRRK2‐targeted therapy in PD. The novel drugs developed will need to address important safety concerns, especially those connected to long‐term side effects, and the interactions of LRRK2 within its environment. In this regard, important information to acquire will be the relationship between LRRK2 and GCase. In fact, given that GBA1 and LRRK2 variants represent common genetic risk factors in PD and that personalized treatments are under development for both of these genes, understanding how LRRK2 variants modulate GCase activity will be invaluable for deciding whether LRRK2 therapy can be used in GBA1‐PD and vice versa. Future clinical trials will need to account for these concerns to enhance the safety and effectiveness of future personalized therapy in PD.
Conclusions
In many ways, LRRK2‐PD should be thought of as a subtype of PD mostly overlapping with iPD but with also some distinctive pathological, pathophysiologic, and clinical characteristics. The underlying mechanisms of LRRK2‐PD are likely multiple. While inflammatory response emerges as one of the main functions of LRRK2, such protein is also arguably involved in several intracellular mechanisms, such as the endolysosomal pathway, synaptic transmission, and ciliogenesis. The progressive understanding of the relationship between LRRK2 and PD set the ground for the development of multiple therapeutic approaches that could open the doors to a novel personalized medicine in PD. Clinical trials with LRRK2‐targeting treatments will still need to address several efficacy and safety concerns, and trial design challenges. Further research on the LRRK2 mechanisms involved in PD development will largely benefit from an enhancement of open science and genetic testing. This will promote a broader availability of data and increase the appropriateness and effectiveness of personalized treatment for PD.
Author Contributions
Yuri L. Sosero: conception, drafting, and review of the manuscript. Ziv Gan‐Or: conception and review of the manuscript.
Conflict of Interest
ZGO received consultancy fees from Lysosomal Therapeutics Inc. (LTI), Idorsia, Prevail Therapeutics, Inceptions Sciences (now Ventus), Ono Therapeutics, Denali, Handl Therapeutics, Neuron23, Bial Biotech, Guidepoint, Lighthouse, and Deerfield. YLS has no conflicts of interest to report.
Acknowledgments
ZGO is supported by the Fonds de recherche du Quebec – Santé (FRQS) Chercheurs‐boursiers award, and is a William Dawson Scholar. YLS is supported by Healthy Brains, Healthy Lives (HBHL) Graduate Student Fellowship, and the FRQS Doctoral training award, in collaboration with Parkinson Canada.
Funding Information
No funding information provided.
Funding Statement
This work was funded by Fonds de recherche du Quebec – Santé; Healthy Brains, Healthy Lives.
References
- 1. Dorsey ER, Sherer T, Okun MS, Bloem BR. The emerging evidence of the Parkinson pandemic. J Parkinsons Dis. 2018;8(s1):S3‐S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. von Linstow CU, Gan‐Or Z, Brundin P. Precision medicine in Parkinson's disease patients with LRRK2 and GBA risk variants – Let's get even more personal. Transl Neurodegener. 2020;9(1):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bandres‐Ciga S, Diez‐Fairen M, Kim JJ, Singleton AB. Genetics of Parkinson's disease: an introspection of its journey towards precision medicine. Neurobiol Dis. 2020;137:104782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ross OA, Soto‐Ortolaza AI, Heckman MG, et al. Association of LRRK2 exonic variants with susceptibility to Parkinson's disease: a case‐control study. Lancet Neurol. 2011;10(10):898‐908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nukada H, Kowa H, Saitoh T, Tazaki Y, Miura S. A big family of paralysis agitans (author's transl). Rinsho Shinkeigaku. 1978;18(10):627‐634. [PubMed] [Google Scholar]
- 6. Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, Obata F. A new locus for Parkinson's disease (PARK8) maps to chromosome 12p11.2‐q13.1. Ann Neurol. 2002;51(3):296‐301. [DOI] [PubMed] [Google Scholar]
- 7. Paisán‐Ruíz C, Jain S, Evans EW, et al. Cloning of the gene containing mutations that cause PARK8‐linked Parkinson's disease. Neuron. 2004;44(4):595‐600. [DOI] [PubMed] [Google Scholar]
- 8. Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal‐dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44(4):601‐607. [DOI] [PubMed] [Google Scholar]
- 9. Healy DG, Falchi M, O'Sullivan SS, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2‐associated Parkinson's disease: a case‐control study. Lancet Neurol. 2008;7(7):583‐590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gilks WP, Abou‐Sleiman PM, Gandhi S, et al. A common LRRK2 mutation in idiopathic Parkinson's disease. Lancet. 2005. Jan 29‐Feb 4;365(9457):415‐416. [DOI] [PubMed] [Google Scholar]
- 11. Tan EK, Shen H, Tan LC, et al. The G2019S LRRK2 mutation is uncommon in an Asian cohort of Parkinson's disease patients. Neurosci Lett. 2005;384(3):327‐329. [DOI] [PubMed] [Google Scholar]
- 12. Gopalai AA, Lim JL, Li HH, et al. LRRK2 N551K and R1398H variants are protective in Malays and Chinese in Malaysia: A case‐control association study for Parkinson's disease. Mol Genet Genomic Med. 2019;7(11):e604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tan EK, Peng R, Teo YY, et al. Multiple LRRK2 variants modulate risk of Parkinson disease: a Chinese multicenter study. Hum Mutat. 2010;31(5):561‐568. [DOI] [PubMed] [Google Scholar]
- 14. Heckman MG, Soto‐Ortolaza AI, Aasly JO, et al. Population‐specific frequencies for LRRK2 susceptibility variants in the Genetic Epidemiology of Parkinson's Disease (GEO‐PD) Consortium. Mov Disord. 2013;28(12):1740‐1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Greggio E, Zambrano I, Kaganovich A, et al. The Parkinson disease‐associated leucine‐rich repeat kinase 2 (LRRK2) is a dimer that undergoes intramolecular autophosphorylation. J Biol Chem. 2008;283(24):16906‐16914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sen S, Webber PJ, West AB. Dependence of leucine‐rich repeat kinase 2 (LRRK2) kinase activity on dimerization. J Biol Chem. 2009;284(52):36346‐36356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gilsbach BK, Kortholt A. Structural biology of the LRRK2 GTPase and kinase domains: implications for regulation. Front Mol Neurosci. 2014;7:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Taylor M, Alessi DR. Advances in elucidating the function of leucine‐rich repeat protein kinase‐2 in normal cells and Parkinson's disease. Curr Opin Cell Biol. 2020;63:102‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cookson MR. The role of leucine‐rich repeat kinase 2 (LRRK2) in Parkinson's disease. Nat Rev Neurosci. 2010;11(12):791‐797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mata IF, Davis MY, Lopez AN, et al. The discovery of LRRK2 p.R1441S, a novel mutation for Parkinson's disease, adds to the complexity of a mutational hotspot. Am J Med Genet B Neuropsychiatr Genet. 2016;171(7):925‐930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sheng Z, Zhang S, Bustos D, et al. Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci Transl Med. 2012;4(164):164ra1. [DOI] [PubMed] [Google Scholar]
- 22. Shu L, Zhang Y, Sun Q, Pan H, Tang B. A comprehensive analysis of population differences in LRRK2 variant distribution in Parkinson's disease. Front Aging Neurosci. 2019;11:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang Y, Sun Q, Yi M, et al. Genetic analysis of LRRK2 R1628P in Parkinson's disease in Asian populations. Parkinsons Dis. 2017;2017:8093124‐8093126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Farrer MJ, Stone JT, Lin CH, et al. Lrrk2 G2385R is an ancestral risk factor for Parkinson's disease in Asia. Parkinsonism Relat Disord. 2007;13(2):89‐92. [DOI] [PubMed] [Google Scholar]
- 25. Sosero YL, Yu E, Krohn L, et al. LRRK2 p.M1646T is associated with glucocerebrosidase activity and with Parkinson's disease. Neurobiol Aging. 2021;103:142.e1‐142.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ryan KJ, White CC, Patel K, et al. A human microglia‐like cellular model for assessing the effects of neurodegenerative disease gene variants. Sci Transl Med. 2017;9(421):eaai7635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li YI, Wong G, Humphrey J, Raj T. Prioritizing Parkinson's disease genes using population‐scale transcriptomic data. Nat Commun. 2019;10(1):994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Iwaki H, Blauwendraat C, Leonard HL, et al. Genomewide association study of Parkinson's disease clinical biomarkers in 12 longitudinal patients' cohorts. Mov Disord. 2019;34(12):1839‐1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dan X, Wang C, Ma J, et al. MAPT IVS1+124 C>G modifies risk of LRRK2 G2385R for Parkinson's disease in Chinese individuals. Neurobiol Aging. 2014;35(7):1780.e7‐1780.e10. [DOI] [PubMed] [Google Scholar]
- 30. Lai D, Alipanahi B, Fontanillas P, et al. Genomewide association studies of LRRK2 modifiers of Parkinson's disease. Ann Neurol. 2021;90(1):76‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Trinh J, Gustavsson EK, Vilariño‐Güell C, et al. DNM3 and genetic modifiers of age of onset in LRRK2 Gly2019Ser parkinsonism: a genome‐wide linkage and association study. Lancet Neurol. 2016;15(12):1248‐1256. [DOI] [PubMed] [Google Scholar]
- 32. Yu WJ, Cheng L, Li NN, Wang L, Tan EK, Peng R. Interaction between SNCA, LRRK2 and GAK increases susceptibility to Parkinson's disease in a Chinese population. eNeurologicalSci. 2015;1(1):3‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. MacLeod DA, Rhinn H, Kuwahara T, et al. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson's disease risk. Neuron. 2013;77(3):425‐439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fernández‐Santiago R, Garrido A, Infante J, et al. α‐synuclein (SNCA) but not dynamin 3 (DNM3) influences age at onset of leucine‐rich repeat kinase 2 (LRRK2) Parkinson's disease in Spain. Mov Disord. 2018;33(4):637‐641. [DOI] [PubMed] [Google Scholar]
- 35. Brown EE, Blauwendraat C, Trinh J, et al. Analysis of DNM3 and VAMP4 as genetic modifiers of LRRK2 Parkinson's disease. Neurobiol Aging. 2021;97:148.e17‐148.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nalls MA, Blauwendraat C, Vallerga CL, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson's disease: a meta‐analysis of genome‐wide association studies. Lancet Neurol. 2019;18(12):1091‐1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Iwaki H, Blauwendraat C, Makarious MB, et al. Penetrance of Parkinson's Disease in LRRK2 p.G2019S Carriers Is Modified by a Polygenic Risk Score. Mov Disord. 2020;35(5):774‐780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bloem BR, Okun MS, Klein C. Parkinson's disease. Lancet. 2021;397(10291):2284‐2303. [DOI] [PubMed] [Google Scholar]
- 39. Kalia LV, Lang AE, Hazrati LN, et al. Clinical correlations with Lewy body pathology in LRRK2‐related Parkinson disease. JAMA Neurol. 2015;72(1):100‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Henderson MX, Sengupta M, Trojanowski JQ, Lee VMY. Alzheimer's disease tau is a prominent pathology in LRRK2 Parkinson's disease. Acta Neuropathol Commun. 2019;7(1):183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ling H, Kara E, Bandopadhyay R, et al. TDP‐43 pathology in a patient carrying G2019S LRRK2 mutation and a novel p.Q124E MAPT. Neurobiol Aging. 2013;34(12):2889.e5‐2889.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wider C, Dickson DW, Wszolek ZK. Leucine‐rich repeat kinase 2 gene‐associated disease: redefining genotype‐phenotype correlation. Neurodegener Dis. 2010;7(1–3):175‐179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Alcalay RN, Mejia‐Santana H, Mirelman A, et al. Neuropsychological performance in LRRK2 G2019S carriers with Parkinson's disease. Parkinsonism Relat Disord. 2015;21(2):106‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Marras C, Schüle B, Munhoz RP, et al. Phenotype in parkinsonian and nonparkinsonian LRRK2 G2019S mutation carriers. Neurology. 2011;77(4):325‐333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Koros C, Simitsi A, Stefanis L. Genetics of Parkinson's disease: genotype‐phenotype correlations. Int Rev Neurobiol. 2017;132:197‐231. [DOI] [PubMed] [Google Scholar]
- 46. Tijero B, Gómez Esteban JC, Somme J, et al. Autonomic dysfunction in parkinsonian LRRK2 mutation carriers. Parkinsonism Relat Disord. 2013;19(10):906‐909. [DOI] [PubMed] [Google Scholar]
- 47. Trinh J, Gustavsson EK, Guella I, et al. The role of SNCA and MAPT in Parkinson disease and LRRK2 parkinsonism in the Tunisian Arab‐Berber population. Eur J Neurol. 2014;21(11):e91‐e92. [DOI] [PubMed] [Google Scholar]
- 48. Ruiz‐Martínez J, Gorostidi A, Goyenechea E, et al. Olfactory deficits and cardiac 123I‐MIBG in Parkinson's disease related to the LRRK2 R1441G and G2019S mutations. Mov Disord. 2011;26(11):2026‐2031. [DOI] [PubMed] [Google Scholar]
- 49. Gaig C, Vilas D, Infante J, et al. Nonmotor symptoms in LRRK2 G2019S associated Parkinson's disease. PLoS One. 2014;9(10):e108982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hu MT. REM sleep behavior disorder (RBD). Neurobiol Dis. 2020;143:104996. [DOI] [PubMed] [Google Scholar]
- 51. Tolosa E, Vila M, Klein C, Rascol O. LRRK2 in Parkinson disease: challenges of clinical trials. Nat Rev Neurol. 2020;16(2):97‐107. [DOI] [PubMed] [Google Scholar]
- 52. Fernández‐Santiago R, Iranzo A, Gaig C, et al. Absence of LRRK2 mutations in a cohort of patients with idiopathic REM sleep behavior disorder. Neurology. 2016;86(11):1072‐1073. [DOI] [PubMed] [Google Scholar]
- 53. Khan NL, Jain S, Lynch JM, et al. Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson's disease: clinical, pathological, olfactory and functional imaging and genetic data. Brain. 2005;128(Pt 12):2786‐2796. [DOI] [PubMed] [Google Scholar]
- 54. Goldwurm S, Zini M, Di Fonzo A, et al. LRRK2 G2019S mutation and Parkinson's disease: a clinical, neuropsychological and neuropsychiatric study in a large Italian sample. Parkinsonism Relat Disord. 2006;12(7):410‐419. [DOI] [PubMed] [Google Scholar]
- 55. Marras C, Alcalay RN, Caspell‐Garcia C, et al. Motor and nonmotor heterogeneity of LRRK2‐related and idiopathic Parkinson's disease. Mov Disord. 2016;31(8):1192‐1202. [DOI] [PubMed] [Google Scholar]
- 56. Nishioka K, Kefi M, Jasinska‐Myga B, et al. A comparative study of LRRK2, PINK1 and genetically undefined familial Parkinson's disease. J Neurol Neurosurg Psychiatry. 2010;81(4):391‐395. [DOI] [PubMed] [Google Scholar]
- 57. Alcalay RN, Mirelman A, Saunders‐Pullman R, et al. Parkinson disease phenotype in Ashkenazi Jews with and without LRRK2 G2019S mutations. Mov Disord. 2013;28(14):1966‐1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gan‐Or Z, Leblond CS, Mallett V, Orr‐Urtreger A, Dion PA, Rouleau GA. LRRK2 mutations in Parkinson disease; a sex effect or lack thereof? A meta‐analysis Parkinsonism Relat Disord. 2015;21(7):778‐782. [DOI] [PubMed] [Google Scholar]
- 59. Kestenbaum M, Alcalay RN. Clinical Features of LRRK2 Carriers with Parkinson's Disease. Adv Neurobiol. 2017;14:31‐48. [DOI] [PubMed] [Google Scholar]
- 60. Shu L, Zhang Y, Pan H, et al. Clinical heterogeneity among LRRK2 variants in Parkinson's disease: a meta‐analysis. Front Aging Neurosci. 2018;10:283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Trinh J, Zeldenrust FMJ, Huang J, et al. Genotype‐phenotype relations for the Parkinson's disease genes SNCA, LRRK2, VPS35: MDSGene systematic review. Mov Disord. 2018;33(12):1857‐1870. [DOI] [PubMed] [Google Scholar]
- 62. Hakimi M, Selvanantham T, Swinton E, et al. Parkinson's disease‐linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures. J Neural Transm (Vienna). 2011;118(5):795‐808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jankovic J, Tan EK. Parkinson's disease: etiopathogenesis and treatment. J Neurol Neurosurg Psychiatry. 2020;91(8):795‐808. [DOI] [PubMed] [Google Scholar]
- 64. Rui Q, Ni H, Li D, Gao R, Chen G. The role of LRRK2 in neurodegeneration of Parkinson disease. Curr Neuropharmacol. 2018;16(9):1348‐1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Panagiotakopoulou V, Ivanyuk D, De Cicco S, et al. Interferon‐γ signaling synergizes with LRRK2 in neurons and microglia derived from human induced pluripotent stem cells. Nat Commun. 2020;11(1):5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ho DH, Nam D, Seo M, Park SW, Seol W, Son I. LRRK2 inhibition mitigates the neuroinflammation caused by TLR2‐specific α‐synuclein and alleviates neuroinflammation‐derived dopaminergic neuronal loss. Cell. 2022;11(5):861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Langston RG, Beilina A, Reed X, et al. Association of a common genetic variant with Parkinson's disease is mediated by microglia. Sci Transl Med. 2022;14(655):eabp8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wallings RL, Tansey MG. LRRK2 regulation of immune‐pathways and inflammatory disease. Biochem Soc Trans. 2019;47(6):1581‐1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ahmadi Rastegar D, Dzamko N. Leucine rich repeat kinase 2 and innate immunity. Front Neurosci. 2020;14:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cook DA, Kannarkat GT, Cintron AF, et al. LRRK2 levels in immune cells are increased in Parkinson's disease. NPJ Parkinsons Dis. 2017;3:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lee HS, Lobbestael E, Vermeire S, Sabino J, Cleynen I. Inflammatory bowel disease and Parkinson's disease: common pathophysiological links. Gut. 2021;70(2):408‐417. [DOI] [PubMed] [Google Scholar]
- 72. Hui KY, Fernandez‐Hernandez H, Hu J, et al. Functional variants in the LRRK2 gene confer shared effects on risk for Crohn's disease and Parkinson's disease. Sci Transl Med. 2018;10(423):eaai7795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Fava VM, Manry J, Cobat A, et al. A missense LRRK2 variant is a risk factor for excessive inflammatory responses in leprosy. PLoS Negl Trop Dis. 2016;10(2):e0004412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Muñoz‐Delgado L, Macías‐García D, Periñán MT, et al. Peripheral inflammatory immune response differs among sporadic and familial Parkinson's disease. NPJ Parkinsons Dis. 2023;9(1):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Jeong GR, Lee BD. Pathological functions of LRRK2 in Parkinson's disease. Cell. 2020;9(12):2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mir R, Tonelli F, Lis P, et al. The Parkinson's disease VPS35[D620N] mutation enhances LRRK2‐mediated Rab protein phosphorylation in mouse and human. Biochem J. 2018;475(11):1861‐1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Purlyte E, Dhekne HS, Sarhan AR, et al. Rab29 activation of the Parkinson's disease‐associated LRRK2 kinase. EMBO J. 2018;37(1):1‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zimprich A, Benet‐Pagès A, Struhal W, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late‐onset Parkinson disease. Am J Hum Genet. 2011;89(1):168‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Eguchi T, Kuwahara T, Sakurai M, et al. LRRK2 and its substrate Rab GTPases are sequentially targeted onto stressed lysosomes and maintain their homeostasis. Proc Natl Acad Sci U S A. 2018;115(39):E9115‐e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Henry AG, Aghamohammadzadeh S, Samaroo H, et al. Pathogenic LRRK2 mutations, through increased kinase activity, produce enlarged lysosomes with reduced degradative capacity and increase ATP13A2 expression. Hum Mol Genet. 2015;24(21):6013‐6028. [DOI] [PubMed] [Google Scholar]
- 81. Hockey LN, Kilpatrick BS, Eden ER, et al. Dysregulation of lysosomal morphology by pathogenic LRRK2 is corrected by TPC2 inhibition. J Cell Sci. 2015;128(2):232‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ho PW, Leung CT, Liu H, et al. Age‐dependent accumulation of oligomeric SNCA/alpha‐synuclein from impaired degradation in mutant LRRK2 knockin mouse model of Parkinson disease: role for therapeutic activation of chaperone‐mediated autophagy (CMA). Autophagy. 2020;16(2):347‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Nguyen M, Krainc D. LRRK2 phosphorylation of auxilin mediates synaptic defects in dopaminergic neurons from patients with Parkinson's disease. Proc Natl Acad Sci U S A. 2018;115(21):5576‐5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Erb ML, Moore DJ. LRRK2 and the endolysosomal system in Parkinson's disease. J Parkinsons Dis. 2020;10(4):1271‐1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Williams ET, Chen X, Moore DJ. VPS35, the retromer complex and Parkinson's disease. J Parkinsons Dis. 2017;7(2):219‐233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ludtmann MHR, Kostic M, Horne A, Gandhi S, Sekler I, Abramov AY. LRRK2 deficiency induced mitochondrial Ca2+ efflux inhibition can be rescued by Na+/Ca2+/Li+ exchanger upregulation. Cell Death Dis 2019;10(4):265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wang C, Yang T, Liang M, Xie J, Song N. Astrocyte dysfunction in Parkinson's disease: from the perspectives of transmitted α‐synuclein and genetic modulation. Transl Neurodegener. 2021;10(1):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Gan‐Or Z, Dion PA, Rouleau GA. Genetic perspective on the role of the autophagy‐lysosome pathway in Parkinson disease. Autophagy. 2015;11(9):1443‐1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Pang SY, Lo RCN, Ho PW, et al. LRRK2, GBA and their interaction in the regulation of autophagy: implications on therapeutics in Parkinson's disease. Transl Neurodegener. 2022;11(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Roosen DA, Cookson MR. LRRK2 at the interface of autophagosomes, endosomes and lysosomes. Mol Neurodegener. 2016;11(1):73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Manzoni C, Lewis PA. LRRK2 and autophagy. Adv Neurobiol. 2017;14:89‐105. [DOI] [PubMed] [Google Scholar]
- 92. Wallings R, Manzoni C, Bandopadhyay R. Cellular processes associated with LRRK2 function and dysfunction. FEBS J. 2015;282(15):2806‐2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Orenstein SJ, Kuo SH, Tasset I, et al. Interplay of LRRK2 with chaperone‐mediated autophagy. Nat Neurosci. 2013;16(4):394‐406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Plowey ED, Cherra SJ 3rd, Liu YJ, Chu CT. Role of autophagy in G2019S‐LRRK2‐associated neurite shortening in differentiated SH‐SY5Y cells. J Neurochem. 2008;105(3):1048‐1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Boecker CA, Goldsmith J, Dou D, Cajka GG, Holzbaur ELF. Increased LRRK2 kinase activity alters neuronal autophagy by disrupting the axonal transport of autophagosomes. Curr Biol. 2021;31(10):2140‐54.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Lara Ordóñez AJ, Fasiczka R, Naaldijk Y, Hilfiker S. Rab GTPases in Parkinson's disease: a primer. Essays Biochem. 2021;65(7):961‐974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Wauters F, Cornelissen T, Imberechts D, et al. LRRK2 mutations impair depolarization‐induced mitophagy through inhibition of mitochondrial accumulation of RAB10. Autophagy. 2020;16(2):203‐222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Dhekne HS, Yanatori I, Gomez RC, et al. A pathway for Parkinson's Disease LRRK2 kinase to block primary cilia and Sonic hedgehog signaling in the brain. Elife. 2018;7:e40202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lara Ordónez AJ, Fernández B, Fdez E, et al. RAB8, RAB10 and RILPL1 contribute to both LRRK2 kinase‐mediated centrosomal cohesion and ciliogenesis deficits. Hum Mol Genet. 2019;28(21):3552‐3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Schaub JR, Stearns T. The Rilp‐like proteins Rilpl1 and Rilpl2 regulate ciliary membrane content. Mol Biol Cell. 2013;24(4):453‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Gonzalez‐Reyes LE, Verbitsky M, Blesa J, et al. Sonic hedgehog maintains cellular and neurochemical homeostasis in the adult nigrostriatal circuit. Neuron. 2012;75(2):306‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Howlett EH, Jensen N, Belmonte F, et al. LRRK2 G2019S‐induced mitochondrial DNA damage is LRRK2 kinase dependent and inhibition restores mtDNA integrity in Parkinson's disease. Hum Mol Genet. 2017;26(22):4340‐4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Verma M, Callio J, Otero PA, Sekler I, Wills ZP, Chu CT. Mitochondrial calcium dysregulation contributes to dendrite degeneration mediated by PD/LBD‐associated LRRK2 mutants. J Neurosci. 2017;37(46):11151‐11165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Panicker N, Ge P, Dawson VL, Dawson TM. The cell biology of Parkinson's disease. J Cell Biol. 2021;220(4):e202012095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Ge W, Li D, Gao Y, Cao X. The roles of lysosomes in inflammation and autoimmune diseases. Int Rev Immunol. 2015;34(5):415‐431. [DOI] [PubMed] [Google Scholar]
- 106. Gan‐Or Z, Amshalom I, Kilarski LL, et al. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology. 2015;84(9):880‐887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Grabowski GA, Gatt S, Horowitz M. Acid beta‐glucosidase: enzymology and molecular biology of Gaucher disease. Crit Rev Biochem Mol Biol. 1990;25(6):385‐414. [DOI] [PubMed] [Google Scholar]
- 108. Gegg ME, Burke D, Heales SJ, et al. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann Neurol. 2012;72(3):455‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Alcalay RN, Levy OA, Waters CC, et al. Glucocerebrosidase activity in Parkinson's disease with and without GBA mutations. Brain. 2015;138(Pt 9):2648‐2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Krohn L, Öztürk TN, Vanderperre B, et al. Genetic, structural, and functional evidence link TMEM175 to synucleinopathies. Ann Neurol. 2020;87(1):139‐153. [DOI] [PubMed] [Google Scholar]
- 111. Ysselstein D, Nguyen M, Young TJ, et al. LRRK2 kinase activity regulates lysosomal glucocerebrosidase in neurons derived from Parkinson's disease patients. Nat Commun. 2019;10(1):5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Kedariti M, Frattini E, Baden P, et al. LRRK2 kinase activity regulates GCase level and enzymatic activity differently depending on cell type in Parkinson's disease. NPJ Parkinsons Dis. 2022;8(1):92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Ysselstein D, Young TJ, Nguyen M, et al. Evaluation of strategies for measuring lysosomal glucocerebrosidase activity. Mov Disord. 2021;36(12):2719‐2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Liu G, Boot B, Locascio JJ, et al. Specifically neuropathic Gaucher's mutations accelerate cognitive decline in Parkinson's. Ann Neurol. 2016;80(5):674‐685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Swan M, Doan N, Ortega RA, et al. Neuropsychiatric characteristics of GBA‐associated Parkinson disease. J Neurol Sci. 2016;370:63‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Cilia R, Tunesi S, Marotta G, et al. Survival and dementia in GBA‐associated Parkinson's disease: the mutation matters. Ann Neurol. 2016;80(5):662‐673. [DOI] [PubMed] [Google Scholar]
- 117. Gan‐Or Z, Liong C, Alcalay RN. GBA‐associated Parkinson's disease and other synucleinopathies. Curr Neurol Neurosci Rep. 2018;18(8):44. [DOI] [PubMed] [Google Scholar]
- 118. Omer N, Giladi N, Gurevich T, et al. A possible modifying effect of the G2019S mutation in the LRRK2 gene on GBA Parkinson's disease. Mov Disord. 2020;35(7):1249‐1253. [DOI] [PubMed] [Google Scholar]
- 119. Ortega RA, Wang C, Raymond D, et al. Association of dual LRRK2 G2019S and GBA variations with parkinson disease progression. JAMA Netw Open. 2021;4(4):e215845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Yahalom G, Greenbaum L, Israeli‐Korn S, et al. Carriers of both GBA and LRRK2 mutations, compared to carriers of either, in Parkinson's disease: risk estimates and genotype‐phenotype correlations. Parkinsonism Relat Disord. 2019;62:179‐184. [DOI] [PubMed] [Google Scholar]
- 121. Schneider SA, Alcalay RN. Neuropathology of genetic synucleinopathies with parkinsonism: review of the literature. Mov Disord. 2017;32(11):1504‐1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Heckman MG, Soto‐Ortolaza AI, Contreras MYS, et al. LRRK2 variation and dementia with Lewy bodies. Parkinsonism Relat Disord. 2016;31:98‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB Consortium. Neurology. 2017;89(1):88‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Jennings D, Huntwork‐Rodriguez S, Henry AG, et al. Preclinical and clinical evaluation of the LRRK2 inhibitor DNL201 for Parkinson's disease. Sci Transl Med. 2022;14(648):eabj2658. [DOI] [PubMed] [Google Scholar]
- 125. Senkevich K, Rudakou U, Gan‐Or Z. New therapeutic approaches to Parkinson's disease targeting GBA, LRRK2 and Parkin. Neuropharmacology. 2022;202:108822. [DOI] [PubMed] [Google Scholar]
- 126. Tolosa E, Gaig C, Santamaría J, Compta Y. Diagnosis and the premotor phase of Parkinson disease. Neurology. 2009;72(7 Suppl):S12‐S20. [DOI] [PubMed] [Google Scholar]
- 127. Zhao HT, John N, Delic V, et al. LRRK2 antisense oligonucleotides ameliorate α‐synuclein inclusion formation in a Parkinson's disease mouse model. Mol Ther Nucleic Acids. 2017;8:508‐519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Korecka JA, Talbot S, Osborn TM, et al. Neurite collapse and altered ER Ca(2+) control in human Parkinson disease patient iPSC‐derived neurons with LRRK2 G2019S mutation. Stem Cell Reports. 2019;12(1):29‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Lesniak RK, Nichols RJ, Montine TJ. Development of mutation‐selective LRRK2 kinase inhibitors as precision medicine for Parkinson's disease and other diseases for which carriers are at increased risk. Front Neurol. 2022;13:1016040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Herzig MC, Kolly C, Persohn E, et al. LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum Mol Genet. 2011;20(21):4209‐4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Fuji RN, Flagella M, Baca M, et al. Effect of selective LRRK2 kinase inhibition on nonhuman primate lung. Sci Transl Med. 2015;7(273):273ra15. [DOI] [PubMed] [Google Scholar]
- 132. Whiffin N, Armean IM, Kleinman A, et al. The effect of LRRK2 loss‐of‐function variants in humans. Nat Med. 2020;26(6):869‐877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Härtlova A, Herbst S, Peltier J, et al. LRRK2 is a negative regulator of Mycobacterium tuberculosis phagosome maturation in macrophages. EMBO J. 2018;37(12):e98694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Shutinoski B, Hakimi M, Harmsen IE, et al. Lrrk2 alleles modulate inflammation during microbial infection of mice in a sex‐dependent manner. Sci Transl Med. 2019;11(511):eaas9292. [DOI] [PubMed] [Google Scholar]
- 135. Liu W, Liu X, Li Y, et al. LRRK2 promotes the activation of NLRC4 inflammasome during Salmonella Typhimurium infection. J Exp Med. 2017;214(10):3051‐3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Alcalay RN, Hsieh F, Tengstrand E, et al. Higher urine bis(monoacylglycerol)phosphate levels in LRRK2 G2019S mutation carriers: implications for therapeutic development. Mov Disord. 2020;35(1):134‐141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Klein C, Hattori N, Marras C. MDSGene: closing data gaps in genotype‐phenotype correlations of monogenic Parkinson's disease. J Parkinsons Dis. 2018;8(s1):S25‐s30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Di Maio R, Hoffman EK, Rocha EM, et al. LRRK2 activation in idiopathic Parkinson's disease. Sci Transl Med. 2018;10(451):eaar5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Gaig C, Ezquerra M, Martí MJ, et al. Screening for the LRRK2 G2019S and codon‐1441 mutations in a pathological series of parkinsonian syndromes and frontotemporal lobar degeneration. J Neurol Sci. 2008;270(1–2):94‐98. [DOI] [PubMed] [Google Scholar]
- 140. Gaig C, Martí MJ, Ezquerra M, Cardozo A, Rey MJ, Tolosa E. G2019S LRRK2 mutation causing Parkinson's disease without Lewy bodies. BMJ Case Rep. 2009;2009:bcr0820080632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Gomez A, Ferrer I. Involvement of the cerebral cortex in Parkinson disease linked with G2019S LRRK2 mutation without cognitive impairment. Acta Neuropathol. 2010;120(2):155‐167. [DOI] [PubMed] [Google Scholar]
- 142. Rajput A, Dickson DW, Robinson CA, et al. Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology. 2006;67(8):1506‐1508. [DOI] [PubMed] [Google Scholar]
- 143. Ross OA, Toft M, Whittle AJ, et al. Lrrk2 and Lewy body disease. Ann Neurol. 2006;59(2):388‐393. [DOI] [PubMed] [Google Scholar]
- 144. Giasson BI, Covy JP, Bonini NM, et al. Biochemical and pathological characterization of Lrrk2. Ann Neurol. 2006;59(2):315‐322. [DOI] [PubMed] [Google Scholar]
- 145. Silveira‐Moriyama L, Guedes LC, Kingsbury A, et al. Hyposmia in G2019S LRRK2‐related parkinsonism: clinical and pathologic data. Neurology. 2008;71(13):1021‐1026. [DOI] [PubMed] [Google Scholar]
- 146. Poulopoulos M, Cortes E, Vonsattel JP, et al. Clinical and pathological characteristics of LRRK2 G2019S patients with PD. J Mol Neurosci. 2012;47(1):139‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Vilas D, Sharp M, Gelpi E, et al. Clinical and neuropathological features of progressive supranuclear palsy in Leucine rich repeat kinase (LRRK2) G2019S mutation carriers. Mov Disord. 2018;33(2):335‐338. [DOI] [PubMed] [Google Scholar]
- 148. Wszolek ZK, Pfeiffer RF, Tsuboi Y, et al. Autosomal dominant parkinsonism associated with variable synuclein and tau pathology. Neurology. 2004;62(9):1619‐1622. [DOI] [PubMed] [Google Scholar]
- 149. Giordana MT, D'Agostino C, Albani G, et al. Neuropathology of Parkinson's disease associated with the LRRK2 Ile1371Val mutation. Mov Disord. 2007;22(2):275‐278. [DOI] [PubMed] [Google Scholar]
- 150. Covy JP, Yuan W, Waxman EA, Hurtig HI, Van Deerlin VM, Giasson BI. Clinical and pathological characteristics of patients with leucine‐rich repeat kinase‐2 mutations. Mov Disord. 2009;24(1):32‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Hasegawa K, Kowa H. Autosomal dominant familial Parkinson disease: older onset of age, and good response to levodopa therapy. Eur Neurol. 1997;38(Suppl 1):39‐43. [DOI] [PubMed] [Google Scholar]
- 152. Hasegawa K, Stoessl AJ, Yokoyama T, Kowa H, Wszolek ZK, Yagishita S. Familial parkinsonism: study of original Sagamihara PARK8 (I2020T) kindred with variable clinicopathologic outcomes. Parkinsonism Relat Disord. 2009;15(4):300‐306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Martí‐Massó JF, Ruiz‐Martínez J, Bolaño MJ, et al. Neuropathology of Parkinson's disease with the R1441G mutation in LRRK2. Mov Disord. 2009;24(13):1998‐2001. [DOI] [PubMed] [Google Scholar]
- 154. Puschmann A, Englund E, Ross OA, et al. First neuropathological description of a patient with Parkinson's disease and LRRK2 p.N1437H mutation. Parkinsonism Relat Disord. 2012;18(4):332‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Takanashi M, Funayama M, Matsuura E, et al. Isolated nigral degeneration without pathological protein aggregation in autopsied brains with LRRK2 p.R1441H homozygous and heterozygous mutations. Acta Neuropathol Commun. 2018;6(1):105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Blauwendraat C, Pletnikova O, Geiger JT, et al. Genetic analysis of neurodegenerative diseases in a pathology cohort. Neurobiol Aging. 2019;76:214.e1‐241.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Fung HC, Chen CM, Hardy J, Hernandez D, Singleton A, Wu YR. Lack of G2019S LRRK2 mutation in a cohort of Taiwanese with sporadic Parkinson's disease. Mov Disord. 2006;21(6):880‐881. [DOI] [PubMed] [Google Scholar]
- 158. Yonova‐Doing E, Atadzhanov M, Quadri M, et al. Analysis of LRRK2, SNCA, Parkin, PINK1, and DJ‐1 in Zambian patients with Parkinson's disease. Parkinsonism Relat Disord. 2012;18(5):567‐571. [DOI] [PubMed] [Google Scholar]
- 159. Li Q, Jing Y, Lun P, Liu X, Sun P. Association of gender and age at onset with glucocerebrosidase associated Parkinson's disease: a systematic review and meta‐analysis. Neurol Sci. 2021;42(6):2261‐2271. [DOI] [PubMed] [Google Scholar]
- 160. Balestrino R, Tunesi S, Tesei S, Lopiano L, Zecchinelli AL, Goldwurm S. Penetrance of glucocerebrosidase (GBA) mutations in Parkinson's disease: a kin cohort study. Mov Disord. 2020;35(11):2111‐2114. [DOI] [PubMed] [Google Scholar]
- 161. Jesús S, Huertas I, Bernal‐Bernal I, et al. GBA variants influence motor and non‐motor features of Parkinson's disease. PLoS One. 2016;11(12):e0167749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Thaler A, Gurevich T, Bar Shira A, et al. A “dose” effect of mutations in the GBA gene on Parkinson's disease phenotype. Parkinsonism Relat Disord. 2017;36:47‐51. [DOI] [PubMed] [Google Scholar]
- 163. Gan‐Or Z, Mirelman A, Postuma RB, et al. GBA mutations are associated with rapid eye movement sleep behavior disorder. Ann Clin Transl Neurol. 2015;2(9):941‐945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Brockmann K, Srulijes K, Pflederer S, et al. GBA‐associated Parkinson's disease: reduced survival and more rapid progression in a prospective longitudinal study. Mov Disord. 2015;30(3):407‐411. https://www.denalitherapeutics.com/investors/press‐release?id=8141&type=api, https://www.parkinson.org/advancing‐research/our‐research/pdgeneration [DOI] [PubMed] [Google Scholar]