

Abstract

β-Hydroxyketones (aldols) are prepared from the alkylation-deprotection of O-silylated aryl cyanohydrins with epoxides. Key to the success of the method was the suppression of an in situ cyclic imidate formation that occurs upon initial opening of the epoxide ring.
Introduction
The aldol addition is among the most important carbon-carbon bond forming reactions in chemical synthesis.1 The β-hydroxycarbonyl product scaffold is of such great significance that alternative methods for its preparation have been investigated, such as the “non-aldol aldol” epoxide rearrangements developed by Jung.2,3,4 Though significant, this approach is limited by its inability to form a new carbon-carbon bond.
Another alternative to the formation of aldols is the reaction of an acyl anion equivalent with an epoxide (Scheme 1). Like the aldol addition, this strategy results in the formation of a new carbon-carbon bond, now introduced between the carbonyl and the α-carbon of the final product. This distinction can facilitate useful alternatives in developing synthetic strategies for the formation of complex molecules. A significant advantage of this approach lies in its use of an epoxide. The epoxide stereochemistry is predictably transmitted to the products, and many excellent enantio- and diastereoselective methods for the preparation of epoxides are known.5
Scheme 1.

Aldols from acyl anion equivalents.
Perhaps the best-known acyl anion equivalents are the anions formed from 1,3-dithianes, which are known to react with a variety of electrophiles including epoxides.6 Cyanohydrin anions can also serve as acyl anion equivalents. These intermediates play a role in the classic benzoin condensation of benzaldehydes7 and modern variations involving acylsilanes.8,9,10,11 Anions formed from deprotonation of protected cyanohydrins have been successfully reacted with a variety of electrophiles including alkyl halides,12 carbonyls,13 and α,β-unsaturated carbonyls.14,15 Surprisingly, the reaction of these useful acyl anion equivalents with epoxides as a route to the formation of β-hydroxycarbonyls has not been explored. Reported attempts to react protected cyanohydrins with epoxides are scarce and have been largely unsuccessful.16,17,18,19,20
Herein we report a new method for the formation of aldols via the reaction of silyl-protected cyanohydrins with epoxides (Scheme 2). In this method, a protected cyanohydrin is deprotonated with an appropriate base and the resulting anion reacted with an epoxide. Subsequent removal of the protecting group triggers the expulsion of cyanide to furnish an aldol product. The ease of preparation of protected cyanohydrins from aldehydes and the variety of well-established, procedures for the stereoselective preparation of epoxides establishes this method as a useful alternative approach to the formation of aldol products.
Scheme 2.

Formation of aldols via the reaction of protected cyanohydrin anions with epoxides.
Results and Discussion
We began by treating TMS-protected mandelonitrile with LiHMDS in THF, followed by introduction of 1,2-epoxybutane. The reaction failed to provide the desired adduct, giving 3-hydroxypentanenitrile as the major product. This was presumably formed via decomposition of the cyanohydrin substrate and subsequent reaction of the epoxide with the in situ liberated cyanide. Repeating the reaction in toluene completely suppressed the formation of 3-hydroxypentanenitrile, and the desired adduct was formed as a minor product along with considerable amounts of benzaldehyde and various benzoin derivatives.
Given the complications arising from decomposition of the substrate, we next investigated reactions of the more robust TBS-protected mandelonitrile. Similar treatment of this substrate with LiHMDS in THF followed by 1,2-epoxybutane gave a 1:1 mixture of the desired adduct and a cyclic imidate, each as a 1:1 mixture of diastereomers (Scheme 3). The formation of a cyclic imidate was not surprising, as the intramolecular attack of an alkoxide onto a nitrile is well known.21 Indeed, Rychnovsky17 proposed that this side-reaction likely contributed to the low adduct yield in the reaction of an acetonide-protected cyanohydrin with an epoxide.
Scheme 3.

Formation of desired adduct and cyclic imidate.
This experiment was repeated several times and the ratio of adduct to imidate varied considerably, at times as high as 4:1 and as low as 1:10. The variability in product distribution was largely correlated to differences in how the reaction work-up was performed. When the reaction mixture was removed and added to saturated aqueous ammonium chloride, the product distribution favored the desired adduct. However, when the quenching solution was instead added into the reaction flask, the product distribution greatly favored the undesired imidate.
The following set of equilibria was proposed (Scheme 4) to explain this observation. After initial attack of the carbanion on the epoxide, the two anionic species (acyclic adduct anion and cyclic imidate anion) establish an equilibrium that favors the desired acyclic adduct. This is rationalized by comparing the pKa of their conjugate acids (roughly 16 and 25, respectively). As the quenching acid is introduced, the conjugate acids (acyclic adduct and cyclic imidate) establish a second equilibrium favoring the more stable imidate. This is rationalized by the formation of the stronger σ bond in the imidate replacing the weaker π bond in the nitrile adduct. Quenching the reaction mass via drop-wise addition to a large excess of acid shuts down the pathway that allows interconversion of the two neutral species via their conjugate bases, preserving the initial ratio of anions in the final products.
Scheme 4.

Equilibrium of adduct and imidate via their conjugate bases during acid quench.
Thus, when TBS-protected mandelonitrile was reacted with LiHMDS and 1,2-epoxybutane in toluene and quenched by drop-wise addition of the reaction mass to a large excess of vigorously stirred saturated aqueous ammonium chloride a 13:1 mixture of desired adduct and cyclic imidate was observed. Subsequent treatment of product mixture with TBAF in THF provided the expected aldol in 77% isolated yield (two steps) after column chromatography (Scheme 5).
Scheme 5.

Successful formation of aldol using after deprotection with TBAF.
With the problematic substrate decomposition and product cyclization now under control, we looked to optimize the alkylation conditions with respect to solvent. The results are summarized in Table 1. The reaction solvent had a significant influence on the product distribution. Hexane greatly favored formation of the undesired imidate, likely due to its extreme hydrophobicity which adversely affected the aqueous workup. Surprisingly, THF offered only a modest preference for the desired acyclic adduct. We suspect this is due to the solvent’s hygroscopic nature, introducing trace amounts of water prior to workup. Both diethyl ether and toluene gave satisfactory results, each greatly favoring the desired adduct.
Table 1.
Solvent optimization using TBS-protected mandelonitrile and 1,2-epoxybutane.
Determined by 1H-NMR
Isolated yield
We next explored the influence of the silyl group on the reaction. The results are summarized in Table 2. The TMS, TES, and TBS derivatives of mandelonitrile were reacted with LiHMDS and 1,2-epoxybutane using toluene as the solvent. Yields improved as the stability of the silyl group toward base was increased, with only the TBS group providing satisfactory results.
Table 2.
Silyl group optimization using silyl-protected mandelonitriles and 1,2-epoxybutane in toluene.

| |
|---|---|
|
| |
| R | Yield (%)a |
| TMS | trace |
| TES | 51 |
| TBS | ll |
Isolated yield
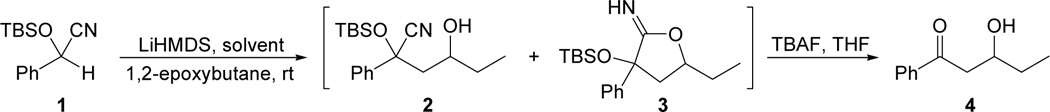
The scope of the method with respect to epoxide was demonstrated by reacting TBS-protected mandelonitrile with several epoxides. The results are summarized in Table 3. Mono-, di-, and trialkyl substituted epoxides were well-tolerated (entries 1, 2, 8, 9). The reaction was also compatible with ether (entry 6) and remote alkene (entry 3) functionalities. Epoxides with adjacent halogen (entry 7), alkene (entry 4), or arene (entry 5) groups failed to provide any of the desired aldol product.
Table 3.
Epoxide scope – reaction of TBS-protected mandelonitrile with various epoxides.
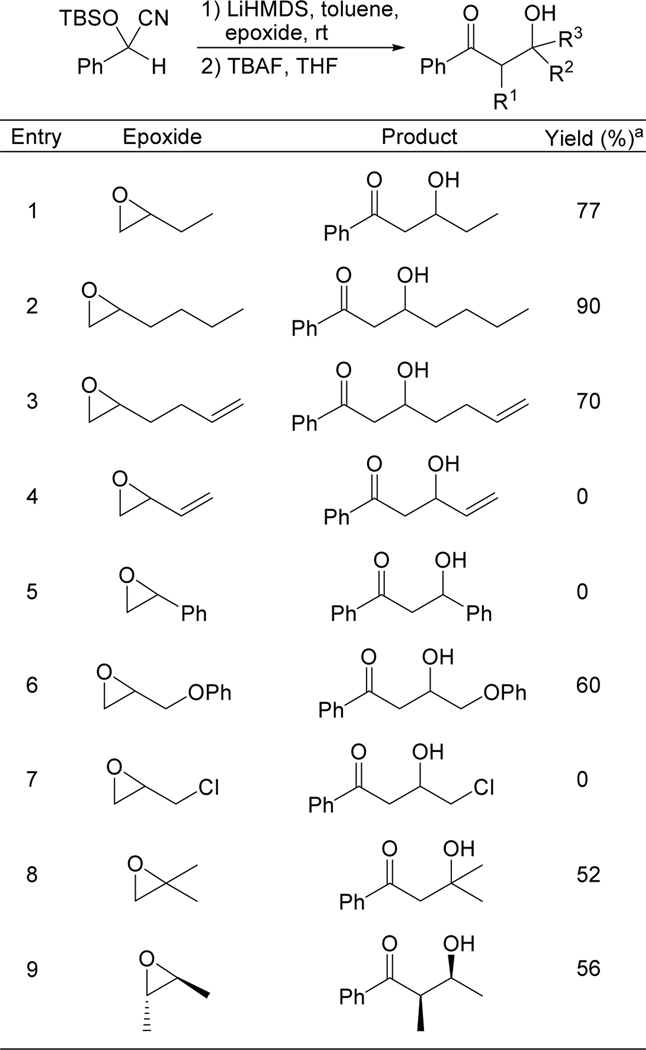
|
Isolated yield
The scope of the method with respect to cyanohydrin was also explored. Various TBS-protected aryl cyanohydrins were reacted with 1,2-epoxybutane under our optimized conditions. The results are summarized in Table 4. Alkyl (entry 2), halogen (entry 3), ether (entry 5), and amine (entry 6) substituents were each well-tolerated. The more sterically demanding naphthyl group (entry 8) also showed no inhibition of the reaction. Cyanohydrins with nitrile (entry 4) and trifluoromethyl (entry 7) substituents gave complex product mixtures with no aldol products observed.
Table 4.
Cyanohydrin scope – reaction of TBS-protected aryl cyanohydrins with 1,2-epoxybutane.
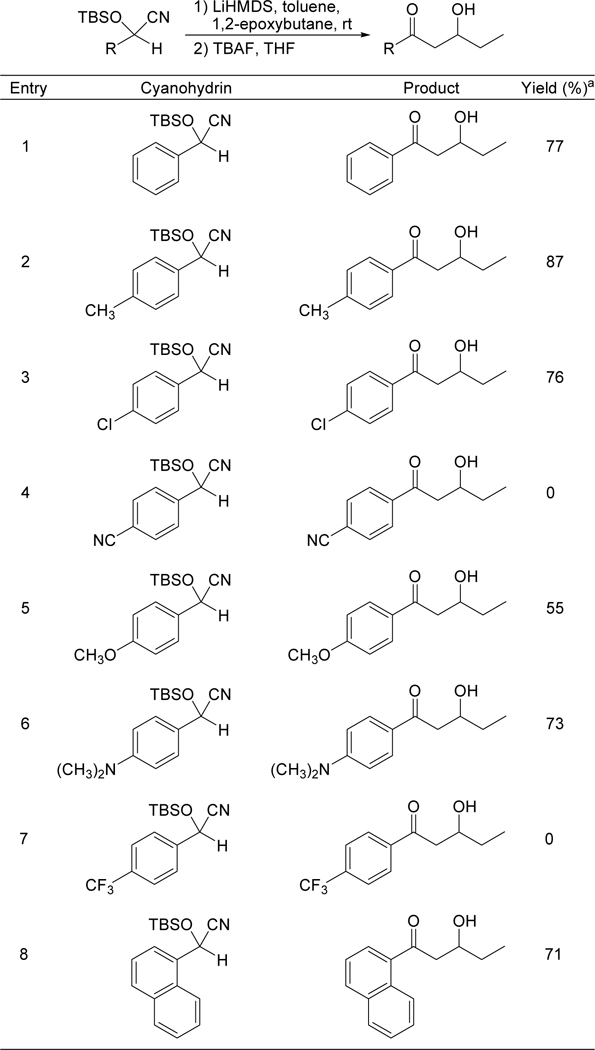
|
Isolated yield
In conclusion, a new method for the preparation of aldols via the reaction of silyl-protected aryl cyanohydrins with epoxides has been developed. The specific workup procedure employed was critical to mitigating the formation of an undesired cyclic imidate, and the use of a TBS protecting group in toluene or ether as solvent gave superior results. The method was effective for the formation of several aldols from a variety of epoxides and cyanohydrins.
Typical Procedure:
A solution of 1.0 M LiHMDS in THF (1.2 mL, 1.2 equiv) was added drop-wise with stirring to a solution of silyl-protected cyanohydrin (1.0 mmol) in 5 mL of dry ether or toluene under argon. Epoxide (2.0 equiv) was immediately added dropwise and the reaction stirred for an additional five minutes. The reaction mass was removed via syringe and added dropwise below the surface of 10 mL of vigorously stirred saturated aqueous NH4Cl. The mixture was extracted with CH2Cl2 (3 × 10 mL) and the combined organic layers dried over anhydrous Na2SO4. The dried solution was concentrated in vacuo and the crude product mixture dissolved in 5 mL of dry THF. A solution of 1.0 M TBAF in THF (1.2 mL, 1.2 equiv) was added drop-wise and stirred for 20 minutes. Water (5 mL) was added and the mixture extracted with ethyl acetate (2 × 5 mL). The combined organic layers were dried over anhydrous sodium sulfate and solvents removed in vacuo. The residue was purified by flash chromatography (silica gel, 20% ethyl acetate/hexane) to give the aldol product.
Supplementary Material
Acknowledgments
This project was supported in part by SC EPSCoR/IDeA [17-RE10], the Ronald E. McNair Postbaccalaureate Achievement Program [P217A130111], and Winthrop University. Research reported in this publication was also supported by the National Institute of General Medical Sciences of the National Institutes of Health [P20GM103499]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We also thank Michael Walla, University of South Carolina, for acquisition of HRMS data.
Footnotes
Declaration of Competing Interest
The authors declare no known competing interests that could have influenced this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Modern Aldol Reactions; Mahrwald R, Ed.; Wiley-VCH: Weinheim: 2004; pp 1–61. [Google Scholar]
- 2.Jung ME; D’Amico DC J. Am. Chem. Soc 1993, 115, 12208–12209. [Google Scholar]
- 3.Jung ME; Marquez R. Tetrahedron Lett. 1999, 40, 3129–3132. [Google Scholar]
- 4.Jung ME; van den Heuvel A. Tetrahedron Lett. 2002, 43, 8169–8172. [Google Scholar]
- 5.Xia Q-H; Ge H-Q; Ye C-P; Liu Z-M; Su K-X Chem. Rev. 2005, 105, 1603–1662. [DOI] [PubMed] [Google Scholar]
- 6.Corey EJ; Seebach D. Angew. Chem., Int. Ed. 1965, 4, 1075–1077. [Google Scholar]
- 7.Wiberg KB J. Am. Chem. Soc. 1954. 76, 5371–5375. [Google Scholar]
- 8.Tarr JC; Johnson JS J. Org. Chem. 2010, 75, 3317–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Linghu X; Bausch CC; Johnson JS J. Am. Chem. Soc. 2005, 127, 1833–1840. [DOI] [PubMed] [Google Scholar]
- 10.Nicewicz DA; Yates CM; Johnson JS J. Org. Chem. 2004, 69, 6548–6555. [DOI] [PubMed] [Google Scholar]
- 11.Nicewicz DA; Yates CM; Johnson JS Angew. Chem. Int. Ed. 2004, 43, 2652–2655. [DOI] [PubMed] [Google Scholar]
- 12.Stork G; Maldanado LJ Am. Chem. Soc. 1971, 93, 5286–5287. [Google Scholar]
- 13.Hünig S; Wehner G. Chem. Ber. 1979, 112, 2062–2067. [Google Scholar]
- 14.Reich HJ; Biddle MM J. Org. Chem. 2005, 70, 3375–3382. [DOI] [PubMed] [Google Scholar]
- 15.Hünig S; Öller M. Chem. Ber. 1981, 114, 959–967. [Google Scholar]
- 16.Deuchert K; Hertenstein U; Hünig S; Wehner G. Chem. Ber. 1979, 112, 2045–2061. [Google Scholar]
- 17.Rychnovsky SD; Swenson SS J. Org. Chem. 1997, 62, 1333–1340. [Google Scholar]
- 18.Hijikuro I; Doi T; Takahashi TJ Am. Chem. Soc. 2001, 123, 3716–3722. [DOI] [PubMed] [Google Scholar]
- 19.Garcia GA; Munoz H; Tamariz J. Synth. Commun. 1983, 13, 569–574. [Google Scholar]
- 20.Malanco FL; Maldonado LA Synth. Commun. 1976, 6, 515–519. [Google Scholar]
- 21.Wovkulich PM; Baggiolini EG; Hennessy BM; Uskokovic MR; Mayer E; Norman AW J. Org. Chem. 1983, 48, 4433–4436. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.