

Abstract
Parainfluenza viruses, members of the enveloped, negative-sense, single stranded RNA Paramyxoviridae family, impact global child health as the cause of significant lower respiratory tract infections. Parainfluenza viruses enter cells by fusing directly at the cell surface membrane. How this fusion occurs via the coordinated efforts of the two molecules that comprise the viral surface fusion complex, and how these efforts may be blocked, are the subjects of this chapter. The receptor binding protein of parainfluenza forms a complex with the fusion protein of the virus, remaining stably associated until a receptor is reached. At that point, the receptor binding protein actively triggers the fusion protein to undergo a series of transitions that ultimately lead to membrane fusion and viral entry. In recent years it has become possible to examine this remarkable process on the surface of viral particles and to begin to understand the steps in the transition of this molecular machine, using a structural biology approach. Understanding the steps in entry leads to several possible strategies to prevent fusion and inhibit infection.
1. Introduction to parainfluenza virus infection and entry
Acute respiratory infection is the leading cause of mortality in children under 5 years of age, accounting for nearly one-fifth of childhood deaths worldwide and 2–3 million childhood deaths annually (Nair et al., 2010). The enveloped human parainfluenza viruses (HPIVs) types 1, 2, and 3, human metapneumovirus (Williams et al., 2004), rhinovirus (Miller et al., 2007), and respiratory syncytial virus (RSV) cause the majority of childhood lower respiratory tract disease in the United States. The HPIVs 1 and 3 belong to the Respirovirus genus within the Paramyxovirinae subfamily of the Paramyxoviridae family of negative-stranded RNA viruses, while HPIV2 belongs to the Rubulavirus genus. Although infection by the HPIVs causes major respiratory diseases including croup, bronchiolitis and pneumonia, affecting millions of infants and young children as well as older and immune compromised adults (Chemaly et al., 2012; Henrickson et al., 2004; Lo et al., 2012; Maziarz et al., 2010; Nair et al., 2010; Roghmann et al., 2003; Seo et al., 2014; Shah et al., 2016; Weinberg et al., 2009) there are currently no effective vaccines or antiviral treatments (Englund et al., 2013; Schmidt et al., 2011; van Asten et al., 2012). HPIV3 accounts for the vast majority of HPIV infections following transplantation, causing pneumonia with a 35% acute mortality rate and up to a 50% mortality rate at 6 months (Fontana and Strasfeld, 2019; Lo et al., 2013; Nichols et al., 2001). A survey study estimated that HPIV recently accounted for 7% of all hospitalizations for fever and/or acute respiratory illness in children under 5 years in the United States (Weinberg et al., 2009) and half of these result from HPIV3 infection, with most of the remainder caused by HPIV1.
HPIVs enter cells by fusing directly with the cell plasma membrane. Virions bear two surface glycoproteins that cooperate to mediate viral entry into host cells: the receptor-binding protein hemagglutinin-neuraminidase (HN), and the fusion (F) protein. The series of cooperative steps that mediate entry have been elucidated in detail for HPIV3 since it was first demonstrated, using HPIV3 as the prototype, that for most Paramyxoviridae—with certain exceptions—virus-induced membrane fusion requires active participation of both HN and F (Marcink et al., 2020b; Moscona and Peluso, 1991; Porotto et al., 2003, 2011a, 2012c; Xu et al., 2013). HN stabilizes the F protein before receptor is engaged, to prevent viral inactivation (Porotto et al., 2012c). However, once host cell receptors have been engaged, HN switches to new roles: the HN stalk communicates with two sites in the HN head, thereby activating the trimeric fusion (F) protein, inducing a conformational change in F that allows for its insertion into the host cell membrane (Chang and Dutch, 2012; Moscona, 2005; Moscona and Peluso, 1993; Palermo et al., 2007, 2009; Porotto et al., 2003, 2007a). The F protein then undergoes further structural rearrangements that drive viral-host membrane fusion and viral entry into the target cell (Chang and Dutch, 2012; Harrison, 2008; Plattet and Plemper, 2013; White et al., 2008). This step requires ongoing interaction between HN and F through a series of transient intermediates and does not, by itself, set HPIV3 F inexorably on the path to fusion (Porotto et al., 2011a). Following entry and replication, HN promotes viral egress by cleaving sialic acid residues on the host cell membrane to allow the release of new viral particles and spread of virus (Huberman et al., 1995; Porotto et al., 2001).
The efficiency of F-triggering by HN critically influences the degree of fusion, and thus the extent of viral entry (Palermo et al., 2009; Porotto et al., 2003). The binding, triggering, and receptor cleaving functions of HN are in a balance that directly determines the outcome of infection (Porotto et al., 2005). The individual functions of the HN/F protein complex must maintain a balance to allow for coordinated execution of these steps. Our analysis of the series of steps carried out by the HN-F fusion complex leads us to propose a new paradigm for how the HN-F complex—viewed in its entirety as a fusion machine—promotes viral entry (see Fig. 1A–D). Studies of other paramyxoviruses have confirmed the finding that the receptor binding protein is essential to the F-mediated fusion process (Bagai and Lamb, 1995; Horvath et al., 1992; Hu et al., 1992, reviewed in Lamb, 1993), with several fascinating exceptions (e.g. RSV (Chaiwatpongsakorn et al., 2011; San-Juan-Vergara et al., 2011), hMPV (Chang et al., 2012; Schildgen et al., 2011; Schowalter et al., 2009)), and reveal that each paramyxovirus solves the problem of entry using variations on this theme. By imaging interactions between viral glycoproteins at high resolution in situ as the interacting proteins progress through their roles in the cell, it is now possible to investigate the entry mechanism in detail. For HPIV3, cryo-ET was applied to analyze intact virions and follow the process by which HN activates F to mediate fusion in complex with receptors on a target cell membrane. This review follows the steps of viral entry for HPIV, organized around the structural information available for each step in the process.
Fig. 1.
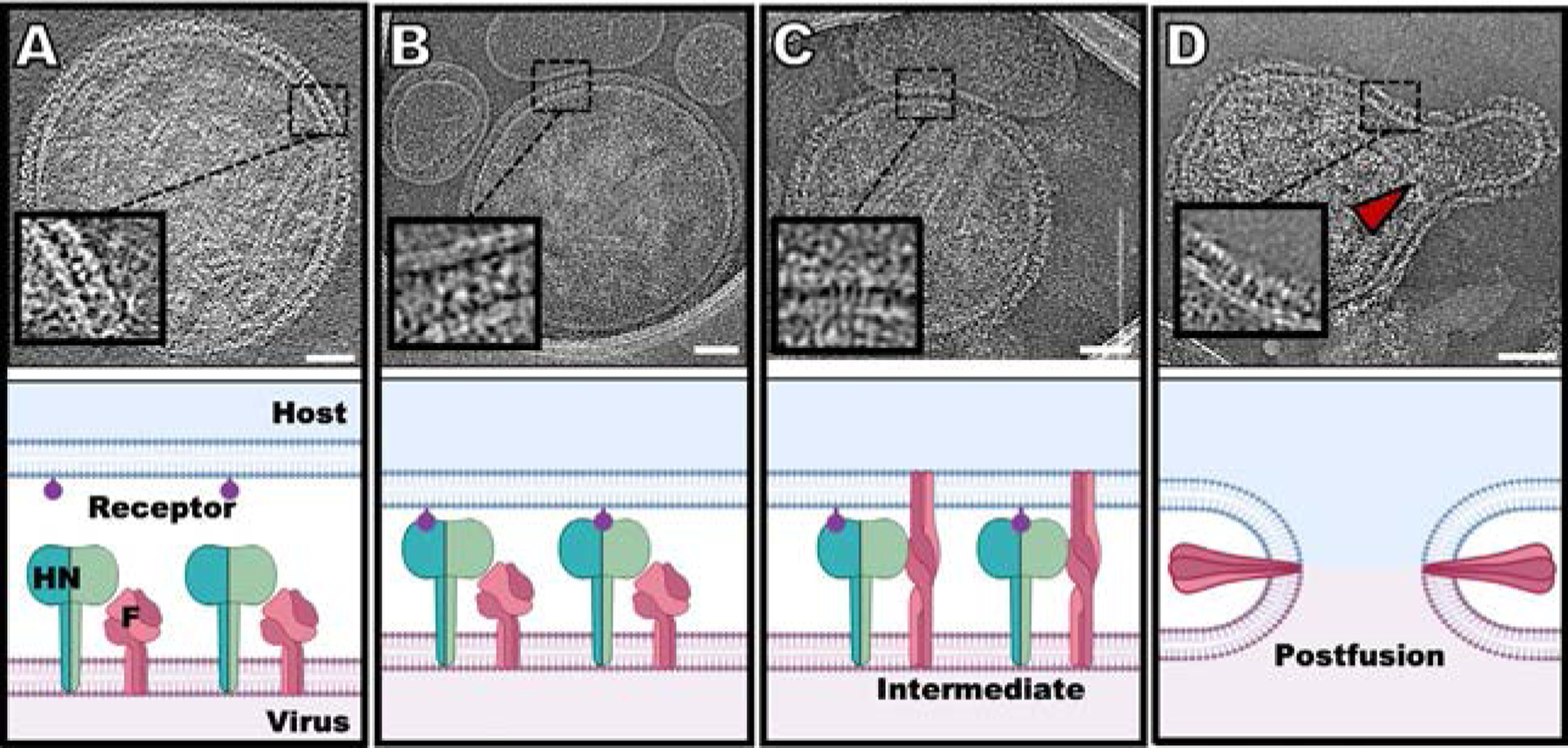
Schematic diagram of the steps in HPIV3 entry, with snapshots of cryo-electron tomographic images at each step (24). (A) HN (green) and F (dark pink) can be found densely packed on the viral surface (light pink). (B) Sialic acid (purple) binding to HN occurs in the presence of a surrogate host target membrane (erythrocyte membrane fragment) (blue). (C) Upon triggering of F by HN, F undergoes a large conformational change from a pre-fusion globular structure to a transient extended structure that crosses both membranes. (D) F folds back onto itself, pulling the viral and cell membranes toward each other, in a process that ultimately results in a merged membrane. Scale bars: (A–D) 50nm. Adapted from Marcink, T.C., Wang, T., des Georges, A., Porotto, M., Moscona A., 2020. Human parainfluenza virus fusion complex glycoproteins imaged in action on authentic viral surfaces. PLoS Pathog. 16 (9), e1008883, with permission.
2. HN’s structure and receptor binding
The four activities of HN—stabilization of pre-fusion F, receptor binding, F-triggering, and receptor cleaving—are regulated within a dimeric type II membrane protein consisting of an N-terminal cytoplasmic domain, a membrane-spanning region, a stalk region, and a globular head. The stalk confers specificity for the homologous F in the fusion activation process (Bose et al., 2011; Deng et al., 1995; Melanson and Iorio, 2006; Porotto et al., 2003, 2005; Sergel et al., 1993; Stone-Hulslander and Morrison, 1999; Tanabayashi and Compans, 1996; Yuan et al., 2011; Yuasa et al., 1995). HN receptor interaction is a critical prelude to F-triggering and viral entry. The primary binding site, which has both receptor binding and receptor cleaving (neuraminidase) activities, is located on the globular head for HPIV3 (and other paramyxoviruses for which structural information is available) (Crennell et al., 2000; Lawrence et al., 2004; Yuan et al., 2005; Zaitsev et al., 2004). HPIV3 HN can bind receptors that contain either α2–3 or α2–6-linked terminal sialic acids, and the specificity of clinical strains of HPIV3 for sialic acid receptors in human lung remains to be fully elucidated (Dirr et al., 2015; Fukushima et al., 2015; Moscona and Peluso, 1996; Suzuki et al., 2001; Tong et al., 2018; Zhang et al., 2005).
The oligomeric state of HN on the viral surface has been unclear until recently. Dimeric crystal structures of HPIV3 and Newcastle Disease virus (NDV; a related paramyxovirus) have been solved, and the only tetrameric crystal structure of HN was obtained in solution for PIV5 (Crennell et al., 2000; Streltsov et al., 2015; Yuan et al., 2005). The dimer of dimers interaction in PIV5 is relatively weak compared to the dimer itself, and the interface domains between dimers are not well conserved across other viral HN proteins. Our recent cryo-electron tomographic studies of HPIV3 HN have shown that HN is present as a dimer on the viral surface (Marcink et al., 2020b) (see Fig. 2A–D). Subtomogram averaged densities were incompatible with the tetrameric PIV5 crystal structures, and showed dimeric HN resting in a position superior to F, in a pre-receptor engaged state (Marcink et al., 2020b) (see Fig. 2D).
Fig. 2.
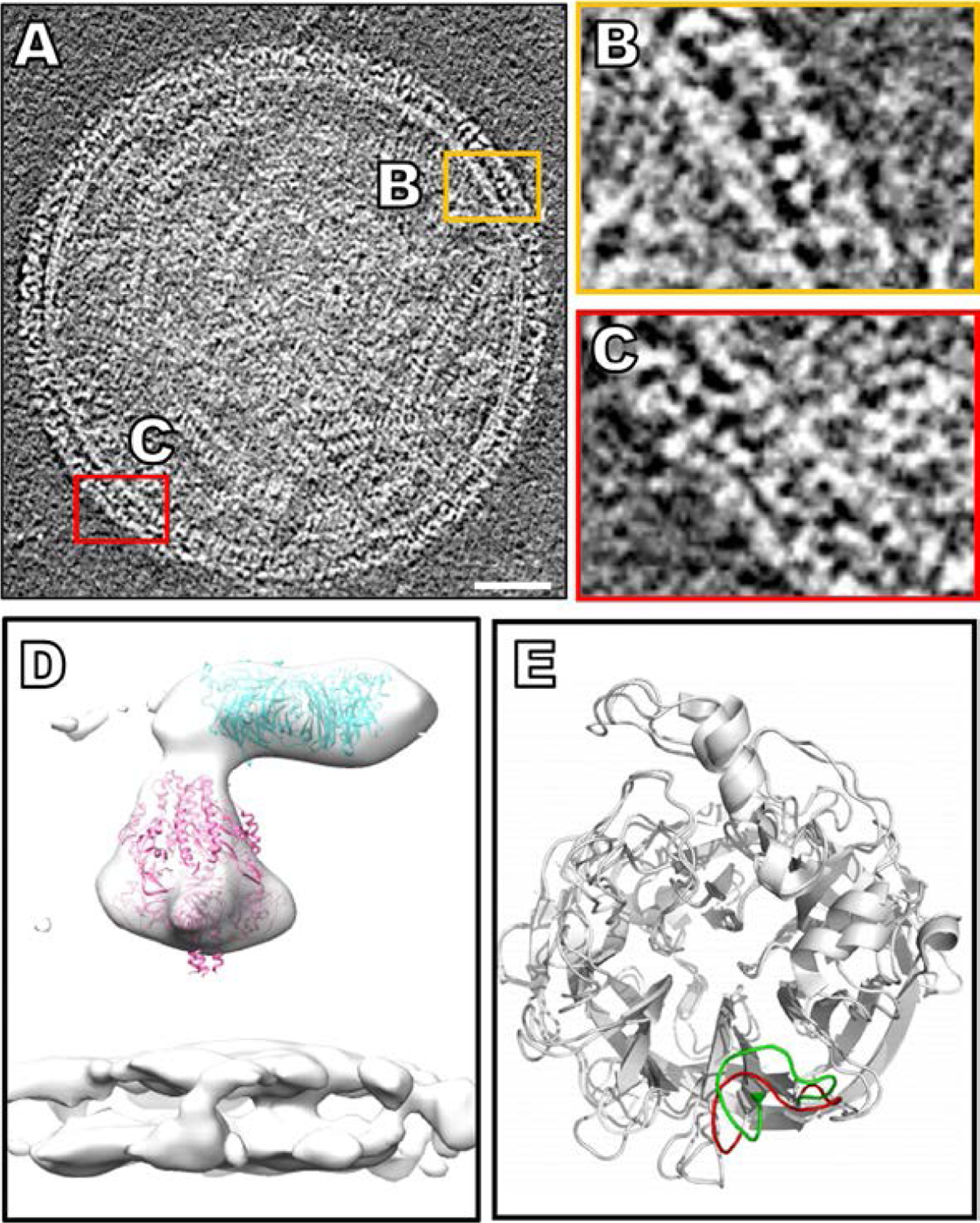
The HN visualized on the virion surface and examined by cryo-electron tomography. (A) Contrast inverted cryo-ET central slice of HPIV3 before receptor engagement (Marcink et al., 2020b). (B and C) Enlarged regions of the surface glycoproteins with HN and F in tight apposition. (D) Sub-volume average of surface glycoproteins with crystal structure of the HN dimer (PDB ID: 4MZA) and the cryo-EM structure of pre-fusion F (PDB ID: 6MJZ) in green and pink (respectively), fitted into the sub-volume average of the HPIV3 viral surface. (E) A flexible loop on HN that adopts an open conformation (red) in direct vicinity of the active site of the apo-form of the protein and closes (green) upon inhibitor binding (Winger and von Itzstein, 2012). Panels (A–D): Adapted from Marcink, T.C., Wang, T., des Georges, A., Porotto, M., Moscona A., 2020. Human parainfluenza virus fusion complex glycoproteins imaged in action on authentic viral surfaces. PLoS Pathog. 16 (9), e1008883, with permission. Panel (E): From Winger, M., von Itzstein, M., 2012. Exposing the flexibility of human parainfluenza virus hemagglutinin-neuraminidase. J. Am. Chem. Soc. 134 (44), 18447–52, with permission.
A second receptor binding site, located in HN’s dimer interface, is critical for activating F (Palmer et al., 2014; Porotto et al., 2007a, 2012a,b; Xu et al., 2013). The residues that form this binding/F-activating site II affect HN dimerization and HN-F interaction, both of which in turn impact HN’s ability to activate F. Specific residues at binding site II determine the fitness for viruses to infect host tissues (Iketani et al., 2018). The dimer interface, and its modulation of HN-F interaction, is critical to infection in the host; even a subtle change at the dimer interface of the globular domain can impact the HN-F fusion machine, and markedly alter host infection (Xu et al., 2013).
The HPIV3 HN molecule has significant flexibility. For example, the loop around D216 that forms part of the primary binding site can assume multiple conformations. Molecular dynamics analyses have shown that this D216 loop is in an open conformation in the apo state, but when a substrate or inhibitor binds, the loop closes and engages the sialic acid substrate (El-Deeb et al., 2017; Pascolutti et al., 2018; Winger and von Itzstein, 2012) (see Fig. 2E). The side chain of R212, also part of the primary HPIV3 binding site, undergoes a significant reorientation upon substrate binding as well (Dirr et al., 2015; Lawrence et al., 2004; Pascolutti et al., 2018), consistent with our recent observations that mutations at this site alter receptor avidity of clinical strains (unpublished data). However for PIV5 (a related paramyxovirus formerly known as simian virus 5; a tractable model for molecular studies with no established human disease association), the loop corresponding to the HPIV3 D216 region (residues 186–190 in PIV5) appears flexible but does not appear to close upon substrate binding (Yuan et al., 2005) It is not known whether, or how, these movements of the HPIV3 primary binding site may affect the second binding/activating site and/or the HN stalk; this is an area of great interest. Ultimately, this first binding step in which HN’s primary binding site engages sialic acid receptor initiates the cascade of events that follow, leading to activation of F and fusion.
3. F: Primed for action in its pre-fusion state
The F proteins of HPIVs are trimers and contain a short stalk region with a large globular head, together making up a tree-like domain (Stewart-Jones et al., 2018; Yin et al., 2006). F is a type I transmembrane (TM) protein, with its N-terminus outside the viral particle. It is synthesized as an inactive F0 precursor which is cleaved by host cell proteases into its active form composed of two subunits, F1 and F2, linked by a disulfide bridge between their heptad repeat N-terminal domains (HRNs) (Scheid and Choppin, 1978) (Fig. 3). This cleavage is generally thought to depend on the intracellular protease furin for glycoprotein maturation (Ortmann et al., 1994) and generates the hydrophobic fusion peptide that will pierce the target membrane during the fusion process, after the activation step exposes the peptide at the surface of the molecule. Uncleaved F proteins are unable to promote fusion; the intracellular F cleavage step is essential.
Fig. 3.
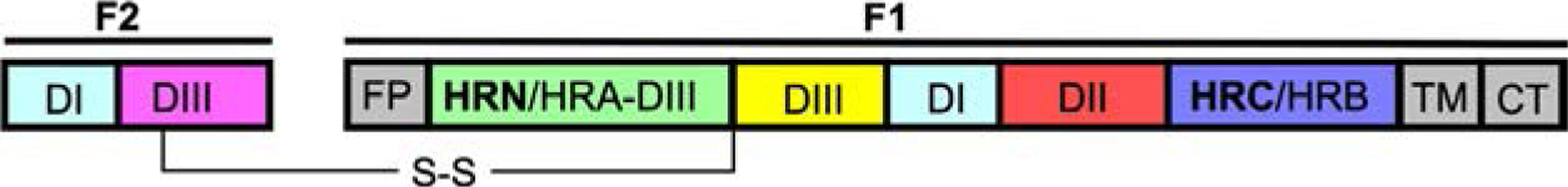
Linear map of the HPIV fusion protein with fusion peptide (FP), transmembrane domain (TM), N-terminal heptad repeat (HRN/HRA-DIII) and C-terminal heptad repeat (HRC/HRB) domains.
The C- and N-terminal HR domains of each F monomer are 7-mer repeats in which every seventh residue is either a leucine, isoleucine or valine, and the entire structure is an amphipathic α-helix. Along with the C-terminal HR domain (HRC/HRB) and N-terminal HR domain (HRN/HRA DIII), two other domains (DI, DII) comprise the large internal cavity of the pre-fusion F (Fig. 3). The HPIV F proteins are quite different in structure from other class I viral fusion proteins such as HIV Env and Influenza HA (Gamblin et al., 2004). They, however, share with other class I fusion proteins—such as SARS CoV-2 S (Wrapp et al., 2020), Ebola virus GP (Lee et al., 2008), and HIV Env (Lee et al., 2016)—the strategy for membrane fusion, which involves reorientation from a small pre-fusion state to an extended intermediate state after activation. In the pre-fusion state, the F is in a metastable pre-fusion conformation with the hydrophobic fusion peptide buried in the interior of the molecule. The intermediate state that is elicited after activation of F is thermodynamically unfavorable and collapses unto itself, forming a six-helix bundle composed of the interacting HRC and HRN domains, and bringing the viral and host membranes together (Marcink et al., 2020b).
The TM regions of F have been understudied but may play an important role in the conformational transitions of F. For PIV5, F’s TM adopts different conformations depending on membrane composition. In negative-curvature membranes, the transmembrane domain adopts a β-strand conformation at the termini, which could further increase the curvature of the membrane, enhancing fusion promotion (Lee et al., 2018; Yao et al., 2015). The central core of the transmembrane domain remains α-helical, stabilizing the trimer during this process.
4. HN interaction with F and HN’s role in the activation of F
HN and F are associated prior to receptor binding, and during this time HN stabilizes F, preventing its premature activation (Farzan et al., 2011a; Palermo et al., 2009; Porotto et al., 2012c). Bimolecular fluorescence complementation (BiFC) in living cells was used to visualize HN-F complexes at physiological temperatures as a representation of viral surfaces and to identify the location and timing of HN-F interaction during the fusion process (Porotto et al., 2012a) (see Fig. 4A). HPIV3 HN and F interact before receptor engagement and remain engaged during the fusion process, separating only after complete membrane merger (Porotto et al., 2012a). Dimer interface mutations that alter the putative site II impact this HN-F interaction on cell surfaces, modulating fusion and infection (Xu et al., 2013) (see Fig. 4B). These HN dimer interface mutations alter the surface area between the two HN monomers thereby altering the flexibility of the HN dimer, affecting both receptor avidity and activation of F. Mutations that lead to tight, stable dimers increase the interaction between HN and F and result in more active F-triggering. These findings highlight the correlation between the flexibility of the HN dimer interface and HN’s ability to activate F (Xu et al., 2013).
Fig. 4.
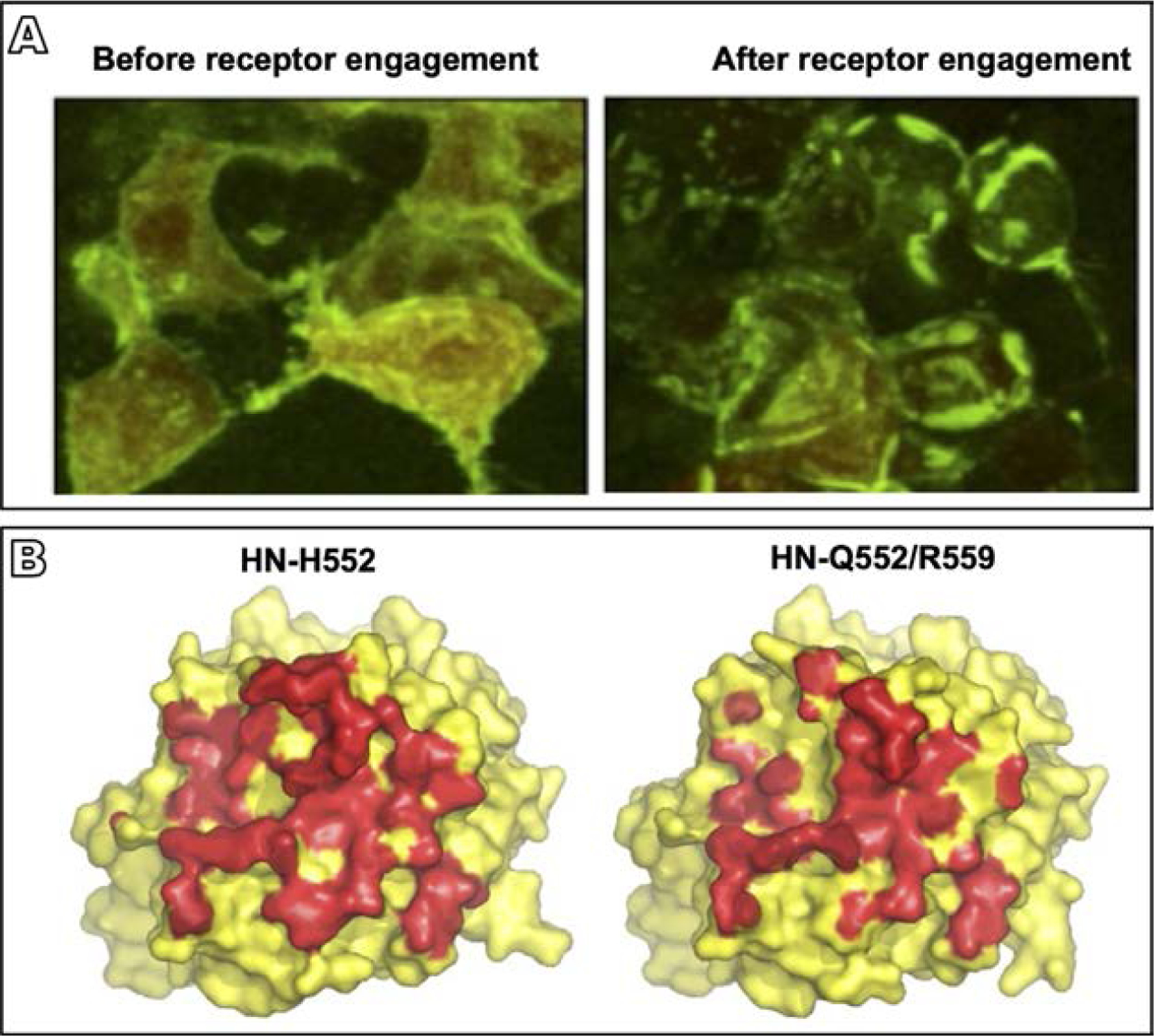
HPIV3 HN-F interaction and the role of the HN dimer interface in this process. (A) 293T cells co-transfected with constructs encoding HN and F, each with their corresponding bimolecular fluorescence complementation tag, were treated overnight with zanamivir to prevent HN-receptor binding (Porotto et al., 2012a). Evenly distributed fluorescence was visualized across the cell surface in the absence of receptor engagement (left panel), but after the zanamivir was removed and receptor engagement was permitted, HN-F interaction occurred in clusters at the sites of cell-cell contact (right panel) (B) Surface rendering of the crystal structure of the HN monomer globular head viewed from above for HN with the indicated mutated site II residues (Xu et al., 2013). HN residues in contact with the opposing monomer in the dimer interface are shown in red. Introduction of the double mutation Q552/R559 in site II results in the loss of about 800 of 3700Å2 buried interface area in the dimer (right hand side), revealing the effect of alterations in this site to the dimer structure. Adapted from Porotto, M., Palmer, S.G., Palermo, L.M., Moscona, A., 2012a. Mechanism of fusion triggering by human parainfluenza virus type III: communication between viral glycoproteins during entry. J. Biol. Chem. 287 (1), 778–93, with permission; Xu, R., Palmer, S.G., Porotto, M., Palermo, L.M., Niewiesk, S., Wilson, I.A., et al., 2013. Interaction between the hemagglutinin-neuraminidase and fusion glycoproteins of human parainfluenza virus type III regulates viral growth in vivo, MBio. 4 (5), e00803–13, with permission.
F triggering occurs at 37°C but not at 4°C (Porotto et al., 2011a,b), allowing the use of temperature to examine the receptor-engaged intermediate states of the HN-F complex prior to initiation of fusion. The receptor engagement induced HN to trigger F at 37°C, activating fusion, and the F-interacting peptide then binds to the exposed heptad repeat region of F, blocking F’s folding at an extended stage and trapping the intermediates (Porotto et al., 2010b). Blocking F’s refolding after activation by HN allows for imaging of HN and F interactions in the presence of different receptors and inhibitors that target distinct parts of the complex. This makes it possible to unravel the mechanism whereby HN interacts with F during the fusion process.
To examine the HN-F complex prior to receptor engagement on authentic viral surfaces instead of cell surfaces, first, negative stain imaging electron microscopy was used to show that HN and F were colocalized before receptor engagement, consistent with the BiFC data in cells and with biochemical evidence showing HN and F interacting before receptor engagement (Porotto et al., 2012a). Cryo-electron tomography showed that in regions of the virus whose surface contained two layers of density, HN was present in a canopy over the pre-fusion F in close association with the F (Gui et al., 2015). Recent cryo-electron tomography with subtomogram averaging of unperturbed virions extended the resolution of these observations to show that HN rests above F in what appears to be a possible mechanism for protecting it from activation (Marcink et al., 2020b). In virions that were isolated from infected cell supernatant fluid and attached directly to the cryo-EM grid without any disruptive purification steps, this HN canopy extends across the entirety of the virus’s surface, closely associated with pre-fusion F in the absence of target cell receptors (Marcink et al., 2020b).
Upon receptor engagement, HN turns to its activating role. In addition to the residues at the HN dimer interface, specific residues on the stalk domain have been implicated in triggering F (Bishop et al., 2008; Bose et al., 2011; Porotto et al., 2003; Wang and Iorio, 1999). In the cryo-EM tomography studies, after HN binds receptor, its height decreases by 25Å, from 163 to 138Å (Marcink et al., 2020b). This decrease in height could alter HN’s stalk’s interaction with F and thereby participate in the process of F activation. However, precisely how the stalk domain of paramyxovirus HN activates F remains to be characterized. We propose that the interaction of the globular head of HN with its receptor provides a critical signal, via the dimer interface residues, to the stalk. The stalk initiates F activation, possibly in a mechanism related to the observed length change.
5. The transient intermediate state of F
Before F is activated by HN, the F trimer is anchored to the viral membrane through the transmembrane domain. Adjacent to this transmembrane domain lies the HRC domain. The HRN domain and fusion peptide are packaged within the pre-fusion head region. Once F is activated, the HRN domain and fusion peptide unfold and extend into the host membrane in a “pre-hairpin” intermediate state. This transient intermediate state is extremely unstable and short-lived. This instability leads F to refold, bringing the HR regions at the C- and N-terminals together, forming a six-helix bundle that is associated with the subsequent membrane fusion.
The intermediate states of class I fusion proteins have rarely been structurally characterized. A set of important investigations have focused on characterizing the influenza HA and HIV gp41 intermediate states (Benhaim et al., 2020; Benton et al., 2020; Das et al., 2018; Frey et al., 2008; Gao et al., 2020; Herschhorn et al., 2016; Liu et al., 2017; Sadler et al., 2008; Xu and Wilson, 2011). Two studies have captured an intermediate state of influenza’s HA by cryo-EM; these experiments were carried out using soluble HA in the presence (Gao et al., 2020) or absence of (Benton et al., 2020) antibodies, so that the native intermediate state remains unknown (Gao et al., 2020). For PIV, negative stain electron microscopy was used to capture images of the PIV5 F protein intermediate state, triggered by heat and captured with a fusion-inhibitory peptide (Kim et al., 2011) (see Fig. 5A). Molecular dynamics analysis was used to model the intermediate state of F, confirming that the densities corresponding to the negative stain images were the transient intermediates of F.
Fig. 5.

The PIV F in its transient intermediate state during execution of fusion. (A) Examination of the pre-hairpin intermediate of PIV5 F by thin-sectioning and negative stain EM (Kim et al., 2011). Yellow arrows show intermediate F while the blue arrows show pre-fusion F distant from a region of interaction. (B) HPIV3 Contrast-inverted images where viral particles can be observed attached to target erythrocyte fragment membranes using cryo-ET (Marcink et al., 2020b). (C) Upper panel shows an enlarged region of interactions between the viral and target erythrocyte fragment membranes where elongated densities linking both membranes are visible. Insets include cyan lines where distance plot measurements were taken. Lower panel shows density line plots revealing a repeating 20–35Å-wide density. Scale bars: (B) 50nm. Panel (A): Adapted from Kim, Y.H., Donald, J.E., Grigoryan, G., Leser, G.P., Fadeev, A.Y., Lamb, R.A., et al., 2011. Capture and imaging of a prehairpin fusion intermediate of the paramyxovirus PIV5. Proc. Natl. Acad. Sci. U. S. A. 108 (52), 20992–7, with permission. Panels (B and C): From Marcink, T.C., Wang, T., des Georges, A., Porotto, M., Moscona A., 2020. Human parainfluenza virus fusion complex glycoproteins imaged in action on authentic viral surfaces. PLoS Pathog. 16 (9), e1008883, with permission.
For HPIV3, our laboratory studied virions in native conditions to characterize the intermediate state of F during interaction with a host target. Using red blood cell fragments that contain sialic acid receptor moieties as a host cell membrane surrogate, viruses were allowed to bind receptors on these targets and captured the extended conformation of F using fusion inhibitory peptides that interfere with HRC-HRN binding during six-helix bundle formation. Once the pre-fusion F was activated and the intermediate state locked, palisade-like patches of intermediate F densities were noted (see Fig. 5B). These densities were similar in diameter to the intermediate state of influenza HA (Benton et al., 2020; Gao et al., 2020; Marcink et al., 2020b). A small molecule that blocks HN-receptor-interaction (zanamivir, Greengard et al., 2000) was used to show that if HN-receptor engagement is disrupted after F was activated by HN, the target cell did not detach, confirming that F was inserted into the host membrane. These palisade-like patches of the intermediate state of F clustered near the site of viral–host interaction, suggesting coordinated activation of F at the site of fusion (see Fig. 5C).
6. Final stages of fusion and entry
After the insertion of the fusion peptide into the target membrane during formation of the transient intermediate, the subsequent refolding of F is driven by HRN-HRC interaction. However, the fusion peptide itself and its interaction with host membranes may impact this refolding process. Fusion peptide domains of the F protein are important for interaction with the host membrane. These domains resemble SNARE proteins creating coiled coils and zippering during fusion (Mostafavi et al., 2017), promoting refolding of the intermediate F to the post-fusion state. The fusion peptide domain of F for PIV5 was studied extensively by NMR and shown to adopt different orientations depending on the lipid composition of the membrane (Donald et al., 2011; Yao and Hong, 2013, 2014). Solid and solution-state NMR studies revealed that the fusion peptide adopts a β-sheet conformation in high curvature lipids and an α-helical conformation in the extended intermediate and post-fusion states. PIV5 and HIV seem to share a similar conformational dependence on the membrane-surface charge, further pointing to the similarities between class I fusion proteins after activation. This dynamic nature of the fusion peptide allows F to take cues from the surrounding environment to change from β-sheet to α-helical conformation at the right time.
Even after F activation, refolding and during the final fusion process itself, HN continues to regulate the fusion process. For HPIV3, the presence of HN is crucial until merger of the viral and cellular membranes. Using a mutated HN that lacks receptor-cleaving (neuraminidase) activity and thus constitutively engages its sialic acid receptor, receptor engagement was specifically disrupted at precise times during fusion of cells expressing the HN/F fusion complex, using the small molecule zanamivir described above. Only when HN continuously engages its receptor can F proceed through the fusion process. Even after insertion of the fusion peptide into the target membrane, F still requires the activating signal from HN to complete fusion (Porotto et al., 2011a). It will be important to determine how these findings about cell-cell fusion mediated by the fusion complex correlate to the individual virion entry process.
7. Fitness requirements of the HN/F fusion complex in human lung
Mechanisms that affect respiratory virus-cell interplay in the context of humans are finely tuned to the environment of the host (Maziarz et al., 2010; Shah et al., 2016). To understand natural viral infection and the mechanisms of HPIV entry that are authentic to human lungs, it is essential to analyze field strains in airway tissues. It has been shown for HPIV3, for example, that conclusions drawn from laboratory strains can be misleading with regard to the receptor interaction and viral fusion properties that govern entry and fitness in vivo (Maziarz et al., 2010; Shah et al., 2016). Analysis of clinical strains suggests that the HN-F fusion pairs of circulating HPIV-3 viruses maintain a balance of properties that result in an inverse correlation between fusion in cultured cells and growth in authentic tissues and in vivo; the fusion complex operates under specific constraints that govern viability in the different environments of cell cultures and human beings. For example, enhanced activation of F for fusion or entry, while effective for a virus in laboratory conditions, is a disadvantage in the natural host. An enhanced F-triggering produces inactive viral particles in the natural host, in which the F protein may have been triggered before contacting target cells (Palermo et al., 2009) and an overly active HN-F interaction may lead to premature, and therefore ineffective, activation of the fusion process (Farzan et al., 2011a). For these reasons, we emphasize efforts to study mechanisms of entry using viral complexes that are fit for infection of lung, where the HN-F fusion complex is less receptor-avid and F-activation is more tightly regulated (Iketani et al., 2018; Palmer et al., 2014).
8. Inhibitors of entry
There is no specific antiviral therapy available for the treatment of HPIV infections (Hodson et al., 2011; Krilov, 2011; Maziarz et al., 2010; Nichols et al., 2001; Stankova et al., 2007). The potential benefit of antiviral treatment, especially for very young infants and immunocompromised hosts, is clear (Moscona, 2005; Moss et al., 2011). Several features of the viral entry mechanism are attractive targets for inhibitors. Binding, fusion, and each step of cell entry are critical stages which could be targeted to prevent HPIV infection and disease (Chibanga et al., 2019; Falsey, 2012; Marcink et al., 2020a; Moscona, 2005; Moscona et al., 2010; Outlaw et al., 2019). One potential strategy for HPIV targets the removal of lung epithelial sialic acid receptor for HPIV, thereby preventing viral entry. A recombinant sialidase, first developed as an antiviral agent for influenza, cleaves sialic acids from the host cell surface, inactivating the host cell receptor recognized by HPIV (Chen et al., 2011; Guzman-Suarez et al., 2012; Triana-Baltzer et al., 2011; Waghmare et al., 2015). The molecule (DAS181; Fludase) is an engineered neuraminidase molecule (Moscona et al., 2010) and has shown promise in proof-of-concept treatment of immunocompromised humans (Drozd et al., 2013). DAS181 has entered clinical trials at this time for the treatment of HPIV in hematopoietic stem cell transplant patients, and additional studies in high-risk populations are underway. However, significant concerns about the potential for resistance to this compound arise from the fact that treatment of HPIV-infected cells with neuraminidase elicits variant viruses with HN mutations that confer resistance to receptor depletion. Such variants have been found in immunocompromised individuals with long-term HPIV3 infection (unpublished data). There are several new strategies under exploration that take advantage of new mechanistic understanding of the fusion complex to develop targeted antivirals.
8.1. Antivirals targeting HN’s role in receptor binding
Blocking receptor binding of HN is a potentially attractive antiviral strategy (Chibanga et al., 2019). Zanamivir has been shown to block receptor engagement (Greengard et al., 2000; Porotto et al., 2004), in a proof of concept for binding inhibitors. Early crystal structures showed zanamivir binding in the active site (Guillon et al., 2014; Lawrence et al., 2004) with no significant structural distortions outside the active site. Suramin, a trypanocidal drug, was shown in vitro to work in synergy with zanamivir (Bailly et al., 2016); STD-NMR showed that suramin and zanamivir simultaneously bind to HN but suramin binds more avidly and is more potent at inhibiting receptor binding in vitro (Bailly et al., 2016). The two activities at the primary binding site of HN, neuraminidase and receptor binding, can be separated by their pH optima; neuraminidase activity is high at low pH values, optimal at pH 5, and negligible at neutral pH, while receptor binding occurs at neutral pH (Mishin et al., 2010; Tappert et al., 2013). Antivirals may target one of the other function of HN, e.g., blocking receptor interaction without blocking neuraminidase activity. STD-NMR studies have shown that at low pH, Neu2en-based hPIV HN inhibitors target the binding function of HN and, to a lesser extent, the neuraminidase activity (Pascolutti et al., 2018). Binding inhibitors are of interest especially for their potential in combination with compounds that work by other mechanisms, where the ability of HN to evade receptor blockade (see above) would be mitigated.
8.2. Hijacking HN to trigger F prematurely
Activation of F by HN requires precise timing to effectively attach and fuse with a host cell. It is possible to subvert the normal process whereby HN activates F and cause HN to activate the F protein’s fusion mechanism before it reaches the target host cell, thus incapacitating F before it can mediate viral entry. After premature activation, F assumes the irreversible post-fusion state and is permanently inactivated (Bottom-Tanzer et al., 2019; Moscona, 2005) (see Fig. 6A). Several pre-activating compounds were identified that induce HN to inactivate F, without altering HN’s neuraminidase and receptor binding functions, and explored the mechanism by cryo-electron tomography (Bottom-Tanzer et al., 2019; Farzan et al., 2011b; Marcink et al., 2020a) (see Fig. 6B). In a virtual screen for molecules that bind to HN’s site II, docking studies revealed a set of candidates, with one of them (PAC-3066, Bottom-Tanzer et al., 2019) being highly effective at binding. The pre-activating compounds did not block HN’s receptor binding or neuraminidase activity but instead bound to HN’s site II that is involved in triggering F. Cryo-electron tomography showed a large structural rearrangement in F before and after the addition of the HN-binding pre-activating compound. 2D projections show F in a pre-fusion state with HN in a position superior to the head of F, but in the presence of the pre-activating compound, only HN and post-fusion F densities can be seen (Marcink et al., 2020a) (see Fig. 6C, D). Compounds that act by this strategy induce HN to destabilize the F protein to the point where it folds to its post-fusion state asynchronously with membrane merger. This mechanism of action will be unlikely to elicit resistance, since if resistance to this destabilizing strategy is elicited, the resulting virus will be difficult to activate by HN and likely unfit (Marcink et al., 2020a).
Fig. 6.
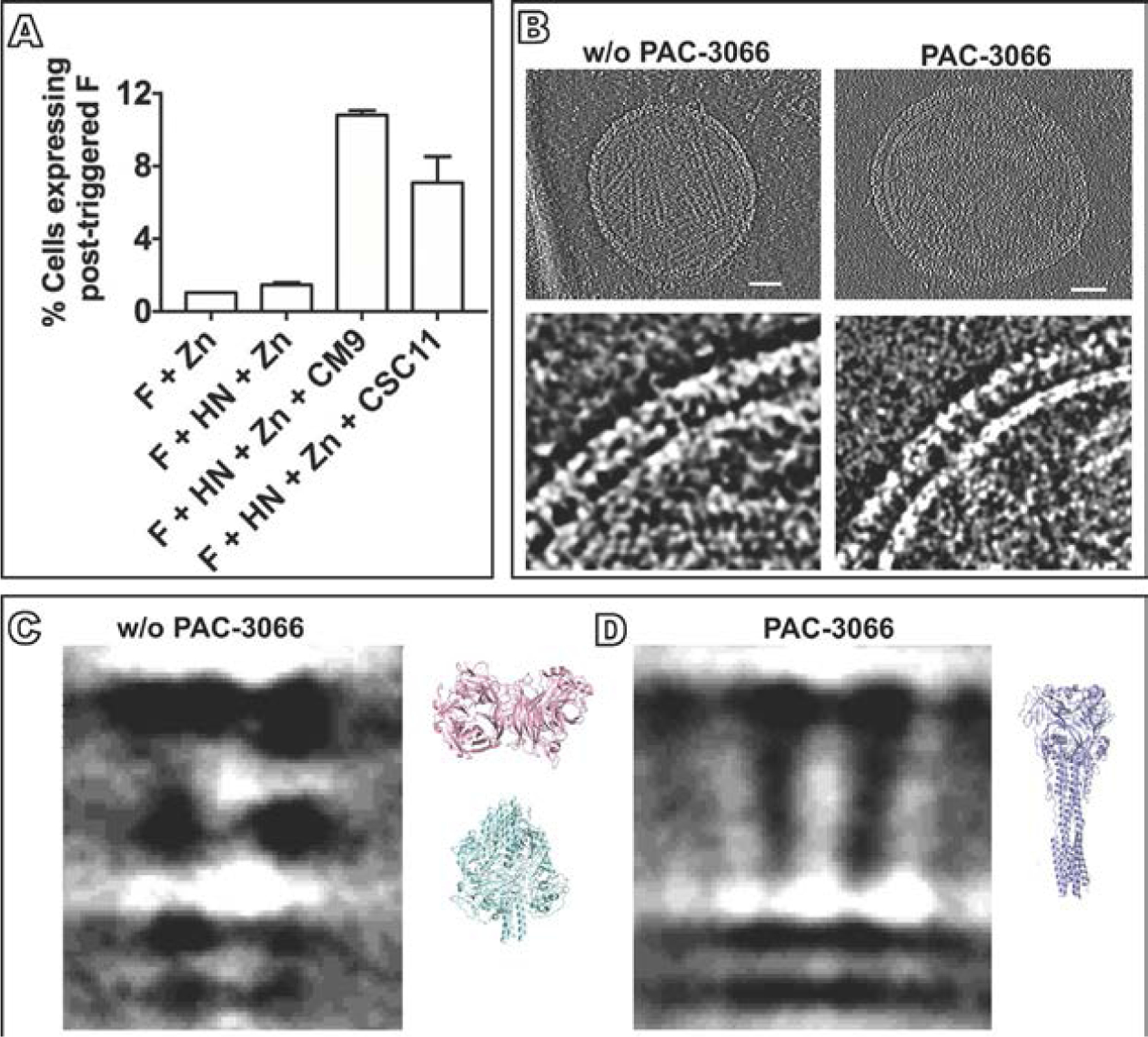
Structural analysis of the mechanism of action of a small molecule that stimulates HN to trigger F. (A) Cells cotransfected with HN and F or transfected with F only were incubated in the presence of zanamivir alone to block HN-receptor binding, or with zanamivir together with CM9 or CSC11, two molecules that interact with HN to induce it to trigger F (Bottom-Tanzer et al., 2019). The cells were stained with monoclonal antibodies specific for the post-activated state of F. The proportions of activated F are shown as percentages of cells expressing post-triggered F on the y axis (±SD). (B) Overview of HPIV3 without and with PAC-3066, a next-generation small molecule that stimulates HN to activate F. Bars, 50nm (Marcink et al., 2020a). Lower panels show the viral surface glycoproteins without and with PAC-3066. (C) Cryo-electron 2D-subtomogram averages from a subset of viral surface glycoproteins in the absence of PAC-3066, where pre-fusion F can be identified (left) and with PAC3066, showing post-fusion F (Marcink et al., 2020a). Structural comparison coordinates of HN (PDB accession number 4MZA) and pre-fusion F (PDB accession number 6MJZ) are shown in pink and cyan, respectively. (D) Subtomogram averages where post-fusion F can be identified after PAC-3066 incubation. Structural comparison coordinates of the post-fusion F (PDB accession number 1ZTM) are shown in light blue. Panel (A): Adapted from Bottom-Tanzer, S.F., Rybkina, K., Bell, J.N., Alabi, C.A., Mathieu, C., Lu. M., et al., 2019. Inhibiting human parainfluenza virus infection by preactivating the cell entry mechanism. MBio 10 (1), with permission. Panels (B and C): From Marcink, T.C., Yariv, E., Rybkina, K., Mas, V., Bovier, F.T., des Georges, A., et al., 2020. Hijacking the fusion complex of human parainfluenza virus as an antiviral strategy, mBio 11 (1), with permission.
8.3. Antibodies that target F’s conformation or HN-F interaction
Structures of neutralizing antibodies complexed with F have been solved for members of the paramyxovirus family ( Jones et al., 2019; McLellan et al., 2013; Stewart-Jones et al., 2018; Tian et al., 2017). For HPIV3, the cryo-EM structure of pre-fusion F was complexed with a prefusion-specific neutralizing antibody (PIA174) that binds the apex of the pre-fusion F trimer (Stewart-Jones et al., 2018) (see Fig. 7A). Pre-fusion F structures for PIV1–4 were solved via stabilizations achieved by cavity space-filling mutations along with strategically placed disulfide bonds to stabilize the pre-fusion F (see Fig. 7B). Using these stabilized F proteins, neutralizing antibodies were raised for all four PIV serotypes. A more recent study scanned for neutralizing antibodies in peripheral blood, tonsils, and spleens of healthy children and adults, selecting for neutralizing antibodies against HPIV3 (Boonyaratanakornkit et al., 2021). Most of these neutralizing antibodies were found bound to the apex of pre-fusion F in a 1:1 ratio. The most potent antibody (PI3-E12 Fab) was solved in complex with pre-fusion F by cryo-EM with a similar binding site at the apex of F to that previously identified (Stewart-Jones et al., 2018). For PIV5, synthetic antibodies have been designed to target both HN and F. A large number of unique synthetic antibodies to sites on F and HN were assessed for their neutralization properties, and those most neutralizing were imaged by cryo-EM. Three synthetic antibodies bound to the pre-fusion stalk of F while two bound to the sides of the pre-fusion F head. All but two of the top eight synthetic antibodies imaged by cryo-EM seem to block the stalk interaction of HN with F (Boonyaratanakornkit et al., 2021). These pre-fusion F targeted synthetic antibodies may block HN and F interaction or stabilize F in the pre-fusion form, and may be important tools for ongoing structural studies.
Fig. 7.
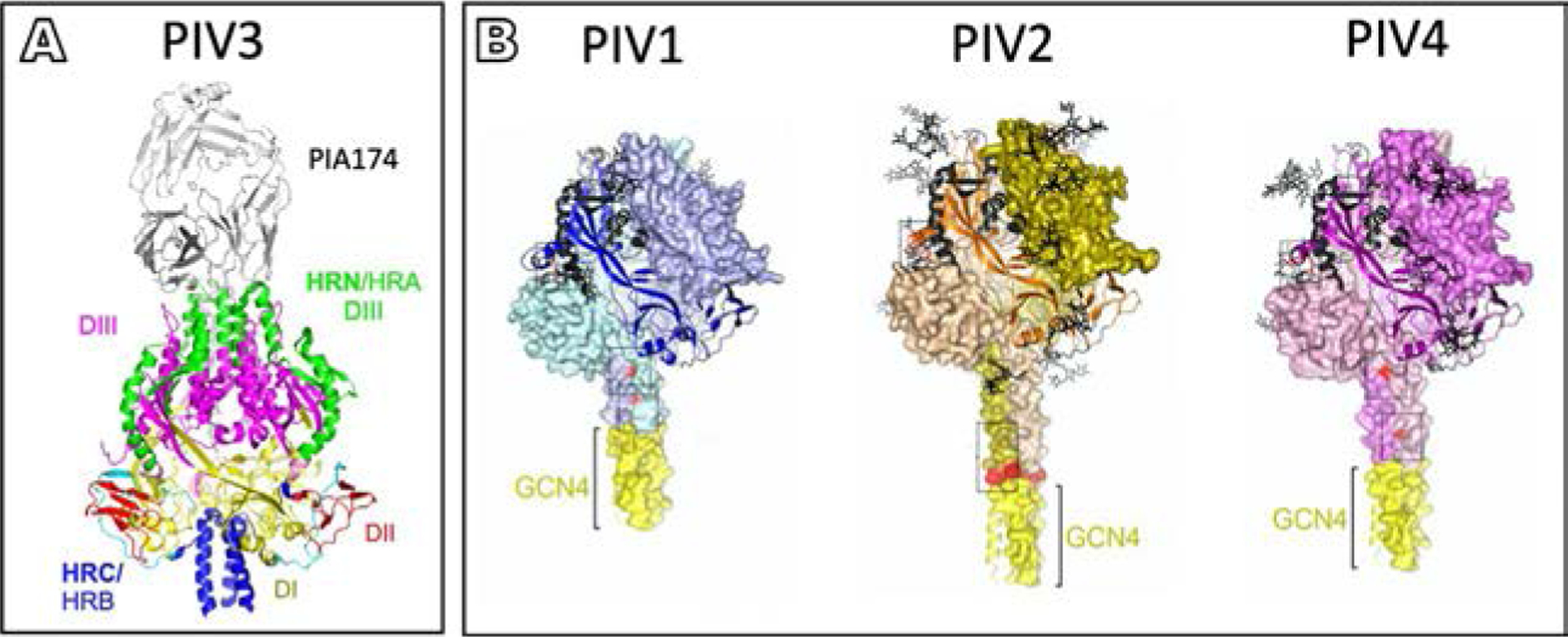
Neutralizing antibodies targeting pre-fusion F. (A) Structure-based design of prefusion-stabilizing mutations in PIV3 F with PIA174 antibody positioned at the apex of F (Stewart-Jones et al., 2018). HPIV3 F trimer domain organization showing DI (yellow), DII (red), HRN/HRA-DIII (green), F1-DIII (magenta), and HRC/HRB (blue). See Fig. 3 for description of domains. (B) Models of stabilized PIV1, PIV2, and PIV4, respectively, based on PIV5 pre-fusion F coordinates (PDB ID 4WSG) (Stewart-Jones et al., 2018). Boxes indicate regions of stabilizing mutations that were incorporated into F to generate pre-fusion F specific antibodies. Adapted from Stewart-Jones, G.B.E., Chuang, G.Y., Xu, K., Zhou, T., Acharya, P., Tsybovsky, Y., et al., 2018. Structure-based design of a quadrivalent fusion glycoprotein vaccine for human parainfluenza virus types 1–4. Proc. Natl. Acad. Sci. U. S. A. 115 (48), 12265–70, with permission.
8.4. Blocking fusion at F’s transitional intermediate state
Finally, the fusion protein is an exciting antiviral target, since it undergoes a series of structural transitions as it mediates viral entry. Each of these steps is a potential target for antiviral development. F must form a transient intermediate to draw the viral and cell membranes together, and the conserved coiled-coil motif required for this step can be specifically targeted by fusion-inhibitory peptides that bind to specific sequences on the F protein (Porotto et al., 2007b, 2009, 2010b). These fusion-inhibitory peptides act by binding to the exposed HRN domains of the F protein trimer in the extended pre-hairpin state, preventing F from advancing to the next step in fusion. Furthermore, attaching lipids enhances the effectiveness of these peptides (Porotto et al., 2010a). Through crystallographic structure analysis of the binding interactions, these peptides have been improved upon readily, allowing for faster and enhanced antiviral development (Outlaw et al., 2019, 2020a,b) (see Fig. 8A). Membrane targeting of fusion-inhibitory peptides by use of a lipid tag allows efficient inhibition of the transient intermediate of fusion and also increases serum half-life and tissue biodistribution of the peptides (Outlaw et al., 2021; Pessi et al., 2012; Porotto et al., 2010a,b). Recently, fusion inhibitory peptides have been developed which target both HPIV and RSV and were effective in blocking HPIV infection in vivo (Outlaw et al., 2019, 2020b, 2021). These peptide inhibitors mimic the C-terminal region of HPIV and bind to the intermediate state of F, blocking the refolding of F and thereby preventing viral fusion (see Fig. 8B). These studies offer molecular-level insight into the mechanism whereby which a single peptide can target multiple viral targets, suggesting that lipopeptide fusion inhibitory peptides are promising candidates for development of multitargeted entry inhibitors for enveloped viruses (Outlaw et al., 2019, 2020b, 2021).
Fig. 8.
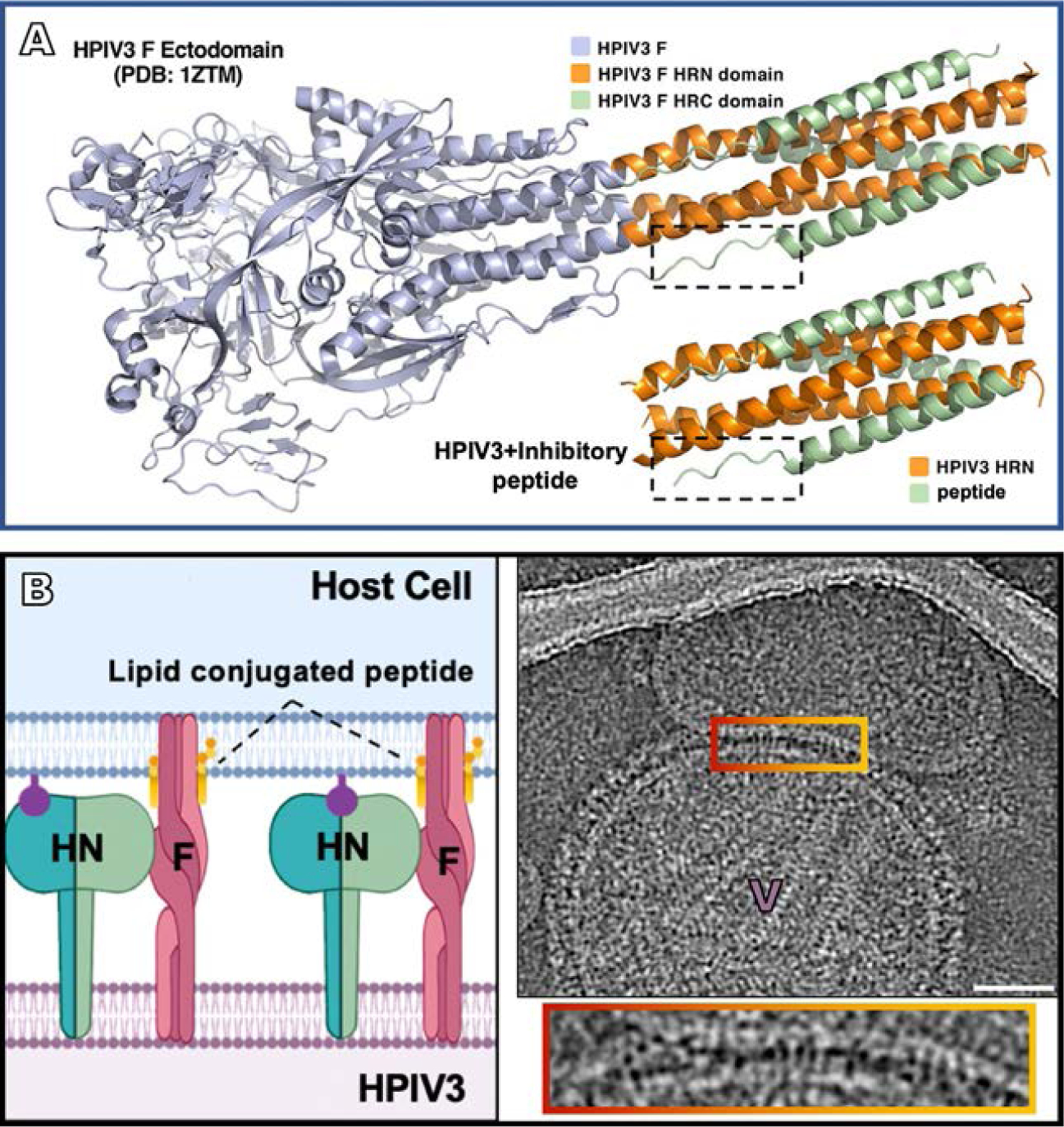
Blocking fusion at the intermediate state of F. (A) HPIV3 F ectodomain in post-fusion conformation (PDB 1ZTM), with the inhibitory peptide shown coassembled with HPIV3-HRN underneath (PDB 6NRO) (Outlaw et al., 2020a). This inhibitory peptide mimics the post-fusion state blocking F in the intermediate state. (B left panel) Schematic of lipid-conjugated inhibitory peptide inserting into the target cell membrane via their lipid tails and “locking” the extended F in its transient intermediate state, preventing refolding to the post-fusion conformation (Marcink et al., 2020b). (B right panel) Contrast-inverted images where viral particles can be observed attached to target erythrocyte fragment membranes. Enlarged region of interaction between the viral and target erythrocyte fragment membranes show elongated densities linking both membranes. Panel (A): Adapted from Outlaw, V.K., Kreitler, D.F., Stelitano, D., Porotto, M., Moscona, A., Gellman, S.H., 2020. Effects of single alpha-to-beta residue replacements on recognition of an extended segment in a viral fusion protein. ACS Infect. Dis., with permission. Panel (B): From Marcink, T.C., Wang, T., des Georges, A., Porotto, M., Moscona A., 2020. Human parainfluenza virus fusion complex glycoproteins imaged in action on authentic viral surfaces. PLoS Pathog. 16 (9), e1008883, with permission.
9. Future directions
Recent advances make it possible to image interactions between viral glycoproteins in situ as the interacting proteins progress through their roles in the cell, and examine the entry process in more mechanistic detail than ever before. For HPIV3, cryo-ET was applied to dissect the stepwise process by which HN triggers F-mediated fusion when forming a complex with receptors on a target cell membrane. The surrogate target cell membranes used for this strategy—human erythrocyte fragments—were chosen because they contain the sialic acid receptors used by HPIV3 as well as a reasonably authentic lipid composition similar to typical plasma membranes. However, now, by combining correlative light and electron microscopy (CLEM) with focused ion beam scanning electron microscopy (FIB-SEM) it is feasible to image the HN-F fusion complex interactions with receptors during entry in the context of cellular ultrastructure in authentic target cells.
In future experiments, determining high resolution structures of fusion/entry complexes on authentic viral surfaces will permit analysis of the HN stalk region, which has been shown to be integral for F activation. This view will be necessary to understanding the relationship between HN and F prior to receptor engagement and to answering outstanding questions about how the activation signal is transmitted from the head of HN, via the stalk, to F. The use of recombinant viruses bearing specific mutations in HN should permit a detailed assessment of the effect on the fusion complex of altering sites on HN that modulate fusion (Palermo et al., 2007; Porotto et al., 2003, 2005, 2006, 2007a; Xu et al., 2013). By sequentially imaging HN-F complexes with an altered primary sialic acid binding site on HN’s globular head, or secondary binding site at HN’s dimer interface that may transmit the activation signal, one can determine how these regions modulate the complex. Combining these measurements with molecular dynamics analyses should further explain how engagement of receptor by HN at the primary binding site transmits the activation signal through the stalk of HN to trigger F and the rest of entry.
Acknowledgments
Work from our laboratory cited in this review was partly supported by NIH/National Institute of Allergy and Infectious Diseases (NIAID) RO1AI031971 and RO1AI114736 to A.M. and by the Sharon Golub fund.
References
- Bagai S, Lamb R, 1995. Quantitative measurement of paramyxovirus fusion: differences in requirements of glycoproteins between SV5 and human parainfluenza virus 3 or Newcastle disease virus. J. Virol 69, 6712–6719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailly B, Dirr L, El-Deeb IM, Altmeyer R, Guillon P, von Itzstein M, 2016. A dual drug regimen synergistically blocks human parainfluenza virus infection. Sci. Rep 6, 24138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benhaim MA, Mangala Prasad V, Garcia NK, Guttman M, Lee KK, 2020. Structural monitoring of a transient intermediate in the hemagglutinin fusion machinery on influenza virions. Sci. Adv 6 (18), eaaz8822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton DJ, Gamblin SJ, Rosenthal PB, Skehel JJ, 2020. Structural transitions in influenza haemagglutinin at membrane fusion pH. Nature 583 (7814), 150–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop KA, Hickey AC, Khetawat D, Patch JR, Bossart KN, Zhu Z, et al. , 2008. Residues in the stalk domain of the hendra virus g glycoprotein modulate conformational changes associated with receptor binding. J. Virol 82 (22), 11398–11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonyaratanakornkit J, Singh S, Weidle C, Rodarte J, Bakthavatsalam R, Perkins J, et al. , 2021. Protective antibodies against human parainfluenza virus type 3 infection. MAbs 13 (1), 1912884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose S, Welch BD, Kors CA, Yuan P, Jardetzky TS, Lamb RA, 2011. Structure and mutagenesis of the parainfluenza virus 5 hemagglutinin-neuraminidase stalk domain reveals a four-helix bundle and the role of the stalk in fusion promotion. J. Virol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottom-Tanzer SF, Rybkina K, Bell JN, Alabi CA, Mathieu C, Lu M, et al. , 2019. Inhibiting human parainfluenza virus infection by preactivating the cell entry mechanism. MBio 10 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaiwatpongsakorn S, Epand RF, Collins PL, Epand RM, Peeples ME, 2011. Soluble respiratory syncytial virus fusion protein in the fully cleaved, pretriggered state is triggered by exposure to low-molarity buffer. J. Virol 85 (8), 3968–3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang A, Dutch RE, 2012. Paramyxovirus fusion and entry: multiple paths to a common end. Viruses 4 (4), 613–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang A, Masante C, Buchholz UJ, Dutch RE, 2012. Human metapneumovirus (HMPV) binding and infection are mediated by interactions between the HMPV fusion protein and heparan sulfate. J. Virol 86 (6), 3230–3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemaly RF, Hanmod SS, Rathod DB, Ghantoji SS, Jiang Y, Doshi A, et al. , 2012. The characteristics and outcomes of parainfluenza virus infections in 200 patients with leukemia or recipients of hematopoietic stem cell transplantation. Blood 119 (12), 2738–2745. [DOI] [PubMed] [Google Scholar]
- Chen YB, Driscoll JP, McAfee SL, Spitzer TR, Rosenberg ES, Sanders R, et al. , 2011. Treatment of parainfluenza 3 infection with DAS181 in a patient after allogeneic stem cell transplantation. Clin. Infect. Dis 53 (7), e77–e80. [DOI] [PubMed] [Google Scholar]
- Chibanga VP, Dirr L, Guillon P, El-Deeb IM, Bailly B, Thomson RJ, et al. , 2019. New antiviral approaches for human parainfluenza: inhibiting the haemagglutinin-neuraminidase. Antiviral Res. 167, 89–97. [DOI] [PubMed] [Google Scholar]
- Crennell S, Takimoto T, Portner A, Taylor G, 2000. Crystal structure of the multifunctional paramyxovirus hemagglutinin- neuraminidase. Nat. Struct. Biol 7 (11), 1068–1074. [DOI] [PubMed] [Google Scholar]
- Das DK, Govindan R, Nikic-Spiegel I, Krammer F, Lemke EA, Munro JB, 2018. Direct visualization of the conformational dynamics of single influenza hemagglutinin trimers. Cell 174 (4), 926–937.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng R, Wang Z, Mirza A, Iorio R, 1995. Localization of a domain on the paramyxovirus attachment protein required for the promotion of cellular fusion by its homologous fusion protein spike. Virology 209, 457–469. [DOI] [PubMed] [Google Scholar]
- Dirr L, El-Deeb IM, Guillon P, Carroux CJ, Chavas LM, von Itzstein M, 2015. The catalytic mechanism of human parainfluenza virus type 3 haemagglutinin-neuraminidase revealed. Angew. Chem. Int. Ed. Engl 54 (10), 2936–2940. [DOI] [PubMed] [Google Scholar]
- Donald JE, Zhang Y, Fiorin G, Carnevale V, Slochower DR, Gai F, et al. , 2011. Transmembrane orientation and possible role of the fusogenic peptide from parainfluenza virus 5 (PIV5) in promoting fusion. Proc. Natl. Acad. Sci. U. S. A 108 (10), 3958–3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drozd DR, Limaye AP, Moss RB, Sanders RL, Hansen C, Edelman JD, et al. , 2013. DAS181 treatment of severe parainfluenza type 3 pneumonia in a lung transplant recipient. Transpl. Infect. Dis 15 (1), E28–E32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Deeb IM, Guillon P, Dirr L, von Itzstein M, 2017. Exploring inhibitor structural features required to engage the 216-loop of human parainfluenza virus type-3 hemagglutinin-neuraminidase. MedChemComm 8 (1), 130–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund JA, Karron RA, Cunningham CK, Larussa P, Melvin A, Yogev R, et al. , 2013. Safety and infectivity of two doses of live-attenuated recombinant cold-passaged human parainfluenza type 3 virus vaccine rHPIV3cp45 in HPIV3-seronegative young children. Vaccine 31 (48), 5706–5712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falsey AR, 2012. Current management of parainfluenza pneumonitis in immunocompromised patients: a review. Infect Drug Resist 5, 121–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farzan S, Palermo LM, Yokoyama CC, Orefice G, Fornabaio M, Sarkar A, et al. , 2011a. Premature activation of the paramyxovirus fusion protein before target cell attachment: corruption of the viral fusion machinery. J. Biol. Chem [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farzan SF, Palermo LM, Yokoyama CC, Orefice G, Fornabaio M, Sarkar A, et al. , 2011b. Premature activation of the paramyxovirus fusion protein before target cell attachment with corruption of the viral fusion machinery. J. Biol. Chem 286 (44), 37945–37954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana L, Strasfeld L, 2019. Respiratory virus infections of the stem cell transplant recipient and the hematologic malignancy patient. Infect. Dis. Clin. North Am 33 (2), 523–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey G, Peng H, Rits-Volloch S, Morelli M, Cheng Y, Chen B, 2008. A fusion-intermediate state of HIV-1 gp41 targeted by broadly neutralizing antibodies. Proc. Natl. Acad. Sci. U. S. A 105 (10), 3739–3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima K, Takahashi T, Ueyama H, Takaguchi M, Ito S, Oishi K, et al. , 2015. Amino acid substitutions contributing to alpha2,6-sialic acid linkage binding specificity of human parainfluenza virus type 3 hemagglutinin-neuraminidase. FEBS Lett 589 (11), 1278–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamblin SJ, Haire LF, Russell RJ, Stevens DJ, Xiao B, Ha Y, et al. , 2004. The structure and receptor binding properties of the 1918 influenza hemagglutinin. Science 303 (5665), 1838–1842. [DOI] [PubMed] [Google Scholar]
- Gao J, Gui M, Xiang Y, 2020. Structural intermediates in the low pH-induced transition of influenza hemagglutinin. PLoS Pathog. 16 (11), e1009062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greengard O, Poltoratskaia N, Leikina E, Zimmerberg J, Moscona A, 2000. The anti-influenza virus agent 4-GU-DANA (zanamivir) inhibits cell fusion mediated by human parainfluenza virus and influenza virus HA. J. Virol 74 (23), 11108–11114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui L, Jurgens EM, Ebner JL, Porotto M, Moscona A, Lee KK, 2015. Electron tomography imaging of surface glycoproteins on human parainfluenza virus 3: association of receptor binding and fusion proteins before receptor engagement. MBio 6 (1), e02393–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillon P, Dirr L, El-Deeb IM, Winger M, Bailly B, Haselhorst T, et al. , 2014. Structure-guided discovery of potent and dual-acting human parainfluenza virus haemagglutinin-neuraminidase inhibitors. Nat. Commun 5, 5268. [DOI] [PubMed] [Google Scholar]
- Guzman-Suarez BB, Buckley MW, Gilmore ET, Vocca E, Moss R, Marty FM, et al. , 2012. Clinical potential of DAS181 for treatment of parainfluenza-3 infections in transplant recipients. Transpl. Infect. Dis 14 (4), 427–433. [DOI] [PubMed] [Google Scholar]
- Harrison SC, 2008. Viral membrane fusion. Nat. Struct. Mol. Biol 15 (7), 690–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrickson KJ, Hoover S, Kehl KS, Hua W, 2004. National disease burden of respiratory viruses detected in children by polymerase chain reaction. Pediatr. Infect. Dis. J 23 (Suppl. 1), S11–S18. [DOI] [PubMed] [Google Scholar]
- Herschhorn A, Ma X, Gu C, Ventura JD, Castillo-Menendez L, Melillo B, et al. , 2016. Release of gp120 restraints leads to an entry-competent intermediate state of the HIV-1 envelope glycoproteins. MBio 7 (5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodson A, Kasliwal M, Streetly M, MacMahon E, Raj K, 2011. A parainfluenza-3 outbreak in a SCT unit: sepsis with multi-organ failure and multiple co-pathogens are associated with increased mortality. Bone Marrow Transplant 46 (12), 1545–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath CM, Paterson RG, Shaughnessy MA, Wood R, Lamb RA, 1992. Biological activity of paramyxovirus fusion proteins: factors influencing formation of syncytia. J. Virol 66, 4564–4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Ray R, Compans RW, 1992. Functional interactions between the fusion protein and hemagglutinin-neuraminidase of human parainfluenza viruses. J. Virol 66, 1528–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huberman K, Peluso R, The MA, 1995. Hemagglutinin-neuraminidase of human parainfluenza virus type 3: role of the neuraminidase in the viral life cycle. Virology 214, 294–300. [DOI] [PubMed] [Google Scholar]
- Iketani S, Shean RC, Ferren M, Makhsous N, Aquino DB, des Georges A, et al. , 2018. Viral entry properties required for fitness in humans are lost through rapid genomic change during viral isolation. MBio 9 (4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones HG, Battles MB, Lin CC, Bianchi S, Corti D, McLellan JS, 2019. Alternative conformations of a major antigenic site on RSV F. PLoS Pathog. 15 (7), e1007944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YH, Donald JE, Grigoryan G, Leser GP, Fadeev AY, Lamb RA, et al. , 2011. Capture and imaging of a prehairpin fusion intermediate of the paramyxovirus PIV5. Proc. Natl. Acad. Sci. U. S. A 108 (52), 20992–20997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krilov LR, 2011. Respiratory syncytial virus disease: update on treatment and prevention. Expert Rev. Anti Infect. Ther 9 (1), 27–32. [DOI] [PubMed] [Google Scholar]
- Lamb R, 1993. Paramyxovirus fusion: a hypothesis for changes. Virology 197, 1–11. [DOI] [PubMed] [Google Scholar]
- Lawrence MC, Borg NA, Streltsov VA, Pilling PA, Epa VC, Varghese JN, et al. , 2004. Structure of the haemagglutinin-neuraminidase from human parainfluenza virus type III. J. Mol. Biol 335 (5), 1343–1357. [DOI] [PubMed] [Google Scholar]
- Lee JE, Fusco ML, Hessell AJ, Oswald WB, Burton DR, Saphire EO, 2008. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature 454 (7201), 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Ozorowski G, Ward AB, 2016. Cryo-EM structure of a native, fully glycosylated, cleaved HIV-1 envelope trimer. Science 351 (6277), 1043–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Yao H, Kwon B, Waring AJ, Ruchala P, Singh C, et al. , 2018. Conformation and trimer association of the transmembrane domain of the parainfluenza virus fusion protein in lipid bilayers from solid-state NMR: insights into the sequence determinants of trimer structure and fusion activity. J. Mol. Biol 430 (5), 695–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Pan J, Cai Y, Grigorieff N, Harrison SC, Chen B, 2017. Conformational states of a soluble, uncleaved HIV-1 envelope trimer. J. Virol 91 (10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo MS, Lee GM, Gunawardane N, Burchett SK, Lachenauer CS, Lehmann LE, 2012. The impact of RSV, adenovirus, influenza, and parainfluenza infection in pediatric patients receiving stem cell transplant, solid organ transplant, or cancer chemotherapy. Pediatr. Transplant [DOI] [PubMed] [Google Scholar]
- Lo MS, Lee GM, Gunawardane N, Burchett SK, Lachenauer CS, Lehmann LE, 2013. The impact of RSV, adenovirus, influenza, and parainfluenza infection in pediatric patients receiving stem cell transplant, solid organ transplant, or cancer chemotherapy. Pediatr. Transplant 17 (2), 133–143. [DOI] [PubMed] [Google Scholar]
- Marcink TC, Yariv E, Rybkina K, Mas V, Bovier FT, des Georges A, et al. , 2020a. Hijacking the fusion complex of human parainfluenza virus as an antiviral strategy. MBio 11 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcink TC, Wang T, des Georges A, Porotto M, Moscona A, 2020b. Human parainfluenza virus fusion complex glycoproteins imaged in action on authentic viral surfaces. PLoS Pathog. 16 (9), e1008883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maziarz RT, Sridharan P, Slater S, Meyers G, Post M, Erdman DD, et al. , 2010. Control of an outbreak of human parainfluenza virus 3 in hematopoietic stem cell transplant recipients. Biol. Blood Marrow Transplant 16 (2), 192–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLellan JS, Chen M, Leung S, Graepel KW, Du X, Yang Y, et al. , 2013. Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science 340 (6136), 1113–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melanson VR, Iorio RM, 2006. Addition of N-glycans in the stalk of the Newcastle disease virus HN protein blocks its interaction with the F protein and prevents fusion. J. Virol 80 (2), 623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EK, Lu X, Erdman DD, Poehling KA, Zhu Y, Griffin MR, et al. , 2007. Rhinovirus-associated hospitalizations in young children. J Infect Dis 195 (6), 773–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishin VP, Watanabe M, Taylor G, Devincenzo J, Bose M, Portner A, et al. , 2010. N-linked glycan at residue 523 of human parainfluenza virus type 3 hemagglutinin-neuraminidase masks a second receptor-binding site. J. Virol 84 (6), 3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscona A, 2005. Entry of parainfluenza virus into cells as a target for interrupting childhood respiratory disease. J. Clin. Invest 115 (7), 1688–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscona A, Peluso RW, 1991. Fusion properties of cells persistently infected with human parainfluenza virus type 3: participation of hemagglutinin-neuraminidase in membrane fusion. J. Virol 65, 2773–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscona A, Peluso RW, 1993. Relative affinity of the human parainfluenza virus type 3 hemagglutinin-neuraminidase for sialic acid correlates with virus-induced fusion activity. J. Virol 67 (11), 6463–6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscona A, Peluso RW, 1996. Analysis of human parainfluenza virus 3 receptor binding variants: evidence for the use of a specific sialic acid-containing receptor. Microb. Pathog 20 (3), 179–184. [DOI] [PubMed] [Google Scholar]
- Moscona A, Porotto M, Palmer S, Tai C, Aschenbrenner L, Triana-Baltzer G, et al. , 2010. A recombinant sialidase fusion protein effectively inhibits human parainfluenza viral infection in vitro and in vivo. J Infect Dis 202 (2), 234–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss RB, Steigbigel RT, Sanders RL, Fang F, 2011. Perspective: emerging challenges in the treatment of influenza and parainfluenza in transplant patients. Adv. Virol 2011, 910930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafavi H, Thiyagarajan S, Stratton BS, Karatekin E, Warner JM, Rothman JE, et al. , 2017. Entropic forces drive self-organization and membrane fusion by SNARE proteins. Proc. Natl. Acad. Sci. U. S. A 114 (21), 5455–5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair H, Nokes DJ, Gessner BD, Dherani M, Madhi SA, Singleton RJ, et al. , 2010. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet 375 (9725), 1545–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols WG, Corey L, Gooley T, Davis C, Boeckh M, 2001. Parainfluenza virus infections after hematopoietic stem cell transplantation: risk factors, response to antiviral therapy, and effect on transplant outcome. Blood 98 (3), 573–578. [DOI] [PubMed] [Google Scholar]
- Ortmann D, Ohuchi M, Angliker H, Shaw E, Garten W, Klenk HD, 1994. Proteolytic cleavage of wild type and mutants of the F protein of human parainfluenza virus type 3 by two subtilisin-like endoproteases, furin and Kex2. J. Virol 68 (4), 2772–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outlaw VK, Bottom-Tanzer S, Kreitler DF, Gellman SH, Porotto M, Moscona A, 2019. Dual inhibition of human parainfluenza type 3 and respiratory syncytial virus infectivity with a single agent. J. Am. Chem. Soc 141 (32), 12648–12656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outlaw VK, Kreitler DF, Stelitano D, Porotto M, Moscona A, Gellman SH, 2020a. Effects of single alpha-to-beta residue replacements on recognition of an extended segment in a viral fusion protein. ACS Infect. Dis [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outlaw VK, Lemke JT, Zhu Y, Gellman SH, Porotto M, Moscona A, 2020b. Structure-guided improvement of a dual HPIV3/RSV fusion inhibitor. J. Am. Chem. Soc 142 (5), 2140–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outlaw VK, Cheloha RW, Jurgens EM, Bovier FT, Zhu Y, Kreitler DF, et al. , 2021. Engineering protease-resistant peptides to inhibit human parainfluenza viral respiratory infection. J. Am. Chem. Soc 143 (15), 5958–5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palermo LM, Porotto M, Greengard O, Moscona A, 2007. Fusion promotion by a paramyxovirus hemagglutinin-neuraminidase protein: pH modulation of receptor avidity of binding sites I and II. J. Virol 81 (17), 9152–9161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palermo L, Porotto M, Yokoyama C, Palmer S, Mungall B, Greengard O, et al. , 2009. Human parainfluenza virus infection of the airway epithelium: the viral hemagglutinin-neuraminidase regulates fusion protein activation and modulates infectivity. J. Virol 83 (13), 6900–6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer SG, DeVito I, Jenkins SG, Niewiesk S, Porotto M, Moscona A, 2014. Circulating clinical strains of human parainfluenza virus reveal viral entry requirements for in vivo infection. J. Virol 88 (22), 13495–13502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascolutti M, Dirr L, Guillon P, Van Den Bergh A, Ve T, Thomson RJ, et al. , 2018. Structural insights into human parainfluenza virus 3 hemagglutinin-neuraminidase using unsaturated 3- N-substituted sialic acids as probes. ACS Chem. Biol 13 (6), 1544–1550. [DOI] [PubMed] [Google Scholar]
- Pessi A, Langella A, Capito E, Ghezzi S, Vicenzi E, Poli G, et al. , 2012. A general strategy to endow natural fusion-protein-derived peptides with potent antiviral activity. PLoS One 7 (5), e36833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plattet P, Plemper RK, 2013. Envelope protein dynamics in paramyxovirus entry. MBio 4 (4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porotto M, Greengard O, Poltoratskaia N, Horga M-A, Moscona A, 2001. Human parainfluenza virus type 3 HN-receptor interaction: the effect of 4-GU-DANA on a neuraminidase-deficient variant. J. Virol 76, 7481–7488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porotto M, Murrell M, Greengard O, Moscona A, 2003. Triggering of human parainfluenza virus 3 fusion protein(F) by the hemagglutinin-neuraminidase (HN): an HN mutation diminishing the rate of F activation and fusion. J. Virol 77 (6), 3647–3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porotto M, Murrell M, Greengard O, Lawrence MC, McKimm-Breschkin JL, Moscona A, 2004. Inhibition of parainfluenza virus type 3 and Newcastle disease virus hemagglutinin-neuraminidase receptor binding: effect of receptor avidity and steric hindrance at the inhibitor binding sites. J. Virol 78 (24), 13911–13919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porotto M, Murrell M, Greengard O, Doctor L, Moscona A, 2005. Influence of the human parainfluenza virus 3 attachment protein’s neuraminidase activity on its capacity to activate the fusion protein. J. Virol 79 (4), 2383–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porotto M, Fornabaio M, Greengard O, Murrell MT, Kellogg GE, Moscona A, 2006. Paramyxovirus receptor-binding molecules: engagement of one site on the hemagglutinin-neuraminidase protein modulates activity at the second site. J. Virol 80 (3), 1204–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porotto M, Fornabaio M, Kellogg GE, Moscona A, 2007a. A second receptor binding site on human parainfluenza virus type 3 hemagglutinin-neuraminidase contributes to activation of the fusion mechanism. J. Virol 81 (7), 3216–3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porotto M, Carta P, Deng Y, Kellogg GE, Whitt M, Lu M, et al. , 2007b. Molecular determinants of antiviral potency of paramyxovirus entry inhibitors. J. Virol 81 (19), 10567–10574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porotto M, Yokoyama C, Orefice G, Kim H-S, Moscona A, 2009. Kinetic dependence of paramyxovirus entry inhibition. J. Virol 83 (13), 6947–6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porotto M, Yokoyama C, Palermo LM, Mungall B, Aljofan M, Cortese R, et al. , 2010a. Viral entry inhibitors targeted to the membrane site of action. J. Virol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porotto M, Rockx B, Yokoyama CC, Talekar A, Devito I, Palermo LM, et al. , 2010b. Inhibition of Nipah virus infection in vivo: targeting an early stage of paramyxovirus fusion activation during viral entry. PLoS Pathog. 6 (10), e1001168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porotto M, Devito I, Palmer SG, Jurgens EM, Yee JL, Yokoyama CC, et al. , 2011a. Spring-loaded model revisited: paramyxovirus fusion requires engagement of a receptor binding protein beyond initial triggering of the fusion protein. J. Virol 85 (24), 12867–12880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porotto M, Yi F, Moscona A, LaVan DA, 2011b. Synthetic protocells interact with viral nanomachinery and inactivate pathogenic human virus. PLoS One 6 (3), e16874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porotto M, Palmer SG, Palermo LM, Moscona A, 2012a. Mechanism of fusion triggering by human parainfluenza virus type III: communication between viral glycoproteins during entry. J. Biol. Chem 287 (1), 778–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porotto M, Salah Z, Devito I, Talekar A, Palmer SG, Xu R, et al. , 2012b. The second receptor binding site of the globular head of the Newcastle disease virus (NDV) hemagglutinin-neuraminidase activates the stalk of multiple paramyxovirus receptor binding proteins to trigger fusion. J. Virol 86 (10), 5730–5741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porotto M, Salah ZW, Gui L, Devito I, Jurgens EM, Lu H, et al. , 2012c. Regulation of paramyxovirus fusion activation: the hemagglutinin-neuraminidase protein stabilizes the fusion protein in a pretriggered state. J. Virol 86 (23), 12838–12848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roghmann M, Ball K, Erdman D, Lovchik J, Anderson LJ, Edelman R, 2003. Active surveillance for respiratory virus infections in adults who have undergone bone marrow and peripheral blood stem cell transplantation. Bone Marrow Transplant. 32 (11), 1085–1088. [DOI] [PubMed] [Google Scholar]
- Sadler K, Zhang Y, Xu J, Yu Q, Tam JP, 2008. Quaternary protein mimetics of gp41 elicit neutralizing antibodies against HIV fusion-active intermediate state. Biopolymers 90 (3), 320–329. [DOI] [PubMed] [Google Scholar]
- San-Juan-Vergara H, Sampayo-Escobar V, Reyes N, Cha B, Pacheco-Lugo L, Wong T, et al. , 2011. Cholesterol-rich microdomains as docking platforms for respiratory syncytial virus in normal human bronchial epithelial cells. J. Virol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheid A, Choppin P, 1978. Two disulfide-linked polypeptide chains constitute active F protein of paramyxoviruses. Virology 80, 51–66. [DOI] [PubMed] [Google Scholar]
- Schildgen V, van den Hoogen B, Fouchier R, Tripp RA, Alvarez R, Manoha C, et al. , 2011. Human Metapneumovirus: lessons learned over the first decade. Clin. Microbiol. Rev 24 (4), 734–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt AC, Schaap-Nutt A, Bartlett EJ, Schomacker H, Boonyaratanakornkit J, Karron RA, et al. , 2011. Progress in the development of human parainfluenza virus vaccines. Expert Rev. Respir. Med 5 (4), 515–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schowalter RM, Chang A, Robach JG, Buchholz UJ, Dutch RE, 2009. Low-pH triggering of human metapneumovirus fusion: essential residues and importance in entry. J. Virol 83 (3), 1511–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo S, Xie H, Campbell AP, Kuypers JM, Leisenring WM, Englund JA, et al. , 2014. Parainfluenza virus lower respiratory tract disease after hematopoietic cell transplant: viral detection in the lung predicts outcome. Clin. Infect. Dis 58 (10), 1357–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergel T, McGinnes LW, Peeples ME, Morrison TG, 1993. The attachment function of the Newcastle disease virus hemagglutinin-neuraminidase protein can be separated from fusion promotion by mutation. Virology 193, 717–726. [DOI] [PubMed] [Google Scholar]
- Shah DP, Shah PK, Azzi JM, Chemaly RF, 2016. Parainfluenza virus infections in hematopoietic cell transplant recipients and hematologic malignancy patients: a systematic review. Cancer Lett. 370 (2), 358–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankova J, Carret AS, Moore D, McCusker C, Mitchell D, Davis M, et al. , 2007. Long-term therapy with aerosolized ribavirin for parainfluenza 3 virus respiratory tract infection in an infant with severe combined immunodeficiency. Pediatr. Transplant 11 (2), 209–213. [DOI] [PubMed] [Google Scholar]
- Stewart-Jones GBE, Chuang GY, Xu K, Zhou T, Acharya P, Tsybovsky Y, et al. , 2018. Structure-based design of a quadrivalent fusion glycoprotein vaccine for human parainfluenza virus types 1–4. Proc. Natl. Acad. Sci. U. S. A 115 (48), 12265–12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone-Hulslander J, Morrison TG, 1999. Mutational analysis of heptad repeats in the membrane-proximal region of Newcastle disease virus HN protein. J. Virol 73 (5), 3630–3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streltsov VA, Pilling P, Barrett S, McKimm-Breschkin JL, 2015. Catalytic mechanism and novel receptor binding sites of human parainfluenza virus type 3 hemagglutinin-neuraminidase (hPIV3 HN). Antiviral Res. 123, 216–223. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Portner A, Scroggs RA, Uchikawa M, Koyama N, Matsuo K, et al. , 2001. Receptor specificities of human respiroviruses. J. Virol 75 (10), 4604–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabayashi K, Compans R, 1996. Functional interactions of paramyxovirus glycoproteins: identification of a domain in Sendai virus HN which promotes cell fusion. J. Virol 70, 6112–6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tappert MM, Porterfield JZ, Mehta-D’Souza P, Gulati S, Air GM, 2013. Quantitative comparison of human parainfluenza virus hemagglutinin-neuraminidase receptor binding and receptor cleavage. J. Virol 87 (16), 8962–8970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian D, Battles MB, Moin SM, Chen M, Modjarrad K, Kumar A, et al. , 2017. Structural basis of respiratory syncytial virus subtype-dependent neutralization by an antibody targeting the fusion glycoprotein. Nat. Commun 8 (1), 1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong J, Fu Y, Meng F, Kruger N, Valentin-Weigand P, Herrler G, 2018. The sialic acid binding activity of human parainfluenza virus 3 and mumps virus glycoproteins enhances the adherence of group B streptococci to HEp-2 cells. Front. Cell. Infect. Microbiol 8, 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triana-Baltzer GB, Sanders RL, Hedlund M, Jensen KA, Aschenbrenner LM, Larson JL, et al. , 2011. Phenotypic and genotypic characterization of influenza virus mutants selected with the sialidase fusion protein DAS181. J. Antimicrob. Chemother 66 (1), 15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Asten L, van den Wijngaard C, van Pelt W, van de Kassteele J, Meijer A, van der Hoek W, et al. , 2012. Mortality attributable to 9 common infections: significant effect of influenza a, respiratory syncytial virus, influenza B, norovirus, and parainfluenza in elderly persons. J Infect Dis 206 (5), 628–639. [DOI] [PubMed] [Google Scholar]
- Waghmare A, Wagner T, Andrews R, Smith S, Kuypers J, Boeckh M, et al. , 2015. Successful treatment of parainfluenza virus respiratory tract infection with DAS181 in 4 immunocompromised children. J. Pediatric Infect. Dis. Soc 4 (2), 114–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Iorio RM, 1999. Amino acid substitutions in a conserved region in the stalk of the Newcastle disease virus HN glycoprotein spike impair its neuraminidase activity in the globular domain. J. Gen. Virol 80 (Pt. 3), 749–753. [DOI] [PubMed] [Google Scholar]
- Weinberg GA, Hall CB, Iwane MK, Poehling KA, Edwards KM, Griffin MR, et al. , 2009. Parainfluenza virus infection of young children: estimates of the population-based burden of hospitalization. J. Pediatr 154 (5), 694–699. [DOI] [PubMed] [Google Scholar]
- White JM, Delos SE, Brecher M, Schornberg K, 2008. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol 43 (3), 189–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JV, Harris PA, Tollefson SJ, Halburnt-Rush LL, Pingsterhaus JM, Edwards KM, et al. , 2004. Human metapneumovirus and lower respiratory tract disease in otherwise healthy infants and children. N. Engl. J. Med 350 (5), 443–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winger M, von Itzstein M, 2012. Exposing the flexibility of human parainfluenza virus hemagglutinin-neuraminidase. J. Am. Chem. Soc 134 (44), 18447–18452. [DOI] [PubMed] [Google Scholar]
- Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, et al. , 2020. Cryo-EM Structure of the 2019-nCoV Spike in the prefusion Conformation. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Wilson IA, 2011. Structural characterization of an early fusion intermediate of influenza virus hemagglutinin. J. Virol 85 (10), 5172–5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Palmer SG, Porotto M, Palermo LM, Niewiesk S, Wilson IA, et al. , 2013. Interaction between the hemagglutinin-neuraminidase and fusion glycoproteins of human parainfluenza virus type III regulates viral growth in vivo. MBio 4 (5), e00803–e00813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H, Hong M, 2013. Membrane-dependent conformation, dynamics, and lipid interactions of the fusion peptide of the paramyxovirus PIV5 from solid-state NMR. J. Mol. Biol 425 (3), 563–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H, Hong M, 2014. Conformation and lipid interaction of the fusion peptide of the paramyxovirus PIV5 in anionic and negative-curvature membranes from solid-state NMR. J. Am. Chem. Soc 136 (6), 2611–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H, Lee MW, Waring AJ, Wong GC, Hong M, 2015. Viral fusion protein transmembrane domain adopts beta-strand structure to facilitate membrane topological changes for virus-cell fusion. Proc. Natl. Acad. Sci. U. S. A 112 (35), 10926–10931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HS, Wen X, Paterson RG, Lamb RA, Jardetzky TS, 2006. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 439 (7072), 38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan P, Thompson TB, Wurzburg BA, Paterson RG, Lamb RA, Jardetzky TS, 2005. Structural studies of the parainfluenza virus 5 hemagglutinin-neuraminidase tetramer in complex with its receptor, sialyllactose. Structure 13 (5), 803–815. [DOI] [PubMed] [Google Scholar]
- Yuan P, Swanson KA, Leser GP, Paterson RG, Lamb RA, Jardetzky TS, 2011. Structure of the Newcastle disease virus hemagglutinin-neuraminidase (HN) ectodomain reveals a four-helix bundle stalk. Proc. Natl. Acad. Sci. U. S. A 108 (36), 14920–14925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuasa T, Kawano M, Tabata N, Nishio M, Kusagawa S, Komada H, et al. , 1995. A cell fusion-inhibiting monoclonal antibody binds to the presumed stalk domain of the human parainfluenza type 2 virus hemagglutinin-neuraminidase protein. Virology 206 (2), 1117–1125. [DOI] [PubMed] [Google Scholar]
- Zaitsev V, von Itzstein M, Groves D, Kiefel M, Takimoto T, Portner A, et al. , 2004. Second sialic acid binding site in Newcastle disease virus hemagglutinin-neuraminidase: implications for fusion. J. Virol 78 (7), 3733–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Bukreyev A, Thompson CI, Watson B, Peeples ME, Collins PL, et al. , 2005. Infection of ciliated cells by human parainfluenza virus type 3 in an in vitro model of human airway epithelium. J. Virol 79 (2), 1113–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]