

Abstract
The human microbiome is intimately related to cancer biology and plays a vital role in the efficacy of cancer treatments, including immunotherapy. Extraordinary evidence has revealed that several microbes influence tumor development through interaction with the host immune system, i.e., immuno–oncology–microbiome (IOM). This review focuses on the intratumoral microbiome in IOM and describes the available data and computational methods for discovering biological insights of microbial profiling from host bulk, single-cell, and spatial sequencing data. Critical challenges in data analysis and integration are discussed. Specifically, the microorganisms associated with cancer and cancer treatment in the context of IOM are collected and integrated from the literature. Lastly, we provided our perspectives for future directions in IOM research.
Keywords: immuno-oncology-microbiome, intratumoral microbiome, cancer treatment and diagnosis, immunotherapy, computational methods
SIGNIFICANCE OF EXPLORING IMMUNO–ONCOLOGY–MICROBIOME
The human microbiome is the collection of all microorganisms, which are non-negligible components of the human body, residing on or within human tissues and biofluids, such as the skin, oral mucosa, lung, and gastrointestinal tract [1]. Human microbes are believed to play a broad role in cancer diagnosis, pathogenesis, and treatment by interacting with the host immune system [2]. The immune-mediated interactions among immune cells, tumors, and microbes in the tumor microenvironment (TME; see Glossary) are defined as immuno–oncology–microbiome (IOM) [2]. In the context of IOM, the interactions between the host microbiome and tumor mainly include two categories [2, 3] (Figure 1, Key Figure): (i) interactions involving gut microbes, which affect both local and distant tumor growth and survival by impacting host immune system, and (ii) interactions involving intratumoral microbes, which either reside in the TME or tumor/immune cells to influence tumor progression and antitumor immunity. As polymorphic microbes become new cancer hallmarks, research into the intricate relationship in IOM is receiving increasing attention [4].
Figure 1. Key Figure. Schematic overview of computational studies of immuno–oncology-microbiome (IOM).
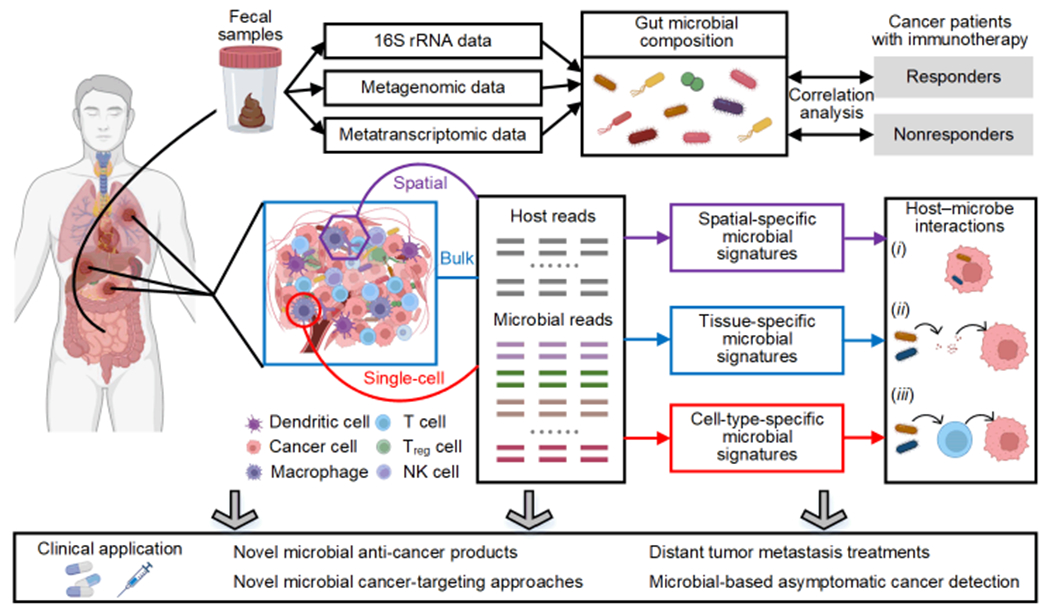
To analyze the effects of gut microbiota on host tumors, researchers can acquire the gut microbial composition using the 16S ribosomal RNA (rRNA) data, metagenomic data, or metatranscriptomic data from stool samples. Correlation analysis between gut microbial composition and cancer patients undergoing immunotherapy can help researchers understand clinical response heterogeneity. For host tumor tissues, microbial profiling can be derived from existing host sequencing data, including bulk sequencing, single-cell sequencing, and spatial transcriptome data. The computational analyses of these data can enable researchers to obtain the tissue-specific, cell-type-specific, or spatial-specific microbial signatures for further IOM studies and clinical applications.
The gut microbes can manipulate and infiltrate the gut epithelial barrier, relocate to other tissues and organs through the blood, and influence the host immune context [2]. They exhibit broad effects on primary lymphoid organs [5, 6], TME [2, 7, 8], adaptive immune response [9, 10], and inflammatory responses in the intestines and other organs [11]. Existing computational analyses of gut microbes mainly investigate the associations between the gut microbial diversity/composition and cancer types/immune responses [12–14] through 16S ribosomal RNA sequencing, metagenomic shotgun sequencing, flow cytometry, immunohistochemistry, and cytokine assays from host stool samples. With decades of development, the analyses of gut microbes are now becoming more mature [15, 16], and projects have been created for archiving gut microbiome data related to cancer, such as the Human Microbiome Project [17] and MetaHIT [18].
On the other hand, intratumoral microbes have been observed in multiple cancer types, such as colorectal [19], pancreatic [20], breast [21, 22], and lung cancer [23–25]. They not only reside in tumor cells, contributing to tumor progression, metastasis, oncogenesis, and drug resistance, but also interact with immune cells in the TME to promote or inhibit immune responses and activities [2, 25, 26]. The role and functions of intratumoral microbes have been confirmed to be largely tumor-specific [24, 27, 28], yet the intratumoral microbiome’s direct causal roles and underlying mechanisms remain unclear [29]. As such, attention should be focused on analyzing data directly derived from TME: (i) microbial profiling extracted from host whole-genome sequencing (WGS), whole-exome sequencing (WES), and whole-transcriptome sequencing (RNA-seq), known as “microbial profiling derived from bulk sequencing”, (ii) microbial profiling extracted from host single-cell RNA sequencing (scRNA-seq) data, known as “microbial profiling derived from single-cell sequencing”, and (iii) microbial profiling extracted from host spatially resolved transcriptomics (SRT) data, known as “microbial profiling derived from spatial sequencing”. Specific challenges and limitations lead to an increasing need for computational methods development to discover novel insight into modes of action, functionalities, and causal relations of intratumor microbes to specific cancer types, as well as for the successful prediction and diagnosis of cancers. Here, we provide a timely review of computational methods for the intratumoral microbiome in IOM, alongside the challenges and future perspectives.
ANALYSIS OF INTRATUMORAL MICROBIAL PROFILING DERIVED FROM BULK SEQUENCING
To leverage the large databases of clinically-annotated samples with bulk data, such as The Cancer Genome Atlas (TCGA), computational tools and pipelines have been developed to analyze the IOM involving intratumoral microbes [23, 30–32]. The pre-processing for deriving microbial profiling from host bulk sequencing data involves four steps: (i) Identify microbial reads from WGS, WES, or RNA-seq data of tumor/normal tissues. This step aligns sequencing reads to host reference genomes using a short read aligner Mapping and Assembly with Quality (MAQ) [33] or the ultrafast RNA-seq aligner Spliced Transcripts Alignment to a Reference (STAR) [34]. The unmapped reads are considered candidate microbial reads. (ii) Characterize microbial taxonomic profiles. Metagenomic analysis is performed for the taxonomic profiling of microbes by mapping candidate microbial reads from the previous step to all known microbial genomes using Kraken2 [35], Metagenomic Phylogenetic Analysis (MetaPhlAn2) [36], or SHOGUN [37]. The remaining unmapped reads can be assembled as novel microbial genomes using MetaVelvet [38] or MEGAHIT [39]. An existing bioinformatics tool, PathSeq [40], can provide an integrative analysis of the first two steps. (iii) Remove contaminants. Due to the low microbial biomass in host sequencing samples [32], contamination in the laboratory environment and the sequencing process significantly impact downstream analysis. The contaminants can be removed using decontam [41] or SourceTracker [42]. (iv) Normalize decontaminated microbial data. The decontaminated microbial data are re-normalized, alleviating batch effects while preserving biological signals. Commonly used normalization methods include log-ratio transformation, log upper quartile, cumulative sum scaling, and variance stabilization [43, 44]. Similar pipelines can also be applied for microbiome profiling from single-cell and spatial data.
Several studies have been conducted using the above methods to investigate the effect of tumor-associated microbes on host tumors. For example, Poore et al. analyzed microbial profiling from 4,831 WGS and 13,285 RNA-seq datasets across 10,481 patients and 33 cancer types from the TCGA compendium [30]. The read counts at the genus taxonomic level of each dataset were accessible. These bulk sequencing-derived microbial profiling were used to discover tumor-type specific microbial signatures [30]. Pan-cancer analyses showed that Fusobacterium was overabundant in gastrointestinal (GI) cancers compared with non-GI cancers in both primary tumor tissue and adjacent solid-tissue normal samples. In addition, Dohlman et al. extracted microbial reads by retrieving raw WGS and WES sequencing data of different cancer types from the TCGA database [32]. Combined with the matched host mRNA expression data, they investigated host genes whose transcriptional patterns highly correlated with the abundance of identified tumor-associated microbial species [32]. They found that pathways significantly and consistently enriched by these genes were related to immune system activation, such as antigen presentation and natural killer cell-mediated cytotoxicity [32]. They also showcased that microbes that are equally prevalent across cancer types and blood samples are generally contaminants [32].
The qualitative comparison of existing computational methods used to analyze microbial profiling derived from host data have been summaried in Table 1. Nevertheless, microbiome profiling and analysis from host bulk samples are still facing challenges. First, the selection of microbial reference genome is critical, which may cause significant differences in sequence mapping as the species/strains and the version of microbial genomes included in the reference varies. Secondly, the existing pipeline is designed for microbiota mapping suitable for discovery-driven questions, e.g., what are the signature microbes involved in cancer tissue. While for individual species/strain mapping, the pipeline needs to be optimized with a more flexible sequence alignment threshold due to the extremely short reference genome. Such adjustment may bring more false positives in quantifying the species/strain’s abundance. Lastly, reliable and systematic benchmarking standards and protocols for evaluating and comparing results from different sequencing technologies and protocols are still lacking.
Table 1.
The qualitative comparison of different computational methods used to analyze microbial profiling derived from host data.
| Tools | Description | Strength | Limitation | Code | Refs |
|---|---|---|---|---|---|
| STAR | An RNA-seq aligner based on sequential maximum mappable seed search in suffix arrays | Fast mapping speed; high alignment precision and sensitivity; good default performance | Memory intensive | C++ | [34, 72, 73] |
| MAQ | A short-read aligner based on hash table and mapping quality scores | Accurate and feature-rich short reads alignment | Unsuitable for the alignment of longer reads where indels may occur frequently; single-threaded software; slow running speed | C++ | [33, 74, 75] |
| Kraken2 | A taxon binning tool based on the exact alignment of K-mers | High accuracy at genus and species level; fast running speed | Unsuitable for running in the personal computer environment due to the RAM limitation | C++ | [35, 76] |
| MetaPhlAn2 | A taxon profiling tool based on the alignment of unique taxonomic marker genes | High accuracy at genus and species level; efficient memory usage | Limited identification of microbial eukaryotes | Python | [36, 76–78] |
| SHOGUN | A microbiome quantification framework including contaminating read filtering and relative abundance profiling | A modular, accurate, and scalable pipeline; data analysis and transformation steps can be run individually or together in an automated workflow | Computationally intensive taxonomy assignments | Python | [30, 37] |
| MetaVelvet | A short-read de novo assembler for metagenomic data based on the de Bruijn graph | High sensitivity for sequence diversity | Low assembly length statistics | C++ | [38, 79] |
| MEGAHIT | An NGS de novo assembler for large and complex metagenomic data based on succinct de Bruijn graph | Efficient memory usage and fast running speed | Suboptimal assembly of genomes of high abundance population members on very large datasets | C++ | [39, 76, 79, 80] |
| decontam | Simple statistical methods to identify and remove contaminant sequences in marker-gene and metagenomic data | Easy integration with existing metagenomic sequencing workflows | Auxiliary data from DNA quantitation and negative control data are required | R | [41, 81] |
| Source Tracker | A Bayesian approach to estimate the proportion of contaminants in a given community | Directly estimate source proportions; model the uncertainty of known and unknown source environments | Limited for discerning sources with similar bacterial communities; high running time, only applicable to datasets between small and medium in size with few sources | R | [42, 82, 83] |
| Shortread | A Bioconductor package for input, quality assessment and exploration of high-throughput sequence data | Suitable for removing low complexity reads, low-quality reads, and PCR duplicates tagged with the same unique molecular identifier and cellular barcode | Lack of sophisticated and flexible programming frameworks | R | [84, 85] |
| Trimmomatic | A flexible trimmer for Illumina sequence data | A more flexible and efficient preprocessing tool, which could correctly handle paired-end data | Relatively slow and overly time-consuming | Java | [86, 87] |
| umi-tools | A software package modeling sequencing errors in unique molecular identifiers (UMI) | Easily be integrated into existing pipelines for analysis of sequencing techniques utilizing UMIs | High UMI preprocessing runtime cost | Python | [86, 88] |
| Wilcoxon rank-sum test | A nonparametric method used to test the differences between two populations | It can be used for the comparison of a non-normally distributed, but at least ordinally scaled, parameter in two unpaired samples | The correlation obtained by statistical tests is typically a “spurious correlation” | R/Python | [45, 46, 52, 89, 90] |
| Spearman correlation | A method used to test whether there is a monotonous relationship between two variables | It is preferable when variables feature heavy-tailed distributions or when outliers are present | R/Python | [46, 52, 90, 91] |
ANALYSIS OF INTRATUMORAL MICROBIAL PROFILING DERIVED FROM SINGLE-CELL SEQUENCING
The primary advantage of microbial profiling analysis from single-cell sequencing compared to bulk data analyses is the ability of cell barcodes to pair microbes with corresponding somatic cells. Deriving the sequencing reads of microbial profiling from the host single-cell sequencing data can help researchers uncover cell-type-specific microbial signatures and infer crosstalk between microbes, immune cells, and tumor cells [45, 46].
Currently, studies using host single-cell data to discover the heterogeneity of microbial diversities and abundances in different cancer types are limited. Robinson et al. developed cell-type-specific intracellular microbes to extract microbial reads from 21 human scRNA-seq datasets of cancer patients across three cancer types (i.e., merkel cell carcinoma, colorectal carcinoma, and non-small cell lung carcinoma) and produced a list of candidate cell-type-specific intracellular microbial taxa (from class-level to species-level) [45]. A Wilcoxon rank-sum test on the cell-type-specific microbial taxa abundance revealed that tumor samples from patients receiving immunotherapy exhibited more abundant bacterial taxa in the immune cells than in tumor cells [45]. Then, Ghaddar et al. developed a single-cell analysis of host-microbiome interactions (SAHMI) to extract microbial reads from human scRNA-seq data for two pancreatic cancer cohorts, including 41 pancreatic ductal adenocarcinomas (PDA) tumor samples and 14 normal pancreatic tissues samples [46]. They identified host-cell-associated bacteria in a subset of tumors by examining microbial reads paired with host somatic cell barcodes. In addition, by investigating differentially expressed genes (DEGs) in cells associated with bacteria, the strongest bacteria-associated DEGs are linked to PDA or microbiome-related inflammation, indicating that microbes can be involved in growth and inflammatory processes in PDA [46]. Specifically, Table 2 presents the host sequencing datasets that have been used for mining microbial profiling.
Table 2.
Overview of host sequencing datasets that have been used for mining microbial profiling.
| Cancer typea | Sample counts | Cancer stage | Survival time | Gender | Source | Refs |
|---|---|---|---|---|---|---|
| Bulk sequencing data | ||||||
|
| ||||||
| COAD | 1,006 | Yes | Yes | Yes | TCGA (https://portal.gdc.cancer.gov/) | - |
| READ | 372 | Yes | Yes | Yes | ||
| KIRC | 1,140 | Yes | Yes | Yes | ||
| THCA | 880 | Yes | Yes | Yes | ||
| STAD | 1,091 | Yes | Yes | Yes | ||
| BRCA | 1,497 | Yes | Yes | Yes | ||
| HNSC | 907 | Yes | Yes | Yes | ||
| LUAD | 951 | Yes | Yes | Yes | ||
|
| ||||||
| Single-cell sequencing data | ||||||
|
| ||||||
| Pancreatic ductal adenocarcinomas | 24 | Yes | NA | Yes | Genome Sequence Archive under project CRA001160 | [92] |
| Merkel cell carcinoma | 2 | Yes | NA | Yes | NCBI BioProject PRJNA483959 (patient 2586-4), PRJNA484204 (patient 9245-3) | [93] |
| Colorectal carcinoma | 6 | Yes | NA | Yes | ArrayExpress EMTAB-8410 | [94] |
| Non-small cell lung carcinoma | 13 | Yes | NA | Yes | NCBI BioProject PRJNA591860 | [95] |
Cancer type of different samples.
COAD, colon adenocarcinoma; READ, rectum adenocarcinoma; KIRC, kidney renal clear cell carcinoma; THCA, thyroid carcinoma; STAD, stomach adenocarcinoma; BRCA, breast invasive carcinoma; HNSC, head and neck squamous cell carcinoma; LUAD, lung adenocarcinoma.
IOM analysis of microbial profiling derived from single-cell sequencing faces similar limitations and difficulties as the bulk data, such as the direct mapping of human papillomavirus (HPV) from the host scRNA-seq data [47]. Choosing the viral reference genome and annotation files wisely and optimizing tool parameters are crucial to the mapping results, considering the much lower virus read depths involved in single cells than bulk samples. Additional inherent limitations of data at the single-cell level include: (i) It is difficult to determine the condition and cellular localization of microbes extracted from host sequencing data. For example, we cannot determine whether detected microbial nucleic acids come from living, lysed, intra-, or extracellular microorganisms [30, 46]. (ii) The decontamination step remains challenging in analyzing microbial profiling derived from host data. Removal of contaminants in silico cannot replace gold-standard wet experiments, such as sterile processing, sterile-certified reagents, and negative blanks of reagents [46]. Many technical operations in decontamination limit the analysis of individual-specific, region-specific, low-abundance, or difficult-to-detect microbes [30, 46]. (iii) The low-biomass microorganisms mined from host data will also impact the subsequent analysis, especially for single-cell data. Pan-cancer analysis of intratumoral microbiome abundance estimation showed only one bacterial cell per 147 tumor cells in the TME [4]. The low microbial biomass issue will significantly impact the capture of microbial profiling for a specific species mining from host tissue samples infected by microbes, requiring additional reliability validation for data mining. (iV) The mechanisms for capturing microbial nucleic acids in scRNA-seq data are still debatable. Several explanations have been discussed, including more polyadenylation than previously believed in prokaryotic transcripts and L-form switching in microbes [46]. More efforts are needed to investigate the origin of microbial acids in host sequencing techniques.
ANALYSIS OF INTRATUMORAL MICROBIAL PROFILING DERIVED FROM SPATIAL SEQUENCING
The emerging SRT provides the spatial information that bulk and single-cell RNA-sequencing approaches cannot deliver [48]. Particularly, SRT defines the organization of tissue functional niches and crosstalk that modulate cellular function in human tumor studies [48, 49]. For example, Shi et al. found that antibiotic treatment can disrupt the spatial networks in the gut microbiome of mice [50]. It reported an altered spatial association with the most significant fold change between Oscillibacter and Veillonella, which have been linked to altered inflammatory responses and metabolic activities in the host [50]. Another study developed a novel spatial meta-transcriptomic analysis method that captures intratumoral microbes and host transcriptomic data with spatial coordinates [51]. By examining tumor tissue samples from 12 patients with early-stage lung cancer, they found specific species or strains are significantly enriched in tumor cells compared with other cell types, and the bacterial burden is strongly positively associated with the expression of oncogenic β-catenin [51]. Therefore, mining microbial nucleic acids from SRT data of host tissue samples is a promising research aspect for tumor-immune-microbiome crosstalk in the TME with direct evidence of locations it can provide.
So far, mature and popularized computational tools have yet to be established for host spatial microbiome data analysis. As the SRT data analysis is still in its infancy, deriving microbiome profiling from host SRT becomes more challenging in building connections between microbes and functional spatially variable genes in the context of tissue architecture. Moreover, current computational tools in IOM studies, including bulk, single-cell, and spatial data, mainly rely on basic statistical tests for exploring host-microbiome interactions, such as the Wilcoxon rank-sum test [45]. Yet, the correlation between microbial signatures and host tissue type or cell type obtained by statistical tests could be a “spurious correlation” [52]. This spurious correlation between microorganisms and the host is typically not caused by the intrinsic biological association between them but is generated from the involvement of confounding factors (a coincidence or the presence of a certain third, unseen factor). As an emerging method for investigating the scRNA-seq [53], STR data [54], as well as host-microbiome data [55], deep learning, such as graph neural network (GNN), shows great potential in representing host gene and microbial signatures and building their relations, simultaneously. It is an ideal way to identify reliable tumor-associated immuno-microbiome interactions by examining genes or pathways associated with immunity and metabolism.
INTEGRATED ANALYSIS OF IOM DATA
Unlike conventional microbial analysis that investigates the different diversity and composition of gut microbes between healthy and diseased individuals, IOM studies provide a more systematic and mechanistic way to explore the intricate interactions between microbes and cancer hallmarks (Figure 2). Computational mining of microbiome from host bulk, single-cell, and SRT data of host tissue samples can help researchers investigate the identity and spatial distribution of the cell-/cancer-associated microbes, the host cell types with which they interact, and the specific host genes that can be regulated by intracellular microbes. Hence, computational analyses that leverage complementary information of different kinds of data will provide great opportunities for studying intracellular and extracellular microbe-microbe interactions, as well as microbe-host interactions. In addition, analysis of host antitumor response is a critical aspect in IOM studies, which is also not involved in conventional microbial analysis. The integrated analysis of various host and microbial data types is a trend that can bring unique features to solve IOM problems. Specifically, three categories of data integration can be expected.
Figure 2. Roadmap for computationally studying IOM.
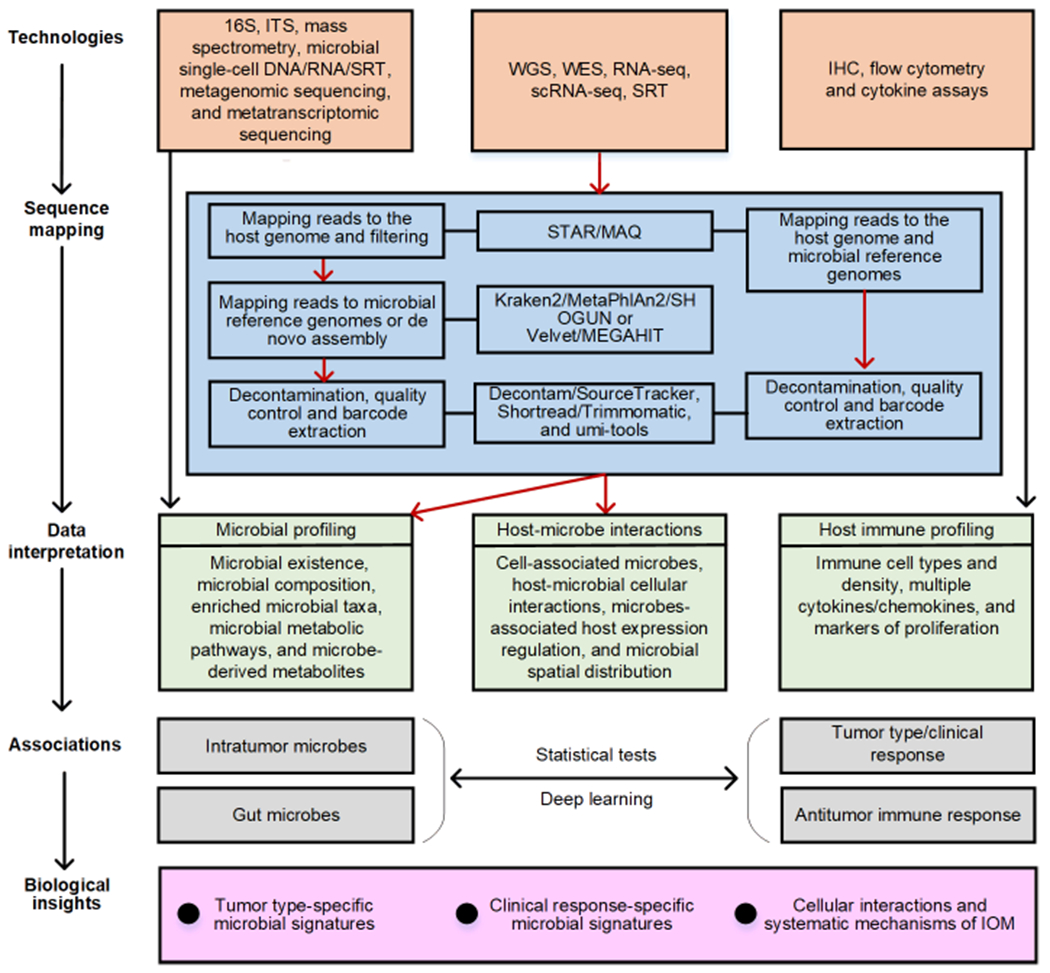
The tasks in IOM studies are to investigate tumor type-specific and clinical response-specific microbial signatures as well as cellular interactions and systematic mechanisms of IOM. By using various sequencing technologies on host fecal and tissue samples, researchers can obtain data interpretation of IOM, including microbial profiling, host-microbe interactions, and host immune profiling. Computational methods, such as statistical tests and deep learning, reveal the associations between microbes and tumor-type/clinical response or host antitumor immune response by using the collected data interpretation. The processes highlighted in red arrows represent the microbial profiling mining of IOM analysis involving intratumoral microbes, which fills in the missing part of current IOM studies (i.e., the effect of the intratumoral microbiota in the TME).
(i) Integration of different microbial sequencing data. Newsome et al. collected fecal samples from 65 non-small cell lung cancer (NSCLC) patients (undergoing immune checkpoint inhibitor/inhibition therapy). These fecal samples were investigated in terms of microbial composition and their transcriptome activities using 16S rRNA gene amplicon sequencing and metatranscriptomic sequencing [56]. They revealed that the genus Ruminococcus is the strongest associated taxa in responders, and the responders’ enriched microbial pathways are carbon fixation pathways in the prokaryotes [56].
(ii) Integration of microbiome data with host data. As the 16S rRNA gene amplicon sequencing identifies the composition of intratumoral microbes, the resulting microbial composition can guide the size of chosen microbial reference genomes when computationally mining microbial profiling from host SRT data, enabling a less time-consuming and precise alignment. In a recent study, spatial transcriptomics and scRNA-seq were modified to capture microbial rRNA with spatial coordinates in host SRT data and microbial 16S rRNA with host cell barcodes at the single-cell level from oral squamous cell carcinoma (OSCC) and colorectal cancer (CRC) patients [57]. The results showed that bacteria are enriched in the TME niches with immune and epithelial cell functions and promote tumor growth rather than being distributed randomly in the TME. In addition, they found that cell-associated bacteria affect the expression of host genes involved in inflammation, metastasis, cell dormancy, and DNA repair pathways [57].
(iii) Integration of gut and intratumoral microbiome and host data. Co-analyzing data from multiple sequencing technologies on both fecal and tumor tissue samples enables us to explore the TME-intestinal transmission of microbes and metabolites and reveal the systematic interactions of IOM [9]. Uribe-Herranz et al. analyzed 16S rRNA gene amplicon sequencing and mass spectrometry data from fecal and tumor tissue samples of mice with melanoma and lung/cervical cancer [9]. They revealed that vancomycin treatment could eliminate gram-positive bacteria and decrease SCFA concentrations in the gut, enhancing the host’s antitumor immune response. Additionally, by applying immunohistochemistry (IHC), flow cytometry, and cytokine assays, researchers can examine the microbes-associated immune cell types, the density of immune cells and markers of antigen processing and presentation in TME, and the frequency of immune cells in the host systemic circulation to reveal the modulation role of microorganisms on the antitumor immune response.
Integrating different types of microbiome and host data not only inherits the challenges in bulk, single-cell, and spatial data mentioned above but more for data harmonization. The proper alignment of data types from multiple samples requires the careful removal of sample bias. The sequencing and modality bias still exist even for data profiled from the same data. More importantly, how to align microbial profiling in spatial spots and single cells is still unclear, as the microbiome obtained from scRNA-seq data is intracellular only while SRT can capture microbes residing in the TME. All these things make the challenge for integrated analysis squared.
CLINICAL APPLICATION OF IOM STUDIES
Designing robust computational methods in IOM studies enables the identification of tumor-associated microbial signatures and reveals more undiscovered IOM. These outcomes can aid the understanding of the underlying mechanisms of tumor development and progression, as well as the heterogeneity in the clinical response of patients. For example, the microbiota can promote lung cancer development via γδ T cell [24] (Figure 3A), and the commensal microbiome, such as Bifidobacterium longum, Collinsella aerofaciens, and Enterococcus faecium, may have a mechanistic impact on antitumor immunity in metastatic melanoma patients [12] (Figure 3B). Moreover, the modulation of pulmonary microbiota using antibiotic treatment can promote immunosurveillance against lung metastases, shedding light on new precise treatment designs for cancer prevention [25] (Figure 3C). Studies have showcased that fecal microbiota transplantation (FMT) promotes the immunotherapy response in refractory melanoma patients by modulating the gut microbiome [58, 59]. Enterococcus gallinarum MRx0518 treatment can cause the up-regulation of genes and metagenes associated with anti-tumor activity in solid tumors [60], showing potent immunostimulatory activity and anti-tumorigenic efficacy in a clinical trial (NCT03934827). Another clinical trial (NCT03829111) suggested that CBM588, a bifidogenic live bacterial product, appears to improve the overall survival of patients with metastatic renal cell carcinoma who were receiving nivolumab plus ipilimumab [61]. Microbial modulation in immunotherapy can be further reinforced by investigating the mechanism of action, long-term efficacy, and stability of gut microbiome modulation in the cancer treatment [62].
Figure 3. Clinical application of IOM studies.

Outcomes from computational methods in IOM studies help researchers to understand the IOM and guide more precise cancer treatments. (A) IOM can help researchers understand the mechanisms of tumor development. For example, the dysregulation of local microbiota can promote lung cancer development via γδ T cells [24]. (B) Microbes may contribute to the heterogeneity in the clinical response of cancer patients receiving the same treatments. For example, the differential composition of the commensal microbiome of metastatic melanoma patients may affect the effect of the immunotherapy [12]. (C) IOM can guide cancer treatment. An example is the modulation of pulmonary microbiota by antibiotic treatment, which promotes immunosurveillance against melanoma metastases to the lung [25]. (D) Microbial therapies provide a new opportunity for treating cancers. For example, tumor cell lysis triggered by oncolytic T-VEC releases TDA, GM-CSF, and new viral particles, which can enhance the activation of dendritic cells and initiate a systemic antitumor adaptive immune response in advanced melanoma patients [64]. Areg: amphiregulin; NK cell: natural killer cell; T-VEC: talimogene laherparepvec; TDA: tumor-derived antigens; GM-CSF: granulocyte-macrophage colony-stimulating factor.
Given that current cancer treatments cannot address refractory metastatic cancers, drug-resistant cancers, and cancers that evade immune clearance, “bugs as drugs” (such as microbial therapies) may provide solutions to these unresolvable clinical needs [2, 63]. Microbial therapies, including oncolytic viral therapy and bacterial antitumor therapy, treat cancer by exploiting tumor-specific infectious microbes [2, 63]. For oncolytic viral treatment, talimogene laherparepvec (T-VEC), a modified herpesvirus, is used to treat advanced melanoma by destroying tumor cells and triggering tumor-specific immune responses [64] (Figure 3D). For bacterial antitumor therapy, attenuated recombinant Listeria monocytogenes bacteria have been proven to induce long-lasting tumor-specific cytolytic T lymphocyte (CTL) responses by efficiently delivering recombinantly-expressed tumor antigens [63].
Recently, researchers have engineered genetically attenuated, auxotrophic, and inducible versions of Escherichia, Bifidobacterium, Listeria, Shigella, Clostridium, Lactococcus, Vibrio, and Salmonella species, which exhibited antitumor efficacy in preclinical models [65]. For example, attenuated Salmonella expressing aquatic flagellin has been demonstrated to destroy tumor cells by activating the human immune system [65]. Bifidobacterium bifidum strains can induce an antitumor host immune response, thus improving the efficacy of checkpoint inhibitors in mice [66]. Moreover, Clostridium bacteria can lyse tumor cells growing in hypoxic environments, and Clostridium novyi spores have been used to treat patients with solid tumors [67]. These known bacterial–host interactions can be valuable resources for evaluating computational predictions from bulk and single-cell data. Thus, we summarized a list of microorganisms linked to cancer and cancer treatment in the context of the IOM according to published literature (Table 3). These gathered microorganisms contribute to overcoming the issue that the lack of valid benchmark standards for evaluating and comparing results from different sequencing technologies and computational approaches.
Table 3.
A list of microorganisms linked with cancer and cancer treatment in the context of the IOM based on published literature.
| Cancer type | Microbes associated with cancer | Microbes associated with cancer treatment | Refs | |||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Microbe | Taxa | Type | Microbe | Taxa | Type | |||
|
| ||||||||
| Stomach cancer, gastric mucosa-associated lymphoid tissue (MALT) lymphoma, and cancer of the oesophagus | Helicobacter pylori $ | s | Bacteria | [96, 97] | ||||
|
| ||||||||
| Lung cancer | Prevotella | g | Bacteria | Clostridia | c | Bacteria | [9, 24, 98–100] | |
| Veillonella | g | Bacteria | Enterococcushirae * | s | Bacteria | |||
| Haemophilus | g | Bacteria | Firmicutes | p | Bacteria | |||
| Streptococcus | g | Bacteria | ||||||
| Leuconostocaceae | f | Bacteria | ||||||
| Sphingomonadaceae | f | Bacteria | ||||||
| Alphaproteobacteria | c | Bacteria | ||||||
| Herbaspirillum | g | Bacteria | ||||||
| Staphylococcus | g | Bacteria | ||||||
| Delftia | g | Bacteria | ||||||
| Burkholderiales | o | Bacteria | ||||||
| Proteobacteria | p | Bacteria | ||||||
|
| ||||||||
| Gastrointestinal tumor | Fusobacterium | g | Bacteria | - | [23, 30] | |||
| Candida | g | Fungi | ||||||
|
| ||||||||
| Liver hepatocellular carcinoma (LIHC) | Orthohepadnavirus | g | Viruses | - | [30] | |||
| Hepatotoxic microcystis | g | Bacteria | ||||||
|
| ||||||||
| Pancreatic adenocarcinoma (PDAC) | Sachharopolyspora | g | Bacteria | Sachharopolyspora | g | Bacteria | [20, 101] | |
| Pseudoxanthomonas | g | Bacteria | Pseudoxanthomonas | g | Bacteria | |||
| Streptomyces | g | Bacteria | Streptomyces | g | Bacteria | |||
| Bacillusclausii | s | Bacteria | Bacillus clausii | s | Bacteria | |||
| Malassezia | g | Fungi | ||||||
|
| ||||||||
| Pancreatic ductal adenocarcinoma (PDA) | Proteobacteria | p | Bacteria | Malassezia | g | Fungi | [20, 27, 102] | |
| Actinobacteria | p | Bacteria | Gammaproteobacteria * | c | Bacteria | |||
| Fusobacteria | p | Bacteria | ||||||
| Verruco microbia | p | Bacteria | ||||||
| Malassezia | g | Fungi | ||||||
|
| ||||||||
| Breast cancer | Fusobacterium nucleatum | s | Bacteria | - | [21] | |||
|
| ||||||||
| Colorectal Cancer (CRC) | Fusobacterium nucleatum | s | Bacteria | E. coli * | s | Bacteria | [8, 19, 30, 103–112] | |
| Faecali bacterium | g | Bacteria | Comamonas * | g | Bacteria | |||
| Fusobacterium nucleatum | s | Bacteria | Fusobacterium nucleatum | s | Bacteria | |||
| Bacteroi des | g | Bacteria | Bifidobacterium pseudolongum | s | Bacteria | |||
| Enterotoxigenic B. fragilis | s | Bacteria | Lactobacillus johnsonii | s | Bacteria | |||
| Campylobacterjejuni | s | Bacteria | Olsenella | g | Bacteria | |||
| Genotoxic pks+ E. coli | s | Bacteria | Bacteroides vulgatus * | s | Bacteria | |||
| Clostridiumramosum * | s | Bacteria | ||||||
|
| ||||||||
| Melanomao | Bifidobacteria | g | Bacteria | Bifidobacterium pseudolongum | s | Bacteria | [7, 8, 12, 113–116] | |
| Bifidobacteriumlongum | s | Bacteria | Bifidobacteria | g | Bacteria | |||
| Enterococcus faecium | s | Bacteria | Akkermansia muciniphila | s | Bacteria | |||
| Collinsella aerofaciens | s | Bacteria | Collinsella aerofaciens | s | Bacteria | |||
| Bacteroides | g | Bacteria | Enterococcus faecium | s | Bacteria | |||
| Parabacteroides | g | Bacteria | Olsenella | g | Bacteria | |||
| Lactobacillus johnsonii | s | Bacteria | ||||||
| Bifidobacterium longum | s | Bacteria | ||||||
| Bacteroides * | g | Bacteria | ||||||
| Burkholderiales * | o | Bacteria | ||||||
| Bifidobacterium * | g | Bacteria | ||||||
|
| ||||||||
| Cancer at anogenital sites, cancer of the upper aerodigestive tract, and cancer of the skin | Human papillomavirus $ | s | Viruses | - | - | [97] | ||
|
| ||||||||
| Cervical cancer | Human papillomavirus $ | s | Viruses | Clostridia | c | Bacteria | [9, 97, 117] | |
| Firmicutes | p | Bacteria | ||||||
|
| ||||||||
| Cervical squamous cell carcinoma (CESC) | Alphapa pillomavirus | g | Viruses | - | [30] | |||
|
| ||||||||
| Burkitt lymphoma, immunosuppression-related non-Hodgkin lymphoma, extranodal NK/T-cell lymphoma, Hodgkin lymphoma, and nasopharyngeal carcinoma | Epstein- Barr virus $ | s | Viruses | - | [97] | |||
|
| ||||||||
| Hepatocellular carcinoma, cholangiocarcinoma, and non-Hodgkin lymphoma | Hepatitis B virus $ | s | Viruses | - | [97] | |||
|
| ||||||||
| Hepatocellular carcinoma, lymphoid malignancies, leukaemias, and cancer of the thyroid | Hepatitis C virus $ | s | Viruses | - | [97] | |||
|
| ||||||||
| Kaposi sarcoma, primary effusion lymphoma, and multiple myeloma | Kaposisarcom a herpesvirus $ | s | Viruses | - | [97] | |||
|
| ||||||||
| Kaposi sarcoma, non-Hodgkin lymphoma, Hodgkin lymphoma, cervical and anogenital cancers, cancer of the skin, cancer of the conjunctiva, Cancer of the lung, and Cancer of the liver | Human immuno deficiency virus I $ | s | Viruses | - | [97] | |||
|
| ||||||||
| T-cell malignancies, cutaneous T-cell lymphoma, B- and T-cell lymphomas, and non-lymphomatous tumours | Human T-cell leukemi a virus type I $ | - | Viruses | - | [97] | |||
|
| ||||||||
| Cancer of the urinary bladder and cancers of the female genital tract | Schistosoma haematobium $ | s | Eukaryota | - | [90] | |||
|
| ||||||||
| Cholangiocarcinoma and hepatocellular carcinoma | Opisthorchis viverrini $ | s | Eukaryota | - | [90] | |||
|
| ||||||||
| Clonorc hissinensis $ | s | Eukaryota | ||||||
|
| ||||||||
| Bladder cancer | - | Bifidobacterium pseudolongum | s | Bacteria | [8] | |||
| Lactobacillus johnsonii | s | Bacteria | ||||||
| Olsenella | g | Bacteria | ||||||
|
| ||||||||
| Liver cancer | - | Clostridium scindens | s | Bacteria | [118] | |||
|
| ||||||||
| Epithelial tumor | - | Akkermansia muciniphila * | s | Bacteria | [119] | |||
|
| ||||||||
| Prostate cancer | - | Akkermansia muciniphila | s | Bacteria | [120] | |||
|
| ||||||||
| Sarcoma | - | Enterococcus hirae * | s | Bacteria | [99, 100] | |||
Microorganisms associated with cancer drug treatment. P, Phylum; c, Class; o, Order; f, Family; g, Genus; s, Species.
Microorganisms labeled as human carcinogens (“oncomicrobes”) by the International Association for Cancer Registries (IACR).
CONCLUDING REMARKS
Microorganisms in the gastrointestinal tract and TME niches contribute to tumor development and progression by interacting with the host immune system. Although it has been deeply investigated and developed, the gut microbiome only measures microbiome cohort and indirect connections to cancers, which is insufficient to characterize IOM computationally. With the development of sequencing techniques, investigating the intratumoral microbiome from host samples is receiving more attention. Current analyses of host bulk, single-cell, and spatial intratumoral microbial profiling provide a collection of decontaminated microbial compositions of tumor tissues. Microbial profiling mined from host intratumoral samples shows two strengths: (i) it can identify cell-type-specific intracellular microbes that match with host data in the same tissue, providing new insights into the investigation of the multi-omic IOM in host tissue samples; and (ii) it is easier to obtain this data than it is to obtain clinical biopsies.
Yet, computational methods and tools are critically needed to conquer issues in intratumoral microbiome prediction. Several questions are to be answered using computational strategies in elucidating IOM (see Outstanding Questions). It is also worth noting that new sequencing technologies, such as microbial single-cell DNA sequencing [68], microbial single-cell RNA sequencing [69], and microbial SRT sequencing [70], provide a different perspective on data for investigating IOM. When these technologies are applied to patient fecal samples, individual-microbe scale data provided by them help researchers elucidate the mechanistic interactions of IOM. Similar to the single-cell multimodal omics (scMulti-omics) technologies used in the host somatic cells [71], microbial scMulti-omics technologies can measure multiple molecular types from a single microbe (e.g., genomics, transcriptomics, and proteomics), thus enabling researchers to explore the links between microbial transcriptional regulatory mechanisms, tumor, and the host immune system. Although these technologies still need to be developed for general use in microbiological analyses or IOM research, we anticipate that more experimental data will be generated for in-depth functional analysis soon.
OUTSTANDING QUESTIONS.
How should computational methods for better profiling microbial communities and investigating the IOM at a higher level of taxonomic resolution (such as at the strain level) be developed?
What should advanced computational tools be generated to effectively mine microbes from the spatially resolved transcriptomics data to investigate IOM interactions?
What is the best practice for revealing undiscovered IOM mechanisms by integrating microbial data from bulk sequencing, single-cell sequencing, and spatially resolved transcriptomics data of host samples?
How should more sophisticated computational methods be designed for elucidating and interpreting the intrinsic biological associations among microorganisms, the host immune system, and transformed cells?
What is the sensitive and reliable method to apply scMulti-omics sequencing technologies to single-cell microbe to support IOM research?
Moreover, the promising trends for studying the cellular interactions and systematic mechanisms of IOM mainly include three aspects: (i) the analysis of microbial profiling from host single-cell and spatial sequencing data, (ii) the co-analysis of data from multiple sequencing technologies on different samples, and (iii) the utilization of deep learning methods to investigate biological associations of IOM. Overall, the computational analysis of IOM using high-throughput sequencing data is paving the way we understand host-microbiome relationships and interactions and how microbes are involved in TME and cancer treatment.
HIGHLIGHTS.
The gut and intratumoral microbiota significantly affect cancer development and progression by interacting with the host’s immune system, i.e., the immuno–oncology–microbiome (IOM).
Available microbial data in IOM studies is mined from existing host bulk sequencing and single-cell sequencing datasets to provide unprecedented opportunities for investigating IOM in various cancer types.
The development of rigorous computational methods for characterizing and elucidating IOM is urgently needed to guide researchers to unravel new biological insights and develop precision cancer therapeutics.
A reliable benchmarking system is needed to model IOM interactions, analyze IOM data, and evaluate computational predictions.
In-depth functional analyses of IOM mechanisms are necessary to develop novel therapeutic strategies targeting microbiota to improve cancer treatment outcomes.
ACKNOWLEDGMENTS
This work was supported by awards R01-GM131399 from the National Institute of General Medical Sciences of the National Institutes of Health. This work was supported by the Pelotonia Institute for Immuno-Oncology (PIIO) at the Ohio State University. The content is solely the responsibility of the authors and does not necessarily represent the official views of the PIIO. We want to thank Yihua Wang, Yuhan Sun, and Yuli Zhang from Shandong University for collecting materials. Figures 1 and 3 are created with BioRender.com.
GLOSSARY
- Bulk sequencing
examines the sequence information of bulk samples, usually containing multiple cells.
- Deep learning
is a type of machine learning algorithm that uses multiple layers to extract higher-level features from the raw input progressively.
- Graph neural network (GNN)
is a class of neural networks for processing data best represented by graph data structures. They were popularized by their use in supervised learning on the properties of various molecules.
- Metagenomic shotgun sequencing
a method in which nucleic acid from a sample is sequenced to identify and characterize microorganisms present in the sample, is being evaluated and used with increasing frequency for clinical microbiology diagnostic.
- Single-cell RNA sequencing (scRNA-seq)
examines the expression profiles of individual cells with optimized next-generation sequencing technologies, providing a higher resolution of cellular diversity.
- Spatially resolved transcriptomics (SRT)
is an overarching term for a range of methods designed for assigning cell types (identified by the mRNA readouts) to their locations in the histological sections. This method can also be used to determine the subcellular localization of mRNA molecules.
- The tumor microenvironment (TME)
is the environment around a tumor, including the surrounding blood vessels, immune cells, fibroblasts, signaling molecules, and the extracellular matrix (ECM).
- The Cancer Genome Atlas (TCGA):
a landmark cancer genomics program, molecularly characterized over 20,000 primary cancer and matched normal samples spanning 33 cancer types.
- Whole-genome sequencing (WGS):
also known as full genome sequencing, complete genome sequencing, or entire genome sequencing, is the process of determining the entirety, or nearly the entirety, of the DNA sequence of an organism’s genome at a single time.
- Whole-exome sequencing (WES):
also known as exome sequencing, is a genomic technique for sequencing all of the protein-coding regions of genes in a genome (known as the exome).
- Whole-transcriptome sequencing (RNA-seq)
is a sequencing technique that uses next-generation sequencing (NGS) to reveal the presence and quantity of RNA in a biological sample at a given moment.
- Wilcoxon rank-sum test
is a nonparametric test of the null hypothesis that, for randomly selected values X and Y from two populations, the probability of X being greater than Y is equal to the probability of Y being greater than X.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CODE AVAILABILITY
We provided a pipeline for analyzing microbial profiling mined from host single-cell sequencing datasets on GitHub at https://github.com/OSU-BMBL/IOM.
REFERENCE
- 1.Aldars-García L et al. (2021) Systematic review: The gut microbiome and its potential clinical application in inflammatory bowel disease. Microorganisms 9 (5), 977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sepich-Poore GD et al. (2021) The microbiome and human cancer. Science 371 (6536), eabc4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Toker J et al. (2020) The microbiome in immuno-oncology. Immunotherapy, 325–334. [DOI] [PubMed] [Google Scholar]
- 4.Lythgoe MP et al. (2022) Polymorphic microbes: a new emerging hallmark of cancer. Trends Microbiol. [DOI] [PubMed] [Google Scholar]
- 5.Peled JU et al. (2020) Microbiota as predictor of mortality in allogeneic hematopoietic-cell transplantation. N Engl J Med 382 (9), 822–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schluter J et al. (2020) The gut microbiota is associated with immune cell dynamics in humans. Nature 588 (7837), 303–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Y et al. (2019) Gut microbiota dependent anti-tumor immunity restricts melanoma growth in Rnf5−/−mice. Nat Commun 10 (1), 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mager LF et al. (2020) Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 369 (6510), 1481–1489. [DOI] [PubMed] [Google Scholar]
- 9.Uribe-Herranz M et al. (2020) Gut microbiota modulate dendritic cell antigen presentation and radiotherapy-induced antitumor immune response. The Journal of clinical investigation 130 (1), 466–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y et al. (2022) Effects of Gut Microbiota on Host Adaptive Immunity Under Immune Homeostasis and Tumor Pathology State. Frontiers in Immunology 13, 844335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim CH (2021) Control of lymphocyte functions by gut microbiota-derived short-chain fatty acids. Cell Mol Immunol 18 (5), 1161–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matson V et al. (2018) The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 359 (6371), 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gopalakrishnan V et al. (2018) Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359 (6371), 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kartal E et al. (2022) A faecal microbiota signature with high specificity for pancreatic cancer. Gut 71 (7), 1359–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee KA et al. (2021) The gut microbiome: what the oncologist ought to know. British Journal of Cancer 125 (9), 1197–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cammarota G et al. (2020) Gut microbiome, big data and machine learning to promote precision medicine for cancer. Nature Reviews Gastroenterology & Hepatology 17 (10), 635–648. [DOI] [PubMed] [Google Scholar]
- 17.Turnbaugh PJ et al. (2007) The human microbiome project. Nature 449 (7164), 804–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ehrlich SD and Consortium M (2011) MetaHIT: The European Union Project on metagenomics of the human intestinal tract. In Metagenomics of the human body, pp. 307–316, Springer. [Google Scholar]
- 19.Hamada T et al. (2018) Fusobacterium nucleatum in colorectal cancer relates to immune response differentially by tumor microsatellite instability status. Cancer immunology research 6 (11), 1327–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aykut B et al. (2019) The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature 574 (7777), 264–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parhi L et al. (2020) Breast cancer colonization by Fusobacterium nucleatum accelerates tumor growth and metastatic progression. Nat Commun 11 (1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fu A et al. (2022) Tumor-resident intracellular microbiota promotes metastatic colonization in breast cancer. Cell 185 (8), 1356–1372. e26. [DOI] [PubMed] [Google Scholar]
- 23.Dohlman AB et al. (2022) A pan-cancer mycobiome analysis reveals fungal involvement in gastrointestinal and lung tumors. Cell 185 (20), 3807–3822. e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin C et al. (2019) Commensal microbiota promote lung cancer development via γδ T cells. Cell 176 (5), 998–1013. e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le Noci V et al. (2018) Modulation of pulmonary microbiota by antibiotic or probiotic aerosol therapy: a strategy to promote immunosurveillance against lung metastases. Cell reports 24 (13), 3528–3538. [DOI] [PubMed] [Google Scholar]
- 26.Pushalkar S et al. (2018) The Pancreatic Cancer Microbiome Promotes Oncogenesis by Induction of Innate and Adaptive Immune SuppressionMicrobiome Influences Pancreatic Oncogenesis. Cancer discovery 8 (4), 403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geller LT et al. (2017) Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 357 (6356), 1156–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nejman D et al. (2020) The human tumor microbiome is composed of tumor type–specific intracellular bacteria. Science 368 (6494), 973–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu J and Zhang Y (2022) Intratumor microbiome in cancer progression: current developments, challenges and future trends. Biomark Res 10 (1), 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poore GD et al. (2020) Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 579 (7800), 567–574. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Narunsky-Haziza L et al. (2022) Pan-cancer analyses reveal cancer-type-specific fungal ecologies and bacteriome interactions. Cell 185 (20), 3789–3806. e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dohlman AB et al. (2021) The cancer microbiome atlas: a pan-cancer comparative analysis to distinguish tissue-resident microbiota from contaminants. Cell Host Microbe 29 (2), 281–298. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H et al. (2008) Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res 18 (11), 1851–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dobin A et al. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29 (1), 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wood DE and Salzberg SL (2014) Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol 15 (3), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Truong DT et al. (2015) MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Meth 12 (10), 902–903. [DOI] [PubMed] [Google Scholar]
- 37.Hillmann B et al. (2018) Evaluating the information content of shallow shotgun metagenomics. Msystems 3 (6), e00069–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Namiki T et al. , MetaVelvet: an extension of Velvet assembler to de novo metagenome assembly from short sequence reads, Proceedings of the 2nd ACM conference on bioinformatics, computational biology and biomedicine, 2011, pp. 116–124. [Google Scholar]
- 39.Li D et al. (2015) MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31 (10), 1674–1676. [DOI] [PubMed] [Google Scholar]
- 40.Kostic AD et al. (2011) PathSeq: software to identify or discover microbes by deep sequencing of human tissue. Nat Biotechnol 29 (5), 393–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davis NM et al. (2018) Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6 (1), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knights D et al. (2011) Bayesian community-wide culture-independent microbial source tracking. Nat Meth 8 (9), 761–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paulson JN et al. (2013) Differential abundance analysis for microbial marker-gene surveys. Nat Meth 10 (12), 1200–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weiss S et al. (2017) Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 5 (1), 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robinson W et al. (2021) CSI-Microbes: Identifying cell-type specific intracellular microbes from single-cell RNA-seq data. bioRxiv, 2020.05.14.096230. [Google Scholar]
- 46.Ghaddar B et al. (2022) Tumor microbiome links cellular programs and immunity in pancreatic cancer. Cancer Cell 40 (10), 1240–1253. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bost P et al. (2020) Host-viral infection maps reveal signatures of severe COVID-19 patients. Cell 181 (7), 1475–1488. e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hunter MV et al. (2021) Spatially resolved transcriptomics reveals the architecture of the tumor-microenvironment interface. Nat Commun 12 (1), 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ji AL et al. (2020) Multimodal analysis of composition and spatial architecture in human squamous cell carcinoma. Cell 182 (2), 497–514.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shi H et al. (2020) Highly multiplexed spatial mapping of microbial communities. Nature 588 (7839), 676–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wong-Rolle A et al. (2022) Spatial meta-transcriptomics reveal associations of intratumor bacteria burden with lung cancer cells showing a distinct oncogenic signature. Journal for ImmunoTherapy of Cancer 10 (7), e004698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haig BD (2003) What is a spurious correlation? Understanding Statistics: Statistical Issues in Psychology, Education, and the Social Sciences 2 (2), 125–132. [Google Scholar]
- 53.Wang J et al. (2021) scGNN is a novel graph neural network framework for single-cell RNA-Seq analyses. Nature Communications 12 (1), 1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chang Y et al. (2022) Define and visualize pathological architectures of human tissues from spatially resolved transcriptomics using deep learning. Comput Struct Biotechnol J 20, 4600–4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sudhakar P et al. (2021) Computational Biology and Machine Learning Approaches to Understand Mechanistic Microbiome-Host Interactions. Frontiers in Microbiology 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Newsome RC et al. (2022) Interaction of bacterial genera associated with therapeutic response to immune checkpoint PD-1 blockade in a United States cohort. Genome Med 14 (1), 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Niño JLG et al. (2022) Effect of the intratumoral microbiota on spatial and cellular heterogeneity in cancer. Nature, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Davar D et al. (2021) Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 371 (6529), 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Baruch EN et al. (2021) Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 371 (6529), 602–609. [DOI] [PubMed] [Google Scholar]
- 60.Lythgoe M et al. (2021) 543P Neoadjuvant MRx0518 treatment is associated with significant gene and metagene signature changes in solid tumours. Ann Oncol 32, S607. [Google Scholar]
- 61.Dizman N et al. (2022) Nivolumab plus ipilimumab with or without live bacterial supplementation in metastatic renal cell carcinoma: a randomized phase 1 trial. Nat Med 28 (4), 704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lythgoe MP et al. (2022) The potential of fecal microbiota transplantation in oncology. Trends Microbiol 30 (1), 10–12. [DOI] [PubMed] [Google Scholar]
- 63.Forbes NS et al. (2018) White paper on microbial anti-cancer therapy and prevention. Journal for immunotherapy of cancer 6 (1), 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ferrucci PF et al. (2021) Talimogene laherparepvec (T-VEC): an intralesional cancer immunotherapy for advanced melanoma. Cancers 13 (6), 1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kramer MG et al. (2018) Bacterial therapy of cancer: promises, limitations, and insights for future directions. Frontiers in microbiology 9, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee S-H et al. (2021) Bifidobacterium bifidum strains synergize with immune checkpoint inhibitors to reduce tumour burden in mice. Nature Microbiology 6 (3), 277–288. [DOI] [PubMed] [Google Scholar]
- 67.Janku F et al. (2021) Intratumoral Injection of Clostridium novyi-NT Spores in Patients with Treatment-refractory Advanced Solid TumorsPhase I Study of Intratumoral Clostridium novyi-NT. Clin Cancer Res 27 (1), 96–106. [DOI] [PubMed] [Google Scholar]
- 68.Zheng W et al. (2022) High-throughput, single-microbe genomics with strain resolution, applied to a human gut microbiome. Science 376 (6597), eabm1483. [DOI] [PubMed] [Google Scholar]
- 69.Dohn R et al. (2021) mDrop-seq: Massively parallel single-cell RNA-seq of Saccharomyces cerevisiae and Candida albicans. Vaccines 10 (1), 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dar D et al. (2021) Spatial transcriptomics of planktonic and sessile bacterial populations at single-cell resolution. Science 373 (6556), eabi4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ma A et al. (2020) Integrative methods and practical challenges for single-cell multi-omics. Trends Biotechnol 38 (9), 1007–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Baruzzo G et al. (2017) Simulation-based comprehensive benchmarking of RNA-seq aligners. Nat Meth 14 (2), 135–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alser M et al. (2021) Technology dictates algorithms: recent developments in read alignment. Genome Biol 22 (1), 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li H and Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25 (14), 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Prasad VVS and Loshma G (2011) HPC-MAQ: a parallel short-read reference assembler. CCSEA 2011 2011, 84. [Google Scholar]
- 76.Meyer F et al. (2022) Critical Assessment of Metagenome Interpretation: the second round of challenges. Nat Meth 19 (4), 429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zeng H et al. (2021) Physiological and metagenomic strategies uncover the rhizosphere bacterial microbiome succession underlying three common environmental stresses in cassava. J Hazard Mater 411, 125143. [DOI] [PubMed] [Google Scholar]
- 78.Lind AL and Pollard KS (2021) Accurate and sensitive detection of microbial eukaryotes from whole metagenome shotgun sequencing. Microbiome 9 (1), 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vollmers J et al. (2017) Comparing and evaluating metagenome assembly tools from a microbiologist’s perspective-not only size matters! PLoS ONE 12 (1), e0169662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Quince C et al. (2017) Shotgun metagenomics, from sampling to analysis. Nat Biotechnol 35 (9), 833–844. [DOI] [PubMed] [Google Scholar]
- 81.Cao Q et al. (2021) Effects of rare microbiome taxa filtering on statistical analysis. Frontiers in microbiology 11, 607325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xu Y et al. (2022) Application of fast expectation-maximization microbial source tracking to discern fecal contamination in rivers exposed to low fecal inputs. Journal of Microbiology, 1–8. [DOI] [PubMed] [Google Scholar]
- 83.Shenhav L et al. (2019) FEAST: fast expectation-maximization for microbial source tracking. Nat Meth 16 (7), 627–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Morgan M et al. (2009) ShortRead: a bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics 25 (19), 2607–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McKenna A et al. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20 (9), 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen S et al. (2018) fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34 (17), i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bolger AM et al. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30 (15), 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Smith T et al. (2017) UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res 27 (3), 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wysock BM et al. (1995) Statistical procedures for corrosion studies. Journal-American Water Works Association 87 (7), 99–112. [Google Scholar]
- 90.Parab S and Bhalerao S (2010) Choosing statistical test. International journal of Ayurveda research 1 (3), 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.De Winter JC et al. (2016) Comparing the Pearson and Spearman correlation coefficients across distributions and sample sizes: A tutorial using simulations and empirical data. Psychol Methods 21 (3), 273. [DOI] [PubMed] [Google Scholar]
- 92.Peng J et al. (2019) Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res 29 (9), 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Paulson K et al. (2018) Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA. Nat Commun 9 (1), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lee H-O et al. (2020) Lineage-dependent gene expression programs influence the immune landscape of colorectal cancer. Nat Genet 52 (6), 594–603. [DOI] [PubMed] [Google Scholar]
- 95.Maynard A et al. (2020) Therapy-induced evolution of human lung cancer revealed by single-cell RNA sequencing. Cell 182 (5), 1232–1251.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Suzuki M et al. (2009) Helicobacter pylori CagA phosphorylation-independent function in epithelial proliferation and inflammation. Cell Host Microbe 5 (1), 23–34. [DOI] [PubMed] [Google Scholar]
- 97.Group IW (2012) IARC Working Group on the Evaluation of Carcinogenic Risks to Humans: Biological agents. Volume 100 B. A review of human carcinogens. IARC Monogr Eval Carcinog Risks Hum 100 (pt B), 1. [PMC free article] [PubMed] [Google Scholar]
- 98.Tsay J-CJ et al. (2021) Lower airway dysbiosis affects lung cancer progression. Cancer discovery 11 (2), 293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Viaud S et al. (2013) The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 342 (6161), 971–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Daillère R et al. (2016) Enterococcus hirae and Barnesiella intestinihominis facilitate cyclophosphamide-induced therapeutic immunomodulatory effects. Immunity 45 (4), 931–943. [DOI] [PubMed] [Google Scholar]
- 101.Riquelme E et al. (2019) Tumor microbiome diversity and composition influence pancreatic cancer outcomes. Cell 178 (4), 795–806.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pushalkar S et al. (2018) The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer discovery 8 (4), 403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pleguezuelos-Manzano C et al. (2020) Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature 580 (7802), 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dejea CM et al. (2018) Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 359 (6375), 592–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Barrett M et al. (2020) Mutagenesis by microbe: The role of the microbiota in shaping the cancer genome. Trends in Cancer 6 (4), 277–287. [DOI] [PubMed] [Google Scholar]
- 106.Yu T et al. (2017) Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell 170 (3), 548–563.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.He Z et al. (2019) Campylobacter jejuni promotes colorectal tumorigenesis through the action of cytolethal distending toxin. Gut 68 (2), 289–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rubinstein MR et al. (2013) Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe 14 (2), 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nanjundappa RH et al. (2017) A gut microbial mimic that hijacks diabetogenic autoreactivity to suppress colitis. Cell 171 (3), 655–667.e17. [DOI] [PubMed] [Google Scholar]
- 110.Scott TA et al. (2017) Host-microbe co-metabolism dictates cancer drug efficacy in C. elegans. Cell 169 (3), 442–456.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.García-González AP et al. (2017) Bacterial metabolism affects the C. elegans response to cancer chemotherapeutics. Cell 169 (3), 431–441.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wallace BD et al. (2010) Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science 330 (6005), 831–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Snyder A et al. (2014) Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 371 (23), 2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Li Y et al. (2020) Prebiotic-induced anti-tumor immunity attenuates tumor growth. Cell reports 30 (6), 1753–1766. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Vétizou M et al. (2015) Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 350 (6264), 1079–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sivan A et al. (2015) Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350 (6264), 1084–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lowy DR and Schiller JT (2017) Preventing cancer and other diseases caused by human papillomavirus infection: 2017 Lasker-DeBakey Clinical Research Award. JAMA 318 (10), 901–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ma C et al. (2018) Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 360 (6391), eaan5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Routy B et al. (2018) Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359 (6371), 91–97. [DOI] [PubMed] [Google Scholar]
- 120.Daisley BA et al. (2020) Abiraterone acetate preferentially enriches for the gut commensal Akkermansia muciniphila in castrate-resistant prostate cancer patients. Nat Commun 11 (1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]