

Abstract
Two HeLa variants defective in the mismatch repair protein hPMS2 were isolated by selection for methylation tolerance. Neither variant expressed detectable hPMS2 protein as determined by western blotting. Cell extracts were defective in correcting a single base mispair and were unable to perform mismatch repair-dependent processing of a methylated DNA substrate. Correction of the repair defect and restoration of sensitivity to a methylating agent was achieved by introducing a wild-type copy of chromosome 7 on which the hPMS2 gene is located. Loss of hPMS2 function in the HeLa variants was associated with a 5-fold increase in mutation frequency in the supF gene of the pZ189 shuttle vector. Wild-type levels of mutagenesis were restored by the transferred chromosome 7. Comparisons of mutational spectra identified multiple base substitutions, frameshifts and, to a lesser extent, single base pair changes as the types of mutation which are selectively increased in a hPMS2-defective background. The location of multiple mutations and frameshifts indicates that misalignment-mediated mutagenesis could underlie most of these events. Thus the mutator phenotype associated with loss of hPMS2 most likely arises because of the failure to correct replication slippage errors. Our data also suggest that a considerable fraction of mutagenic intermediates are recognized by the hMutSβ complex and processed via the hMLH1/hPMS2 heterodimer.
INTRODUCTION
Mismatch repair (MMR) is the main post-replicative pathway for the correction of replication errors. Defects in this pathway are responsible for the increased cancer susceptibility in hereditary non-polyposis colon cancer (HNPCC) patients. The early appearance of cancer of the colon, endometrium or ovary in HNPCC individuals is most likely the consequence of the mutator phenotype of MMR-defective cells. Although the genes involved in the MMR pathway are highly conserved from prokaryotes to eukaryotes, there is an apparent redundancy of MMR complexes in human cells. Mismatch recognition is performed in human cells by two complexes, hMutSα and hMutSβ. The first is a heterodimer of hMSH2 and hMSH6, while the second is formed by hMSH2 and hMSH3. Following recognition, the hMutLα heterodimer, formed by hMLH1 and hPMS2, is loaded onto the mismatched DNA to direct its correction (for reviews see 1,2).
In yeast, a second complex, formed by MLH1 and MLH3, plays an important role in the repair of insertion deletion mispairs via the MSH2/MSH3 pathway (3). The human protein hMLH3, which also forms a dimer with hMLH1, has recently been identified (4). The existence of a choice between the hMLH3/hMLH1 complex and hMutLα raises the possibility of an alternative branch of the human MMR pathway which is independent of hPMS2.
The role of the various mammalian MMR proteins in the repair of different mismatches has been derived from both biochemical and genetic approaches. Thus the purified hMutSα complex preferentially binds to duplex DNA containing single base mismatches and loops of 1 base while the hMutSβ complex recognizes ≥2 base loops. There is some overlap between recognition by the two complexes at the level of 1 base loops (5,6). Loss of MMR results in increased mutation rates at both coding sequences and in non-coding regions such as microsatellites. Tumor cell lines with defects in either hMutSα or hMutLα exhibit large increases in mutation rate at the HPRT gene and microsatellite instability. Many of these cell lines are, however, mutated in more than one MMR gene and it is therefore difficult to infer the precise role of each repair protein in the correction of specific classes of DNA mismatches. Analysis of mutational spectra in hMSH2-defective LoVo cells indicates that frameshift and transition mutations are preferentially increased (7). The hMLH1-defective cell line HCT116 also displays elevated rates of frameshifts and transitions (8,9). In contrast, mutants in hMSH6 (DLD1 and HEC1-B cells) preferentially accumulate transitions and transversions (7,8,10,11).
More limited information is available on the consequences of loss of hPMS2 function and none for hMLH3. Mutations in hPMS2 are found in a minority of HNPCC families and are apparently rare in MMR-defective tumors in general. The only available human tumor cell line with a hPMS2 defect is the endometrial carcinoma-derived HEC1-A (12). In this cell line the presence of a truncating hPMS2 mutation together with a second mutation in hMSH6 is associated with a strong mutator phenotype. This is seen as a very large increase in the rate of frameshifts and base substitutions at the HPRT gene (11). In contrast, tissues of the PMS2–/– mouse carrying an integrated supF gene show only a limited increase in mutation frequency unless the lengths of two runs of identical nucleotides inside the gene are experimentally increased (13).
To better characterize the role of defective hPMS2 function on mutation frequency and on the types of mutation which accumulate in human cells, we have isolated hPMS2-defective HeLa cell variants. Two hPMS2-deficient clonal variants, Clone 7 and Clone 12, were derived by selecting for tolerance to a methylating agent (14,15). Loss of functional hPMS2 is associated with an increased mutation frequency in a target supF gene replicated episomally in either Clone 7 or Clone 12, as well as in HEC1-A cells. A comparison of the mutational spectra of HeLa and Clone 7 cells indicates that the main qualitative changes in supF associated with loss of this single MMR protein are an increase in multiple base substitutions and frameshifts. Single base substitution mutations are affected to a minor extent. Since an hMSH6 defect does not detectably affect spontaneous mutation of supF in the same vector (16), we conclude that the hMutLα heterodimer is required for correction of a significant portion of mismatches recognized by the hMutSβ complex.
MATERIALS AND METHODS
Chemicals
N-methyl-N-nitrosourea (MNU) (Sigma Chemical Co., St Louis, MO) was dissolved in dimethyl sulfoxide and diluted in PBS/20 mM HEPES (pH 7.4) to the required concentrations immediately before use. Anti-hMSH6 and anti-hMSH2 antisera were a kind gift of Prof. J. Jiricny (University of Zurich, Switzerland). Antibodies against hMLH1 (clone G168-728) and hPMS2 (mAb A16-4) were obtained from Pharmingen (Los Angeles, CA). The restriction enzyme DpnI and G418 was obtained from New England Biolabs (Beverly, MA) and Gibco Life Technologies (Gaithersburg, MD), respectively. Isopropyl-β-d-thiogalactoside (IPTG), 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal), colcemid, phytoemaglutinin and cytochalasin B were all purchased from Sigma.
Cell culture, survival determination and selection for methylation tolerance
HeLa cells, the HEC1-A cell line and the methylation-tolerant cell lines were cultured in DMEM (Gibco Life Technologies) supplemented with 10% fetal calf serum and penicillin (100 µg/ml) (complete medium). When needed, G418 was added to the medium at 1 mg/ml. All cultures were incubated at 37°C in 5% CO2 and at 95% relative humidity.
Survival after treatment with the methylating agent MNU was determined by clonogenic assay as previously described (17). Cells at clonal density (100–400/60 mm dish) were treated 18 h after seeding with MNU (30 min at 37°C in PBS/20 mM HEPES, pH 7.4). Cultures were then washed, fed with complete medium and 1–2 weeks later surviving colonies were fixed with methanol, stained with Giemsa and counted.
Microcell transfer
Single human chromosomes were transferred by microcell fusion as already described (18). Briefly, 8 × 105 A9 murine cell lines carrying a single human chromosome 7 were seeded in 12 flasks containing complete medium supplemented with 800 µg/ml G418. Two days later colcemid (60 ng/ml) was added to induce micronuclei. After an additional 2 days, cultures were incubated with cytocalasin B (20 µg/ml) and microcells were recovered by centrifugation, resuspended in 10 ml DMEM serum-free medium and filtered through polycarbonate filters. Microcells were resuspended in DMEM serum-free medium containing 100 µg/ml phytohemaglutinin, seeded on the recipient cell line, treated for 1 min with 47% polyethylene glycol and washed several times with DMEM serum-free medium. After 24 h G418 (1 mg/ml) was added to the medium and the hybrids were isolated after 14 days under selective G418 conditions.
Plasmid methylation and in vitro repair synthesis by cell-free extracts
Plasmid methylation and in vitro repair synthesis were performed as already described (19,20). Briefly, plasmid DNA was methylated with 0.24 mM MNU to obtain around one O6-methylguanine (O6-meGua) per molecule and incubated with cell extracts (300 µg) for 1 h at 37°C in the presence of radiolabeled [α-32P]dATP precursor. The reaction mixture was spotted onto 3MM paper squares and washed three times with Na2HPO4, once with distilled water and once with 70% ethanol. The radioactivity was determined by liquid scintillation counting of the dried filters.
Western blotting
Cell extracts were prepared from 2 × 107 cells resuspended in a buffer containing 50 mM Tris–HCl pH 7.5, 1 mM EDTA, 10 mM DTT and 0.2% Triton X-100. Cell extracts (20 µg) were denatured and separated on 7.5% SDS–polyacrylamide gels together with a prestained low molecular weight marker (Bio-Rad, Hercules, CA). Proteins were transferred to nitrocellulose membranes (Bio-Rad) using a Trans-Blot cell apparatus (Bio-Rad) at 30 mA overnight at 4°C. The remaining protein binding sites of the nitrocellulose paper were blocked by immersion in TBS/Tween (10 mM Tris–HCl, pH 7.5, 150 mM NaCl, 0.05% Tween 20) for 30 min in the presence of 0.1% gelatin and an additional 30 min in the presence of 3–7% skimmed powdered milk. The blocked filter was incubated with primary antibody (1 µg/ml for the monoclonal antibodies anti-PMS2 and anti-hMLH1 or 1:1000 diluted antiserum for hMSH6 and hMSH2) for 1 h at room temperature. After washing with TBS/Tween, the appropriate secondary antibody was added for an additional 1 h. ECL detection reagents (Amersham Italia, Milano, Italy) were used to develop the blots.
DNA transfection, plasmid recovery, shuttling into bacteria and mutagenesis analysis
The plasmid pZ189 contains the SV40 origin of replication, SV40 large T antigen and the supF target gene, which encodes a suppressor tRNA. The tRNA is able to counteract the lacZam mutation carried by Escherichia coli bacterial strain MBM7070. When the target gene supF is mutated white or light blue colonies are produced. Exponentially growing cells, plated in complete medium at a density of 1 × 106 cells/dish were transfected 1 day after seeding with 500 ng of plasmid DNA by the calcium phosphate precipitation procedure. Two days later, plasmid molecules were recovered by the Hirt method and digested with DpnI in order to eliminate non-replicated plasmids. DNA was shuttled into E.coli MBM7070 by electroporation with a Gene Pulser apparatus (Bio-Rad). Bacteria were plated on LB Amp/IPTG/X-Gal Petri dishes and incubated overnight at 37°C. White or light blue colonies were streaked three times to confirm their mutant phenotype. DNA was extracted with Qiagen columns and analyzed on agarose gels for detection of large deletions or rearrangements. Mutants carrying no gel alterations were sequenced with AmpliTaqFS on an ABI Prism 310 automatic sequencer (Perkin Elmer, Milano, Italy). Mutants carrying the same genetic alteration(s) were eliminated from the mutational analysis when derived from the same transfection experiment.
Statistical analysis
The Adams and Skopek algorithm uses a Monte Carlo method to simulate a P value for the standard hypergeometric test for a contingency table (21). Unlike the χ2 test, which can also be applied to contingency tables and requires that all cells contain five or more events, the hypergeometric test is appropriate when applied to sparse data sets, as often found in mutational spectra analysis. The Cariello program (22) uses a random number generator to produce a large number of simulated spectra based on the hypergeometric probability of the experimentally observed input spectra. The degree to which the simulated spectra differ from the input spectra is used to estimate the probability that the two input spectra were derived from the same population. A P value ≤0.05 leads to rejection of the null hypothesis and the conclusion that the input spectra are different.
RESULTS
Characterization of methylation-tolerant MMR-defective HeLa clones
MNU-resistant clones isolated from HeLa cells were found to be defective in MMR. Clones 7 and 12 showed similar increases in MNU resistance (close to 80-fold in D37) (Fig. 1A; 15). Cell-free extracts from either clone were unable to repair a single base mismatch in an in vitro assay (14). Complementation analysis with extracts of known MMR mutants identified defects in the hMutLα complex of both Clones 7 and 12 (14). Western analysis indicated that neither clone expressed hPMS2 protein detectable by an antibody which recognizes the C-terminus of the protein. The levels of hMLH1, MSH2 and hMSH6 were normal (Fig. 1B). The MMR defect of Clones 7 and 12 therefore resides in the hPMS2 protein. Consistent with their previously reported double defect, no detectable hPMS2 or hMSH6 was detected in extracts of HEC1-A cells (Fig. 1B).
Figure 1.
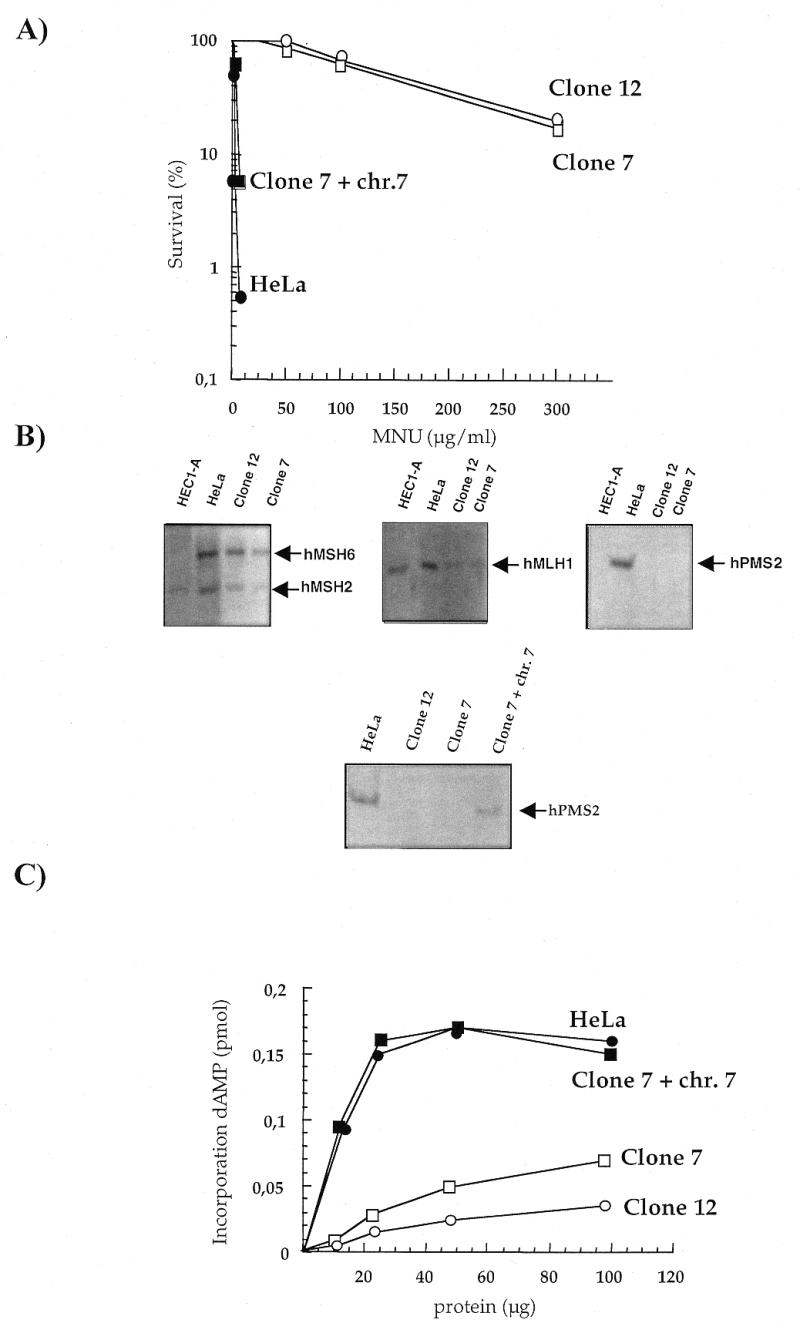
(A) Survival of HeLa and methylation-tolerant derivatives after a 30 min exposure to increasing concentrations of MNU. HeLa (closed circle), Clone 7 (open square), Clone 12 (open circle) and Clone 7 + chromosome 7 (closed square). (B) Identification of the MMR defects by western blotting. Cell extracts were separated on SDS–polyacrylamide gels and immune complexes with hMSH6, hMSH2, hMLH1 and hPMS2 were visualized after transfer to a nitrocellulose membrane as described in Materials and Methods. (C) MMR synthesis on MNU-treated plasmids. The incorporation of dAMP by increasing amounts of extracts from HeLa (closed circle), Clone 7 (open square), Clone 12 (open circle) and Clone 7 + chromosome 7 (closed square) was determined following incubation with methylated plasmid for 30 min at 37°C.
An extra copy of chromosome 7, on which the hPMS2 gene is located, was inserted into Clone 7. The presence of a functional chromosome 7 reversed the methylation-tolerant phenotype (Fig. 1A). The efficiency of MMR was measured by ‘repair synthesis’ performed by cell extracts on a methylated plasmid. This assay measures aberrant processing of DNA O6-meGua and is dependent on a functional MMR (14). Thus, while extracts of MMR-defective Clones 7 and 12 cells did not perform ‘repair synthesis’ on the methylated substrate, the presence of an extra copy of chromosome 7 fully restored MMR in Clone 7 (Fig. 1C).
The majority of the recipient cells contained a single extra copy of chromosome 7 as measured by FISH analysis (data not shown). This resulted in about half the level of hPMS2 of the parental HeLa cells as measured by western analysis (Fig. 1B). This degree of hPMS2 expression is sufficient to restore wild-type levels of MMR. This is in agreement with correction of defective MMR by transfer of either chromosome 2 or 3, which indicates that half of the wild-type level of hMSH2/6 or hMLH1 is sufficient to restore efficient repair.
The absence of detectable hPMS2 protein in Clones 7 and 12 and reversal of the phenotype by transfer of chromosome 7 into Clone 7 indicate that the defect in these clones is due to an alteration in the hPMS2 gene. Furthermore, expression of 50% of the hPMS2 protein in Clone 7 is sufficient to achieve wild-type levels of MMR and almost a full reversion of methylation tolerance.
Mutation in MMR-proficient and hPMS2-defective human cell lines
Spontaneous mutation frequencies were measured after passage of the shuttle vector pZ189 through MMR-proficient and MMR-deficient cell lines. pZ189 was transfected into HeLa cells, the hPMS2-defective derivatives Clones 12 and 7, the MMR corrected Clone 7 + chromosome 7 and the endometrial carcinoma cell line HEC1-A. The plasmid was allowed 48 h to replicate and DNA was recovered and used to transform competent E.coli MBM7070. Mutation frequency was determined by the ratio between white and light blue colonies (which contain a mutant supF gene) and wild-type blue colonies. Three independent experiments were performed for each cell line and the results are shown in Table 1. The spontaneous mutation frequencies of the pZ189 vector replicated in the MMR-defective cell lines were all significantly increased in comparison to the MMR-proficient HeLa cells, with a 5-fold increase in both Clones 7 and 12. Furthermore, in accordance with the correction of the MMR defect in Clone 7 + chromosome 7, the mutation frequency of supF in these cells was reduced to approximately the value observed in the parental HeLa cells. It is interesting to note that a mutation frequency (5 × 10–4) of the same order of magnitude as Clone 7 and Clone 12 was obtained when the vector was passaged through hPMS2–/hMSH6– HEC1-A cells. Although it is hard to identify the reference cell line for HEC1-A cells, this slightly increased value would be consistent with the presence of both hPMS2 and hMSH6 MMR mutations in these cells.
Table 1. Spontaneous mutagenesis in MMR-proficient and MMR-deficient cells.
| HeLa | Clone 12 | Clone 7 | Clone 7 + chromosome 7 | HEC1-A | |
|---|---|---|---|---|---|
| Number of mutants/total colonies | |||||
| Experiment I | 8/103 000 | 17/71 500 | 21/80 000 | 5/53 000 | 28/55 000 |
| Experiment II | 24/301 600 | 17/58 000 | 13/35 000 | 7/62 500 | 18/33 500 |
| Experiment III | 40/821 800 | 16/57 600 | 33/157 000 | 37/75 000 | |
| Total | 72/1 226 400 | 50/170 000 | 67/209 300 | 12/115 500 | 83/163 500 |
| supF mutant | |||||
| Frequency | 0.58 × 10–4 | 2.9 × 10–4 | 3.2 × 10–4 | 1 × 10–4 | 5 × 10–4 |
| Range | 0.5–0.8 | 2.3–2.9 | 2.1–3.7 | 0.9–1.1 | 4.9–5.4 |
| Increase versus HeLa | 5 | 5.5 | 1.7 | ||
In conclusion, replication of the pZ189 vector in several MMR-deficient cells leads to an increased mutation frequency.
Molecular analysis of mutations in the pZ189 shuttle vector
Mutant colonies derived from replication of the plasmid in Clone 12 cells (36 colonies) and in the parental HeLa cell line (44 colonies) were further analyzed to identify the type of mutations (Table 2). Alterations in gel mobility were observed in a large fraction of mutant plasmids recovered from HeLa cells. This value was significantly lower in pZ189 passaged through Clone 12 (27/44 versus 11/36, respectively; Fisher’s exact test, P < 0.008).
Table 2. Frequency of spontaneous supF mutants after passage in hPMS2-defective and hPMS2-proficient cells.
| Mutant | Mutation frequency (×10–5)a | Fold-increase | |||
|---|---|---|---|---|---|
| HeLa | Clone 12 | HeLa | Clone 12 | ||
| Mutants analyzed | 44 (100%) | 36 (100%) | 5.87 | 29.4 | 5 |
| Mutants with altered mobility | 27 (61%) | 11 (30%) (P < 0.008) | 3.60 | 8.98 | 2.5 |
| Mutants sequenced | 17 | 25 | |||
| Mutations fully characterized | |||||
| Single base substitutions | 7 (15.8%) | 9 (24.4%) | 0.93 | 7.35 | 7.8 |
| Tandemb | 1 (2.2%) | 0 | 0.13 | ||
| Multiplec | 2 (4.4%) | 11 (30%) (P < 0.003) | 0.27 | 8.98 | 33.2 |
| Small deletions (>10, <100 bp) | 6 (13.4%) | 0 (P < 0.03) | 0.80 | ||
| Frameshifts | 1 (2.2%) | 5 (14%) (P = 0.08) | 0.13 | 4.08 | 31.4 |
| Multiple mutations | 2 | 11 | |||
| Base substitutions (%) | 4 (55%) | 14 (55%) | |||
| Insertion/deletions (%) | 3 (45%) | 13 (45%) | |||
aMutation frequency is defined as the ratio of white or light blue colonies to the total.
bMutants with two adjacent mutations.
cMutants with more than two mutations in the supF gene.
Molecular analysis of the supF gene of mutant plasmids of apparently normal size was performed by automatic sequencing. The comparison of the mutational spectra of parental HeLa and Clone 12 cells indicates that loss of hPMS2 is associated with several differences in the distribution of the mutants among different classes. The major difference is an increase in the fraction of multiple mutations in plasmids recovered from Clone 12 cells compared to those from HeLa cells (11/36 versus 2/44; Fischer’s exact text, P < 0.003). An increase in the proportion of frameshifts was also observed in pZ189 replicated in Clone 12, although this difference did not reach statistical significance. The increases in these two mutational classes accompanied a concomitant significant decrease in the frequency of small deletions (between 10 and 100 bp) (0/36 in Clone 12 versus 6/44 in HeLa cells; Fischer’s exact text, P < 0.03).
The mutator phenotype of Clone 12 most affected (>30-fold increase) multiple mutations, which were equally divided into base substitutions and frameshifts. The relative distribution in these two classes was, however, unaffected by loss of MMR (Table 2). Frameshift mutations, in particular those leading to loss of one base, also accumulated in the vector replicated in the hPMS2-defective cells, while a more moderate increase in mutation frequency affected single base substitutions.
Tables 3 and 4 present the types of spontaneous mutations found in the supF gene of pZ189 replicated in HeLa and Clone 12, respectively. A rigorous statistical test, which takes into account both the mutation type and its position (20), indicated a significant difference between the spectra derived from MMR+ and MMR– cells (P < 0.002). A paired comparison of mutants divided into three classes of mutations by Fisher’s exact test confirmed the previous analysis (Table 5).
Table 3. Spontaneous mutational spectrum at the supF locus in PZ189 replicated in HeLa cells.
| Mutant | Mutation | Sequencea | Position |
|---|---|---|---|
| Single bp substitutions | |||
| 2 | GC→TA | GCCGTCA | 144 |
| 5 | GC→TA | TTCGAAG | 156 |
| 29 | GC→CG | TTTGATA | 43 |
| 32 | GC→CG | GTTCGAA | 163 |
| 40 | GC→CG | GCAGACT | 129 |
| 58 | GC→CG | ACTCTAA | 133 |
| 65 | GC→TA | AATCTGC | 139 |
| Multiple mutations | |||
| 3 | +T | GCA+TGAC | 128 |
| +A | CCC+AACC | 177 | |
| 37 | –C | TCCCGAG | 110 |
| GC→TA | AATCTGC | 139 | |
| GC→AT | CGTCATC | 146 | |
| GC→AT | CATCGAC | 149 | |
| GC→TA | AATCCTT | 168 | |
| Tandem mutations | |||
| 24 | GC→CG | AATCCTT | 168 |
| GC→TA | ATCCTTC | 169 | |
| Frameshifts | |||
| 4 | –C | CACCACC | 179 |
| Small deletions | |||
| 14 | –14 bp | CCT<>GAG | 97–110 |
| 28 | –77 bp | TTC<>GCA | 11–88 |
| 34 | –29 bp | GCG<> TCA | 115–144 |
| 41 | –11 bp | GGT<>CCC | 162–172 |
| 66 | –82 bp | GAC<>AAA | 153–235 |
| 71 | –10 bp | TGG<>CGG | 104–113 |
aThe mutated base(s) is underlined.
Table 4. Spontaneous mutational spectrum at the supF locus in PZ189 replicated in hMPS2-defective Clone 12 cells.
| Mutant | Mutation | Sequencea | Position |
|---|---|---|---|
| Single bp substitutions | |||
| 8 | GC→AT | AAGGGAG | 123 |
| 11 | AT→TA | CCAAAGG | 120 |
| 13 | GC→TA | TTCCCGA | 109 |
| 18 | GC→CG | TTCGAAT | 164 |
| 27 | GC→CG | TTTGATA | 43 |
| 28 | GC→TA | AAAGGGA | 122 |
| 30 | GC→CG | GCAGACT | 129 |
| 33 | GC→AT | ATCCTTC | 169 |
| 45 | GC→TA | CTTCCCC | 172 |
| Multiple mutations | |||
| 1 | GC→CG | TTTGATA | 43 |
| GC→AT | TTCCCGA | 63 | |
| AT→TA | TCTAAAT | 135 | |
| 2 | GC→CG | TTTGATA | 43 |
| GC→AT | TTCCCGA | 63 | |
| GC→CG | CCCGATA | 65 | |
| AT→TA | TCTAAAT | 135 | |
| 3 | GC→CG | CTGCCGT | 142 |
| +C | TCC+CTTC | 169–170 | |
| 6 | +G | AAG+GCAT | 88–89 |
| +G | GCA+GTTA | 90–91 | |
| +T | CGA+TATC | 165–166 | |
| 12 | +AC | TGG+ACGGT | 103–104 |
| GC→AT | ATCCTTC | 169 | |
| 19 | –A | CCAAAGG | 119–121 |
| –C | TCCCCCA | 172–176 | |
| 21 | TA→CG | CTGTGGT | 98 |
| GC→TA | CCACCAC | 178 | |
| 23 | +G | GGT+GGGG | 102–105 |
| –C | TCCCCCA | 172–176 | |
| 24 | GC→CG | TTTGATA | 43 |
| +T | TTC+TCCG | 63 | |
| –G | CCCGATA | 65 | |
| 25 | –G | CCCGAGC | 111 |
| +T | AGC+TGGC | 114–115 | |
| 46 | GC→TA | CCACCAC | 178 |
| GC→TA | CCACCAT | 181 | |
| Frameshifts | |||
| 9 | –GA | TAT–GATGC | 48–49 |
| 20 | +AG | GAA+AGGGT | 159 |
| 22 | –C | TCCCCCA | 172–176 |
| 29 | –C | TCCCCCA | 172–176 |
| 35 | –C | TCCCCCA | 172–176 |
aThe mutated base(s) is underlined.
Table 5. Comparison of the distribution of mutations in different classes.
| HeLa | Clone 12 | |
|---|---|---|
| Total number of mutants | 17 | 25 |
| Total number of mutations | 22 | 41 |
| Base pair substitutions | 11a | 23 |
| Frameshifts | 4 | 18 (P = 0.055) |
| Small deletions | 6 | 0 (P < 0.0011) |
| Base pair substitutions | ||
| Transitions | 2 (15%) | 6 (26%) |
| GC:AT | 2 (15%) | 5 (22%) |
| AT:GC | 0 | 1 (4%) |
| Transversions | 11 (85%) | 17 (74%) |
| GC:TA | 6 (46%) | 6 (26%) |
| GC:CG | 5 (39%) | 8 (35%) |
| AT:CG | 0 | 0 |
| AT:TA | 0 | 3 (13%) |
| Frameshifts | ||
| Insertions | 2 (50%) | 9 (50%) |
| Deletions | 2 (50%) | 9 (50%) |
aThe tandem mutation was considered a single event.
Transversions strongly predominated over transitions in both HeLa and Clone 12 cells (Table 5). In accordance with previous reports, the vast majority of base substitutions were targeted at GC base pairs in both MMR+ and MMR– cells. A similar distribution between gain and loss of bases among frameshift mutants (~50% in each class) was also found. When the Cariello comparison was applied by mutational class, the two spectra were indistinguishable when either the types of base pair substitutions or the frameshifts alone were considered (P = 0.6 and 0.5, respectively, for base pair substitutions and frameshifts). In contrast, the two spectra appeared again to be significantly different when these classes of mutation were considered together (P < 0.04). Thus the loss of hPMS2 confers on the spontaneous mutation spectrum a complex pattern of changes which affect more than one mutational class.
DISCUSSION
Loss of hPMS2 function in the two methylation-tolerant variants of HeLa cells was associated with a MMR defect and an increase in spontaneous mutagenesis in the supF gene. The mutation frequency was 5-fold higher compared to the parental HeLa cells. A similar mutator phenotype was observed in the hPMS2/hMSH6-defective cell line HEC1-A. Since their isolation, the hPMS2 deficient Clones 7 and 12 have not been extensively cultured. For this reason, they are likely to provide a better reflection of the effects of a MMR defect than comparisons between unrelated tumor cell lines. Although the mutator effects are modest, they do not simply reflect the use of an episomal target gene. Similar moderate increases in mutation are observed in a supF gene integrated in several tissues of PMS2–/– mice (13).
Loss of hPMS2 function in either Clone 12 or HEC1-A cells is associated with much larger increases in mutation rates at the HPRT gene (5.4 and 290 × 10–7/cell/generation respectively, versus 0.27–0.5 × 10–7 in MMR-proficient cells). This increase is largely due to the accumulation of frameshifts and single base substitutions (11,15; our unpublished results). Local DNA sequences can strongly influence the frequency of mutations and therefore the apparent magnitude of the mutator phenotype associated with loss of MMR. Although the particular secondary structure of this tRNA gene should facilitate misalignment of the primer/template during DNA replication, the short runs of mononucleotides present in the supF gene are not particularly susceptible to slippage replication errors. Significantly, an increase in the length of two mononucleotide runs in the supF gene from four and five G and C residues to seven and eight, respectively, led to a 100-fold increase in mutation frequency in several tissues of PMS2–/– mice (13). Similarly, only when a 29 bp mononucleotide poly(G) run was introduced in the tRNA sequence of supF was an increase in single base deletions observed in the hMSH6-defective MT1 cells (16). Thus, unless modified, supF is not a particularly good target gene to identify the mutator phenotype associated with loss of MMR.
Our results clearly show that the spectrum of mutations observed in the supF gene replicated in hPMS2-defective Clone 12 is significantly different from that in wild-type HeLa cells. Despite the limited overall increase in mutation frequency, there are major differences within specific classes of mutation. In particular, multiple mutations, which include both base substitutions and frameshifts, are considerably over-represented in the mutational spectrum of the hPMS2-defective cell line. Spontaneous multiple mutations are generally rare events in intrachromosomal genes such as HPRT or APRT, whereas they constitute a detectable fraction of spontaneous mutations in the supF gene replicated episomally in mammalian cells (23,24). The mechanism of formation of these mutations in the supF gene is not clear. Spontaneous and UV-induced multiple mutations in the supF gene are strongly reduced when the vector is replicated in Xeroderma pigmentosum cells (24). These results led to the hypothesis that nucleotide excision repair introduces nicks which direct an error-prone polymerase to fill in gaps in the DNA at sites of lesion excision (25). Our data indicate that MMR and nucleotide excision repair have opposite effects on the formation of these mutations. If the origin of these mutations is a localized loss of fidelity of DNA replication, triggered either by the presence of some spontaneous DNA damage in the template or the consequence of strong pause sites for the polymerases due to special features of the DNA secondary structure, it is not surprising that loss of MMR leads to an increased frequency of these mutations. The increase in multiple mutations observed in the hPMS2-defective cell line involves both frameshift and base substitutions. Inspection of the sequences flanking mutations of either class suggests that misalignment-mediated mutagenesis (26) could explain more than 50% of these events. Looping out of a base on the template strand (either coding or non-coding) and insertion of a base complementary to the base 5′ of the mutation could explain 14 of 27 mutations (52%) present in the multiple mutations of Clone 12 (mutants 2, 3, 12, 19, 21, 23 and 46). A similar misalignment of primer and template DNA is compatible with the formation of mismatches responsible for four of seven mutations (57%) present in the multiple mutants of HeLa cells (mutants 3 and 37). A comparable percentage of frameshifts within multiple mutations in MMR-proficient and MMR-deficient cells can be explained by the slippage hypothesis of Streisinger and Owen (27). We consider that loss of MMR is therefore most likely associated with an increased number of multiple mutations because it fails to correct slipped-mispaired replication errors.
Our data also suggest that the mechanism of formation of multiple and single base substitutions is different. First, less than 20% of single base mutations in both MMR-proficient and MMR-deficient HeLa cells can be explained by misalignment-mediated mutagenesis. Furthermore, the more moderate increase in the frequency of single versus multiple base substitutions in the hPMS2-defective clone suggests that MMR plays a less prominent role in avoidance of these mutations.
Deletions are one of the most frequent mutations in this as well as in other vectors replicated in mammalian cells (between 30 and 50%) (23,24,28). No increase in this class of mutations was detected in hPMS2-defective cells. The origin of the deletions has been ascribed to introduction of nicks, gaps or double-strand breaks in the plasmid DNA during transfection in the absence of a proper chromatin structure more than to gapped structures occurring during DNA replication. Analysis of spontaneous deletions of the supF gene (29) identified the involvement of both repeated sequences and palindromic or quasi-palindromic sequences as possible contributors to the specificity of deletions (30). Thus, if deletions mainly arise in this target gene as a consequence of recombination events, our data suggest that hPMS2 is not preventing this recombination process. It is interesting that this same clone showed increased rates of intrachromosomal homologous recombination, exclusively due to gene conversion events, at an integrated vector (15). This result suggests that different modes of recombination control spontaneous deletions in the supF gene replicated episomally and recombination of integrated plasmid sequences.
Loss of hMSH6 and hPMS2 function has a differential impact on spontaneous mutagenesis. A comparison of mutational spectra at the HPRT gene in hMutLα- and hMutSα-defective human tumor cell lines identified striking differences in the types of mutation affected (Table 6). In a spontaneous mutational spectrum of normal diploid fibroblasts base substitutions represent >85% of mutations while frameshifts are a minority (10%) (31). Loss of hMLH1 (HCT116 ) or hPMS2 + hMSH6 (HEC1-A) is associated with a much larger increase in frameshift mutagenesis than loss of hMSH6 alone or hMSH2 (7–9,11). Indeed, mostly transitions and transversions accumulated in the hMSH6-defective DLD1 and HEC1-B cell lines (8,11). A difference in the mutator phenotype can also be detected in the supF gene replicated in hMSH6- and hPMS2-defective cell lines. Loss of hMSH6 in MT1 cells does not alter mutation frequency relative to MMR-proficient TK6 cells (16), suggesting that several types of mutagenic intermediates can be repaired by the MutSβ (hMSH2/hMSH3) complex. The clear supF mutator phenotype in the hPMS2-defective HeLa cells affects base substitutions as well as frameshifts (Table 6). By comparison between MT1/TK6 and hPMS2–/HeLa cells we can infer that a considerable proportion of insertion/deletion mispairs and base mismatches are recognized by MutSβ and are processed via the hMLH1/hPMS2 heterodimer.
Table 6. Relative spontaneous mutation rates in MMR-proficient and MMR-deficient human cell lines.
| HPRT genea | Mutation rate (×10–8) | |||
|---|---|---|---|---|
| Frameshifts | Transitions | Transversions | ||
| Normal human fibroblasts | 0.45 | 2.3 | 1.23 | |
| hMutSα-defective | ||||
| LoVo (hMSH2–) | 20 | 80 | ||
| DLD1 (hMSH6–) | 100 | 880 | 836 | |
| HEC1-B (hMSH6–) | 60 | 170 | 250 | |
| hMutLα-defective | ||||
| HCT116 (hMLH1–) | 634 | 580 | 195 | |
| HEC1-A (hPMS2–/HMSH6–) |
1600 |
840 |
550 |
|
| supF geneb | Mutation frequency (×10–5) | |||
| Frameshifts | Base substitutions | Deletions | ||
| |
|
Multiple |
Single |
|
| HeLa | 0.13 | 0.27 | 0.93 | 0.8 |
| Clone 12 (hPMS2–) | 4.08 | 8.98 | 7.35 | <0.8 |
aSpontaneous mutation rates at the HPRT gene for normal human fibroblasts are taken from Bhattacharyya et al. (8) and the distribution in mutational classes from McGregor et al. (31). Mutation rates for LoVo cells are derived from Malkhosyan et al. (7), for DLD1 and HCT116 from Malkhosyan et al. (7), Bhattacharyya et al. (8) and Ohzeki et al. (9) and for HEC1-A and HEC1-B from Glaab et al. (11).
bMutation frequencies at the supF gene replicated in HeLa and Clone 12 are taken from Table 1.
The yeast MMR system is a good model for its human counterpart. The most recently discovered yeast MMR protein, MLH3, forms an alternative complex with MLH1. The recent discovery of the human homolog of MLH3 (4) and the demonstration that it participates in an alternative to hMutLα increases the number of potential branches in the human MMR pathway. It is therefore necessary to define the complexes which participate in the repair of different mismatches. Our data on the supF gene indicate that hPMS2, and therefore hMutLα, plays an important role in the repair of slipped-mispaired replication errors which include both DNA mismatches and loops of 1 base.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Prof. J. Jiricny for providing the antisera to hMSH6 and hMSH2 and to P. Karran for helpful discussions. This work was partially supported by a grant from the Associazione Italiana Ricerca sul Cancro.
REFERENCES
- 1.Kolodner R.D. and Marsischky,G.T. (1999) Curr. Opin. Genet. Dev., 9, 89–96. [DOI] [PubMed] [Google Scholar]
- 2.Jiricny J. (2000) Nature Genet., 24, 6–8. [DOI] [PubMed] [Google Scholar]
- 3.Flores-Rozas H. and Kolodner,R.D. (1998) Proc. Natl Acad. Sci. USA, 95, 12404–12409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lipkin S.M., Wang,V., Jacoby,R., Banerjee-Basu,S., Baxevanis,A.D., Lynch,H.T., Elliott,R.M. and Collins,F.S. (2000) Nature Genet., 24, 27–35. [DOI] [PubMed] [Google Scholar]
- 5.Palombo F., Iaccarino,I., Nakajima,E., Ikejima,M., Shimada,T. and Jiricny,J. (1996) Curr. Biol., 6, 1181–1184. [DOI] [PubMed] [Google Scholar]
- 6.Genschel J., Littman,S.J., Drummond,J.T. and Modrich,P. (1998) J. Biol. Chem., 273, 19895–19901. [DOI] [PubMed] [Google Scholar]
- 7.Malkhosyan S., McCarty,A., Sawai,H. and Perucho,M. (1996) Mutat. Res., 316, 249–259. [DOI] [PubMed] [Google Scholar]
- 8.Bhattacharyya N., Ganesh,A., Phear,G., Richards,B., Skandalis,A. and Meuth,M. (1995) Hum. Mol. Genet., 4, 2057–2064. [DOI] [PubMed] [Google Scholar]
- 9.Ohzeki S., Tachibana,A., Tatsumi,K. and Kato,T. (1997) Carcinogenesis, 18, 1127–1133. [DOI] [PubMed] [Google Scholar]
- 10.Lettieri T., Marra,G., Aquilina,G., Bignami,M., Crompton,N.E., Palombo,F. and Jiricny,J. (1999) Carcinogenesis, 20, 373–382. [DOI] [PubMed] [Google Scholar]
- 11.Glaab W.E., Risinger,J.I., Umar,A., Barrett,J.C., Kunkel,T.A., Carrett,J.C. and Tindall,K.R. (1998) J. Biol. Chem., 41, 26662–26669. [DOI] [PubMed] [Google Scholar]
- 12.Risinger J.I., Umar,A., Glaab,W.E., Tindall,K.R., Kunkel,T.A. and Barrett,J.C. (1998) Cancer Res., 58, 2978–2981. [PubMed] [Google Scholar]
- 13.Narayanan L., Fritzell,J.A., Baker,S.M., Liskay,R.M. and Glazer,P.M. (1997) Proc. Natl Acad. Sci. USA, 94, 3122–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ceccotti S., Aquilina,G., Macpherson,P., Yamada,M., Karran,P. and Bignami,M. (1996) Curr. Biol., 6, 1528–1531. [DOI] [PubMed] [Google Scholar]
- 15.Ciotta C., Ceccotti,S., Aquilina,G., Humbert,O., Palombo,F., Jiricny,J. and Bignami,M. (1998) J. Mol. Biol., 276, 738–743. [DOI] [PubMed] [Google Scholar]
- 16.Tobi S.E., Levy,D.D., Seidman,M.M. and Kraemer,K.H. (1999) Carcinogenesis, 20, 1293–1301. [DOI] [PubMed] [Google Scholar]
- 17.Aquilina G., Hess,P., Fiumicino,S., Ceccotti,S. and Bignami,M. (1995) Cancer Res., 55, 2569–2575. [PubMed] [Google Scholar]
- 18.Aquilina G., Fiumicino,S., Zijno,A., Martinelli,S., Overkamp,W.J.I., Zdzienicka,M., Oshimura,M., Wild,C.P. and Bignami,M. (1997) Mutat. Res., 385, 115–126. [DOI] [PubMed] [Google Scholar]
- 19.Karran P., Macpherson,P., Ceccotti,S., Dogliotti,E., Griffin,S. and Bignami,M. (1993) J. Biol. Chem., 268, 15878–15886. [PubMed] [Google Scholar]
- 20.Ceccotti S., Dogliotti,E., Gannon,J., Karran,P. and Bignami,M. (1993) Biochemistry, 32, 13664–13672. [DOI] [PubMed] [Google Scholar]
- 21.Adams W.T. and Skopek,T.R. (1987) J. Mol. Biol. ,194, 391–396. [DOI] [PubMed] [Google Scholar]
- 22.Cariello N.F., Piegorsch,W.W., Adams,W.T. and Skopek,T.R. (1994) Carcinogenesis, 15, 2281–2285. [DOI] [PubMed] [Google Scholar]
- 23.Razzaque A., Mizusawa,H. and Seidman,M.M. (1983) Proc. Natl Acad. Sci. USA, 80, 3010–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bredberg A., Kraemer,K.H. and Seidman,M.M. (1986) Proc. Natl Acad. Sci. USA, 83, 8273–8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seidman M.M., Bredberg,A., Seetharam,S. and Kraemer,K.H. (1987) Proc. Natl Acad. Sci. USA, 84, 4944–4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kunkel T.A. and Soni,A. (1988) J. Biol. Chem., 263, 14784–14789. [PubMed] [Google Scholar]
- 27.Streisinger G. and Owen,J. (1985) Genetics, 109, 633–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seidman M.M., Dixon,K., Razzaque,A., Zagursky,R.J. and Berman,M.L. (1985) Gene, 389, 233–237. [DOI] [PubMed] [Google Scholar]
- 29.Moraes E.C., Keyse,S.M. and Tyrrell,R.M. (1990) Carcinogenesis, 11, 283–293. [DOI] [PubMed] [Google Scholar]
- 30.Glickman B.W. and Ripley,L.S. (1984) Proc. Natl Acad. Sci. USA, 81, 512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McGregor W.G., Maher,V.M. and McCormick,J.J. (1991) Somat. Cell Mol. Genet., 17, 463–469. [DOI] [PubMed] [Google Scholar]