

Abstract
Replication of DNA is fraught with difficulty and chromosomes contain many lesions which may block movement of the replicative machinery. However, several mechanisms to overcome such problems are beginning to emerge from studies with Escherichia coli. An important enzyme in one or more of these mechanisms is the RecG helicase, which may target stalled replication forks to generate a four-stranded (Holliday) junction, thus facilitating repair and/or bypass of the original lesion. To begin to understand how RecG might catalyse regression of fork structures, we have analysed what the catalytically active form of the enzyme may be. We have found that RecG exists as a monomer in solution as measured by gel filtration but when bound to junction DNA the enzyme forms two distinct protein–DNA complexes that contain one and two protein molecules. However, mutant inhibition studies failed to provide any evidence that RecG acts as a multimer in vitro. Additionally, there was no evidence for cooperativity in the junction DNA-stimulated hydrolysis of ATP. These data suggest that RecG functions as a monomer to unwind junction DNA, which supports an ‘inchworm’ rather than an ‘active rolling’ mechanism of DNA unwinding. The observed in vivo inhibition of wild-type RecG by mutant forms of the enzyme was attributed to occlusion of the DNA target and correlates with the very low abundance of replication forks within an E.coli cell, even during rapid growth.
INTRODUCTION
The extent to which replication of chromosomal DNA encounters problems is only just beginning to be appreciated. The inherently high levels of processivity of the replicative machinery is well known and it had been assumed until recently that this is translated into a high degree of processivity in the duplication of entire chromosomes. However, in Escherichia coli it is now thought that replication forks assembled at the normal origin of replication, oriC, rarely complete duplication of the chromosome without running into blocks to their progression (1). Chemical damage to the template DNA from both endogenous and environmental sources is known to be a potent block to fork progression. Proteins bound ahead of the fork also hinder advance of the replicative machinery. A major source of such protein blocks are RNA polymerases stalled at lesions in the DNA or at specific template sequences (2,3). This raises the question of how a rapidly growing cell balances the requirement for high levels of gene expression with the need for efficient genome duplication. Failure to do so may lead to genetic instability, with potentially lethal consequences for the cell.
Nucleotide excision repair systems allow removal of many types of DNA damage (4), thus reducing the chances of a replication fork encountering problems. However, when a fork does hit a block to its progression, the cell must repair the damaged fork to allow replication to restart. Translesion synthesis is known to occur via specialised DNA polymerases with the ability to insert nucleotides opposite lesions. Some of these have reduced fidelity, which means that their activity may lead to potentially harmful mutations (5–7). An alternative mechanism may be lesion tolerance by RecA in which gaps in the newly replicated DNA are repaired by strand exchange with the intact sister duplex (8). However, recent studies have revealed that a major route to repairing damaged forks might be via regression of the stalled fork structure to form a four-stranded (Holliday) junction, which may allow repair enzymes to access the site of the original damage (3,9). Even if the original lesion could not easily be repaired, recombination systems exist in E.coli that would allow replication to proceed. The Holliday junction formed by fork regression could be targeted by the RuvABC helicase/endonuclease (9,10). Cleavage of the Holliday junction by RuvC would release a free duplex DNA end which could be processed by the RecBCD complex to generate a single-stranded 3′-tail (11). Formation of a RecA nucleoprotein filament on this single-stranded DNA would lead to strand exchange with a homologous duplex DNA (12), either the newly replicated sister duplex or, in a rapidly dividing E.coli cell, another chromosome. The 3′-end of the invading strand in the resulting D-loop is known to be specifically bound by the primosome assembly factor PriA (13,14), resulting in recruitment of the replicative machinery to generate a replication fork (15), whilst the Holliday junction at the other end of the D-loop could be resolved by RuvABC (16,17). Thus replication would be restarted.
How might a stalled replication fork regress to form a Holliday junction? In vitro RecG helicase specifically targets model replication fork structures to stimulate cleavage by RuvC, a Holliday junction-specific endonuclease, whilst genetic data have shown that recG is necessary for the efficient regression of stalled replication forks in vivo (3). However, if RecG sets up Holliday junctions from stalled forks to be subsequently cleaved by RuvABC, it would be expected that single deletions in either recG or ruv would have the same, probably highly defective, DNA repair phenotype as a recG ruv double mutant. In fact, the double mutant has a much more severe DNA repair defect than either single mutant (18). This, and other in vivo observations, suggests that RecG provides an alternative pathway for replication restart that would not require a Holliday junction formed by regression of a fork to be cleaved by RuvABC (3). Possible mechanisms include template switching to facilitate lesion bypass and reversal of fork regression by RecG once the original blocking lesion has been repaired.
Homologues of RecG have been identified in a wide range of bacteria and also in plants (19) and the possibility exists that analogous enzymes exist in other eukaryotes. Indeed, forks have been shown to regress in eukaryotes (20) and evidence is accumulating that defects associated with this process may be connected with enhanced genetic instability (21–24). However, little is known about how regression might be catalysed by RecG. Current studies offer conflicting views about the mechanisms employed by helicases to unwind DNA (or RNA). The ‘active rolling’ model envisages a multimeric helicase with two DNA-binding sites, each present within different subunits of the multimer, which switch between single-stranded and duplex DNA binding (25). In contrast, the ‘inchworm’ model proposes that both DNA-binding sites are present within the same protein molecule, although this molecule could be a subunit of a larger complex (26,27). Thus the ‘active rolling’ model requires a helicase to be at least dimeric, whereas the ‘inchworm’ model has no such requirement. Although it is possible that different unwinding mechanisms are used by different helicases, the high degree of conservation of the so-called helicase motifs within this class of enzymes (28) suggests that this is unlikely. The structures of PcrA (29), Rep (30) and HCV helicase (31,32) all revealed a monomeric enzyme structure, even when bound to a DNA substrate, which lent support to the ‘inchworm’ model. Biochemical evidence also supports a monomeric functional form of PcrA (33), but other studies have suggested that Rep functions as a dimer (34,35). The situation is even more controversial for HCV helicase (36,37) and for UvrD (38–40), a helicase that shares 40% amino acid sequence identity with Rep, since different studies have provided conflicting data supporting one or the other catalytic model. Unfortunately, the existence of a class of helicases that are known to be hexameric has not clarified this issue since these enzymes are only distantly related to the monomeric/dimeric helicases (28) and the only structure available to date cannot distinguish between the different catalytic models (41). The unwinding mechanism employed by helicases remains the subject of intense debate.
Any study to determine the mechanism of RecG action must therefore begin by addressing the functional form of the enzyme. We therefore undertook to determine whether RecG acts as a monomer or as an oligomer during the unwinding of junction DNA. In this study we show that RecG exists as a monomer in solution but that it can form two complexes when bound to a Holliday junction that contain one or two RecG molecules. Furthermore, RecG mutants unable to hydrolyse ATP inhibit the activity of wild-type RecG in vivo, which could be explained by the formation of non-functional heterodimers. However, this efficient in vivo inhibition could not be reproduced in vitro, which argues against a functional oligomeric form of RecG. This conclusion was supported by the absence of any observed dependence of RecG ATPase activity upon protein concentration. The lack of evidence for a multimeric functional form of RecG suggests that RecG may function as a monomer to unwind junction DNA, which supports an ‘inchworm’ rather than an ‘active rolling’ mechanism.
MATERIALS AND METHODS
Construction of mutant recG genes
A maltose-binding protein (MBP)–RecGΔC32 fusion was constructed using the pMAL-c2 vector (New England Biolabs), in which the E.coli malE gene is expressed via an IPTG-inducible promoter, as follows. pPM112 (42), carrying a truncated recG gene (recGΔC47) missing the final 47 codons, was cleaved with NdeI and the ends filled-in with Klenow enzyme before cutting with HindIII. The recGΔC47 fragment produced was cloned into pBluescript II SK– (Stratagene) to create pPM115. The recGΔC47 BamHI fragment from pPM115 was subcloned into pMAL-c2 to form pPM117 so as to create an in-frame fusion of the malE and recGΔC47 genes. The SstII–HindIII fragment from a pET14b clone containing recGΔC32 (pAM228) (42) was subcloned into pPM117, replacing the SstII–HindIII recGΔC47 fragment. The final construct, pPM123, thus contained the malE gene fused in-frame to the 5′-end of the recGΔC32 gene.
To generate RecGK302A and RecGK302R, the recG SalI–KpnI fragment from pAM210, a derivative of the pT7-7 expression plasmid containing a recG gene with six engineered restriction sites (42), was subcloned into pALTER-1 (Promega). This construct was then used as a template for site-directed mutagenesis using the Altered Sites system (Promega). Codon 302, AAA, was mutated to GCA and to CGT to generate the recGK302A and recGK302R fragments, respectively. These were then subcloned into pAM210 to form pAM239 and pAM240, encoding RecGK302A and RecGK302R, respectively.
Protein purification
Wild-type RecG was purified as described (43). The RecG mutant proteins were expressed using a method for wild-type RecG, except that the host strain was a BL21(DE3) plysS strain carrying ΔrecG::kan (AM1125) (42). RecGK302A was purified using the method described above for the wild-type protein. RecGK302R was purified using this same method but with two more chromatography steps, which proved necessary to remove a persistent contaminating nuclease. After heparin–agarose chromatography, the RecGK302R-containing fractions from the phenyl–Sepharose column were dialysed against buffer A (50 mM Tris–HCl pH 7.5, 1 mM EDTA, 1 mM DTT) plus 0.1 M KCl and then passed through a 1 ml double-stranded DNA–cellulose column (Sigma) in the same buffer. RecGK302R eluted in the column flow-through and was then processed through single-stranded DNA–cellulose, Q-Sepharose and Sephacryl S200 columns as described for the wild-type protein. Finally, the RecGK302R from the gel filtration column was dialysed against buffer A, loaded onto a 2 ml S-Sepharose Fast Flow column (Amersham Pharmacia Biotech) and eluted with a 0–1 M KCl gradient in buffer A. RecGK302R eluted between 0.3 and 0.5 M KCl and was dialysed into buffer A plus 50% glycerol and stored at –80°C.
RecGΔC32 fused to MBP (MBPRecGΔC32) was expressed from pPM123 in E.coli strain DH5α by growing cells in 100 ml of LB broth with 40 µg/ml carbenicillin and 0.2% glucose at 25°C. When the absorbance at 650 nm reached 0.5, IPTG was added to 0.3 mM and incubation was continued for another 3 h. The cells were harvested by centrifugation, resuspended in buffer A and frozen at –80°C. After thawing the cell paste at 37°C, phenylmethylsulfonyl fluoride, pepstatin A and E64 were added to 0.5 mM, 1 and 5 µM, respectively, and the cells were sonicated on ice. Cell debris was removed by centrifugation and the supernatant was loaded onto a 3 ml heparin–agarose column. The column was washed with a 0–1 M KCl gradient in buffer A plus the above protease inhibitors. MBPRecGΔC32 eluted between 0.3 and 0.4 M KCl and was then passed through a 25 ml Superose 12 gel filtration column (Amersham Pharmacia Biotech) in buffer A plus 0.2 M KCl and protease inhibitors. The protein eluted with an apparent Mr of 100 000, which approximated to an expected Mr of 115 000 for a monomer, and was dialysed against buffer A plus 50% glycerol prior to storage at –80°C. Note that an amylose column was not used for purification since MBPRecGΔC32 had only a low affinity for this matrix.
Protein concentrations were performed using the Bradford assay (44) with bovine serum albumin as the standard and are expressed as moles of monomeric protein.
Gel filtration
Wild-type RecG was expressed and partially purified via ammonium sulphate precipitation and heparin–agarose chromatography (43). The protein was reprecipitated with 40% ammonium sulphate, resuspended in 5 ml of buffer A (see above) plus 200 mM KCl and then passed through a 0.45 µm filter unit (Millipore). RecG was present at an approximate concentration of 20 µM in this solution. The enzyme was then run through a 2.6 × 78 cm Sephacryl S200 HR gel filtration column (Amersham Pharmacia Biotech) using buffer A plus 200 mM KCl. Protein molecular weight markers were cytochrome c (12 400), equine myoglobin (17 000), carbonic anhydrase (29 000), chicken ovalbumin (44 000), bovine serum albumin (66 000) and bovine γ-globulin (158 000). The void volume was estimated using blue dextran. Markers were run under the same conditions as used for RecG. Kav was calculated from (Ve – Vo)/(Vt – Vo), where Ve is the elution volume, Vo the void volume and Vt the volume of the packed column bed.
DNA substrates
All DNA junctions were made using the method of Parsons et al. (45) in which one of the strands in each junction was labelled at the 5′-end with [γ32P]ATP. All DNA concentrations are in moles of junction substrate. The synthetic oligonucleotides used to construct the junctions were: (a) 5′-GTCGGATCCTCTAGACAGCTCCATGATCACTGGCACTGGTAGAATTCGGC-3′; (b) 5′-CAACGTCATAGACGATTACATTGCTACATGGAGCTGTCTAGAGGATCCGA-3′; (c) 5′-TGCCGAATTCTACCAGTGCCAGTGATGGACATCTTTGCCCACGTTGACCC-3′; (d) 5′-TGGGTCAACGTGGGCAAAGATGTCCTAGCAATGTAATCGTCTATGACGTT-3′; (e) 5′-TGCCGAATTCTACCAGTGCCAGTGAT-3′; (f) 5′-TAGCAATGTAATCGTCTATGACGTT-3′; (g) 5′-GACGCTGCCGAATTCTGGCTTGCTAGGACATCTTTGCCCACGTTGACCC-3′; (h) 5′-CAACGTCATAGACGATTACATTGCTAGGACATGCTGTCTAGAGACTATCGA-3′; (i) 5′-ATCGATAGTCTCTAGACAGCATGTCCTAGCAAGCCAGAATTCGGCAGCGT-3′. Junction 1, a Holliday junction with 25 bp duplex arms, was made using oligonucleotides (a)–(d). This junction lacked a homologous core and therefore the branch point was fixed at a defined position within the structure. Junction 2 was made using oligonucleotides (a), (b), (e) and (f) and, like junction 1, had a defined structure with no regions of homology around the branch point. Junction 3 was a Holliday junction with a homologous core of 12 bp through which the branch point was free to migrate (46), flanked by 19–20 bp heterologous arms, and was made by annealing oligonucleotides (d) and (g)–(i).
The unlabelled Holliday junction DNA (junction 3) for use in ATPase assays was made by annealing equal amounts of (d) and (g)–(i) as above. Efficient production of four-stranded structures was confirmed by gel electrophoresis through 10% polyacrylamide gels in Tris–borate–EDTA followed by ethidium bromide staining and by gel filtration chromatography through a 25 ml Superose 6 column (Amersham Pharmacia Biotech).
DNA binding and unwinding assays
Band shift assays to measure DNA binding were performed in the presence of EDTA as described (13). DNA unwinding assays were performed as previously described using 5 mM ATP and 5 mM magnesium chloride (43). The concentrations of enzyme and junction DNA were as indicated. All gels were quantified using a Molecular Dynamics PhosphorImager.
To measure the effects of RecGK302A upon the rate of unwinding catalysed by wild-type enzyme (Fig. 4C), the wild-type and mutant proteins were premixed in reaction buffer and incubated on ice for 5 min prior to addition to the reaction containing junction DNA. An aliquot was then immediately removed and deproteinised; this sample was taken as time 0. The reaction was then placed at 37°C and samples subsequently removed at the indicated times.
Figure 4.

In vivo and in vitro effects of ATPase-deficient RecG proteins on wild-type activity. (A) The effects of plasmids encoding wild-type RecG, RecGK302A and RecGK302R on the survival of UV-irradiated E.coli strains. A plasmid vector control (pT7-7) and plasmids encoding wild-type RecG (pGS772), K302A (pAM239) and K302R (pAM240) were transformed into (i) AB1157 wild-type (recG+ruv+), (ii) N3793 ΔrecG263 and (iii) N2057 ruvA60::Tn10 strains and the effect on cell survival upon exposure to UV light determined. (B) Effect of RecGK302A on wild-type RecG ATPase activity. Rates of ATP hydrolysis by 10 nM wild-type enzyme were measured in the presence of increasing concentrations of RecGK302A (indicated in nM) with 250 nM Holliday junction DNA (junction 3). ATP hydrolysis was measured as the amount of inorganic phosphate released per second. Error bars represent standard deviations from the mean. (C) Wild-type RecG helicase activity in the presence of RecGK302A. The rate of dissociation of 0.2 nM Holliday junction DNA (junction 3) by 0.01 nM wild-type RecG was monitored in the absence (circles) or presence of 0.1 (squares) or 1 nM (triangles) RecGK302A.
ATPase assays
The hydrolysis of ATP was detected by measurement of the release of inorganic phosphate with acidic ammonium molybdate and malachite green. The method was essentially that of Lanzetta et al. (47) except that Sterox was omitted. Typically, 80 µl reactions were established in 50 mM Tris–acetate, pH 8.0, 20 mM potassium acetate, 1 mM dithiothreitol, 5 mM magnesium acetate, 5 mM ATP and 100 µg µl–1 bovine serum albumin. Unlabelled Holliday junction DNA (junction 3, see above) was added at the indicated concentrations. This mixture was preincubated at 37°C for 1 min then RecG was added. A 10 µl aliquot was immediately removed and added to 800 µl of the ammonium molybdate/malachite green reagent. This was time 0. The RecG reaction was replaced at 37°C and further 10 µl samples removed at the indicated times. Each timed sample was incubated with the ammonium molybdate/malachite green reagent for 1 min at room temperature and then 100 µl of 34% sodium citrate solution was added. After 20 min at room temperature the absorbance at 660 nm was measured.
The effect of RecGK302A upon the ATPase activity of wild-type RecG was determined by measuring the rate of inorganic phosphate release over 2.5 min at 37°C. Wild-type and mutant enzymes were preincubated on ice for 5 min prior to adding to the reaction mixture containing 250 nM junction 3. ATP and magnesium acetate were both present at 5 mM.
To evaluate the effect of protein concentration upon the ATPase activity of wild-type RecG, we established that the reaction obeyed Michaelis–Menten kinetics. The Km with respect to junction 3 was estimated to be ~2 nM in the presence of saturating ATP and RecG concentrations (data not shown). Likewise, the Km for ATP was estimated at 0.2 mM in the presence of saturating DNA and RecG concentrations (data not shown). The rate of ATP hydrolysis was then measured at saturating ATP and junction DNA concentrations (5 mM and 250 nM, respectively) using the buffer system described above. Time 0 plus four other time points were taken for each reaction, with the length of the time course being increased at lower RecG concentrations to enhance detection of the reduced amounts of phosphate. The rates of ATP hydrolysis measured under these conditions were equal to the kcat values and the values shown are the means of at least two independent experiments. Throughout these experiments the concentration of magnesium acetate was 5 mM.
Determination of sensitivity to UV light
Sensitivity to UV light was determined as described previously (48). AB1157 (49), N3793 (50) and N2057 (51) were used as wild-type, ΔrecG263 and ruvA60::Tn10 strains, respectively. Derivatives of these strains carrying recG wild-type (pGS772) (52) or mutant plasmids (pAM239 and 240, see above), all based upon the pT7-7 expression vector (53), were grown and irradiated in LB medium containing 100 µg/ml ampicillin.
RESULTS
Native molecular weight of RecG and stoichiometry of binding to junction DNA
The RecG polypeptide has a molecular weight of 76 000 (54). To estimate the molecular weight of the native RecG species in solution, we performed gel filtration on partially purified enzyme (Fig. 1A). RecG eluted with an apparent molecular weight of 62 000, which indicated that the majority existed in the monomeric form in solution. Indeed, >95% of RecG eluted as a monomer as measured by this technique (data not shown). However, this alone cannot be used to argue that RecG functions as a monomer. Both Rep and HCV helicases are predominantly monomeric in solution but it has been argued that each forms oligomers when bound to their substrates (37,55). We therefore investigated the stoichiometry of RecG when bound to substrate DNA.
Figure 1.
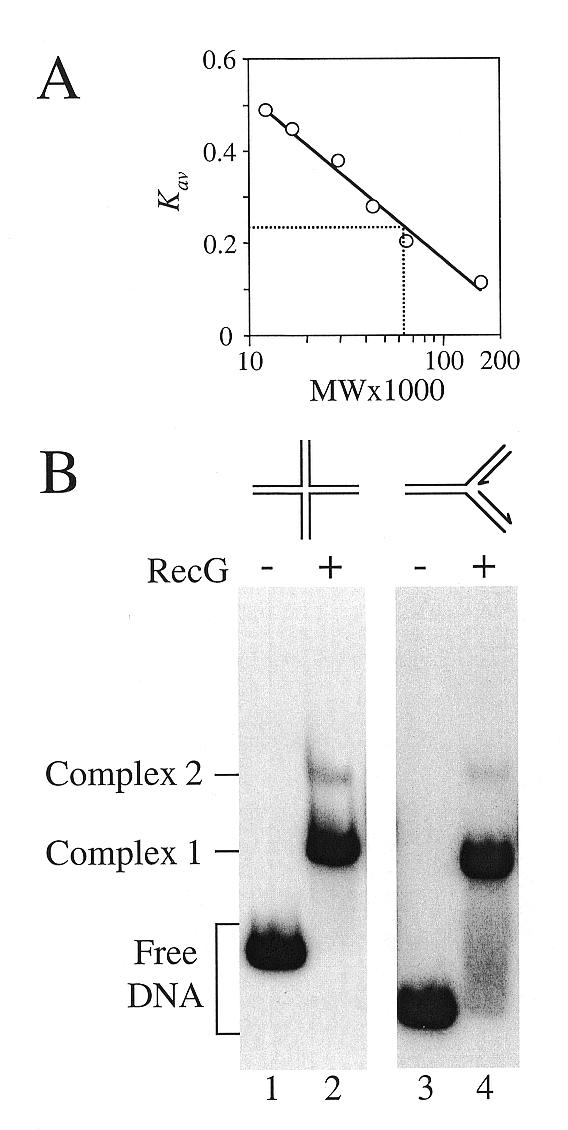
Native molecular weight of RecG and binding stoichiometry to junction DNA. (A) Gel filtration of RecG. The Kav value of RecG was compared to that of protein standards of known size to give an estimated molecular weight of 62 000. (B) Binding reactions containing 50 nM wild-type RecG and 0.2 nM junction 1 (lanes 1 and 2) or junction 2 (lanes 3 and 4) were analysed on band shift gels. The arrows on the fork DNA structure indicate the 3′-termini of the DNA strands. The reactions were electrophoresed on the same gel but for clarity the intervening lanes have been deleted.
Band shift assays have shown that RecG forms two distinct protein–DNA complexes when bound to synthetic Holliday junction structures (42,56,57; Fig. 1B, lane 2). To determine the generality of this observation, we analysed RecG binding to forked DNA structures designed to mimic possible features of damaged replication forks. Two complexes were also observed with such structures, both of which migrated to approximately the same position as those seen with Holliday junction DNA (Fig. 1B, lane 4). However, complex 1 predominated and complex 2 was seen only at higher RecG concentrations (data not shown), which again reflected the pattern seen with Holliday junction DNA (42,56,57).
Thus the pattern of binding of RecG to junction DNA appears to be similar regardless of the junction structure. We utilised Holliday junctions for further analysis of RecG catalysis since their greater stability as compared with synthetic fork structures facilitated many of the in vitro assays.
To determine the stoichiometry of RecG binding to junctions, we used a strategy developed by Lilley and co-workers (58,59) whereby the gene encoding the DNA-binding protein of interest is cloned into a plasmid expression vector fused to the 3′-end of the MBP gene, malE. Overexpression of the fusion product allows purification of a MBP–DNA-binding protein fusion whose molecular weight is increased by 43 000 and partial proteolysis of this fusion protein, or mixing of fusion and wild-type proteins, allows heteromultimer formation to be detected in band shift assays. We attempted to clone wild-type recG into the expression vector pMALc2 so as to produce a MBP–RecG fusion protein. However, all attempts failed at the point when a full-length recG sequence was ligated to the malE gene, which suggested that expression of the fusion product was somehow lethal to the cell. We also attempted to clone a gene for a mutant, non-functional RecG protein defective in ATP hydrolysis and DNA unwinding but which retained the ability to bind junction DNA with an efficiency equal to that of wild-type RecG (RecGK302A, see below). However, this also failed. Finally, we succeeded in cloning a recG gene in which the RecG product had 32 amino acids deleted from the C-terminus (RecGΔC32) (42). We had shown previously that RecGΔC32, fused to an N-terminal 20mer peptide containing six tandem histidine residues, unwound junction DNA very inefficiently but could still form both complexes 1 and 2 (42). We found that MBPRecGΔC32, like HisRecGΔC32, also had little or no junction helicase activity (Fig. 2A) but bound junction DNA with an affinity equal to that of wild-type RecG (Fig. 2B).
Figure 2.
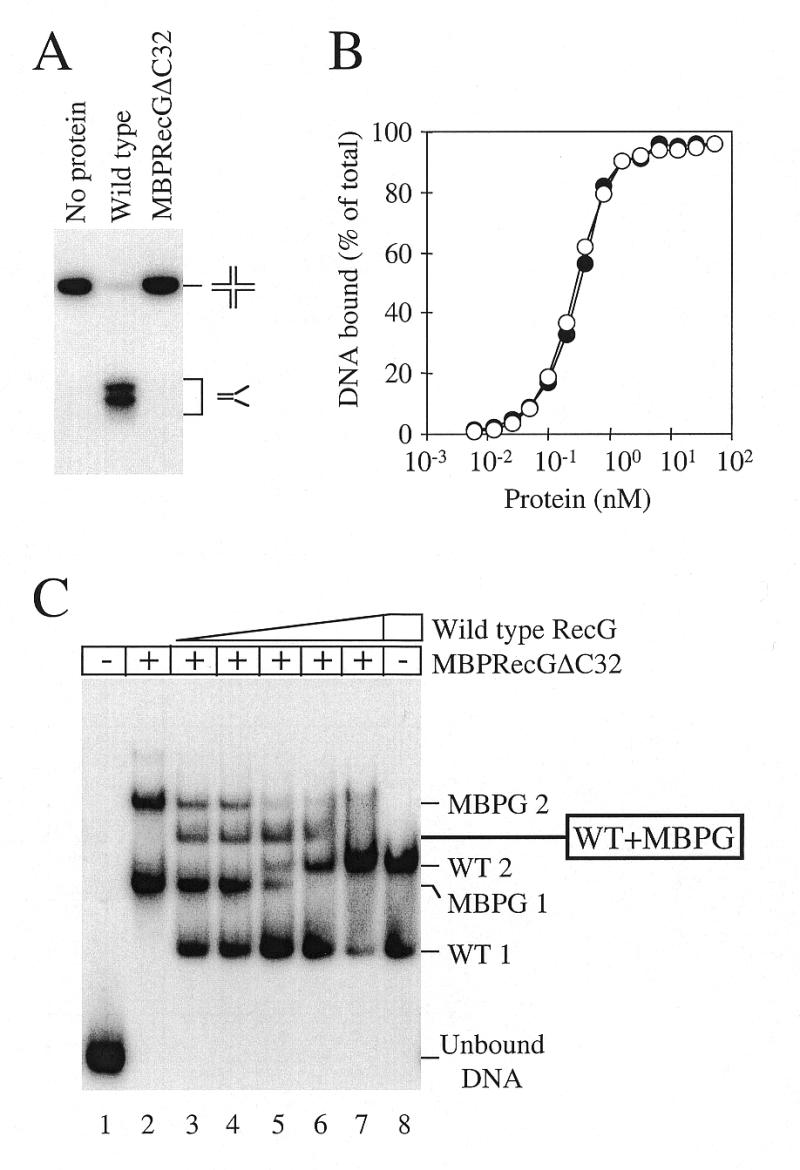
DNA binding and unwinding properties of a MBP–RecGΔC32 fusion. (A) Helicase activity of 100 nM wild-type RecG and 100 nM MBPRecGΔC32 on 0.2 nM junction 3 DNA. Intact junction DNA and flayed duplex products are marked. (B) Comparison of the DNA binding affinities of wild-type RecG (closed circles) and MBPRecGΔC32 (open circles) as measured by band shift assays with 0.2 nM Holliday junction DNA (junction 3). (C) Binding of Holliday junction DNA (junction 3) by a mix of wild-type RecG and MBPRecGΔC32. An aliquot of 50 nM MBPRecGΔC32 fusion polypeptide was bound to 0.2 nM DNA (lanes 2–7), forming two protein–DNA complexes designated MBPG 1 and MBPG 2. Aliquots of 5, 10, 20, 40 and 80 nM wild-type RecG were also added to lanes 3–7, respectively, and 80 nM to lane 8, forming the two protein–DNA complexes marked WT 1 and WT 2. In the presence of both wild-type and MBPRecGΔC32 a novel protein–DNA complex was detected whose mobility suggested it contained one molecule each of wild-type RecG and MBPRecGΔC32, as indicated. Both proteins were omitted from lane 1.
Purified MBPRecGΔC32 protein bound Holliday junction DNA to form two protein–DNA complexes designated MBPG 1 and 2 (Fig. 2C, lane 2). Upon titration of increasing amounts of wild-type RecG, the two protein–DNA complexes normally seen with wild-type RecG, WT 1 and 2, were also observed (Fig. 2C, lanes 3–8). However, a novel protein–DNA species was also detected when both wild-type RecG and MBPRecGΔC32 were present. This complex migrated between complex 2 seen with wild-type RecG and complex 2 formed by MBPRecGΔC32 (Fig. 2C, lanes 3–6). Since no other intermediate protein–DNA complexes were detected, the simplest interpretation was that this novel complex consisted of one molecule each of wild-type RecG and MBPRecGΔC32 bound to a Holliday junction. The corollary of this is that complexes WT 1 and WT 2 formed between wild-type RecG and Holliday junction DNA are one and two RecG molecules bound to a Holliday junction, respectively. Since RecG binds to Holliday junctions and forks with a very similar pattern (Fig. 1B) we conclude that complexes 1 and 2 detected with forked DNA also contain one and two RecG molecules, respectively.
In vitro characterisation of RecG proteins carrying mutations in helicase motif I
The gel filtration and DNA binding data for RecG suggested that the functional form of RecG may be either monomeric or dimeric. If dimeric, it would be predicted that mutant RecG proteins that cannot unwind DNA but can still bind junction DNA would be able to inhibit the function of wild-type RecG by the formation of non-functional heterodimers. If monomeric, no substantial inhibition would be observed. Such an approach has been used previously for the E.coli UvrD helicase (39), the HCV helicase (37) and the bacteriophage T7 helicase-primase (60). We therefore used this mutant inhibition approach to try and identify the catalytically active form of RecG.
Two mutant recG genes were constructed that had the codon for the lysine residue at amino acid position 302 altered to encode either alanine (RecGK302A) or arginine (RecGK302R) (Fig. 3A). This lysine resides within the highly conserved helicase motif I and has been shown to interact with the phosphate tail of ATP in PcrA and Rep (27,30). Moreover, mutation of this residue is known to reduce or abolish ATP hydrolysis and consequently unwinding activity in a range of helicases (61–64). RecGK302A and RecGK302R proteins were overexpressed in a recG deletion strain of E.coli and purified to apparent homogeneity. Both enzymes behaved in an identical manner to wild-type RecG during gel filtration chromatography, which supports a native monomeric state for RecG in solution. The mutant proteins also bound Holliday junction DNA with approximately the same affinity as wild-type RecG (Fig. 3B). However, as expected, both mutant enzymes lacked detectable helicase or ATPase activity on junction structures (Fig. 3C and D).
Figure 3.
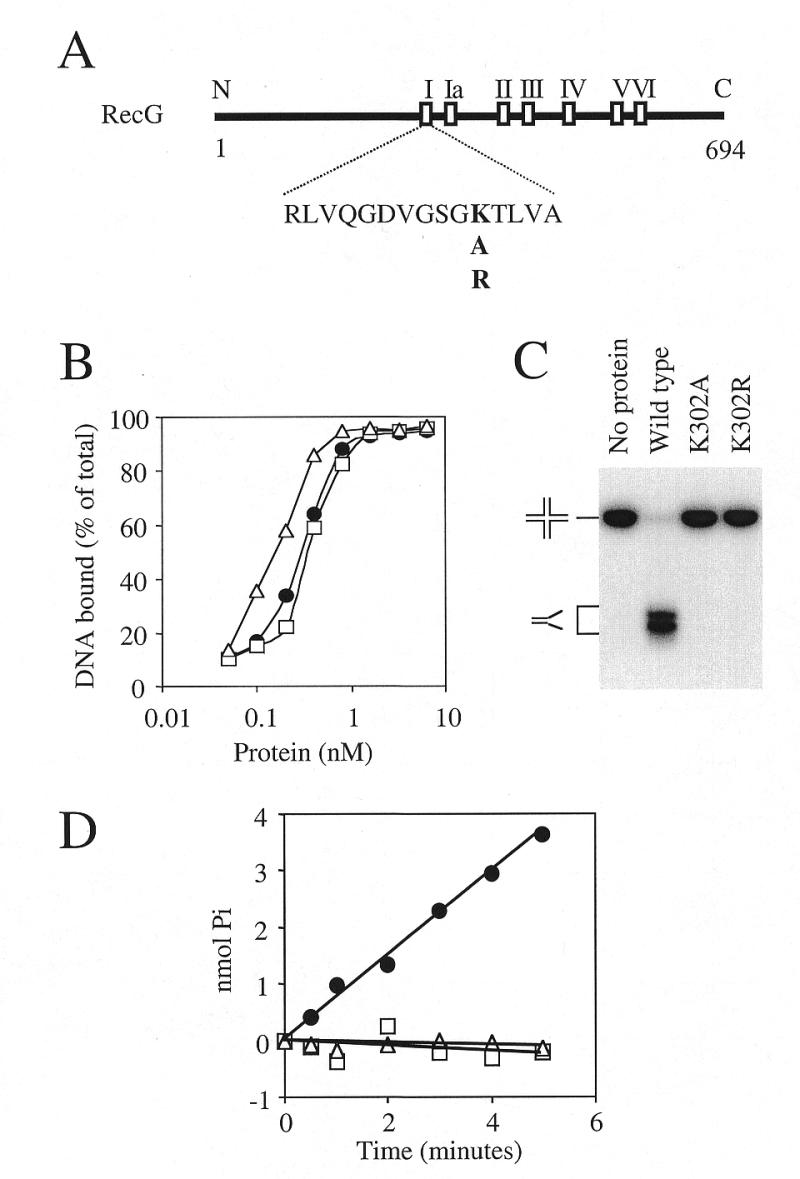
In vitro properties of RecG mutant proteins deficient in ATP hydrolysis. (A) Schematic diagram of the RecG polypeptide with the locations of the seven motifs conserved across a wide range of DNA and RNA helicases. The numbers refer to amino acid residues and the N- and C-termini are also indicated. The amino acid sequence of motif I is shown together with the lysine residue within this sequence which was mutated to alanine (RecGK302A) and arginine (RecGK302R). (B) Comparison of the DNA binding affinities as measured in band shift assays of wild-type RecG (circles), K302A (squares) and K302R (triangles) with 0.2 nM Holliday junction DNA (junction 3). (C) Helicase activity of 100 nM wild-type RecG, K302A and K302R on 0.2 nM Holliday junction DNA (junction 3). The positions of the junction DNA and flayed duplex products after gel electrophoresis are indicated. (D) Rate of ATP hydrolysis by 10 nM wild-type RecG (circles), K302A (squares) and K302R (triangles) in the presence of 100 nM Holliday junction DNA (junction 3). ATP hydrolysis was measured as the release of inorganic phosphate in a 10 µl sample.
In vivo inhibition of wild-type RecG activity by ATPase-deficient mutant enzymes
If the functional form of RecG is oligomeric then RecGK302A and RecGK302R should possess the ability to form non-functional heterodimers with the wild-type enzyme, given that all three proteins are monomeric in solution, and should therefore be able to undergo subunit exchange. Formation of such heterodimers would inhibit RecG helicase activity both in vivo and in vitro. The two mutant versions of recG were assayed for their ability to inhibit the function of wild-type recG in vivo by analysing their effects on survival of UV-irradiated cells. Wild-type recG is needed for efficient DNA repair and probably plays a role in maintaining replication in the face of damage to the template DNA (3,48). Any inhibition of wild-type recG function should therefore manifest itself as a reduction in the ability of the cells to survive exposure to UV.
Plasmids encoding either the mutant proteins or wild-type RecG were introduced into recG+ruv+ (wild-type), ΔrecG263::kan and ruvA60::Tn10 strains and tested for sensitivity to UV light. The plasmid encoding wild-type RecG (pGS772) had no effect upon the survival of the wild-type strain but corrected the DNA repair defect seen in the recG mutant (Fig. 4A, i and ii). pGS772 also slightly enhanced DNA repair in the ruvA strain (Fig. 4A, iii). pAM239 and pAM240, encoding the RecGK302A and K302R proteins, respectively, both gave very different results. In the wild-type strain both plasmids reduced survival, indicating that the two mutant proteins were inhibiting some aspect of DNA repair (Fig. 4A, i). However, in the recG strain neither pAM239 nor pAM240 had any effect when compared with the control strain carrying the vector plasmid (Fig. 4A, ii). These observations indicate that the RecGK302A and K302R proteins were inhibiting DNA repair in the wild-type background in a recG-specific manner. This recG specificity was supported by the observation that the mutant RecG proteins made a ruvA strain extremely sensitive to UV light (Fig. 4A, iii). Previous studies showed that the UV sensitivity of a ruv mutant can be dramatically increased by eliminating RecG activity (18). The RecGK302A and K302R proteins appeared to mimic this effect.
In vitro catalysis by wild-type RecG and inhibition by RecGK302A
The recG-specific inhibition of DNA repair mediated by the helicase-deficient mutant proteins could be explained by their ability to form inactive heteromultimers with wild-type RecG, thus supporting a dimeric functional form of RecG. However, it is also possible that the mutant proteins bound to and thus blocked DNA structures normally targeted by wild-type RecG. To distinguish between these possibilities, the effect of RecGK302A upon the ATPase activity of wild-type enzyme was determined in vitro.
Both RecGK302A and RecGK302R bound junction DNA in an apparently identical manner to wild-type enzyme (Fig. 3B), forming two protein–DNA complexes as seen with wild-type RecG (data not shown). This indicated that both mutant enzymes retained the ability to form a complex on junction DNA that contained two RecG molecules. Thus, if this complex does represent a dimeric functional form of RecG, the mutant enzymes retained the ability to form this putative dimer and would presumably be able to form a heterodimer with wild-type RecG. This assumption also forms the basis for any argument in which the oligomerisation of RecG is used to explain the dominant negative effect seen in vivo (Fig. 4A). The rate of ATP hydrolysis by wild-type enzyme would be expected to be reduced by 50% upon addition of an equal amount of RecGK302A, since half of the wild-type enzyme would be present as inactive heterodimers. However, there was no inhibition of wild-type RecG ATPase activity even in the presence of a 10-fold excess of RecGK302A (Fig. 4B). The reaction conditions used in these experiments also support the formation of Holliday junctions from fork structures by RecG (3) and are therefore likely to allow the formation of RecG species that are active as helicases. These data therefore argue against a dimeric functional form of RecG. However, the results in Figure 4B do not formally exclude this possibility since the individual subunits of a putative RecG dimer may hydrolyse ATP independently of each other but function as a dimer during any unwinding reaction.
This possibility was investigated by determining the effects of RecGK302A upon the ability of wild-type RecG to dissociate Holliday junctions. Again, a dimeric form of RecG would be expected to lead to the inhibition of wild-type RecG by 50% in the presence of an equal amount of RecGK302A. In the absence of the mutant protein, wild-type RecG efficiently unwound Holliday junction DNA (Fig. 4C). The initial rate of dissociation was only reduced to ~70% of the original level by addition of a 10-fold excess of RecGK302A. Neither preincubation of wild-type and mutant enzymes nor the order of their addition if not preincubated had any effect upon the level of inhibition (data not shown). Even when a 100-fold excess of RecGK302A was present, wild-type RecG could still dissociate a significant amount of the junction, although the initial rate was reduced to 25% of the level seen in the absence of the mutant enzyme. However, in this instance RecGK302A (1 nM) was present in excess over not only the wild-type enzyme (0.01 nM) but also the Holliday junction DNA (0.2 nM). This most probably reflects the situation in vivo (Fig. 4A), where higher levels of expression of the plasmid-encoded mutant proteins, in comparison to the single wild-type recG gene on the chromosome, would block access of wild-type RecG to its substrate DNA.
RecGK302A therefore fails to inhibit either the ATPase or the junction dissociation activities of the wild-type enzyme. The lack of any in vitro evidence for RecG dimers, and a plausible explanation for the observed in vivo inhibition, argues that RecG functions as a monomer both as an ATPase and as a helicase.
The effect of protein concentration upon wild-type RecG ATPase activity
The above in vitro studies assumed that RecGK302A retained the ability to form putative dimers with the wild-type enzyme. We therefore utilised a third assay to discriminate between monomeric and dimeric functional forms of RecG which did not rely on this assumption. Several previous studies have analysed whether the ATPase activity of a helicase depends upon the protein concentration (33,37–39). Any increase in kcat with increasing helicase concentration has been used to suggest that oligomerisation stimulates ATPase activity. We measured the kcat for ATP hydrolysis using from 1 to 64 nM wild-type RecG and saturating concentrations of ATP and DNA. However, no dependence upon protein concentration was observed (Fig. 5). The concentration range of RecG used in these measurements is also very effective in unwinding junction DNA (43; Fig. 4C) and must therefore allow formation of the active species of RecG, both for ATP hydrolysis and DNA unwinding. Although the results in Figure 5 could be explained by RecG already being a dimer at 1 nM, the gel filtration data presented above indicated that RecG is predominantly a monomer in solution even when at a concentration of 20 µM. It should also be noted that a monomer RecG–DNA complex predominates in band shift assays and that complexes containing two RecG molecules are only seen at very high protein concentrations (42,56,57; Fig. 1B). These observations argue against significant RecG dimerisation occuring at or below 1 nM. The lack of cooperativity observed here for the wild-type ATPase activity therefore lends support to the view that RecG monomers are functional ATPases and helicases.
Figure 5.

Hydrolysis of ATP by RecG does not display cooperativity. kcat was measured over a range of RecG concentrations, as indicated, in the presence of 250 nM Holliday junction DNA (junction 3). Error bars represent standard deviations from the mean.
DISCUSSION
Recent evidence that the function of RecG might be to unwind damaged replication forks to first generate and then branch migrate Holliday junctions raises the question of how this novel reaction might be accomplished. From previous work it is known that the minimal substrate efficiently recognised by RecG is a branched DNA structure with at least two duplex components (13,57). It follows that there must be multiple interactions between RecG and the DNA substrate to confer such specificity. Furthermore, a helicase reaction must proceed via two DNA-binding sites to allow contact with the substrate to be maintained. Whether a single RecG molecule is capable of this or whether an oligomeric form is required is central to understanding the mechanism of this novel junction-specific helicase. The data presented above have demonstrated that RecG exists predominantly as a monomer in solution but that two molecules of RecG can bind simultaneously to the same DNA junction. However, our failure to find any evidence of cooperativity in RecG ATPase activity or to detect efficient inhibition of wild-type ATPase or unwinding activity by mutant forms of the enzyme all support the conclusion that RecG can act as a monomer to catalyse the unwinding of branched DNA structures. This excludes an ‘active rolling’ mechanism of unwinding, since such a mechanism would require the functional enzyme to have at least two protein subunits (65). Instead, a monomeric RecG helicase is consistent with the ‘inchworm’ model of helicase catalysis and lends support to recent evidence that such a mechanism is responsible for helicase catalysis, at least for some enzymes (27,39).
The presence of the conserved helicase motifs within the C-terminal half of RecG (Fig. 3A), which are known to catalyse translocation along the DNA in PcrA and in Rep (27,30), indicates that this region of the protein is in intimate contact with the substrate DNA. Although the location of other domains of RecG which may interact with the DNA are not known, a previous study suggested that the specificity for junction DNA may reside within the N-terminal half of the protein (42). Any such interactions must therefore be coordinated in a single RecG molecule to promote junction unwinding. It should also be noted that the helicase motifs in both PcrA and Rep interact with single-stranded DNA to catalyse translocation along the DNA substrate. However, there are no stable regions of single-stranded DNA within a Holliday junction (66) nor are there predicted to be any in the fork structure used in Figure 1B. This might suggest that the helicase motifs in RecG do not interact with single-stranded DNA. However, this seems unlikely in view of the high level of sequence conservation within these motifs. An alternative is that any interaction with junction DNA initiates by the transient formation of single-stranded DNA within the junction. The formation of such single-stranded regions in the otherwise completely duplex Holliday junction structure used in many of the experiments described here (junction 3, see Materials and Methods) has been detected biochemically (67). If this process does facilitate the initial binding of RecG, it remains to be seen whether opening of the junction structure to allow RecG binding is spontaneous or is promoted by RecG itself. Once loaded onto the junction, RecG must then proceed to couple its translocation along the DNA to unwinding of the junction. How this is achieved is the subject of current studies.
The finding that mutant RecG proteins inhibit the function of wild-type RecG in vivo but not in vitro suggests that the normal DNA substrate for the wild-type enzyme is blocked by binding of the mutant proteins in vivo. This implies that the target for RecG activity is present at a relatively low concentration in vivo, given that RecG is not an abundant cellular protein (P.McGlynn and R.G.Lloyd, unpublished data) and correlates with the recent finding that RecG may act at damaged replication forks since, although there are likely to be multiple forks in a rapidly growing E.coli cell, their number will be low. This also raises the possibility that RecG might interact with components of the replicative apparatus so as to facilitate targeting of damaged forks. We are currently investigating this possibility.
Acknowledgments
ACKNOWLEDGEMENTS
The authors would like to thank Lynda Harris for superb technical support and Dr Ed Bolt for critical reading of the manuscript. This work was supported by a Medical Research Council programme grant to R.G.L. and Dr Gary Sharples.
REFERENCES
- 1.Sandler S.J. and Marians,K.J. (2000) J. Bacteriol., 182, 9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krasilnikova M.M., Samadashwily,G.M., Krasilnikov,A.S. and Mirkin,S.M. (1998) EMBO J., 17, 5095–5102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McGlynn P. and Lloyd,R.G. (2000) Cell, 101, 35–45. [DOI] [PubMed] [Google Scholar]
- 4.Friedberg E.C., Walker,G.C. and Siede,W. (1995) In Friedberg,E.C., Walker,G.C. and Siede,W. (eds), DNA Repair and Mutagenesis. ASM Press, Washington, DC.
- 5.Tang M., Bruck,I., Eritja,R., Turner,J., Frank,E.G., Woodgate,R., O’Donnell,M. and Goodman,M.F. (1998) Proc. Natl Acad. Sci. USA, 95, 9755–9760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reuven N.B., Tomer,G. and Livneh,Z. (1998) Mol. Cell, 2, 191–199. [DOI] [PubMed] [Google Scholar]
- 7.Wagner J., Gruz,P., Kim,S.R., Yamada,M., Matsui,K., Fuchs,R.P. and Nohmi,T. (1999) Mol. Cell, 4, 281–286. [DOI] [PubMed] [Google Scholar]
- 8.West S.C., Cassuto,E. and Howard-Flanders,P. (1981) Nature, 294, 659–662. [DOI] [PubMed] [Google Scholar]
- 9.Seigneur M., Bidnenko,V., Ehrlich,S.D. and Michel,B. (1998) Cell, 95, 419–430. [DOI] [PubMed] [Google Scholar]
- 10.West S.C. (1997) Annu. Rev. Genet., 31, 213–244. [DOI] [PubMed] [Google Scholar]
- 11.Kowalczykowski S.C. and Eggleston,A.K. (1994) Annu. Rev. Biochem., 63, 991–1043. [DOI] [PubMed] [Google Scholar]
- 12.Cox M.M. (1999) Prog. Nucleic Acid Res. Mol. Biol., 63, 311–366. [DOI] [PubMed] [Google Scholar]
- 13.McGlynn P., Al-Deib,A.A., Liu,J., Marians,K.J. and Lloyd,R.G. (1997) J. Mol. Biol., 270, 212–221. [DOI] [PubMed] [Google Scholar]
- 14.Nurse P., Liu,J. and Marians,K.J. (1999) J. Biol. Chem., 274, 25026–25032. [DOI] [PubMed] [Google Scholar]
- 15.Liu J. and Marians,K.J. (1999) J. Biol. Chem., 274, 25033–25041. [DOI] [PubMed] [Google Scholar]
- 16.Asai T., Bates,D.B. and Kogoma,T. (1994) Cell, 78, 1051–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuzminov A. (1999) Microbiol. Mol. Biol. Rev., 63, 751–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lloyd R.G. (1991) J. Bacteriol., 173, 5414–5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharples G.J., Ingleston,S.M. and Lloyd,R.G. (1999) J. Bacteriol., 181, 5543–5550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zou H. and Rothstein,R. (1997) Cell, 90, 87–96. [DOI] [PubMed] [Google Scholar]
- 21.Yamagata K., Kato,J., Shimamoto,A., Goto,M., Furuichi,Y. and Ikeda,H. (1998) Proc. Natl Acad. Sci. USA, 95, 8733–8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Defossez P.A., Prusty,R., Kaeberlein,M., Lin,S.J., Ferrigno,P., Silver,P.A., Keil,R.L. and Guarente,L. (1999) Mol. Cell, 3, 447–455. [DOI] [PubMed] [Google Scholar]
- 23.Chakraverty R.K. and Hickson,I.D. (1999) Bioessays, 21, 286–294. [DOI] [PubMed] [Google Scholar]
- 24.Lee S.K., Johnson,R.E., Yu,S.L., Prakash,L. and Prakash,S. (1999) Science, 286, 2339–2342. [DOI] [PubMed] [Google Scholar]
- 25.Wong I. and Lohman,T.M. (1992) Science, 256, 350–355. [DOI] [PubMed] [Google Scholar]
- 26.Yarranton G.T. and Gefter,M.L. (1979) Proc. Natl Acad. Sci. USA, 76, 1658–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Velankar S.S., Soultanas,P., Dillingham,M.S., Subramanya,H.S. and Wigley,D.B. (1999) Cell, 97, 75–84. [DOI] [PubMed] [Google Scholar]
- 28.Gorbalenya A.E. and Koonin,E.V. (1993) Curr. Opin. Struct. Biol., 3, 419–429. [Google Scholar]
- 29.Subramanya H.S., Bird,L.E., Brannigan,J.A. and Wigley,D.B. (1996) Nature, 384, 379–383. [DOI] [PubMed] [Google Scholar]
- 30.Korolev S., Hsieh,J., Gauss,G.H., Lohman,T.M. and Waksman,G. (1997) Cell, 90, 635–647. [DOI] [PubMed] [Google Scholar]
- 31.Yao N., Hesson,T., Cable,M., Hong,Z., Kwong,A.D., Le,H.V. and Weber,P.C. (1997) Nature Struct. Biol., 4, 463–467. [DOI] [PubMed] [Google Scholar]
- 32.Kim J.L., Morgenstern,K.A., Griffith,J.P., Dwyer,M.D., Thomson,J.A., Murcko,M.A., Lin,C. and Caron,P.R. (1998) Structure, 6, 89–100. [DOI] [PubMed] [Google Scholar]
- 33.Bird L.E., Brannigan,J.A., Subramanya,H.S. and Wigley,D.B. (1998) Nucleic Acids Res., 26, 2686–2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong I., Chao,K.L., Bujalowski,W. and Lohman,T.M. (1992) J. Biol. Chem., 267, 7596–7610. [PubMed] [Google Scholar]
- 35.Wong I. and Lohman,T.M. (1992) Science, 256, 350–355. [DOI] [PubMed] [Google Scholar]
- 36.Porter D.J., Short,S.A., Hanlon,M.H., Preugschat,F., Wilson,J.E., Willard,D.H.,Jr and Consler,T.G. (1998) J. Biol. Chem., 273, 18906–18914. [DOI] [PubMed] [Google Scholar]
- 37.Levin M.K. and Patel,S.S. (1999) J. Biol. Chem., 274, 31839–31846. [DOI] [PubMed] [Google Scholar]
- 38.Runyon G.T., Wong,I. and Lohman,T.M. (1993) Biochemistry, 32, 602–612. [DOI] [PubMed] [Google Scholar]
- 39.Mechanic L.E., Hall,M.C. and Matson,S.W. (1999) J. Biol. Chem., 274, 12488–12498. [DOI] [PubMed] [Google Scholar]
- 40.Ali J.A., Maluf,N.K. and Lohman,T.M. (1999) J. Mol. Biol., 293, 815–834. [DOI] [PubMed] [Google Scholar]
- 41.Sawaya M.R., Guo,S., Tabor,S., Richardson,C.C. and Ellenberger,T. (1999) Cell, 99, 167–177. [DOI] [PubMed] [Google Scholar]
- 42.Mahdi A.A., McGlynn,P., Lovett,S.D. and Lloyd,R.G. (1997) Nucleic Acids Res., 25, 3875–3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McGlynn P. and Lloyd,R.G. (1999) Nucleic Acids Res., 27, 3049–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bradford M. (1976) Anal. Biochem., 72, 248. [DOI] [PubMed] [Google Scholar]
- 45.Parsons C.A., Kemper,B. and West,S.C. (1990) J. Biol. Chem., 265, 9285–9289. [PubMed] [Google Scholar]
- 46.Connolly B., Parsons,C., Benson,F.E., Dunderdale,H.J., Sharples,G.J., Lloyd,R.G. and West,S.C. (1991) Proc. Natl Acad. Sci. USA, 88, 6063–6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lanzetta P.A., Alvarez,L.J., Reinach,P.S. and Candia,O.A. (1979) Anal. Biochem., 100, 95–97. [DOI] [PubMed] [Google Scholar]
- 48.Lloyd R.G. and Buckman,C. (1991) J. Bacteriol., 173, 1004–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bachmann B.J. (1996) In Neidhardt,F.C., Curtiss,R., Ingraham,J.L., Lin,E.C.C., Low,K.B., Magasanik,B., Reznikoff,W.S., Riley,M., Schaechter,M. and Umbarger,H.E. (eds), Escherichia coli and Salmonella Cellular and Molecular Biology, 2nd Edn. ASM Press, Washington, DC, pp. 2460–2488.
- 50.Al-Deib A.A., Mahdi,A.A. and Lloyd,R.G. (1996) J. Bacteriol., 178, 6782–6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shurvinton C.E., Lloyd,R.G., Benson,F.E. and Attfield,P.V. (1984) Mol. Gen. Genet., 194, 322–329. [DOI] [PubMed] [Google Scholar]
- 52.Lloyd R.G. and Sharples,G.J. (1993) EMBO J., 12, 17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tabor S. and Richardson,C.C. (1985) Proc. Natl Acad. Sci. USA, 82, 1074–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lloyd R.G. and Sharples,G.J. (1991) J. Bacteriol., 173, 6837–6843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chao K.L. and Lohman,T.M. (1991) J. Mol. Biol., 221, 1165–1181. [DOI] [PubMed] [Google Scholar]
- 56.Lloyd R.G. and Sharples,G.J. (1993) Nucleic Acids Res., 21, 1719–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Whitby M.C. and Lloyd,R.G. (1998) J. Biol. Chem., 273, 19729–19739. [DOI] [PubMed] [Google Scholar]
- 58.White M.F. and Lilley,D.M. (1996) J. Mol. Biol., 257, 330–341. [DOI] [PubMed] [Google Scholar]
- 59.Pohler J.R.G., Giraud-Panis,M.E. and Lilley,D.M.J. (1996) J. Mol. Biol., 260, 678–696. [DOI] [PubMed] [Google Scholar]
- 60.Notarnicola S.M. and Richardson,C.C. (1993) J. Biol. Chem., 268, 27198–27207. [PubMed] [Google Scholar]
- 61.Sung P., Higgins,D., Prakash,L. and Prakash,S. (1988) EMBO J., 7, 3263–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pause A. and Sonenberg,N. (1992) EMBO J., 11, 2643–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zavitz K.H. and Marians,K.J. (1992) J. Biol. Chem., 267, 6933–6940. [PubMed] [Google Scholar]
- 64.George J.W., Brosh,R.M.,Jr and Matson,S.W. (1994) J. Mol. Biol., 235, 424–435. [DOI] [PubMed] [Google Scholar]
- 65.Lohman T.M. and Bjornson,K.P. (1996) Annu. Rev. Biochem., 65, 169–214. [DOI] [PubMed] [Google Scholar]
- 66.Ortiz-Lombardia M., Gonzalez,A., Eritja,R., Aymami,J., Azorin,F. and Coll,M. (1999) Nature Struct. Biol., 6, 913–917. [DOI] [PubMed] [Google Scholar]
- 67.West S.C. (1995) Nature, 373, 27–28. [DOI] [PubMed] [Google Scholar]