

Abstract
Background:
Inflammatory bowel disease (IBD) is a chronic intestinal inflammatory condition attributed to a complex interaction between imbalances in the gut microbiome, environmental conditions, and a deregulated immune response. The aim of the study was to investigate the composition of the gut microbiome of Saudi patients with IBD.
Methods:
After obtaining an informed consent, fecal samples were collected from 11 participants with IBD (patients) and 10 healthy individuals (controls). The bacterial components of the microbial population were identified by next-generation sequencing of partial 16S rRNA. Statistically significant dissimilarities were observed between samples for all metrics.
Results:
The key finding was three negative bacterial biomarkers, Paraprevotellaceae, the Muribaculaceae families of Bacteroidetes phylum, and the Leuconostocaceae family of Firmicutes phylum, which had a higher relative abundance in healthy individuals compared to IBD patients. It was also found that primary microbiota signatures at certain genera and species levels, including Prevotella copri, Bifidobacterium adolescentis, Ruminococcus callidus, Coprococcus sp., Ruminococcus gnavus, Dorea formicigenerans, Leuconostoc, Dialister, Catenibacterium, Eubacterium biforme, and Lactobacillus mucosae, were absent in almost all IBD patients, while Veillonella dispar was absent in all healthy individuals.
Conclusions:
The results of this study provide an overview of the variations in microbiota diversity present in Saudi IBD patients compared to healthy controls.
Keywords: Inflammatory bowel disease, microbiota, Saudi Arabia
INTRODUCTION
Inflammatory bowel disease (IBD) is a set of inflammatory conditions involving the gastrointestinal tract. The most common forms are ulcerative colitis (UC) and Crohn’s disease,[1] which are attributed to a dysregulated immune response owing to an imbalance in the gut microbiome triggered by environmental conditions.[2] The symptoms of IBD include rectal bleeding, diarrhea, weight loss, abdominal pain, and extra-intestinal manifestations. UC mainly involves the colonic mucosa, while CD can affect various parts of the digestive tract, from mouth to the anus. Particular types of environmental exposure and a genetic predisposition to IBD may play a role in the impairment of intestinal immunity, which is considered a hallmark of the disease.[3,4,5,6] Such conditions can result in a fall in the quantity of microorganisms in the intestines and a decrease in microbiota diversity, which can subsequently lead to gut inflammation. Several studies have suggested the presence of a link between defective intestinal homeostasis and low biodiversity of the gut of patients with IBD.[6,7,8] In both CD and UC patients, there is often an overall decreased biodiversity resulting in a lower proportion of Firmicutes and an increased proportion of Gammaproteobacteria.[9,10] Altered proportions of gut microbiota are often observed in patients with CD: The Roseburia and Faecalibacterium genera of the Lachnospiraceae and Ruminococcaceae families usually decrease in quantity, and Ruminococcus gnavus generally increases.[10,11] Broadly, then, the microbiome plays a central role in the homeostasis and immunity of the gut, making it a potential focus of novel therapies and remedies toward the treatment of IBD.[1,7]
Studies that have examined the composition of the gut microbiome in Saudi Arabian patients with IBD are scant; thus, this study focuses on the composition of gut microbiota in IBD patients in this region.
MATERIALS AND METHODS
Ethical considerations
This study was approved by the Research Committee at the Unit of Biomedical Ethics complying with international ethical guidelines as set out in the research regulations at the university (reference No. 373-19). An informed consent was obtained from all participants, and the study was carried out according to the Declaration of Helsinki.
Study participants and sample collection
The study participants were recruited from the outpatient IBD clinic at a tertiary care university hospital. Ten healthy individuals were also recruited as controls. Participants were included if they were of Saudi Arabian nationality and agreed to be involved in the study, regardless of age and sex. The mean disease duration was 9.09 ± 7.04 years, (range 1–20 years), and disease extent of CD patients was the ileal region in 33%, colonic in 17%, and ileocolonic in 50%, while the distribution of disease in UC was the rectum in 20%, left-sided colon (up to the splenic flexure) in 40% and pancolitis (extending beyond the hepatic flexure) in 40%. Patients and controls were excluded if they were pregnant, currently breastfeeding, had used biological medication or antibiotic treatments within two months prior to study commencement [Table 1]. The study participants were provided with detailed information regarding the study, including materials for fecal sample collection and a questionnaire for clinical assessment of disease severity. Patients were included upon receipt of a signed consent form. Fecal samples were collected from the participants using iSWAB microbiome kit (Mawi DNA Technologies, Cat.no. ISM-T-1200-R). Upon collection, the fecal samples were stored at 37°C.
Table 1.
Baseline characteristics of the study participants
| Clinical characteristic | IBD patients | Controls |
|---|---|---|
| Age (years) | 28.91±6.4 | 32.4±5.9 |
| Gender | ||
| Female, (%) | 8 (73%) | 5 (50%) |
| Male (%) | 3 (27%) | 5 (50%) |
| Disease duration (years) | 9.1±7.0 | |
| Disease activity in UC patients, n (%) | ||
| Remission | 0 (0%) | |
| Mild | 1 (20%) | |
| Moderate | 0 (0%) | |
| Severe | 4 (80%) | |
| Disease activity in CD patients, n (%) | ||
| Remission | 3 (50%) | |
| Mild | 1 (17%) | |
| Moderate | 0 (0%) | |
| Severe | 2 (33%) | |
| UC Locations, n (%) | ||
| E1: Proctitis | 1 (20%) | |
| E2: left-sided | 2 (40%) | |
| E3: Pancolitis | 2 (40%) | |
| CD Locations, n (%) | ||
| L1: Ileal | 2 (33%) | |
| L2: Colonic | 1 (17%) | |
| L3: Ileocolonic | 3 (50%) | |
| Laboratory finding (mean±SD) | ||
| Hemoglobin (%) | 11.9±1.9 | |
| C-reactive protein (mg/L) | 8.1±9.5 | |
| Albumin (g/dL) | 37.9±6.8 | |
| Treatments, n (%) | ||
| Corticosteroids | 5 (45%) | |
| Prednisone | 4 (80%) | |
| Budesonide | 1 (20%) | |
| 5-aminosalicylic acid (5-ASA) derivatives | 3 (27%) | |
| Immunosuppressants | 4 (36%) | |
| Azathioprine | 4 (100) |
Sampling plan
DNA purification from the fecal samples and ZymoBIOMICS™ Microbial Community Standard DNA was extracted from the human stool samples of 10 healthy controls, 11 IBD patients, and ZymoBIOMICS standard (ZYMO Research, USA) using the Pure link™ microbiome DNA purification kit (Cat.no. A29789; Invitrogen, USA). Sample purity testing was carried out using NanoDrop 7000 Fluorometer (Thermo Fisher Scientific, USA), and sample integrity testing was completed using 1% agarose gel electrophoresis. The result showed an A260:A280 ratio of 1.8:2.1.
DNA isolation and quality assurance
DNA was extracted from stool samples and mock (which helps prevent misreading the analytic result) using PureLink™ microbiome DNA purification kit. The DNA was isolated from 100 μl of stool samples and eluted in 100 μl volume of elution buffer. Results between 0.7 and 18 ng/μl were used to prepare library sequencing.
Bacterial 16S rRNA gene sequencing
A DNA sample was subjected to polymerase chain reaction (PCR) amplification of the V3–V4 regions of bacterial 16S rRNA, in a total volume of 30 μL, performed using the universal primers 341F 5′-ACTCCTACGGGAGGCAGCAG-3′ and 806R 5′-GGACTACHVGGGTWTCTAAT-3′. The PCR program was carried out as follows: initial denaturation at 98°C for 3 min, followed by 30 cycles of 98°C for 45 s, annealing at 55°C for 45 s, extension at 72°C for 45 s, and a final extension at 72°C for 7 min. The PCR products were run on agarose gel (1%) and then gel-purified using a DNA Gel Extraction Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. Next-generation sequencing library construction and high-throughput sequencing using illumine sequencing (MiSeq) were generally used in the microbiome analysis, and these steps were carried out at the Beijing Genomic Institute (BGI) in China to recover ~300 bp (base pair), pair-end reads of the V3 and V4 regions. The ends of each were overlapped to generate high-quality, full-length reads.
Data analysis
A sample size calculation was carried out to determine the probability that the samples were typical.[11] The raw data was analyzed using the Quantitative Insights Into Microbial Ecology 2 (QIIME, v1.80) bioinformatics tool. Trimmed V3–V4 16S rRNA reads, using Trimmomatic software (Version 0.33) were merged into single sequences using the Fast Length Adjustment of SHort reads (FLASH, Version 1.2.11) software. Merged sequences were filtered to detach low-quality sequences. The latter were reads shorter than 110 nucleotides, truncation of a sequence at any site with a mean quality score of <20 over a 50 bp sliding window, or truncated reads lower than 50 bp. The unique sequence group was linked to tags and classified (using operational taxonomic units (OTUs)) with a 97% identity cut-off using the de novo OTU selection. We kept only OTUs with at least 0.01% mean relative abundance as predominant. The relative abundance values ranked OTUs graphically, on x- and y-axes; then the rank curve was rendered using R software (v3.1.1). Taxonomies were assigned by the RDP classifier (Version 2.2)[12] and different databases (GreenGenes, the Ribosomal Database Project, SILVA, the genomic-based 16S rRNA database, and The All-Species Living Tree) with a confidence threshold of 0.7. Chimeric sequences were deleted using Usearch (Version 8.0). Alpha diversity analyses, including observed species, were calculated by Mothur (v1.31.2), using Shannon and Simpson effective. The drawing curve following role-of-three was based on estimating OTU numbers of the extracted tags (in multiples of 500) and detecting the maximum depth (number of reads) permitted to retain all samples in the dataset. Sequences were extracted randomly according to minimum sequence number for all samples, and the extracted sequences formed a new file (OTU table biom). The results were used to calculate beta diversity for measurement of the evolutional development between species: The bigger the index, the greater the evolutionary difference between samples. Beta diversity heat maps were determined using the tool gplots in the R program (v3.1.1) and drawn by the function aheatmap using non-negative matrix factorization algorithms in R. The distance algorithm was Euclidean, and the clustering method was found to be complete. At the phylum level, all species were used to draw the heat map, and taxa of an abundance of less than 0.5% in all samples were classified as “others.” The values were all log-transformed to minimize the dissimilarity degree between the relative abundance values. The representative sequences were aligned against the SILVA databases (Silva_108_core_aligned_seqs) using PyNAST with the “align_seqs.py” script. A representative OTU phylogenetic tree was constructed using the QIIME (v1.80) built-in scripts, including the FastTree method for phylogenetic tree construction. The tags with the highest abundance of each genus were chosen as the corresponding representative sequences. The genus-level phylogenetic tree was obtained in the same way as the OTU phylogenetic tree. Following this, the phylogeny tree was constructed using the R program (v3.1.1).
Statistical analysis
The results were analyzed using a permutation multivariate analysis of variance (PERMANOVA) to test for significance between values. All statistical tests were two-sided and followed by a Benjamini–Hochberg correction procedure. An adjusted P value of <0.05 was set as statistically significant. The Benjamini–Hochberg false discovery rate (FDR) correction was used to correct for multiple hypothesis testing where applicable. Using the Bray–Curtis dissimilarity statistic, weighted and unweighted UniFrac distance metrics were used to identify beta diversity within and between groups,[13] and plotted with principal coordinate analysis (PCoA), which was performed using the “ade4” package and the R program (v3.1.1). The boxplot and rarefaction curve were generated using the R software (v3.1.1). Taxa were filtered using the linear discriminant analysis method. Effect size were used to determine the characteristic most likely to appear different among groups, by coupling standard tests for statistical significance with extra tests encoding biological consistency and effect relevance, which was carried out using the Linear discriminant analysis Effect Size[13] software.[14]
Data availability
The data have been submitted to National Center for Biotechnology Information (NCBI) under BioProject no. PRJNA673073 (https://dataview.ncbi.nlm.nih.gov/object/PRJNA673073) with BioSample identifiers SAMN16591214-SAMN16591234 with accession number SRA16000303-SRA16000323.
RESULTS
Characteristics of the subjects
The study included 11 Saudi IBD patients (six CD and five UC patients) aged 20–41 years, (median age 28.5 years); and 10 healthy controls, aged 20–45 years (median age 34 years). Eight of the patients and five of the controls were females. None of patients or controls were smokers. The mean hemoglobin value for the IBD patients was 11.9 ± 1.9 g/dL, C-reactive protein 8.1 ± 9.5 mg/dL, and the albumin level 37.9 ± 6.8. The detailed characteristics of the participants are shown in Table 1.
Diversity and rarefaction curve
A description of the observed species detected from OTU annotation is shown in Supplementary Table S1 (4.9MB, tif) . Differences in the phylotype richness and evenness (α diversity) were detected between controls and IBD patient groups. Patients with IBD showed lower microbial α-diversity and differed significantly from controls, as reflected by the Shannon diversity index (P < 0.005) and the Simpson diversity index (P < 0.024). The Shannon and Simpson values reflect the species diversity of the communities in terms of both species richness and evenness. However, the Shannon index gives greater weight to sequence richness, while the Simpson index gives greater weight to evenness. With the same species richness, the greater the species evenness reflects greater the community diversity. Alpha diversity per subject indicates a decrease in biodiversity in the patient ostensibly owing to IBD, in terms of Shannon measures. However, almost the converse results were detected in terms of Simpson measures [Figure 1].
Figure 1.
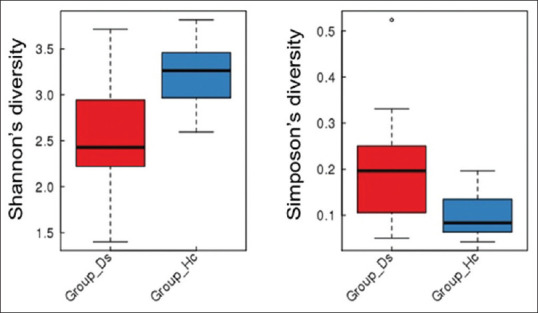
Alpha diversity indices as boxplots to describe richness (P < 0.005) and evenness (P < 0.024) at the healthy (blue) and patient (red) group level. Hc = Control, Ds = IBD patients
To show the difference of OTU distribution in various samples, a principal component analysis (PCA) was used to construct a two-dimensional graph to summarize factors that may be mainly responsible for this difference. The similarity is high if two groups are closely located. Based on the OTU abundance information, each OTU’s relative abundance in each sample was calculated, and the principal coordinate analysis (PCoA) of the OTU was carried out with the relative abundance value [Figure 2]. The PCoA plot partially showed a similar tendency in the distances within and between groups. The diversity of IBD patients was higher toward PCoA 2 direction (PC2), whereas healthy subject diversity was higher toward the PCoA 1 direction (PC1). Overall, the diagram showed that the mean value of the IBD group was localized in the positive directions of PCoA 1 and PCoA 2 (PC1 and PC2). By contrast, the healthy control group was localized negatively. These results indicated that the microbiome signatures of the two groups differed, ostensibly owing to IBD. Rarefaction curves based on the number of OTUs or observed species were drawn [Supplementary Figure S1 (271.2KB, tif) ]. The rarefaction measures indicated that the maximum number of sequences reads it used for further analysis of taxonomy abundance is 36,000 [Supplementary Figure S2 (108.9KB, tif) ]. Also, partial least squares discriminant analysis (PLS-DA) was performed to sharpen the separation between groups of observations, by hopefully rotating the principal component analysis (PCA). A maximum separation among groups is obtained to determine which variables carry the separating information. This shows a clear differentiation between the two groups [Supplementary Figure S3 (68.3KB, tif) ].
Figure 2.
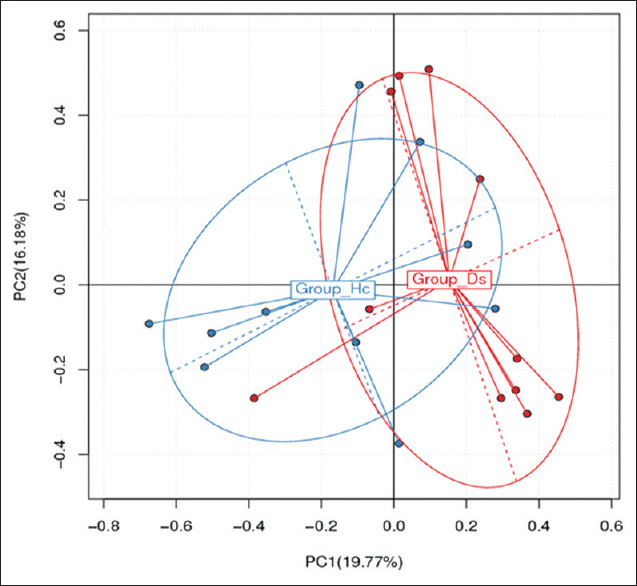
Principal coordinate analysis (PCOS) depends on the total OTUs’ abundance of different samples. Hc: Control; Ds: IBD patients
Gut microbiome at the phyla, family, genera, and species levels
A phylogenetic tree describing taxonomic groups of gut microbiome at the phyla and genera levels is shown in Supplementary Figure S4 (134.7KB, tif) . The results indicate that the most common phyla are Firmicutes (55 genera), Proteobacteria (19 genera), Bacteroidetes (10 genera), Actinobacteria (9 genera), Euryarchaeota (1 genus), Fusobacteria (1 genus), Lentisphaerae (1 genus), Thermi (1 genus), and Verrucomicrobia (1 genus). The results [Figure 3] indicate that the highly abundant OTUs belong to six of the previously mentioned phyla (e.g., Firmicutes, Bacteroidetes, Proteobacteria, Verrucomicrobia, Fusobacteria, and Actinobacteria). The gut microbiota analysis shows a significant change in the proportion of the two most abundant microbial phyla in healthy controls and IBD patients: Firmicutes and Bacteroidetes. By contrast, the IBD group showed greater abundance of Proteobacteria (15% vs. 32%), Verrucomicrobia (0.52% vs. 0.54%), and Fusobacteria (0.006% vs. 0.1%) and lower quantities of the phyla Firmicutes (46% vs. 37%), Bacteroidetes (35% vs. 30%), and Actinobacteria (2.9% vs. 0.6%). The family levels show 19 significant differences between IBD patients and controls. A considerable increase in the abundance of OTUs was found in the families Bacteroidaceae, Porphyromonadaceae, Verrucomicrobiaceae, Enterobacteriaceae, Streptococcoaceae, Rikenellaceae Lactobacillaceae, Alcaligenaceae, Fusobacteraceae, and Veillonellaceae, while a statistically significant reduction was shown in Actinomycetaceae, Bifidobacteriaceae, Paraprevotellaceae, Prevotellaceae, Muribaculoceae, Erysipelotrichaceae, Lachnospiraceae, Ruminococcaceae, and Succinivibrionaceae [Supplementary Figure S5 (378.4KB, tif) ]. The highly abundant phyla included 50 genera [Supplementary Figure S6 (399.3KB, tif) ]. The analysis at the genus level also showed differences between IBD individuals and controls. A significant increase in the abundance of OTUs of the genera Escherichia, Bacteroides, Sutterella, Proteus, and Streptococcus was found, which accounts for more than 1% of the total bacteria. The genera Bifidobacterium, Faecalibacterium, Prevotella, Eubacterium, Coprococcus, Dialister, Roseburia, Ruminococcus, and Blautia, which account for more than 1% of total bacteria, were markedly lower in IBD patients than in healthy controls.
Figure 3.

Relative abundance at the phylum (a) and species (b) level as measured by Metastats at sample and group levels. H = Healthy individuals, P = IBD patients
The results identified 38 bacterial species, where Tenericutes (1 phylotype), Verrucomicrobia (1 phylotype), Actinobacteria (3 phylotypes), Proteobacteria (4 phylotypes), Bacteroidetes (7 phylotypes), and Firmicutes (22 phylotypes) had the greatest abundance. The analysis showed a larger number of species with significantly higher abundance in IBD patients, compared with controls. Of these IBD patient biomes, 22 species significantly decreased, 14 species increased, and two species were absent in comparison with healthy controls [Figure 3]. Compared to healthy controls, the most significant increase in species in IBD patients was Bacteroides fragilis, Escherichia coli, and Lactobacillus salivarius, which account for more than 1% of total bacteria in the gut microbiome. The abundance of OTUs with respect to Faecalibacterium prausnitzii, Bacteroides plebeius, Bacteroides uniformis, Bifidobacterium adolescentis, R. gnavus, Coprococcus eutactus, Prevotella copri, and Ruminococcus bromii, accounts for more than 1% of total bacteria in the microbiome, which was significantly decreased in IBD patients compared to controls.
Significant differences analysis between groups
Linear discriminant analysis Effect Size[13] was used to determine the features (organisms, clades, operational taxonomic units, genes, and functions) most likely to display differences among groups by coupling standard tests for statistical significance with additional tests to encode biological consistency and effect relevance. The key findings indicate three negative bacterial biomarkers, namely Paraprevotellaceae and the Muribaculaceae families of Bacteroidetes and the Leuconostocaceae family of the Firmicutes, which have a higher relative abundance in healthy individuals compared to IBD patients. There was a decrease in Paraprevotellaceae in IBD patients (average relative abundance in control vs. IBD patients of 0.85% vs. 0.004%, respectively), which corresponds with the succinate-producing bacterium. Also, Muribaculaceae was decreased in IBD patients (average relative abundance 0.43% in controls and 0.045% in IBD patients). There was also a marked decrease in IBD patients in terms of the Leuconostocaceae family (average relative abundance, 0.002% and 0.24% in IBD patients and healthy individuals, respectively) [Figure 4]. Also, other primary signatures (or biomarkers) at the genera and species levels were absent in most IBD patients as shown by a linear discriminant analysis (LDA) [Figure 5]. These include P. copri (belonging to the Prevotellaceae family), B. adolescentis (belonging to the Bifidobacteriaceae family), Ruminococcus callidus (belonging to the Ruminococcaceae family), Coprococcus sp., R. gnavus, D. formicigenerans, and Leuconostoc (belonging to the Lachnospiraceae family), Dialister sp. (belonging to the Lachnospiraceae family), Catenibacterium, Eubacterium Biforme (belonging to the Erysipelotrichaceae family), and Lactobacillus mucosae (belonging to the Lactobacillaceae family). By contrast, Veillonella dispar (belonging to Veillonellaceae family) was absent in healthy individuals.
Figure 4.
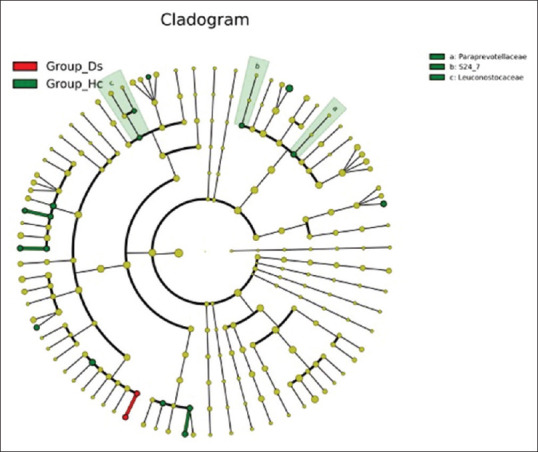
LEfSe tree showing three families statistically significant between healthy individuals and IBD patients. Different colors indicate different groups. LEfSe indicates important microbe biomarkers in the group, and the biomarker name is listed in the upper right corner. Group _Hc = Healthy individuals, group_Ds = IBD patients
Figure 5.
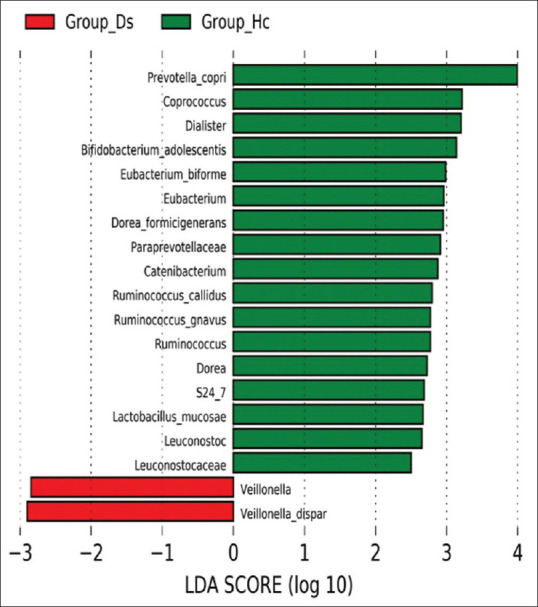
LDA score between healthy (group _Hc) and IBD patients (group_Ds). Distinctive taxa of bacteria with statistically significant differences at the family, genera, and species levels between groups are shown. IBD patients are indicated in red color, and healthy individuals are indicated in green. Group_Hc = Healthy individuals, group_Ds = IBD patients
DISCUSSION
This study suggests that modification in the gut microbiota is associated with individuals diagnosed with IBD, although the data collected on overall intestinal microbiota change in patients with IBD against healthy controls is limited to Saudi Arabia. Also, while the incidence of IBD seems to be increasing in Saudi Arabia,[14] an imbalance in the gut microbiome may contribute to changing epidemiology.[1] In the present study, the next-generation technique was used to investigate gut microbiome composition in Saudi patients with IBD, and these results were compared with internationally published results of various ethnicities and races. Many studies have reported a decrease in biodiversity in patients with IBD, known as α diversity, or species richness and evenness, which is reduced in the fecal microbiome in IBD patients compared with healthy individuals. This decreased diversity has contributed to reduced quantity of the dominant taxa, Firmicutes.[15,16] The results demonstrated a significant difference between the healthy control group and the IBD group, according to PCoA and UniFrac distance analysis; indeed, the data of healthy individuals is consistent with that of Eun et al.[17] The gut microbiota in healthy individuals is dominated by two phyla: Firmicutes (46.2 and 50.0%) and Bacteroidetes (34.60 and 40.00%). The remaining phyla, including Proteobacteria (15.40 and 6.20%) and Actinobacteria (2.90 and 0.85%), tended to be of lower concentrations. However, data of the gut microbiota in IBD patients showed an increase in Proteobacteria (33.00 and 33.81%) and Firmicutes (36.70 and 38.00%) (which includes the Clostridium and Faecalibacterium), although Bacteroidetes (29.80 and 25.41%) (which includes Prevotella) were significantly decreased compared to the controls. The Firmicutes are engaged in short-chain fatty acid and butyrate production. These molecules have a role in gut homeostasis, and the Bacteroidetes are involved in carbohydrate digestion, producing substrates for colonocytes.[18] The reduction of these two domain phyla of gut bacteriome has been shown in various IBD group studies. Sokol et al. (2017) and Quince et al. (2015) found that Actinobacteria (mainly Bifidobacterium) in IBD patients was depleted.[8,19] However, some other studies have found that Bifidobacterium increased in IBD patients.[20]
The present results also show that Fusobacteria significantly increased in the IBD group. By contrast, other studies have indicated that the abundance of Fusobacteria is reduced in IBD patients.[17] At the family level, we observed a low abundance of some bacterial families of Pasteurellaceae, Lachnospiraceae (including the genera of Dorea, Blautia, Anaerostipes, Roseburia, Coprococcus), and Ruminococcaceae (including Ruminococcus and Faecalibacterium). These results broadly correspond with Sokol et al.[21] We also observed an increase in the families of Rikenellaceae, Gemellaceae, Enterobacteriaceae, Eubacteriaceae (including Eubacterium sp.) in IBD patients compared to healthy controls.
As indicated above, it was found that IBD affected the human intestinal microbiome with respect to 16 genera and species biomarkers. The overall key signatures (biomarkers) concerned at the genera and species levels were P. copri (which is affiliated with the Prevotellaceae family), B.adolescentis (which belong to the Bifidobacteriaceae family), R. callidus (which belongs to the Ruminococcaceae family), Coprococcus sp., R. gnavus, D. formicigenerans, and Leuconostoc sp. (which belongs to the Lachnospiraceae family), Dialister sp. (which is affiliated with the Lachnospiraceae family), Catenibacterium sp. and E. Biforme (which belong to the Erysipelotrichaceae family), and L. mucosae (which belongs to the Lactobacillaceae family) were virtually absent in all IBD patients. By contrast, V. dispar (which belongs to the Veillonellaceae family) was absent in healthy individuals, while IBD patients showed a significant increase in this bacterium. The absence of biomarkers at the genera or species levels in IBD patients or controls may not be surprising at the outset, because this finding has been well documented.[22] Whether, the absence is an effect or a cause of intestinal inflammation is a fundamental question that remains to be resolved. A number of gut microflora evolve with a complex polysaccharide-rich diet and dietary fermentation, resulting in the production of short-chain fatty acids (SCFAs). SCFAs include propionate, acetate, and butyrate, which represent a primary energy source for colonic epithelial cells,[23] and preserve them from inflammation.[24,25] The reduction of dietary fiber correlates with IBD development.[26,27] Aside from this, a lack of fiber diets is associated with a low concentration of SCFAs.[27] SCFA-producing bacteria, such as the genera Coprococcus,[28] Dialister,[11] R. callidus,[15] and E. biforme.,[29] decreased in IBD patients. Also, lactic acid bacteria, such as the genus Leuconostoc, reduced in IBD patients; this genus is particularly well adapted to sugary niches and consequently possesses a broad spectrum of biocatalytic properties useful in carbohydrate modification.[30] B. adolescentis is a species of gram-positive, anaerobic, and folate compound producing bacteria that regulate intestinal homeostasis. Folate (also known as vitamin B9) shares in many metabolic processes that are considered essential for cell division. Folate supplementation is required for patients with IBD owing to its capacity to control rectal cell reproduction and promoting the permanence of Foxp3 regulatory T (Treg) cells, decreasing intestinal inflammation;[11] this species is a constant resident of the gastrointestinal tract, vagina, and mouth.[31] R. gnavus produces inflammatory polysaccharide, which is known to reduce the production of inflammatory cytokines, such as TNFα by dendritic cells,[32] while L. mucosae protect the host from mucosal inflammation by downregulation of inflammatory cytokines or stimulation of IL-10, an anti-inflammatory cytokine;[29] R. gnavus and L. Mucosae populations bloom during flares of symptoms in IBD patients. Also, P. Copri and V. disper are gram-negative; P. copri promote dextran sulfate sodium-induced colitis in mice, in association with raised IFN-g production. Thus, P.copri enhance Th1 immune response in experimental colitis, while V. disper plays a role in dysmetabolism of bile acids in IBD,[33] because it is able to degrade cholate and deoxycholate in secondary products; secondary biliary acids are an essential component in pro-inflammatory processes.[34]
In recent years, one study[35] has shown a correspondence between ethnicity or population variations and gut microbiome structures. A number of geographically associated divergences in gut microbiome composition contribute to differences in the host immunity and genetics. In other cases, cultural and behavioral features such as diet, hygiene, parasitic load, environmental exposure, etc., are more significant than genetics. This study has examined the gut microbiome of the remote hunter-gatherer community, which is based on starchy diets, including tubers or cassava, nuts, wild game, plants, and honey, with significant quantities of Prevotella sp., Bacteroides sp., Bifidobacterium sp., Proteobacteria, Ruminobacter sp. Spirochaetes, and Clostridiales, among others.[35] Concurrently, the diets of Western (US/European) urban industrialized societies, and those consuming diets high in protein and fatty food, improved sanitation and hygiene practices, and habitual use of antibiotics and other drugs often show a great abundance in Firmicutes.[36,37,38]
There are many studies demonstrating geographical or ethnicity-specific factors which might influence intestinal microbiota composition of IBD patients. For instance, the American community,[34] Saudi population, both Chinese[39] and Korean communities,[17] and the European community[20,21] showed low abundances of Firmicutes, Bacteroidetes, Lachnospiraceae, Faecalibacterium sp., Ruminococcus sp., and Eubacteriome sp., while Proteobacteria was low abundance in IBD patients. On the other hand, they indicated an increase in abundance of Ruminococcus gnavus in IBD patients in Europe an (France,[21] Vevey in Switzerland[20]) and North American populations,[34] but it was decreasing in Saudi patients. Also, the Chinese[39] showed high abundances of Bifidobacterium sp., but North American,[34] Saudi, and European (France[21]) communities showed low abundance in Bifidobacterium sp.
Many studies have pointed out the geographical or ethnicity-specific divergences in human gut microbiota structures. However, the determination of change in the gut microbiota as a cause or consequence of IBD is a complex problem. Still, it is unclear whether IBD is a cause or consequence of microbiome change, although it may be concluded that microbiome changes and IBD are biomarkers of each other.
We acknowledge that the study we present suffers from some limitations such as the small sample size and absence of mucosa-associated microbiota data. Additionally, we recognize that sampling CD patients with different disease distributions separate from UC patients would have provided us with more insights about the microbiota profile of each disease population. Therefore, larger, well-funded, prospectively designed studies are needed to further validate our observations.
In summary, the results of this study provide an overview of the variations in microbiota diversity present in Saudi IBD patients compared to healthy controls. Further larger studies are needed to validate these findings.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
SUPPLEMENTARY INFORMATION
Statistics of gut 16S rRNA (V3-V4 region) gene sequence datasets of healthy individuals versus IBD patients
The gut microbiomes composition of IBD patients and non-IBD controls was examined. Illumina MiSeq was used to analyze all 21 fecal samples based on the 16S rRNA (V3-V4 region) gene amplicon sequencing. The statistical analysis of the raw data and its processing are shown in Supplementary Figure S1. A total of 8,420,158 clean sequence reads were generated from stool samples of healthy individuals and IBD patients, with average read numbers of 446,343 and 359,702 per sample, respectively. A total of 8,390,357 tag-linked sequences were from stool samples of healthy individuals and IBD patients with average read numbers of 444,870 and 358,332 per sample, respectively. A total of 1,029,852 sequence tags were generated from stool samples of healthy people and IBD patients, with average read numbers of 45,909 and 51,886 per subject, respectively. Sequence tags were assigned to 622 OTUs (operational taxonomic units) across samples with =97% similarity. A summation of 3,728 OTUs for the 21 subjects was generated with 177 OTUs per subject, ranging from 65 to 255 OTUs, and averaging 203 and 135 OTUs for healthy individuals and IBD patients, respectively. The results for the number of observed species (number of OTUs) per subject increased in healthy individuals and decreased in patients with IBD.
Comparison among numbers of clean and tagged sequences and the recovered sequence tags at the sample and group levels for healthy individuals (blue) and patients with IBD. HDB.C: Healthy individuals; HDB.P: Patients with IBD
Number of observed species between healthy individuals and IBD patients as rarefaction measures to describe the maximum depth permitted to retain all subjects in the dataset to study relative taxonomic abundance. The arrow indicates the suitable sample size for analyzing taxonomy abundance (36,000 sequence reads). HDB.C = Control, HDB.P = IBD patients
Partial least square discriminant analysis (PLS-DA) based on OTUs abundance. Hc = Healthy, Ds = IBD patients
Genus level phylogenetic tree of the gut microbiome. Genera of the same phylum are specified by the same color
Average taxonomic composition in samples at family level as measured by Metastats. HDB.C = Healthy individuals, HDB.P= IBD patients
Relative abundance at the genus level as measured by Metastats at sample and group levels. HDB.C = Healthy individuals, HDB.P= IBD patients
OUT taxonomy
Acknowledgements
We would like to sincerely thank Dr. Padhmanand Sudhakar, KU Leuven University, Belgium, for the revision of the methodology and analysis, and Dr. Trevor Rawbone, Cardiff, UK, for English editing and proofreading of the manuscript.
REFERENCES
- 1.Halfvarson J, Brislawn CJ, Lamendella R, Vázquez-Baeza Y, Walters WA, Bramer LM, et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat Microbiol. 2017;2:17004. doi: 10.1038/nmicrobiol.2017.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bain CC, Schridde A. Origin, differentiation, and function of intestinal macrophages. Frontn Immunol. 2018;9:2733. doi: 10.3389/fimmu.2018.02733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alshehri D, Saadah O, Mosli M, Edris S, Alhindi R, Bahieldin A. Dysbiosis of gut microbiota in inflammatory bowel disease: Current therapies and potential for microbiota-modulating therapeutic approaches. Bosn J Basic Med Sci. 2021;21:270–83. doi: 10.17305/bjbms.2020.5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alatawi H, Mosli M, Saadah O, Dulai PS, Al-Hindi RR, Bahieldin A, et al. Primary non-response in inflammatory bowel disease, definition, potential causes, therapeutic drug monitroring and microbiota-a review. Applied Ecology and Environmental Research. 2020;18:5505–25. [Google Scholar]
- 5.Casen C, Vebø HC, Sekelja M, Hegge FT, Karlsson MK, Ciemniejewska E, et al. Deviations in human gut microbiota: A novel diagnostic test for determining dysbiosis in patients with IBS or IBD. Aliment Pharmacol Ther. 2015;42:71–83. doi: 10.1111/apt.13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, et al. Reduced diversity of faecal microbiota in Crohn's disease revealed by a metagenomic approach. Gut. 2006;55:205–11. doi: 10.1136/gut.2005.073817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Putignani L, Del Chierico F, Vernocchi P, Cicala M, Cucchiara S, Dallapiccola B Dysbiotrack Study Group. Gut microbiota dysbiosis as risk and premorbid factors of IBD and IBS along the childhood– adulthood transition. Inflamm Bowel Dis. 2016;22:487–504. doi: 10.1097/MIB.0000000000000602. [DOI] [PubMed] [Google Scholar]
- 8.Sokol H, Seksik P. The intestinal microbiota in inflammatory bowel diseases: Time to connect with the host. Curr Opin Gastroenterol. 2010;26:327–31. doi: 10.1097/MOG.0b013e328339536b. [DOI] [PubMed] [Google Scholar]
- 9.Huttenhower C, Kostic AD, Xavier RJ. Inflammatory bowel disease as a model for translating the microbiome. Immunity. 2014;40:843–54. doi: 10.1016/j.immuni.2014.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gevers D, Kugathasan S, Denson LA, Vázquez-Baeza Y, Van Treuren W, Ren B, et al. The treatment-naive microbiome in new-onset Crohn's disease. Cell Host Microbe. 2014;15:382–92. doi: 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joossens M, Huys G, Cnockaert M, De Preter V, Verbeke K, Rutgeerts P, et al. Dysbiosis of the faecal microbiota in patients with Crohn's disease and their unaffected relatives. Gut. 2011;60:631–7. doi: 10.1136/gut.2010.223263. [DOI] [PubMed] [Google Scholar]
- 12.Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, et al. Ribosomal database project: Data and tools for high throughput rRNA analysis. Nucl Acids Res. 2014;42:D633–42. doi: 10.1093/nar/gkt1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, et al. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–8. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Al-Ghamdi AS, Al-Mofleh IA, Al-Rashed RS, Al-Amri SM, Aljebreen AM, Isnani AC, et al. Epidemiology and outcome of Crohn's disease in a teaching hospital in Riyadh. World J Gastroenterol. 2004;10:1341–4. doi: 10.3748/wjg.v10.i9.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang S, Denman SE, Morrison M, Yu Z, Dore J, Leclerc M, et al. Dysbiosis of fecal microbiota in Crohn's disease patients as revealed by a custom phylogenetic microarray. Inflamm Bowel Dis. 2010;16:2034–42. doi: 10.1002/ibd.21319. [DOI] [PubMed] [Google Scholar]
- 16.Martinez C, Antolin M, Santos J, Torrejon A, Casellas F, Borruel N, et al. Unstable composition of the fecal microbiota in ulcerative colitis during clinical remission. Am J Gastroenterol. 2008;103:643–8. doi: 10.1111/j.1572-0241.2007.01592.x. [DOI] [PubMed] [Google Scholar]
- 17.Eun CS, Kwak MJ, Han DS, Lee AR, Park DI, Yang SK, et al. Does the intestinal microbial community of Korean Crohn's disease patients differ from that of western patients? BMC Gastroenterol. 2016;16:28. doi: 10.1186/s12876-016-0437-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang L, Dong D, Jiang C, Li Z, Wang X, Peng Y. Insight into alteration of gut microbiota in Clostridium difficile infection and asymptomatic C. difficile colonization. Anaerobe. 2015;34:1–7. doi: 10.1016/j.anaerobe.2015.03.008. [DOI] [PubMed] [Google Scholar]
- 19.Quince C, Ijaz UZ, Loman N, Eren AM, Saulnier D, Russell J, et al. Extensive modulation of the fecal metagenome in children with Crohn's disease during exclusive enteral nutrition. Am J Gastroenterol. 2015;110:1718–29. doi: 10.1038/ajg.2015.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mar JS, LaMere BJ, Lin DL, Levan S, Nazareth M, Mahadevan U, et al. Disease severity and immune activity relate to distinct interkingdom gut microbiome states in ethnically distinct ulcerative colitis patients. MBio. 2016;7:e01072–16. doi: 10.1128/mBio.01072-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sokol H, Leducq V, Aschard H, Pham HP, Jegou S, Landman C, et al. Fungal microbiota dysbiosis in IBD. Gut. 2017;66:1039–48. doi: 10.1136/gutjnl-2015-310746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ni J, Wu GD, Albenberg L, Tomov VT. Gut microbiota and IBD: Causation or correlation? Nat Rev Gastroenterol Hepatol. 2017;14:573–84. doi: 10.1038/nrgastro.2017.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ahmad M, Krishnan S, Ramakrishna BS, Mathan M, Pulimood AB, Murthy SN, et al. Butyrate and glucose metabolism by colonocytes in experimental colitis in mice. Gut. 2000;46:493–9. doi: 10.1136/gut.46.4.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Willing B, Halfvarson J, Dicksved J, Rosenquist M, Järnerot G, Engstrand L, et al. Twin studies reveal specific imbalances in the mucosaassociated microbiota of patients with ileal Crohn's disease. Inflamm Bowel Dis. 2009;15:653–60. doi: 10.1002/ibd.20783. [DOI] [PubMed] [Google Scholar]
- 25.Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, et al. T reg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–6. doi: 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- 26.Chiba M, Tsuji T, Nakane K, Komatsu M. High amount of dietary fiber not harmful but favorable for Crohn disease. Perm J. 2015;19:58–61. doi: 10.7812/TPP/14-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pituch-Zdanowska A, Banaszkiewicz A, Albrecht P. The role of dietary fibre in inflammatory bowel disease. Prz Gastroenterol. 2015;10:135–41. doi: 10.5114/pg.2015.52753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parada Venegas D, De la Fuente MK, Landskron G, González MJ, Quera R, Dijkstra G, et al. Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front Immunol. 2019;10:277. doi: 10.3389/fimmu.2019.00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Moreno de LeBlanc A, Del Carmen S, Zurita-Turk M, Santos Rocha C, van de Guchte M, Azevedo V, et al. Importance of IL-10 modulation by probiotic microorganisms in gastrointestinal inflammatory diseases. ISRN Gastroenterol 2011. 2011 doi: 10.5402/2011/892971. 892971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim D, Robyt JF. Properties of Leuconostoc mesenteroides B-512FMC constitutive dextransucrase. Enzyme Microb Technol. 1994;16:1010–5. doi: 10.1016/0141-0229(94)90134-1. [DOI] [PubMed] [Google Scholar]
- 31.Schell MA, Karmirantzou M, Snel B, Vilanova D, Berger B, Pessi G, et al. The genome sequence of Bifidobacterium longum reflects its adaptation to the human gastrointestinal tract. Proc Natl Acad Sci U S A. 2002;99:14422–7. doi: 10.1073/pnas.212527599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Henke MT, Kenny DJ, Cassilly CD, Vlamakis H, Xavier RJ, Clardy J. Ruminococcus gnavus, a member of the human gut microbiome associated with Crohn's disease, produces an inflammatory polysaccharide. Proc Natl Acad Sci U S A. 2019;116:12672–7. doi: 10.1073/pnas.1904099116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Larsen JM. The immune response to Prevotella bacteria in chronic inflammatory disease. Immunology. 2017;151:363–74. doi: 10.1111/imm.12760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duboc H, Rajca S, Rainteau D, Benarous D, Maubert MA, Quervain E, et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut. 2013;62:531–9. doi: 10.1136/gutjnl-2012-302578. [DOI] [PubMed] [Google Scholar]
- 35.Gupta VK, Paul S, Dutta C. Geography, ethnicity or subsistence-specific variations in human microbiome composition and diversity. Front Microbiol. 2017;8:1162. doi: 10.3389/fmicb.2017.01162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mardanov A, Babykin MM, Beletsky AV, Grigoriev AI, Zinchenko VV, Kadnikov VV, et al. Metagenomic analysis of the dynamic changes in the gut microbiome of the participants of the MARS-500 experiment, simulating long term space flight. Acta Nature. 2013;5:116–25. [PMC free article] [PubMed] [Google Scholar]
- 37.Greenhill AR, Tsuji H, Ogata K, Natsuhara K, Morita A, Soli K, et al. Characterization of the gut microbiota of Papua New Guineans using reverse transcription quantitative PCR. PLoS One. 2015;10:e0117427. doi: 10.1371/journal.pone.0117427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sankaranarayanan K, Ozga AT, Warinner C, Tito RY, Obregon-Tito AJ, Xu J, et al. Gut microbiome diversity among Cheyenne and Arapaho individuals from western Oklahoma. Curr Biol. 2015;25:3161–9. doi: 10.1016/j.cub.2015.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang W, Chen L, Zhou R, Wang X, Song L, Huang S, et al. Increased proportions of Bifidobacterium and the Lactobacillus group and loss of butyrate-producing bacteria in inflammatory bowel disease. J Clin Microbiol. 2014;52:398–406. doi: 10.1128/JCM.01500-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparison among numbers of clean and tagged sequences and the recovered sequence tags at the sample and group levels for healthy individuals (blue) and patients with IBD. HDB.C: Healthy individuals; HDB.P: Patients with IBD
Number of observed species between healthy individuals and IBD patients as rarefaction measures to describe the maximum depth permitted to retain all subjects in the dataset to study relative taxonomic abundance. The arrow indicates the suitable sample size for analyzing taxonomy abundance (36,000 sequence reads). HDB.C = Control, HDB.P = IBD patients
Partial least square discriminant analysis (PLS-DA) based on OTUs abundance. Hc = Healthy, Ds = IBD patients
Genus level phylogenetic tree of the gut microbiome. Genera of the same phylum are specified by the same color
Average taxonomic composition in samples at family level as measured by Metastats. HDB.C = Healthy individuals, HDB.P= IBD patients
Relative abundance at the genus level as measured by Metastats at sample and group levels. HDB.C = Healthy individuals, HDB.P= IBD patients
OUT taxonomy
Data Availability Statement
The data have been submitted to National Center for Biotechnology Information (NCBI) under BioProject no. PRJNA673073 (https://dataview.ncbi.nlm.nih.gov/object/PRJNA673073) with BioSample identifiers SAMN16591214-SAMN16591234 with accession number SRA16000303-SRA16000323.