

Abstract
INTRODUCTION:
Plasma-measured tau phosphorylated at threonine 217 (p-tau217) is a potential non-invasive biomarker of Alzheimer’s disease (AD). We investigated whether plasma p-tau217 predicts subsequent cognition and positron emission tomography (PET) markers of pathology in autosomal dominant AD.
METHODS:
We analyzed baseline levels of plasma p-tau217 and its associations with amyloid-PET, tau-PET, and word list delayed recall measured 7.61 years later in non-demented age- and education-matched presenilin-1 E280A carriers (n = 24) and non-carrier (n = 20) family members.
RESULTS:
Carriers had higher plasma p-tau217 levels than non-carriers. Baseline plasma p-tau217 was associated with subsequent amyloid and tau PET pathology levels and cognitive function.
DISCUSSION:
Our findings suggest that plasma p-tau217 predicts subsequent brain pathological burden and memory performance in presenilin-1 E280A carriers. These results provide support for plasma p-tau217 as a minimally invasive diagnostic and prognostic biomarker for AD, with potential utility in clinical practice and trials.
Keywords: Autosomal Dominant Alzheimer’s Disease, Presenilin-1, Dementia, Tau Pathology, Blood Biomarkers
1. Background
In vivo imaging of tau neurofibrillary tangle accumulation via positron emission tomography (PET) has improved the study, diagnosis, and monitoring of early Alzheimer’s disease (AD) (1). Measuring tau via cerebrospinal fluid (CSF) samples has similarly shown utility as an early and specific measure of AD pathology (2). However, there is a critical need for sensitive, cost-effective, and minimally invasive biomarkers of AD. Plasma-based measures of phosphorylated tau (p-tau), which are less costly and invasive compared to PET or CSF, increase early in the disease process and reliably discriminate between AD and other neurodegenerative diseases (3). Further research is needed to determine the utility of plasma p-tau as a preclinical biomarker.
Plasma-measured tau phosphorylated at site threonine 217 (p-tau217) has emerged as a particularly promising and specific AD biomarker (3–6). Growing evidence shows elevated plasma p-tau217 across preclinical to clinical disease stages (5,7–9), particularly in adults with high brain β-amyloid (Aβ) load (5,10–12). Further, p-tau217 may have better diagnostic ability than other plasma biomarkers in early stages (e.g., p-tau181, neurofilament light (NfL), Aβ40, Aβ42) (5,7,13,14). We previously showed in a kindred with autosomal dominant Alzheimer’s disease (ADAD) due to a mutation on the presenilin-1 (PSEN1) gene that increased levels of plasma p-tau217 were able to distinguish carriers from age-matched non-carriers 20 years prior to their estimated age of symptom onset (7).
The association between tau measured through plasma and PET is important to characterize, as tau-PET remains the gold standard for in vivo quantification of tau pathology for research and clinical purposes. Plasma p-tau217 is correlated with concurrent tau-PET in individuals with high Aβ, mild cognitive impairment, and AD (5,15–17). Notably, a recent study found increased plasma p-tau217 in cognitively unimpaired individuals with positive Aβ-PET imaging and negative tau-PET, suggesting that plasma p-tau217 levels become abnormal before accumulation is detectable via tau-PET (15).
Less is known, however, about the association between plasma p-tau217 and subsequent tau-PET accumulation. In sporadic AD (e.g., older adults with high Aβ), one study reported an association between plasma p-tau217 and increasing tau-PET in the entorhinal cortex on average 1.6 years later (15). A second study, similarly, examining measurements one to two years from baseline, reported an association with increasing medial temporal lobe tau-PET (18). Further research into this association is critical to determine whether p-tau217 may serve as an early marker of AD pathology and aid in early detection. If plasma p-tau217 can predict future tau-PET at a longer interval, clinical trials may be able to enroll individuals at an earlier stage.
In this study, we leveraged a cohort of carriers of the PSEN1 E280A mutation for ADAD to examine whether baseline levels of plasma p-tau217 are associated with subsequent PET-based markers of AD pathology in the brain, measured on average 7.61 years following plasma collection. Secondarily, we examined the association between plasma p-tau217 and subsequent cognition. These findings would inform the use of plasma p-tau217 as a biomarker for the selection, monitoring, and evaluation in clinical trials and other investigations.
2. Methods
2.1. Study design and participants
This cohort study included 24 PSEN1 E280A mutation carriers (23 Aβ-pathology positive) and 20 age- and education-matched non-carriers from the same kindred, enrolled in the Massachusetts General Hospital (MGH) COLBOS (Colombia-Boston) longitudinal biomarker study. Participants were recruited from the Alzheimer’s Prevention Initiative (API) registry of familial AD, which currently includes more than 6,000 living members of the kindred and approximately 1,200 mutation carriers (19). Characteristics of this kindred have been well characterized (20–22). Notably, the onset of clinical impairment occurs in midlife, with the median age of onset of mild cognitive impairment (MCI) at 44 years old and dementia at 49 years old (21).
Participants with a diagnosis of dementia at the time of blood sample collection or with a significant medical, psychiatric, or neurological disorder (e.g., stroke, seizures, substance abuse, and other disorders that affect motor, visuospatial or cognitive abilities) were excluded. Neither the participants nor raters were informed of the genetic status of the individuals. This study was approved by the institutional ethics review boards of the University of Antioquia in Medellin, Colombia, and the MGH in Boston, MA, USA. All participants provided written informed consent before inclusion in the study.
Blood sampling was collected at baseline. Neuroimaging and cognitive memory assessment were completed at follow-up (mean = 7.61 ± 4.05 years). All participants were cognitively unimpaired at baseline. At follow up, all non-carriers and 18 carriers were cognitively unimpaired, and six carriers progressed to MCI. Participants were considered cognitively unimpaired if they had a Mini-Mental State Examination (MMSE) (23) score ≥26 and a Functional Assessment Staging Test (FAST) (24) score of 1 or 2. Impaired carriers were defined as having a FAST score of 3.
2.2. Plasma p-tau217 assay
Plasma was collected in the morning (without fasting) at the University of Antioquia in aliquots of 1 mL. Samples were stored at −80°C. Concentrations of plasma p-tau217 were measured using immunoassays at Lilly Research Laboratories, using the MSD platform (Meso Scale Discovery) as previously described [7]. Biotinylated-IBA493 was used as a capture antibody and SULFO-TAG-4G10-E2 (anti-Tau) as the detector. Additional details of the plasma P-tau217 analysis are described in Palmqvist et al., 2020, Supplemental Material (7).
2.3. Clinical and cognitive assessments
Clinical assessments were performed at the University of Antioquia. Participants underwent a clinical interview and were administered the MMSE, FAST, and a Spanish version of the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) word list, which has been adapted for this Colombian population (25). Other cognitive tests were performed in these participants as part of a Spanish neuropsychological test battery used by the Grupo de Neurociencias de Antioquia – Colombia (26) (data not shown in this article). Cognitive measures were administered in Spanish by a neuropsychologist, or a psychologist trained in neuropsychological assessment. Neurological examinations were performed by a neurologist or by a general practitioner trained in assessing neurodegenerative disorders.
2.4. Image acquisition and processing
All participants in this study traveled from Colombia to Boston (USA) for positron emission tomography (PET) at the MGH. PET data were acquired on a Siemens ECAT HR+ (3D mode; 63 image planes; 15.2 cm axial field of view; 5.6 mm transaxial resolution; 2.4 mm slice interval).
11C-Pittsburgh compound B (11C-PiB) PET was acquired with an 8.5 to 15 mCi bolus injection followed immediately by a 60-minute dynamic acquisition in 69 frames (12×15 seconds, 57×60 seconds). 11C-PiB PET data were quantified as the distribution volume ratio (DVR) with cerebellar grey as a reference region; regional time-activity curves were used to compute regional DVRs for each region of interest (ROI) using the Logan graphical method applied to data obtained between 40 and 60 minutes after injection (27). 11C-PiB retention was assessed using a large cortical ROI aggregate that included frontal, lateral temporal, and retrosplenial cortices as described previously (28).
[F18] Flortaucipir (FTP) was acquired between 80 and 100 minutes after a 9.0 to 11.0 mCi bolus injection in 4 separate 5-minute frames. [F18] FTP-specific binding was expressed in FreeSurfer ROIs as the standardized uptake value ratio (SUVR) to the cerebellum. The spatially transformed SUVR PET data were smoothed with an 8mm Gaussian kernel to account for individual anatomic differences (29). SUVR values were represented graphically on vertices at the pial surface. A priori ROIs were inferior temporal cortex, entorhinal cortex, and precuneus (30,31).
Partial volume correction was applied using the extended Muller-Gartner method implemented in FreeSurfer for both PiB and FTP (32).
2.5. Genotyping
Genomic DNA was extracted from the blood by standard protocols, and PSEN1 E280A characterization was done at the University of Antioquia using methods previously described (33). Genomic DNA was amplified with the primers PSEN1-S 5′ AACAGCTCAGGAGAGGAATG 3′ and PSEN1-AS 5′ GATGAGACAAGTNCCNTGAA 3′. We used the restriction enzyme BsmI for restriction fragment length polymorphism analysis. Each participant was classified as a PSEN1 E280A carrier or non-carrier.
2.6. Statistical analysis
Analyses and visualizations were performed in R (version 4.0.3) and used a significance threshold of two-tailed p < .05. Group differences in normally distributed continuous variables were compared using independent two-sample t-tests (Levene’s test was used for examining equality of variances). Group differences in non-normally distributed continuous variables were compared using Mann-Whitney U tests. Chi-square tests were used for categorical variables. Spearman correlation was used to test associations between continuous variables in the whole sample (reported in the main text), with and without covariates (age, sex, time between PET and blood measures). Correlations were additionally conducted within each group and presented in the supplementary materials. One potential outlying value was identified (mutation carrier who converted to MCI at follow-up), and correlation analyses were repeated excluding this carrier, with consistent results (see Table S1).
Pearson correlation was used in exploratory analyses of plasma p-tau217 and vertex-wise Aβ- and tau-PET within carriers. PET images were normalized to standard (MNI) space and projected onto the average surface, and vertex-wise values were sampled at the midpoint of the gray matter. Partial volume correction was applied using the extended Muller-Gartner method implemented in FreeSurfer (32). Results were displayed as −log10(p), significant at cluster-wise p < .05 (minimum cluster extent = 100 mm2) after false discovery rate (FDR) correction for multiple comparisons. Clustering and multiple comparisons corrections were performed using FreeSurfer tools.
3. Results
3.1. Baseline sample characteristics and plasma p-tau217 levels
A total of 24 PSEN1 E280A carriers and 20 non-carriers were included in analyses (Table 1). Carriers and non-carriers did not differ in age at baseline (t(42) = 1.71, p = .094), years of education (t(42) = 1.41, p = .167), or sex (χ2 = 0.78, p = .378). Carriers had higher plasma p-tau217 levels than non-carriers (W = 122, p = .005). Neuroimaging occurred on average 7.61 years after plasma sample collection (median = 6.00 years), with a statistically significant longer interval for non-carriers than for carriers (t(42) = 2.07, p = .044).
Table 1.
Baseline demographic and plasma p-tau217 data
| Non-carriers (n=20) | Carriers (n=24) | Test Statistic | p value | 95% CI | |
|---|---|---|---|---|---|
| Age at baseline (years) | 27.6±6.98 | 31.1±6.81 | t(42) = 1.71 | .094 | [−7.78, 0.63] |
| Education (years) | 11.3±4.10 | 9.38±4.63 | t(42) = 1.41 | .167 | [−0.81, 4.56] |
| Sex (Male/Female) | 11/9 | 10/14 | Χ2(1) = 0.78 | .378 | |
| p-tau217 (pg/ml) | 2.53±1.39 | 5.27±4.69 | W = 122 | .005 | [−3.07, −0.42] |
| Time between blood samples and PET scans (years) | 8.95±4.19 | 6.50±3.65 | t(42) = 2.07 | .044 | [−4.83, −0.07] |
Note: Means and standard deviations given for age, education, p-tau217, and follow-up time. Group differences were assessed using t-tests for normally distributed variables and Wilcoxon rank sum test for non-normally distributed variables.
Abbreviations: CI, Confidence Interval.
3.2. Group differences in biomarkers at follow-up assessment
Of the 24 carriers, 18 remained cognitively unimpaired and six converted to MCI at follow-up. Carriers exhibited elevated neuroimaging biomarkers at follow-up (Table 2), namely higher cortical Aβ DVR (W = 11, p < .001) and regional tau-PET SUVR in the entorhinal cortex (W = 51, p < .001), inferior temporal cortex (W = 141, p = .019), and precuneus (W = 86, p < .001). CERAD word list delayed recall (W = 357.5, p = .005) and MMSE scores (W = 366, p = .002) were lower in carriers than non-carriers.
Table 2.
Follow-up neuroimaging and cognitive data
| Non-carriers (n=20) | Carriers (n=24) | Test Statistic | p value | 95% CI | |
|---|---|---|---|---|---|
| 11C PiB-PET (DVR) | 1.11±0.04 | 1.69±0.39 | W = 11 | < .001 | [−0.69, −0.37] |
| Entorhinal cortex FTP (SUVR) | 1.02±0.12 | 1.52±0.45 | W = 51 | <.001 | [−0.58, −0.26] |
| Inferior temporal FTP (SUVR) | 1.21±0.13 | 1.56±0.68 | W = 141 | .019 | [−0.29, −0.01] |
| Precuneus FTP (SUVR) | 1.05±0.13 | 1.72±1.02 | W = 86 | < .001 | [−0.59, −0.11] |
| Mini Mental State Examination | 29.0±0.97 | 26.7±3.50 | W = 366 | .002 | [<0.01, 2.00] |
| CERAD delayed recall | 7.85±1.18 | 5.17±3.21 | W = 357.5 | .005 | [1.00, 4.00] |
Note: Means and standard deviations given. Group differences were assessed using t-tests for normally distributed variables and Wilcoxon rank sum test for non-normally distributed variables.
Abbreviations: CERAD, Consortium to Establish a Registry for Alzheimer’s Disease neuropsychological battery; CI, Confidence Interval; DVR, distribution volume ratio; FTP, flortaucipir; PiB, Pittsburgh Compound B; SUVR, standardized uptake value ratio.
3.3. Associations between plasma p-tau217 levels and age, cortical Aβ, regional tau, and cognition
To assess the utility of plasma p-tau217 as an early AD biomarker, we examined its associations with various concurrent and subsequent markers of AD in the whole sample. Older age at baseline was associated with higher levels of plasma p-tau217, r = 0.41, p = .006, 95% confidence interval (CI) [0.13, 0.63] (Fig. 1A). Higher baseline plasma p-tau217 was associated with lower MMSE (r = −0.57, p < .001, CI[−0.74, −0.33]; Fig.1B) and delayed recall (r = −0.52, p < .001, CI[−0.71, −0.26]; Fig. 1C) scores at follow-up. Higher baseline plasma p-tau217 was also associated with higher subsequent PET measures of cortical Aβ (r = 0.55, p < .001, CI[0.30, 0.73]; Fig. 2A) and tau in all ROIs: entorhinal cortex (r = 0.47, p = .001, CI[0.20, 0.67]; Fig. 2B), inferior temporal cortex (r = 0.44, p = .003, CI[0.16, 0.65]; Fig. 2B), and precuneus (r = 0.53, p < .001, CI[0.28, 0.72]; Fig. 2D). We additionally examined these relationships separately for carriers and non-carriers, finding that associations between p-tau217 and age, PET pathology, and cognition were only significant for carriers (Table S2).
Figure 1.

Plasma p-tau217 associations with age and cognition in the whole sample. Scatterplots with simple regression line and standard error showing the association between plasma p-tau217 (picograms per milliliter) and (A) age at baseline, (B) Mini Mental State Examination Score, and (C) Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) word list delayed recall. Black circles: Non-Carriers; red circles: Carriers (unimpaired), blue circles: Carriers (MCI converters).
Figure 2.

Plasma p-tau217 associations with PET-based pathology in the whole sample. Scatterplots with simple regression line and standard error showing the association between plasma p-tau217 (picograms per milliliter) and (A) mean cortical β amyloid-PET, (B) entorhinal cortex tau-PET, (C) inferior temporal cortex tau-PET, and (D) precuneus tau-PET. DVR = distribution volume ratio, SUVR = standardized uptake value ratio. Black circles: Non-Carriers; red circles: Carriers (unimpaired), blue circles: Carriers (MCI converters).
Consistent results were observed in the whole-group after controlling for age, sex, and time between measurements: MMSE r = −0.42, p = .006, CI[−0.65, −0.13]; delayed recall r = −0.30, p = .056, CI[−0.56, 0.01]; cortical Aβ-PET r = 0.49, p = .001, CI[0.22, 0.69]; entorhinal cortex tau-PET r = 0.39, p = .012, CI[0.09, 0.62]; inferior temporal cortex tau-PET r = 0.31, p = .043, CI[0.01, 0.57]; and precuneus tau-PET r = 0.48, p = .001, CI[0.21, 0.69]. Within-group correlations were not significant after controlling for these covariates (Table S3). There was no significant relationship between p-tau217 and tau-PET ROIs when including Aβ as a covariate, but the negative associations with cognition remained significant (Table S4). Further, cortical Aβ was a significant partial mediator of the relationship between plasma p-tau217 and tau-PET (Table S5).
3.4. Associations between plasma p-tau217 and whole-brain Aβ- and tau-PET in PSEN1 carriers
We assessed the relationship between plasma p-tau217 and vertex-wise PET pathology in mutation carriers. Plasma p-tau217 was positively correlated with Aβ burden in frontal, lateral temporal, parietal, and retrosplenial cortices. Correlations with tau-PET were strongest effects in temporal and parietal regions, consistent with the known anatomy of early tau accumulation in mutation carriers, as well as involvement of frontal regions. (Fig. 3A). When limiting analyses to cognitively unimpaired carriers only (Fig. 3B), Aβ- and tau-PET correlations with plasma p-tau217 were observed to a smaller extent, primarily in the left lateral temporal cortices.
Figure 3.
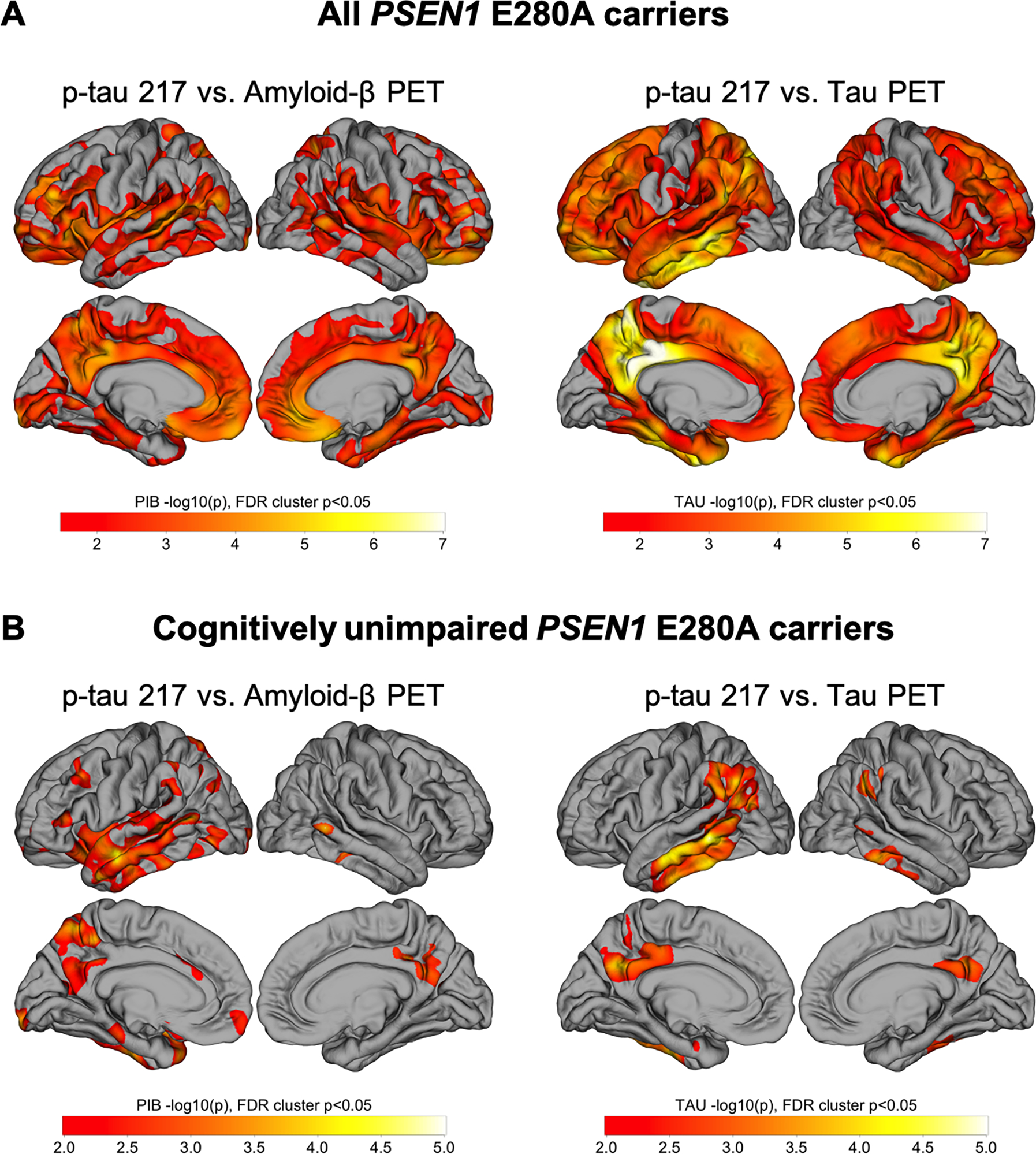
Whole-cortex analysis of Aβ- and tau-PET versus plasma p-tau217. Pearson correlations were performed between p-tau217 concentrations and Aβ- (left) and tau- (right) PET. Results are displayed as −log10(p), significant at cluster p < .05 after false discovery rate (FDR) correction. Correlations performed in (A) all PSEN1 E280A carriers (n = 24) and (B) cognitively unimpaired PSEN1 E280A carriers (n = 18).
Results were attenuated when adjusting for age, sex, and time between measurements in the carrier group, but the regional associations between p-tau217 and tau-PET were similar to unadjusted results (Fig. S1A). When limiting analyses to unimpaired carriers only, the age, sex, and time adjusted associations did not survive FDR correction. Similarly, including cortical Aβ as a covariate attenuated the associations in all carriers, and the associations did not remain significant when limiting to unimpaired carriers only (Fig. S1B). As a supplementary analysis, we also examined the associations between p-tau217 and cortical thickness. Associations with cortical thickness were weaker than those with tau- and amyloid-PET, and none of the thickness results survived multiple comparisons correction with FDR (Fig. S2).
4. Discussion
The primary aim of this study was to examine whether plasma p-tau217 is associated with subsequent PET-based markers of AD pathology in the brain and cognitive performance. We examined this association in a cohort of PSEN1 E280A carriers, who will develop dementia by midlife, and non-carrier family members, using plasma p-tau217 and neuroimaging markers collected on average 7.61 years apart. Consistent with our hypotheses, plasma p-tau217 was elevated in cognitively unimpaired PSEN1 carriers as compared to non-carrier family members. Critically, baseline p-tau217 levels were associated with subsequent Aβ- and tau-PET deposits and lower memory performance. Together, our results suggest that plasma p-tau217 is a promising biomarker for early AD detection and progression.
In our sample, carriers had higher plasma levels of p-tau217 than non-carriers prior to the onset of cognitive impairment, and, within carriers, higher p-tau217 was associated with older age. The median age of onset of MCI in this kindred is 44 years (21), over a decade older than the average age of carriers in this sample at the time of plasma collection. Although these data are not longitudinal, due to the well-characterized clinical trajectory of the mutation carriers and near complete penetrance of the mutation, age serves as a proxy for time until clinical onset and provides a model for disease progression. As such, these associations provide evidence that plasma p-tau217 may be an early marker of preclinical AD and related to disease progression, potentially increasing as clinical onset approaches. Consistent results have been previously reported from these kindred (7) and from studies of sporadic AD, using comparisons of unimpaired older adults with high- versus low-Aβ (15,34). However, longitudinal studies are required to describe the trajectory of plasma p-tau217 across disease stage.
Although converging findings indicate this early change in plasma p-tau217, little has been reported about its associations with subsequent PET-based pathology, the current gold standard in measuring in vivo AD pathology. Prior findings in older adults at risk for sporadic AD found an association between plasma p-tau217 and concurrent Aβ-PET (17) and tau-PET imaging (5,16,17,35); however, only two studies, to our knowledge, have examined the relationship with future tau-PET. In these prior studies, with measurements conducted approximately one to two years apart, found a relationship between plasma p-tau217 and subsequent medial temporal lobe tau-PET (15,18). Our study expands on these results by showing an association with widespread Aβ and tau pathology 7.61 years after plasma collection. Regional analyses revealed an association with mean cortical Aβ and regional tau-PET in three key anatomical regions: interior temporal cortex, entorhinal cortex, and precuneus. Our findings in inferior temporal cortex and entorhinal cortex are consistent with prior findings in plasma-PET investigations (5,15,18,35), and we additionally show this association with precuneus, a region previously shown to be early impacted by AD pathology in this kindred (31,36). Further, our study is the first to conduct whole-brain analyses, revealing the correlations between plasma and tau-PET mirror the known progression of early tau pathology accumulation (30,31). As expected early in the course of the disease, the correlations were limited to the temporal cortices in the cognitively unimpaired carriers. In contrast, when including the carriers with MCI, the spatial extent was much greater, including parietal and frontal cortices.
To note, the associations between p-tau217 and regional tau-PET were no longer significant when controlling for cortical Aβ, which was shown to be a partial mediator of the plasma- tau-PET relationship, and the voxel-wise associations were attenuated when controlling for cortical Aβ. Other findings have shown an association between p-tau217 and Aβ-PET (15,37) and p-tau217 has been shown to do particularly well at discriminating between AD and other neurodegenerative diseases (11,13). Together, these findings suggest that p-tau217 may be reflecting both Aβ- and tau-related processes, though more work is needed to determine the exact pathology that is indicated by p-tau217.
Other plasmatic biomarkers, such as p-tau181, have been associated with markers of neurodegeneration in AD-related areas through MRI, Fluorodeoxyglucose PET (FDG PET), amyloid PET, and tau PET (38,39), and have been able to differentiate AD from other neurodegenerative diseases (40). P-tau181 has also been found to predict tau pathology six years later in temporoparietal regions that are associated with AD (41). However, in several studies, p-tau217 has shown superiority and greater diagnostic accuracy than other biomarkers in plasma and CSF, such as p-tau181 (5,7,13,14).
In addition to pathological markers, elevated plasma p-tau217 was associated with lower subsequent delayed recall and global cognition. Tau-PET burden has been consistently associated with worse cognition in these kindred (31,36,42), though only one prior study has reported an association between plasma p-tau217 and cognition in this kindred (7). In sporadic AD, longitudinal increases in plasma p-tau217 were associated with worse cognition (12). However, another study found that tau-PET had a stronger association with cognition than did plasma p-tau217 (35). More work is needed to clarify the association between plasma p-tau217 and cognition and the extent it can predict declines in various cognitive domains and global cognitive changes.
This study has several strengths and limitations. A primary strength of this study is the kindred with a single variant mutation for autosomal dominant AD, whose clinical trajectory is well-characterized (20,21). Due to the early median age of onset for MCI in this kindred, typical age-related confounds prevalent in studies of older adults are mitigated in this sample. This is particularly important for studies of tau pathology, which can accumulate with age in the absence of other AD pathology (42). Another strength is the 7.61-year interval between plasma collection, at which time all participants were cognitively unimpaired, and neuroimaging measures, at which time only six participants converted to MCI, thereby highlighting the utility of early plasma p-tau217 for predicting pathology prior to conversion to dementia. Despite the advantages provided by studying these kindred, our sample size is relatively small for a biomarker study, and the extent to which these findings can be generalized to sporadic AD is unknown. Recent findings indicate similar in vivo pathology in sporadic and autosomal dominant AD, including CSF measures of p-tau (43). However, future studies in additional autosomal dominant and sporadic AD populations are needed to investigate plasma biomarkers’ generalizability. Additionally, analysis of blood samples was not available at follow-up in our sample. Future studies would benefit from longitudinal collection of both plasma p-tau217 and tau-PET to assess the trajectory of each biomarker, as well as investigate potential differences in the plasma-PET association at varying follow-up intervals.
In sum, our results show that baseline levels of plasma p-tau217 predict subsequent levels of amyloid and tau burden and worse future memory performance in PSEN1 E280A carriers. These findings add to the growing literature suggesting that plasma p-tau217 is an early marker for AD by demonstrating an association between plasma and PET measures of pathology. Our results provide support for plasma p-tau217 as a potential minimally invasive diagnostic and prognostic biomarker of AD pathology and cognition, with promising utility in clinical practice and trials.
Supplementary Material
Highlights.
Non-demented PSEN1 carriers have higher plasma p-tau217 than do age-matched non-carriers.
Higher baseline p-tau217 is associated with greater future amyloid-PET pathology burden.
Higher baseline p-tau217 is associated with greater future tau-PET pathology burden.
Higher baseline p-tau217 is associated with worse future memory performance.
Research in Context.
Systematic review:
We used PubMed to review the literature on plasma p-tau217 in Alzheimer’s disease. Recent studies have reported converging results indicating plasma p-tau217 as a promising and specific biomarker for Alzheimer’s disease; however, the relationship between plasma p-tau217 and future PET pathology has not been widely studied. Relevant citations are appropriately cited in the article.
Interpretation:
Our results demonstrate an association between baseline plasma p-tau217 and subsequent measures of in vivo brain pathology and cognition. These findings add to the growing literature supporting the utility of plasma p-tau217 as a minimally invasive diagnostic and prognostic marker of Alzheimer’s disease.
Future directions:
Future studies should investigate the longitudinal relationships between plasma p-tau217 and tau PET pathology in both autosomal dominant and sporadic Alzheimer’s disease.
Acknowledgments
This paper was made possible by Grants from the National Institute on Aging (NIA) R01 AG054671, RF1AG077627. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH/NIA. The funding source did not have a role in the study design; collection, analysis, or interpretation of data; writing of the report; or in the decision to submit the article for publication. The authors would like to thank the PSEN1 Colombian families for contributing their valuable time and effort, without which this study would not have been possible. We thank the research staff of the Group of Neuroscience of Antioquia for their help coordinating study visits for the Colombia-Boston Study.
Footnotes
Conflict of Interests
Dr. Langella was supported by a grant from the Alzheimer’s Association (AA Research Fellowship). Dr. Vila-Castelar was supported by a grant from the Alzheimer’s Association (AA Research Fellowship). Dr. Quiroz was supported by grants from the National Institute on Aging (R01 AG054671, RF1AG077627), the Alzheimer’s Association, and Massachusetts General Hospital ECOR. Dr. Quiroz reports receiving consulting fees from Biogen. Dr. Hansson is by the Swedish Research Council (2016–00906), the Knut and Alice Wallenberg foundation (2017–0383), the Marianne and Marcus Wallenberg foundation (2015.0125), the Strategic Research Area MultiPark (Multidisciplinary Research in Parkinson’s disease) at Lund University, the Swedish Alzheimer Foundation (AF-939932), the Swedish Brain Foundation (FO2021–0293), The Parkinson foundation of Sweden (1280/20), the Konung Gustaf V:s och Drottning Victorias Frimurarestiftelse, the Skåne University Hospital Foundation (2020-O000028), Regionalt Forskningsstöd (2020–0314) and the Swedish federal government under the ALF agreement (2018-Projekt0279). Dr. Zetterberg is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2018–02532), the European Research Council (#681712 and #101053962), Swedish State Support for Clinical Research (#ALFGBG-71320), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809–2016862), the AD Strategic Fund and the Alzheimer’s Association (#ADSF-21–831376-C, #ADSF-21–831381-C and #ADSF-21–831377-C), the Olav Thon Foundation, the Erling-Persson Family Foundation, Stiftelsen för Gamla Tjänarinnor, Hjärnfonden, Sweden (#FO2019–0228), the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 860197 (MIRIADE), the European Union Joint Programme – Neurodegenerative Disease Research (JPND2021–00694), and the UK Dementia Research Institute at UCL (UKDRI-1003). Mr. Fox-Fuller was supported by a fellowship from the National Institute on Aging (1F31AG062158–01A1). Dr. Dage is an inventor on patents or patent applications of Eli Lilly and Company relating to the assays, methods, reagents and / or compositions of matter used in this work. Dr. Dage has served as a consultant for Karuna Therapeutics and received research support from ADx Neurosciences, Roche Diagnostics and Eli Lilly and Company. Dr. Su reports grants from NIH/NIBIB, The Alzheimer’s Association, The BrightFocus Foundation, NIH/NIA, State of Arizona, personal fees from Green Valley Pharmaceutical LLC, outside the submitted work. Dr. Ramirez-Gomez reports grants from The Alzheimer’s Association and NIH/NIA. Dr. Lopera was supported by an Anonymous Foundation, and the Administrative Department of Science, Technology and Innovation (Colciencias Colombia;111565741185). Drs. Reiman and Lopera are principal investigators of the Alzheimer’s Prevention Initiative (API) Autosomal Dominant AD Trial, which is supported by NIA, philanthropy, Genentech, and Roche. Dr. Blennow is supported by the Swedish Research Council (#2017–00915), the Alzheimer Drug Discovery Foundation (ADDF), USA (#RDAPB-201809–2016615), the Swedish Alzheimer Foundation (#AF-930351, #AF-939721 and #AF-968270), Hjärnfonden, Sweden (#FO2017–0243 and #ALZ2022–0006), the Swedish state under the agreement between the Swedish government and the County Councils, the ALF-agreement (#ALFGBG-715986 and #ALFGBG-965240), the European Union Joint Program for Neurodegenerative Disorders (JPND2019–466-236), the National Institute of Health (NIH), USA, (grant #1R01AG068398–01), and the Alzheimer’s Association 2021 Zenith Award (ZEN-21–848495). Dr. Reiman reports grants from National Institute on Aging (R01 AG031581, P30 AG19610), Banner Alzheimer’s Foundation and the NOMIS Foundation during the conduct of the study. He reports receiving personal fees as a Scientific Advisor to Roche Diagnostics (travel expenses only), MagQ, Avid Radiopharmaceuticals and is a share-holding co-founder of ALZPath, outside the submitted work. In addition, he is the inventor of a patent issued to Banner Health, which involves the use of biomarker endpoints in at-risk persons to accelerate the evaluation of Alzheimer’s disease prevention therapies and is outside the submitted work. Dr. Blennow has served as a consultant or at advisory boards for Abcam, Axon Neuroscience, BioArctic, Biogen, Lilly, MagQu, Novartis, Roche Diagnostics, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB, a GU Venture-based platform company at the University of Gothenburg. The authors report no further potential conflicts of interest.
References
- 1.Chandra A, Valkimadi P, Pagano G, Cousins O, Dervenoulas G, Politis M. Applications of amyloid, tau, and neuroinflammation PET imaging to Alzheimer’s disease and mild cognitive impairment. Hum Brain Mapp. 2019. Sep 14;40(18):5424–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bjerke M, Engelborghs S. Cerebrospinal Fluid Biomarkers for Early and Differential Alzheimer’s Disease Diagnosis. J Alzheimers Dis JAD. 2018;62(3):1199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leuzy A, Cullen NC, Mattsson-Carlgren N, Hansson O. Current advances in plasma and cerebrospinal fluid biomarkers in Alzheimer’s disease. Curr Opin Neurol. 2021. Apr 1;34(2):266–74. [DOI] [PubMed] [Google Scholar]
- 4.Telser J, Risch L, Saely CH, Grossmann K, Werner P. P-tau217 in Alzheimer’s disease. Clin Chim Acta Int J Clin Chem. 2022. Jun 1;531:100–11. [DOI] [PubMed] [Google Scholar]
- 5.Thijssen EH, La Joie R, Strom A, Fonseca C, Iaccarino L, Wolf A, et al. Plasma phosphorylated tau 217 and phosphorylated tau 181 as biomarkers in Alzheimer’s disease and frontotemporal lobar degeneration: a retrospective diagnostic performance study. Lancet Neurol. 2021. Sep;20(9):739–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wennström M, Janelidze S, Nilsson KPR, Netherlands Brain Bank, Serrano GE, Beach TG, et al. Cellular localization of p-tau217 in brain and its association with p-tau217 plasma levels. Acta Neuropathol Commun. 2022. Jan 6;10(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palmqvist S, Janelidze S, Quiroz YT, Zetterberg H, Lopera F, Stomrud E, et al. Discriminative Accuracy of Plasma Phospho-tau217 for Alzheimer Disease vs Other Neurodegenerative Disorders. JAMA. 2020. Aug 25;324(8):772–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brickman AM, Manly JJ, Honig LS, Sanchez D, Reyes-Dumeyer D, Lantigua RA, et al. Plasma p-tau181, p-tau217, and other blood-based Alzheimer’s disease biomarkers in a multi-ethnic, community study. Alzheimers Dement J Alzheimers Assoc. 2021. Aug;17(8):1353–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bayoumy S, Verberk IMW, den Dulk B, Hussainali Z, Zwan M, van der Flier WM, et al. Clinical and analytical comparison of six Simoa assays for plasma P-tau isoforms P-tau181, P-tau217, and P-tau231. Alzheimers Res Ther. 2021. Dec 4;13(1):198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cullen NC, Leuzy A, Janelidze S, Palmqvist S, Svenningsson AL, Stomrud E, et al. Plasma biomarkers of Alzheimer’s disease improve prediction of cognitive decline in cognitively unimpaired elderly populations. Nat Commun. 2021. Jun 11;12(1):3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barthélemy NR, Horie K, Sato C, Bateman RJ. Blood plasma phosphorylated-tau isoforms track CNS change in Alzheimer’s disease. J Exp Med. 2020. Nov 2;217(11):e20200861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mattsson-Carlgren N, Janelidze S, Palmqvist S, Cullen N, Svenningsson AL, Strandberg O, et al. Longitudinal plasma p-tau217 is increased in early stages of Alzheimer’s disease. Brain J Neurol. 2020. Dec 5;143(11):3234–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barthélemy NR, Li Y, Joseph-Mathurin N, Gordon BA, Hassenstab J, Benzinger TLS, et al. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer’s disease. Nat Med. 2020. Mar;26(3):398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pichet Binette A, Palmqvist S, Bali D, Farrar G, Buckley CJ, Wolk DA, et al. Combining plasma phospho-tau and accessible measures to evaluate progression to Alzheimer’s dementia in mild cognitive impairment patients. Alzheimers Res Ther. 2022. Mar 29;14(1):46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Janelidze S, Berron D, Smith R, Strandberg O, Proctor NK, Dage JL, et al. Associations of Plasma Phospho-Tau217 Levels With Tau Positron Emission Tomography in Early Alzheimer Disease. JAMA Neurol. 2021. Feb 1;78(2):149–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mielke MM, Frank RD, Dage JL, Jeromin A, Ashton NJ, Blennow K, et al. Comparison of Plasma Phosphorylated Tau Species With Amyloid and Tau Positron Emission Tomography, Neurodegeneration, Vascular Pathology, and Cognitive Outcomes. JAMA Neurol. 2021. Sep 1;78(9):1108–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mattsson-Carlgren N, Janelidze S, Bateman RJ, Smith R, Stomrud E, Serrano GE, et al. Soluble P-tau217 reflects amyloid and tau pathology and mediates the association of amyloid with tau. EMBO Mol Med. 2021. Jun 7;13(6):e14022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leuzy A, Smith R, Cullen NC, Strandberg O, Vogel JW, Binette AP, et al. Biomarker-Based Prediction of Longitudinal Tau Positron Emission Tomography in Alzheimer Disease. JAMA Neurol. 2022. Feb 1;79(2):149–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reiman EM, Langbaum JBS, Fleisher AS, Caselli RJ, Chen K, Ayutyanont N, et al. Alzheimer’s Prevention Initiative: a plan to accelerate the evaluation of presymptomatic treatments. J Alzheimers Dis JAD. 2011;26 Suppl 3:321–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fuller JT, Cronin-Golomb A, Gatchel JR, Norton DJ, Guzmán-Vélez E, Jacobs HIL, et al. Biological and Cognitive Markers of Presenilin1 E280A Autosomal Dominant Alzheimer’s Disease: A Comprehensive Review of the Colombian Kindred. J Prev Alzheimers Dis. 2019;6(2):112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Acosta-Baena N, Sepulveda-Falla D, Lopera-Gómez CM, Jaramillo-Elorza MC, Moreno S, Aguirre-Acevedo DC, et al. Pre-dementia clinical stages in presenilin 1 E280A familial early-onset Alzheimer’s disease: a retrospective cohort study. Lancet Neurol. 2011. Mar;10(3):213–20. [DOI] [PubMed] [Google Scholar]
- 22.Lopera F, Ardilla A, Martínez A, Madrigal L, Arango-Viana JC, Lemere CA, et al. Clinical features of early-onset Alzheimer disease in a large kindred with an E280A presenilin-1 mutation. JAMA. 1997. Mar 12;277(10):793–9. [PubMed] [Google Scholar]
- 23.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975. Nov;12(3):189–98. [DOI] [PubMed] [Google Scholar]
- 24.Reisberg B Functional assessment staging (FAST). Psychopharmacol Bull. 1988;24(4):653–9. [PubMed] [Google Scholar]
- 25.Aguirre-Acevedo DC, Gómez RD, Moreno S, Henao-Arboleda E, Motta M, Muñoz C, et al. [Validity and reliability of the CERAD-Col neuropsychological battery]. Rev Neurol. 2007. Dec 1;45(11):655–60. [PubMed] [Google Scholar]
- 26.Torres VL, Vila-Castelar C, Bocanegra Y, Baena A, Guzmán-Vélez E, Aguirre-Acevedo DC, et al. Normative data stratified by age and education for a Spanish neuropsychological test battery: Results from the Colombian Alzheimer’s prevention initiative registry. Appl Neuropsychol Adult. 2021. Apr;28(2):230–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Logan J, Fowler JS, Volkow ND, Wolf AP, Dewey SL, Schlyer DJ, et al. Graphical analysis of reversible radioligand binding from time-activity measurements applied to [N-11C-methyl]-(−)-cocaine PET studies in human subjects. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 1990. Sep;10(5):740–7. [DOI] [PubMed] [Google Scholar]
- 28.Amariglio RE, Mormino EC, Pietras AC, Marshall GA, Vannini P, Johnson KA, et al. Subjective cognitive concerns, amyloid-β, and neurodegeneration in clinically normal elderly. Neurology. 2015. Jul 7;85(1):56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson KA, Schultz A, Betensky RA, Becker JA, Sepulcre J, Rentz D, et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol. 2016. Jan;79(1):110–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quiroz YT, Sperling RA, Norton DJ, Baena A, Arboleda-Velasquez JF, Cosio D, et al. Association Between Amyloid and Tau Accumulation in Young Adults With Autosomal Dominant Alzheimer Disease. JAMA Neurol. 2018. May 1;75(5):548–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanchez JS, Hanseeuw BJ, Lopera F, Sperling RA, Baena A, Bocanegra Y, et al. Longitudinal amyloid and tau accumulation in autosomal dominant Alzheimer’s disease: findings from the Colombia-Boston (COLBOS) biomarker study. Alzheimers Res Ther. 2021. Jan 15;13(1):27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greve DN, Svarer C, Fisher PM, Feng L, Hansen AE, Baare W, et al. Cortical surface-based analysis reduces bias and variance in kinetic modeling of brain PET data. NeuroImage. 2014. May 15;92:225–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lendon CL, Martinez A, Behrens IM, Kosik KS, Madrigal L, Norton J, et al. E280A PS-1 mutation causes Alzheimer’s disease but age of onset is not modified by ApoE alleles. Hum Mutat. 1997;10(3):186–95. [DOI] [PubMed] [Google Scholar]
- 34.Janelidze S, Palmqvist S, Leuzy A, Stomrud E, Verberk IMW, Zetterberg H, et al. Detecting amyloid positivity in early Alzheimer’s disease using combinations of plasma Aβ42/Aβ40 and p-tau. Alzheimers Dement J Alzheimers Assoc. 2022. Feb;18(2):283–93. [DOI] [PubMed] [Google Scholar]
- 35.Ossenkoppele R, Reimand J, Smith R, Leuzy A, Strandberg O, Palmqvist S, et al. Tau PET correlates with different Alzheimer’s disease-related features compared to CSF and plasma p-tau biomarkers. EMBO Mol Med. 2021. Aug 9;13(8):e14398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guzmán-Vélez E, Diez I, Schoemaker D, Pardilla-Delgado E, Vila-Castelar C, Fox-Fuller JT, et al. Amyloid-β and tau pathologies relate to distinctive brain dysconnectomics in preclinical autosomal-dominant Alzheimer’s disease. Proc Natl Acad Sci U S A. 2022. Apr 12;119(15):e2113641119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pontecorvo MJ, Lu M, Burnham SC, Schade AE, Dage JL, Shcherbinin S, et al. Association of Donanemab Treatment With Exploratory Plasma Biomarkers in Early Symptomatic Alzheimer Disease: A Secondary Analysis of the TRAILBLAZER-ALZ Randomized Clinical Trial. JAMA Neurol. 2022. Oct 17; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moscoso A, Grothe MJ, Ashton NJ, Karikari TK, Lantero Rodríguez J, Snellman A, et al. Longitudinal Associations of Blood Phosphorylated Tau181 and Neurofilament Light Chain With Neurodegeneration in Alzheimer Disease. JAMA Neurol. 2021. Apr 1;78(4):396–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tissot C, L Benedet A, Therriault J, Pascoal TA, Lussier FZ, Saha-Chaudhuri P, et al. Plasma pTau181 predicts cortical brain atrophy in aging and Alzheimer’s disease. Alzheimers Res Ther. 2021. Mar 29;13(1):69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karikari TK, Pascoal TA, Ashton NJ, Janelidze S, Benedet AL, Rodriguez JL, et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer’s disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 2020. May;19(5):422–33. [DOI] [PubMed] [Google Scholar]
- 41.Moscoso A, Grothe MJ, Ashton NJ, Karikari TK, Rodriguez JL, Snellman A, et al. Time course of phosphorylated-tau181 in blood across the Alzheimer’s disease spectrum. Brain J Neurol. 2021. Feb 12;144(1):325–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol (Berl). 2014. Dec;128(6):755–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morris JC, Weiner M, Xiong C, Beckett L, Coble D, Saito N, et al. Autosomal dominant and sporadic late onset Alzheimer disease share a common in vivo pathophysiology. Brain J Neurol. 2022. May 17;awac181. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.