

Abstract
Epidermal Growth Factor Receptor (EGFR) signaling, and EGFR mutations play key roles in cancer pathogenesis, particularly in the development of drug resistance. For the ~20% of all non-small cell lung cancer (NSCLC) patients that harbor an activating mutation, EGFR tyrosine kinase inhibitors (TKIs) provide initial clinical responses. However, long-term efficacy is not possible due to acquired drug resistance. Despite a gradually increasing knowledge of the mechanisms underpinning the development of resistance in tumors, there has been very little success in overcoming it and it is probable that many additional mechanisms are still unknown. Herein, publicly available RNASeq (RNA sequencing) datasets comparing lung cancer cell lines treated with EGFR TKIs until resistance developed with their corresponding parental cells and protein array data from our own EGFR TKI treated xenograft tumors, were analyzed for differential gene expression, with the intent to investigate the potential mechanisms of drug resistance to EGFR TKIs. Pathway analysis, as well as structural disorder analysis of proteins in these pathways, revealed several key proteins, including DUSP1, DUSP6, GAB2, and FOS, that could be targeted using novel combination therapies to overcome EGFR TKI resistance in lung cancer.
Keywords: EGFR TKI Resistance, Lung Cancer, Bioinformatics
Simple Summary:
Lung cancer is by far the deadliest type of cancer in both men and women. The five-year survival rate for patients with lung cancer is one of the lowest of any cancer at 21%. Lung cancer treatment has historically relied on the use of nonspecific, toxic drugs to treat patient’s tumors, however recent trends have favored the development of less toxic, targeted agents. Despite this advancement, the use of these drugs has only been associated with a 1.5-year average increase in survival. This is because no treatment developed thus far is immune to the development of resistance. There is a great unmet need to develop novel treatments specifically to overcome drug resistance and prevent patient relapse.
Graphical Abstract:

Introduction:
Non-small Cell Lung Cancer:
The five-year survival rate for patients with lung cancer is one of the lowest of any cancer at 22%, while the overall five-year survival for cancer patients is 68%.[1] One of the main reasons lung cancer remains so deadly is that it is often diagnosed in the later stages of the disease.[1] Other cancers that are often diagnosed at later stages, such as, liver (20%), esophagus (20%), and pancreas (11%) have equally low 5-year survival rates.[1] However, lung cancer has a higher prevalence and causes a greater number of deaths than all three of these cancer combined.[1] Even when lung cancer is diagnosed at an early stage and patients are able to receive surgical resection, around 55% of these patients will experience postoperative recurrence of the tumor within 5 years.[2, 3]
Non-small cell lung cancer (NSCLC) accounts for about 80–85% of all lung cancer cases.[4] NSCLC patients are usually initially treated with platinum based chemotherapeutic regimen that all too often fail to cure the disease.[5, 6] The major reason for this outcome is the development of drug resistance.[5] Cancer treatment has historically relied on the use of these nonspecific, toxic drugs to treat patient’s tumors, however recent trends in oncology have favored the development of less toxic, molecularly targeted agents.[7, 8] Despite this advancement, the use of these drugs has only been associated with a 1.5-year average increase in survival.[9] This is because no treatment developed thus far is immune to the development of resistance. There is a great unmet need to develop novel treatments specifically to overcome drug resistance and prevent cancer recurrence. Through their ability to target multiple pathways or multiple elements of the same pathway simultaneously, combination therapies may prevent any single new mutation that arises from conferring drug resistance.[5, 10]
Epidermal Growth Factor Receptor:
EGFR is commonly mutated and/or overexpressed in different types of human cancers, including glioblastoma, brain, lung, breast, and ovarian cancer.[11] EGFR mutations and genetic rearrangements can cause sustained activation of downstream signaling via altered receptor endocytosis and trafficking, disrupted receptor ubiquitination and lysosomal degradation, or ligand-independent receptor activation.[11] Tumors from patients with NSCLC can be screened for the presence of EGFR activating mutations, which commonly occur in exon 19 as an in frame deletion or exon 21 as a point mutation (L858R).[12] About 20% of all NSCLC patients are expected to harbor an EGFR activating mutation.[12] Clinically relevant EGFR activating mutations include, deletions in exon 19 that cause elimination of the amino acid motif, LREA, and point mutations in exon 21 that substitute arginine for leucine at position 858 (L858R).[13] These two mutations account for about 85% of EGFR mutations in the NSCLC and make EGFR constitutively active and oncogenic.[13]
EGFR Tyrosine Kinase Inhibitors:
EGFR inhibitors have been shown to provide clinical benefits over chemotherapy for lung cancer patients with EGFR activating mutations.[14] The number of lung cancer patients who harbor an EGFR mutation and are expected to respond to EGFR targeted therapy is around 14–20%.[15, 16] Monoclonal antibodies targeting EGFR were the first anti-EGFR therapies used in the clinic.[17, 18] Anti-EGFR antibodies bind to the ligand binding regions of EGFR with high affinity and compete with the normal ligands to block EGFR activation, inducing receptor internalization and downregulation, and can recruit effector cells of the immune system that induce a cytotoxic immune response.[17, 18] Examples of clinically approved anti-EGFR antibodies include, Cetuximab, a chimeric mouse/human IgG1 antibody, Panitumumab, a humanized IgG2 mAb and, Nimotuzumab, a humanized IgG1 antibody.[17, 18] Small molecule anti-EGFR agents, EGFR TKIs, are reversibly or irreversibly directed to bind to the catalytic domain of EGFR.[17, 18] First generation (gefitinib,[19] erlotinib,[20] and lapatinib[21]), second generation (afatinib[22]), and most recently third-generation (osimertinib[23]) drugs are clinically approved to treat NSCLC patients.[24] Small molecule EGFR TKIs work by binding to and blocking the ATP-binding site on EGFR preventing tyrosine kinase activity and downstream signaling.[17, 18]
Drug resistance has, in nearly all cases, stifled treatment for EGFR mutated NSCLC.[12, 24] In most cases, patients treated using first or second generation TKIs become completely resistant in around 9–13 months.[12, 24] Recently, third-generation EGFR inhibitors, such as osimertinib, have emerged as potential therapeutics to block the growth of EGFR mutants that do not respond to previous generation EGFR TKIs.[12, 24] However, these have also proven to be susceptible to the development of drug resistance.[12, 24] Each succeeding generation was developed to combat resistance issues of the previous generations. Target alteration, increased ligand production, increased downstream pathway activation, and alternative pathway activation have all been proposed as mechanisms of resistance to EGFR TKIs[12, 24]. The common EGFR mutations that confer primary resistance to EGFR TKIs include, exon 19 mutations L747S/D761Y, exon 20 T790M, and the exon 21 mutation T854A.[25] Non-EGFR mutation dependent mechanisms of primary resistance include, deletion of tumor suppressor gene PTEN (Phosphatase and tensin homolog), PIK3CA (Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha) mutations, activation of the NFκB (nuclear factor kappa-light-chain-enhancer of activated B cells) pathway, polymorphisms of pro-apoptotic protein BIM (Bcl-2-like protein 11), increased expression of HGF (Hepatocyte Growth Factor), increased MET (c-Met proto-oncogene protein)-mediated activation, KRAS (KRAS Proto-Oncogene) mutations, BRAF (B-Raf Proto-Oncogene) mutations, and ALK (Anaplastic lymphoma kinase) translocation.[25] About 50% of NSCLC patients treated with EGFR TKIs will develop resistance due to the T790M mutation, which causes resistance to EGFR-TKIs is by increasing the affinity to ATP (Adenosine triphosphate) over the EGFR TKIs, which are competing with ATP for binding.[25] EGFR TKIs are the standard of care for patients harboring EGFR-sensitizing mutations, but as it stands today, the majority of patients given these treatments go on to develop resistance. Despite an increased understanding of complicated EGFR TKI resistance, platinum-based chemotherapy is the only approved regimen for patients experiencing disease progression on Osimertinib to date.[26]
Numerous cellular pathways have been implicated in EGFR TKI resistance, and this has led to the idea that combination drug therapies targeting both EGFR and also particular resistance pathways would be a promising strategy to overcome EGFR TKI resistance.[27, 28] For example, MET signaling is activated in H1975 cells that express the T790M EGFR mutation suggesting that combination treatment with a MET inhibitor and EGFR TKI may represent a strategy to overcome T790M mediated EGFR TKI resistance.[29] The upregulation of Hh (Hedgehog) signaling was reported to contribute to EGFR-TKI resistance and blockade of Hh signaling sensitized EGFR TKI resistant cells to EGFR TKI therapy.[30] A combination treatment with a MEK1/2 inhibitor and lapatinib was able to overcome EGFR TKI resistance through the inhibition of ERK (Extracellular signal-regulated kinases) and the upregulation of BIM.[31] Also, a STAT3 (Signal transducer and activator of transcription 3) inhibitor along with an EGFR TKI was shown to significantly enhance anti-tumor effects of EGFR TKI treatment.[32] Further, since NF-kB activation acts as a survival mechanism upregulated by EGFR TKI treatment, it has been suggested that NF-kB inhibition along with EGFR TKI treatment could overcome this resistance response.[33] Moreover, lapatinib resistance can be mediated through a p110a protein upregulation and/or mutation-induced PI3K activation and based on this a combination targeted therapy with a p110a-specific PI3K inhibitor was shown to overcome lapatinib resistance.[34] These studies serve to highlight the potential of combination therapies to overcome the vast number of resistance mechanisms cells can undergo after EGFR TKI treatment.
Many different cell line models have been developed to study EGFR TKI resistance in lung cancer.[35–40] In one study, KE Ware et, al. established gefitinib resistant lung cancer cell lines through a chronic adaptation model, in which the cells were exposed to step-wise increasing concentrations of gefitinib until they could routinely be cultured in a concentration of 3μM.[40] This led the authors to discover that these cells undergo an induction of FGFR1 (Fibroblast growth factor receptor 1) -FGF2 (Fibroblast growth factor 2) autocrine signaling pathway, which is enough to confer resistance to gefitinib and drive cancerous growth in presence of the drug. In another study, Kyung-A Song et. al. created gefitinib tolerant lung cancer cell lines by exposing the cells to 50nM gefitinib for 6 days and then withdrawing the drug for the next 3 days.[39] Using this model, the authors show that the gefitinib tolerant cells upregulate expression the anti-apoptotic protein, MCL-1 (Induced myeloid leukemia cell differentiation protein). This allows the cells to survive the short-term apoptotic pressure of gefitinib and allows them time to acquire a secondary resistance mechanism that will allow them to grow in presence of the gefitinib. Matias Casas-Selves et. al. used a loss-of-function, whole genome shRNA screen, to identify the canonical Wnt (Proto-Oncogene Wnt-1) pathway, particularly the poly-ADP-ribosylating enzymes tankyrase 1 and 2 that positively regulate canonical Wnt signaling, as crucial to cell survival during initial exposure to gefitinib.[41] This model exposed the cells to gefitinib for 2 days at concentrations that would normally inhibit expansion by ~70%, followed by 4 days without gefitinib. In one last example, Aaron N Hata et. al. created gefitinib tolerant, intermediate resistant, and late resistant cell pools, which allowed them to study the distinct evolutionary paths that can lead the cells to becoming resistant to gefitinib.[36] The authors found that EGFRT790M-positive clones could arise from the selection of pre-existing clones or from initially EGFRT790M-negative drug-tolerant cells. The authors hypothesized that the initial mechanisms that promote survival of the drug-tolerant cells may be enough to prevent apoptosis but may not fully recapitulate the oncogenic signaling provided by EGFR, so these cells provide a reservoir of cells from which genetic mechanisms of acquired resistance can evolve.
We reasoned that a better understanding of the mechanisms responsible for initial cell survival after exposure to EGFR TKIs may help identify treatments that are able to avoid or reverse development of drug resistance in the clinic. Specifically, we sought to identify additional resistance pathways and mechanisms beyond the well-studied mechanisms of resistance to EGFR TKIs such as mutations in the EGFR receptor (T790M, C797S), activation of EGFR downstream proteins (MEK, AKT, PI3K), activation of alternative tyrosine kinase pathways [HER2, MET, AXL (AXL Receptor Tyrosine Kinase)], and activation of growth factor receptors [FGFR (Fibroblast growth factor receptor), IGF1R (Insulin Like Growth Factor 1 Receptor)].[42] To this end, we compared ten publicly available RNASeq datasets of lung cancer cell lines treated with EGFR TKIs until resistance developed and their corresponding parental cells. These datasets were then analyzed for differential gene expression and a comparison analysis was performed to determine similarities between the datasets. To further explore potential mechanisms of EGFR TKI resistance, a list of genes that were significantly changed in a majority of the datasets was combined with protein array data obtained from a xenograft mouse tumor model of EGFR TKI resistance to form a master list. Pathway analysis was then used to plot known interactions between these genes and EGFR. Lastly, structural disorder analysis was performed on each of these genes to explore potential roles of intrinsic disorder in the predicted EGFR interaction network. The genes and pathways identified herein warrant further validation and biological experimentation because they have the potential to lead to new therapies to overcome drug resistance.
Results:
Dataset Collection and Differential Gene Expression Analysis:
With the intent to investigate the development of drug resistance to EGFR TKIs, ten publicly available RNA-Sequencing datasets were downloaded from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database.[43] All of these datasets contain data comparing cell lines treated with EGFR TKIs and their corresponding untreated or parental cells.[35, 36, 38–40] The EGFR TKI resistant cell lines in each dataset were obtained via different protocols, as each protocol was developed and verified by the researchers that obtained the original RNASeq data (Table 1).
Table 1:
EGFR TKI Resistant Datasets
| Data Set 138 | Data Set 235 | Data Set 340 | Data Set 440 | Data Set 539 | Data Set 636 | Data Set 736 | Data Set 836 | Data Set 936 | Data Set 1036 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Accession | GSE80802 | GSE62118 | GSE79688 | GSE79688 | GSE117610 | GSE75602 | GSE75602 | GSE75602 | GSE75602 | GSE75602 |
| CELLS LINE | HCC4006 | HCC827 | HCC827 | HCC4006 | PC9 | PC9 | PC9 | PC9 | PC9 | PC9 |
| EGFR TKI | Gefitinib | Erlotinib | Gefitinib | Gefitinib | Gefitinib | Gefitinib | Gefitinib | Gefitinib | Gefitinib | Gefitinib |
| MAX [C] | 100nM | 2uM | Up to 3uM | Up to 3uM | 50nM | 1mM | Not Specified | Not Specified | Not Specified | Not Specified |
| LENGTH OF TX | 24 Hours | 47 Days | Not Specified | Not Specified | 6 Days | 24 Hours | Not Specified | Not Specified | 2 Weeks | 2 Weeks |
| # of Samples (N) | Resistant (N=4) Control (N=4) |
Resistant (N=2) Control (N=2) |
Resistant (N=2) Control (N=2) |
Resistant (N=2) Control (N=2) |
Resistant (N=3) Control (N=2) |
Resistant (N=2) Control (N=2) |
Resistant (N=2) Control (N=2) |
Resistant (N=2) Control (N=2) |
Resistant (N=2) Control (N=2) |
Resistant (N=2) Control (N=2) |
| # of DEGS | Up = 677 Down = 868 |
Up = 5,791 Down = 6,161 |
Up = 689 Down = 556 |
Up = 4,859 Down = 4,969 |
Up = 649 Down = 1,233 |
Up = 1,946 Down = 2,477 |
Up = 4,766 Down = 4,646 |
Up = 850 Down = 990 |
Up = 1,501 Down = 1,510 |
Up = 2,217 Down = 1,974 |
Raw RNA-SEQ datasets (.fasta files) were downloaded using the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database. Data was originally obtained by authors of the referenced manuscripts. Each dataset contains .fasta files for control cells and EGFR TKI resistant cells. This table details available information about the EGFR TKI used to make the resistant cell line, length of EGFR TKI treatment used to confer resistance, and the max EGFR TKI concentration that resistant cells were exposed to in their growth media. DEGs – differentially expressed genes (p < 0.05); TX – treatment.
After the FASTA files for each dataset were downloaded, they were analyzed for the differential gene expression using tools available on Galaxy, an open-source, free of charge, web-based platform (Figure 1).[44] Table 1 list the number of significantly expressed DEGs found for each dataset (p < 0.05). Table 2 shows the list of differentially expressed genes (DEGs) that are commonly up or down regulated in 7 or more of the 10 RNASeq datasets.
Figure 1: Workflow for RNA-SEQ data analysis.

Datasets were analyzed for differential gene expression using the indicated tools, which are available on Galaxy, an open-source web-based platform. The raw sequencing data was processed to remove any adaptor, PCR primers and low-quality transcripts using FASTQC and Trimmomatic. The samples were then aligned against human genome using HISAT2. Gene expression measurement was then performed from aligned reads using featureCounts. Differential gene expression was calculated using DESeq2.
Table 2:
Most Common DEGs
| DEG | Expression Pattern | # Of Occurrences* |
|---|---|---|
| DHRS2 | Down | 9 |
| DAB2 | Up | 8 |
| DAPK1 | Up | 8 |
| GPCPD1 | Down | 8 |
| NR2F1 | Down | 8 |
| RIMKLA | Down | 8 |
| AHNAK2 | Up | 7 |
| ATP10D | Up | 7 |
| BIRC3 | Up | 7 |
| CDK14 | Up | 7 |
| CLU | Up | 7 |
| CPM | Up | 7 |
| CYP1B1 | Up | 7 |
| EDIL3 | Up | 7 |
| ERAP2 | Up | 7 |
| FSTL1 | Up | 7 |
| GABARAPL1 | Up | 7 |
| GPRC5B | Up | 7 |
| HEG1 | Up | 7 |
| ITPR2 | Up | 7 |
| MMP14 | Up | 7 |
| PAWR | Up | 7 |
| SPP1 | Up | 7 |
| SQSTM1 | Up | 7 |
| SYNPO | Up | 7 |
| UGCG | Up | 7 |
| ACOX2 | Down | 7 |
| AGR2 | Down | 7 |
| AK4 | Down | 7 |
| ALDOA | Down | 7 |
| C11orf86 | Down | 7 |
| C9orf152 | Down | 7 |
| CA9 | Down | 7 |
| CAMK1D | Down | 7 |
| CASC8 | Down | 7 |
| CEACAM6 | Down | 7 |
| DNAH2 | Down | 7 |
| EEPD1 | Down | 7 |
| ELFN2 | Down | 7 |
| EMP1 | Down | 7 |
| EMP2 | Down | 7 |
| ENTPD6 | Down | 7 |
| F12 | Down | 7 |
| FOS | Down | 7 |
| GGCT | Down | 7 |
| GJB2 | Down | 7 |
| GRHL1 | Down | 7 |
| IGFBP2 | Down | 7 |
| KCNF1 | Down | 7 |
| KLHDC7A | Down | 7 |
| LOXL1 | Down | 7 |
| PDK3 | Down | 7 |
| RAB3D | Down | 7 |
| RP11-466H18.1 | Down | 7 |
| S100A14 | Down | 7 |
| S100A9 | Down | 7 |
| SPDEF | Down | 7 |
| SPNS2 | Down | 7 |
| SPRY1 | Down | 7 |
| SULT1A2 | Down | 7 |
| TGM2 | Down | 7 |
| TMEM205 | Down | 7 |
| TMX4 | Down | 7 |
| WFDC2 | Down | 7 |
Genes were commonly up or down regulated in seven or more of the 10 datasets. DEGs – Differentially expressed genes.
IPA Analysis:
Ingenuity Pathway Analysis (IPA) (Qiagen) was used to identify pathways that could affected by the DEGs.[45] To determine the most significantly affected pathways, we sorted them according to their average Z scores. Z score are statistical measures of the match between the expected relationship direction and the observed gene expression, between datasets. However, IPA comparison analysis of the canonical pathways predicted to be most affected by the differently expressed genes in each dataset showed very little consensus between all of the datasets (Figure 2). This is most likely because all of these EGFR TKI resistant models were produced using different methods, treatment times, and EGFR TKIs, along with the different lung cancer cell lines (Table 1). However, most of the datasets agreed in a few pathways. One such pathway was oxidative phosphorylation. A strong decrease in oxidative phosphorylation was seen in 7 out of 10 of the EGFR TKI resistant cell lines (Figure 2). IPA also identified cholesterol synthesis as strongly downregulated pathway in 7 out of 10 of the datasets (Figure 2). Downstream EGFR signaling molecules, such as PI3K/AKT and phospholipases were predicted to be upregulated in a majority of the datasets (Figure 2). The variability in genetic changes leading to EGFR TKI resistance seen here serves to highlight the many different pathways and genetic endpoints that can cause lung cancer cells to become resistant to EGFR TKIs.
Figure 2: Comparison Analysis.

IPA was used to identify the pathways that are affected by differentially expressed genes in each dataset. A comparison analysis was run to compare the obtained differential gene expression data from each dataset. Affected canonical pathways are sorted according to their average Z scores.
To move forward, we decided to take a closer look at the oxidative phosphorylation (OxPhos) pathway, as well as the phospholipase signaling. To the best of our knowledge, these two pathways are not very well studied in the context of resistance to EGFR TKI therapy. Genes comprising the oxidative phosphorylation pathway were strongly downregulated in 7 out of 10 of the datasets (Figure 2). Further analyses indicated that in the different EGFR TKI resistant cell lines different genes of this pathway contributed to the downregulation. Out of the 82 genes listed in the IPA database under canonical OxPhos in our comparison analysis, only 10 showed up as significantly differently expressed in 4 or more of the datasets (Figure 3A). We next took all 82 of the OxPhos genes in IPA and used the software to plot all known interactions between these genes and EGFR. Eleven of these genes have known interactions with EGFR and of these, ATP5F1A (ATP Synthase F1 Subunit Alpha) was significantly downregulated in 6 of the 10 EGFR TKI resistant cell lines and to our knowledge has no previously known role in EGFR TKI resistance (Figure 3B). Similarly, genes comprising cellular phospholipases were strongly upregulated in 7 out of 10 of the datasets (Figure 2). Further analyses indicated that in the different EGFR TKI resistant cell lines different genes with phospholipase activity contributed to this upregulation (Figure 4A). We next took all 47 phospholipase genes in the IPA database and used the software to plot all known interactions between these genes and EGFR. Nine of these genes have known interactions with EGFR, but to our knowledge have no known role in resistance to EGFR TKI therapy (Figure 4B).
Figure 3: Oxidative Phosphorylation.

A) Expression levels of select genes involved in oxidative phosphorylation from each RNASeq dataset according to IPA. Fold change values were calculated against the control cells for each dataset. B) IPA generated network map showing known connections between genes involved in oxidative phosphorylation and those found in the RNASeq datasets with EGFR.
Figure 4: Phospholipase Signaling.

Expression levels of key phospholipases from each RNASeq dataset according to IPA. Fold change values were calculated against the control cells for each dataset. B) IPA generated network map showing known connections between phospholipases found in the RNASeq datasets with EGFR.
Comparison With an In Vivo Xenograft Tumor Model of EGFR TKI Resistance:
To validate the findings of our meta-analysis of EGFR TKI resistance datasets, we used our previously published in vivo xenograft tumor model of EGFR TKI resistance.[46] We performed an analysis of protein expression and phosphorylation using the Phospho-Explorer Antibody Array (Full Moon Biosystems) on the control vs. lapatinib treated tumor samples. This array is used for broad-scope protein phosphorylation profiling and consists of 1318 antibodies related to multiple signaling pathways and biological processes.[47] When comparing protein expression in control vs lapatinib treated tumors we found a significant downregulation of some known TKI resistance markers such as, VEGFR2 (Vascular endothelial growth factor receptor 2), ABL1 (ABL Proto-Oncogene 1), SRC (Proto-oncogene tyrosine-protein kinase Src), MYC (Figure 5).[42] This could be due to the lapatinib treated tumors still being in the early stages of resistance development. We did find upregulation of MEK1, a signaling protein downstream of multiple growth factor receptors including EGFR (Figure 5). MEK1 is a well-known marker of resistance to EGFR TKIs.[42] To the best of our knowledge, the rest of the significantly differential expressed proteins we found using this array have little or no known connection to EGFR TKI resistance (Figure 5).
Figure 5: Protein Array Analysis of a In Vivo Xenograft Tumor Model of EGFR TKI Resistance.

Mice were inoculated with H1650 human lung cancer cells. Mice began treatment when tumors became palpable (2–3mm diameter) and were treated every day with vehicle control or 50mg/kg lapatinib. Protein was collected from H1650 tumors (N=5), samples were pooled equally, and the PhosphoExplorer antibody array was performed (Full Moon Biosystems). RFU for all significantly differently expressed proteins is shown (N=2). A 2-Way ANOVA and Bonferroni post hoc tests were used to determine significance. * = p<0.05, ** = p<0.01, *** = P<0.001.
We further explored EGFR TKI resistance by combining our own tumor dataset with the publicly available data, we produced a master list containing significantly changed genes that are commonly up or down regulated in 6 or more of the RNASeq datasets and added the significantly changed proteins from the PhosphoExplorer array. We then plotted all known interactions between the list and EGFR (Figure 6A). Known pathways in IPA for cellular signaling/ metabolic processes were then overlaid on this plot and we kept only genes or proteins that directly interacted with our pathways of interest (Figure 6B). Using this data, we can observe how these proteins interact and connect to known mechanisms of EGR TKI resistance, such as STAT3, ERK/MAPK, and NF-kB signaling.[42] We also show how these commonly differentially expressed genes/proteins interact with our candidate pathways (oxidative metabolic processes and phospholipases) while also tracing them back to the EGFR signaling pathway (Figure 6B).
Figure 6: IPA Network Map of RNASeq and Protein Array Data.

A) IPA generated network map showing known connections between the most common genes found in the RNASeq datasets and those found in the protein array with each other and EGFR. B) IPA generated network map showing known connections between the most common genes found in the RNASeq datasets and those found in the protein array with EGFR and known pathways in IPA for cellular signaling/ metabolic processes of interest. C) Gene expression in lapatinib and ketoconazole treated EGFR TKI tolerant cells compared to parental. H1975 cells were treated at seeding with 7.5μM lapatinib and/or 15μM ketoconazole for 72 hours. Average fold change ±SEM, compared to untreated, is shown (N=2). A 2-Way ANOVA and Uncorrected Fisher’s LSD post hoc tests were used to determine significance. * = p<0.05, ** = p<0.01.
To further validate this data we checked expression of the DUSP1 (dual specificity phosphatase 1) and DUSP6 (dual specificity phosphatase 6) genes in our previously published EGFR TKI tolerant cell line model.[46] These genes were found to be upregulated and downregulated respectively in the EGFR TKI resistant cells compared to parental cells, in six or more of the RNASeq datasets and show interactions with EGFR signaling and our candidate pathways (Figure 6B). In our EGFR TKI tolerant cell line model, we found that DUSP1 was upregulated 1.5-fold compared to parental cells and DUSP6 was downregulated almost 2-fold compared to parental cells (Figure 6C). We next looked at expression of these two genes after treatment with our previously published combination therapy of lapatinib and ketoconazole.[46] Interestingly, we found that expression of these genes is completely reversed in EGFR TKI tolerant cells after treatment with the combination therapy. DUSP1 expression is downregulated below parental cell expression and DUSP6 expression is upregulated above parental cell expression in the EGFR TKI tolerant cells after treatment with the combination therapy (Figure 6C). We also produced a gene map showing genes that were commonly up or down regulated in six or more datasets but have no known interactions with the EGFR pathway in the IPA database (Figure 7). Several of these targets may prove useful when designing new therapies to overcome EGFR TKI resistance in lung cancer, however further validation is needed.
Figure 7: Differentially Expressed Genes Without Experimentally Validated Connections to the EGFR Pathway in the IPA Database.

IPA generated gene map showing genes that were commonly up or down regulated in six or more datasets but have no known experimentally validated connections to the EGFR pathway in the IPA database.
Intrinsic Disorder Analysis:
Lastly, we analyzed the structural disorder predisposition of each gene on our master list of commonly significantly changed genes, using a set of disorder predictors from the PONDR family and the IUPred computational platform.[48, 49] The different computational tools use different attributes and models for to calculate a disorder predisposition score for every amino acid residue in a query protein. The per-residue disorder predisposition scores are on a scale from 0 to 1, where values of 0 indicate fully ordered residues, values of 1 indicate fully disordered residues, and values ≥ 0.5 are considered disordered residues. For each protein in this study, the mean disorder predisposition profile generated by averaging the outputs of individual predictors was used to evaluate the mean predisposition for intrinsic disorder as predicted percent of disordered residues (PPIDmean). These values were then plotted against the PONDR® FIT-based percent of predicted disordered residues, PPIDPONDR-FIT. Our analysis revealed that the PPIDmean values ranged from 1.32% to 97.37% with a mean of 26.26% (Figure 8A). Based on their PPIDmean ranged values, proteins were classified as mostly ordered (< 10%), moderately disordered (between 10% and 30%) and highly disordered (≥ 30%). This analysis showed that according to these criteria, the analyzed dataset of 359 proteins contained 30.6%, 37.6%, and 31.8% of highly disordered, moderately disordered and mostly ordered protein, respectively.
Figure 8: Global Intrinsic Disorder Analysis.

A) Plot correlating the mean disorder propensity (MDP) and PONDR® FIT score for selected proteins based on the results of PONDR® FIT analyses. Dashed lines represent boundaries separating ordered (MDP < 10%, PONDR® FIT < 10%) and disordered proteins (MDP ≥ 30%, PONDR® FIT ≥ 30%). B) CH-CDF plot analysis of selected proteins. Q1 (lower-right quadrant) – proteins predicted to be ordered by both predictors, Q2 (lower-left quadrant) – proteins predicted to be ordered by CH but disordered by CDF, Q3 (upper-left quadrant) – proteins predicted to be disordered by both predictors, and Q4 (upper-right quadrant) – proteins predicted to be disordered by CH and ordered by CDF.
Binary disorder predictors, charge-hydropathy (CH) plot and the cumulative distribution function (CDF) plot, are computational tools that classify proteins as wholly ordered or wholly disordered.[48, 49] This allows for classification of proteins based on their position within the CH-CDF plot as ordered (proteins predicted to be ordered by both binary predictors), putative native “molten globules” or hybrid proteins (proteins determined to be ordered/compact by CH, but disordered by CDF), putative native coils and native pre-molten globules (proteins predicted to be disordered by both methods), and proteins predicted to be disordered by CH-plot, but ordered by CDF (native coils and native pre-molten globules) or proteins with compact globular conformations (native molten globules and ordered proteins). Figure 8B presents the results of this analysis and shows that 32% of them are highly disordered, 39.7%have a molten globular or hybrid structure, and 28.2% are mostly ordered. Among the EGFR interacting proteins, F2R (Coagulation Factor II Thrombin Receptor), DUSP1, FSTL1 (Follistatin Like 1), EIF5A (Eukaryotic Translation Initiation Factor 5A), DUSP6, BIRC3 (Baculoviral IAP Repeat Containing 3), and FGFR1, and are predicted to be moderately disordered, possessing PPIDmean of 9.6%, 11.7%, 12.6%, 13.6%, 17.3%, 17.5%, and 24.5%, respectively (Figure 9A–G). On the other hand, ADRA1B (Adrenoceptor Alpha 1B), BMPR2 (Bone Morphogenetic Protein Receptor Type 2), ERRFI1 (ERBB Receptor Feedback Inhibitor 1), GAB2 (Growth Factor Receptor Bound Protein 2-Associated Protein 2), FOS (Fos Proto-Oncogene, AP-1 Transcription Factor Subunit), GATA6 (GATA Binding Protein 6), and EGR1, with their corresponding PPIDmean values of 34.2%, 43,5%, 58.0%, 61.7%, 67.6%, 71.4% and 85.6%, are highly disordered (Figure 9H–N). These intrinsically disordered proteins could play key roles in cellular signaling leading to EGFR TKI resistance.
Figure 9: Intrinsic Disorder Analysis of Selected Proteins.
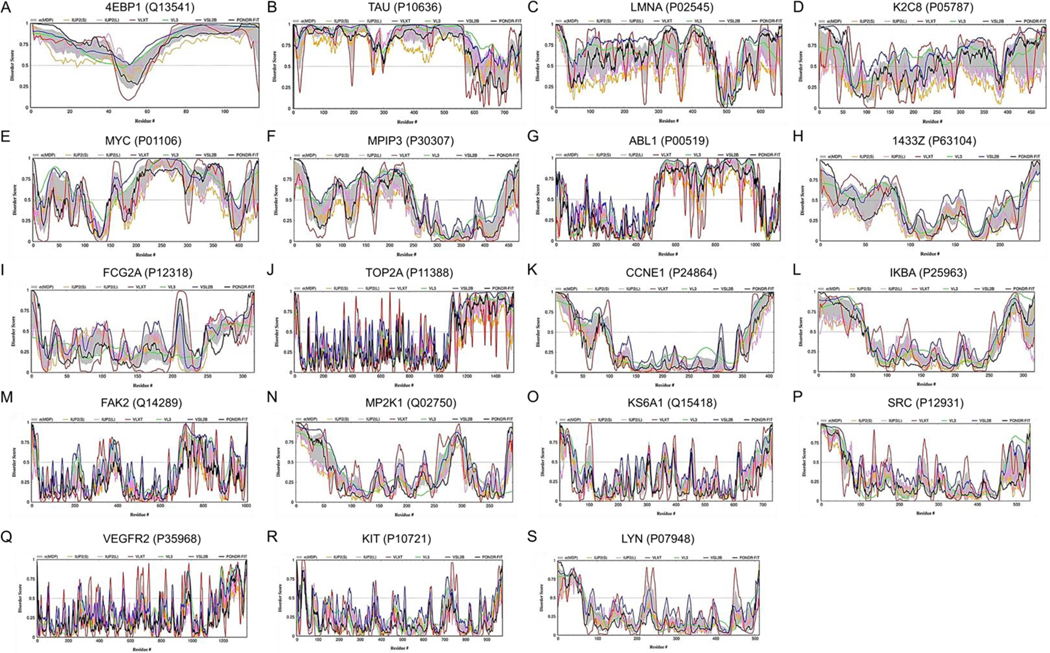
Analysis of per-residue disorder distribution in selected proteins known to interact with EGFR. A. F2R (UniProt ID: P25116; PPID = 9.6%); B. DUSP1 (UniProt ID: P28562; PPID = 11.7%); C. FSTL1 (UniProt ID: Q12841; PPID = 12.6%); D. EIF5A (UniProt ID: P63241; PPID = 13.6%); E. DUSP6 (UniProt ID: Q16828; PPID = 17.3%); F. BIRC3 (UniProt ID: Q13489; PPID = 17.5%); G. FGFR1 (UniProt ID: P11362; PPID = 24.5%); H. ADRA1B (UniProt ID: P35368; PPID = 34.2%); I. BMPR2 (UniProt ID: Q13873; PPID = 43.5%); J. ERRFI1 (UniProt ID: Q9UJM3; PPID = 58.0%); K. GAB2 (UniProt ID: Q9UQC2; PPID = 61.7%); L. FOS (UniProt ID: P01100; PPID = 67.6%); M. GATA6 (UniProt ID: Q92908; PPID = 71.3%); N. EGR1 (UniProt ID: P18146; PPID = 85.8%). Disorder profiles were generated by DiSpi web crawler designed for the rapid prediction and comparison of protein disorder profiles. It aggregates the results from PONDR® VLXT (red lines),[86] PONDR® VL3 (green lines),[89] PONDR® VLS2B (blue lines),[90] PONDR® FIT (black lines),[87] IUPred2_Short (orange lines) and IUPred2_Long (pink lines).[98, 99] Mean disorder scores were calculated by averaging the outputs of individual predictors, and the corresponding distributions of standard deviations are shown by light gray shades. The outputs of the evaluation of the per-residue disorder propensity by these tools are represented as real numbers between 1 (ideal prediction of disorder) and 0 (ideal prediction of order). A threshold of ≥0.5 was used to identify disordered residues and regions in query proteins.
Discussion:
In this study, we have compared publicly available RNASeq datasets for lung cancer cell lines treated with EGFR TKIs and protein array data from our EGFR TKI-treated xenograft tumors, performed pathway analyses, structural disorder analysis of these pathways to dissect the potential molecular mechanisms of drug resistance using the EGFR signaling, and EGFR mutations.
IPA Analysis:
After treatment with EGFR TKIs, the progression of tumor heterogeneity and acquisition of resistance via both genetic and non-genetic mechanisms may depend on the specific EGFR TKI being used.[50] Highly heterogeneous mechanisms mediate resistance to even state-of-the-art third-generation EGFR-TKIs, as such fourth generation EGFR TKIs are under development.[51] In this study, we aimed to use the RNASeq data from ten different EGFR TKI resistant models to attempt to find novel pathways leading to EGFR TKI resistance. By comparing data generated using multiple TKIs and models of varying genetic backgrounds we aimed to find common resistance mechanisms that would be more robust and universal than those that are only described in a single model.
A major finding of our analyses is that one pathway (oxidative phosphorylation) was downregulated in 7 of the 10 EGFR TKI resistant cell lines. Therefore, it may be possible to target EGFR TKI resistance through this strong decrease in OxPhos. It was found that oxidative phosphorylation was downregulated in a group of NSCLC patients with mutated EGFR tumors that also had high mTORC2 (Mammalian target of rapamycin complex 2) activity, leading to a worse prognosis.[52] Novel mTORC2 inhibitors are currently under investigation as cancer treatments, but their translation to the clinic is hindered by off target effects and limited patient response.[53] In lung cancer cells, chronic gefitinib treatment promotes ROS (Reactive oxygen species), which then activates an EMT, leading to gefitinib resistance.[54] Erlotinib resistant lung cancer cells have mitochondrial dysfunction and rely more on glycolysis for survival than non-resistant cells, it has been shown that glucose deprivation can selectively decrease their viability.[55] However, not all the literature agrees. Inhibition of EGFR signaling induced an upregulation of oxidative phosphorylation in NSCLC cell lines.[56] A Glycolysis-to-OxPhos shift has been observed in erlotinib resistant EGFR mutant lung cancer cells that have undergone EMT (Epithelial–mesenchymal transition).[57] Also, in EGFR-TKI-resistant NSCLC cells, OxPhos markers including mitochondria-driven ATP production, mitochondrial membrane potential, and maximal OxPhos capacity were observed to be increased.[58] Inhibition of oxidative phosphorylation in combination with osimertinib could delay or prevent the development of resistance to osimertinib, as osimertinib treatment was shown to induce a strict dependence on mitochondrial OxPhos in EGFR mutant lung cancer cell lines.[59] While OxPhos role in EGFR TKI resistance is pivotal, the reason for the discrepancy among the reports is unclear. This may be attributed to differences in mutation status and cellular context within the many different cell line models have been developed to study EGFR TKI resistance in lung cancer. These include chronic adaptation model, tolerance model, and loss of function model.[35–40] Our study has identified reactivation of oxidative phosphorylation, possibly by upregulation of ATP5F1A, as a potential therapeutic target to overcome EGFR TKI resistance.
Another major finding of our study is that we identified an increase in phospholipase expression and signaling as a potential therapeutic target. While the role phospholipases play in EGFR activation and downstream signaling has been well studied, their contribution to EGFR TKI resistance has not been defined.[60–64] In our study, we have found phospholipase gene expression to be upregulated in 7 of the 10 EGFR TKI resistant cell lines. It is possible that due to the essential roles’ phospholipases play in maintaining EGFR signaling that the upregulation seen in these EGFR TKI resistance cell lines serves to overcome EGFR inhibition by TKIs. More work needs to be done to determine the exact role that phospholipases are performing to contribute to TKI resistance.
Comparison With an In Vivo Xenograft Tumor Model of EGFR TKI Resistance:
To validate the findings of our meta-analysis of EGFR TKI resistance datasets, we used a xenograft tumor model of EGFR TKI resistance and performed a PhosphoExplorer array on the control vs. lapatinib treated tumor samples.[46, 47] We then combined this data with a master list containing significantly changed genes that are commonly up or down regulated in 6 or more of the RNASeq datasets. This allowed us to observe how these commonly differentially expressed genes/proteins interact with our candidate pathways and the EGFR signaling pathway. Using this phospho-protein expression data and the RNASeq data we identified known mechanisms of EGR TKI resistance, such as STAT3, ERK/MAPK, and NF-kB signaling and explored their connections to our candidate processes of oxidative metabolism and phospholipases. For example, LYN (Lck/Yes-Related Novel Protein Tyrosine Kinase) and MYC are downregulated and MAP2K1 and EIF4EBP1 (Eukaryotic Translation Initiation Factor 4E Binding Protein 1) are upregulated in our protein data. LYN is known to be essential to maintain mitochondrial integrity, which is required to support oxidative metabolism.[65] MYC is expected to be downregulated following TKI treatment because it is downstream of EGFR however; it is typically activated in cancer to promote glycolysis when oxidative metabolism is reduced.[66] This deficit in glycolytic signaling may be overcome by the increase observed in MAP2K1 which activates pro glycolytic transcription factors.[67] EIF4EBP1 is a target of the mTOR pathway which is known to promote aerobic glycolysis.[68] In this way, key protein changes observed in our tumor model are consistent with the cellular phenotype most observed in the RNAseq data (reduced OxPhos). This highlights the interplay between different mechanisms of EGFR TKI resistance, both known and predicted, and shows that in most cases resistance is not just conferred by one pathway or gene but in fact multiple pathways work synergistically to enhance cellular resistance to EGFR TKIs.
As further validation of this data, we checked the expression of DUSP1 and DUSP6 in our own model of EGFR TKI tolerance.[44] These genes were found to be commonly up and down regulated respectively in EGFR TKI resistant cells compared to parental cells, in six or more of the RNASeq datasets. Using IPA, we also found that these genes interact with the EGFR signaling pathway and our candidate pathways (OXPHOS and phospholipase signaling). The MAPKs, including ERK, JNK, and p38 are dephosphorylated by DUSP proteins, which are critical regulators of these pathways.[69] Genetic deletion and pharmacological inhibition of DUSP1 was shown to suppress tumor growth in a BCR-ABL fusion protein kinase–induced mouse model of chronic myeloid leukemia (CML), as well as in tumor cells modeling other types of kinase-driven leukemias.[70] This mirrors the data we have shown in this current study, as we have shown that DUSP1 was upregulated in our EGFR TKI tolerant cell line model compared to parental cells and expression was completely reversed in the EGFR TKI tolerant cells after treatment with our combination therapy of lapatinib and ketoconazole. In our previous study we found that the combination therapy was able to re-sensitize EGFR TKI tolerant cells to EGFR TKI therapy.[46] However, another study found that DUSP1 downregulation in NSCLC might be a novel mechanism of gefitinib-acquired resistance and DUSP1 expression correlated with gefitinib sensitivity in a clinical specimens.[69] RNA knockdown of DUSP6 in H441 lung cancer cells was shown to increase ERK activation, whereas overexpression of DUSP6 in H1975 lung cancer cells was shown to reduce ERK activation and promote apoptosis.[71] In this same study, DUSP6 overexpression was also shown to synergize with EGFR TKIs in HCC827 cells. Again, this mirrors the data we have shown in this current study. In our EGFR TKI tolerant cell line model, we found that DUSP6 was downregulated compared to parental cells and expression was completely reversed in the EGFR TKI tolerant cells after treatment with our combination therapy of lapatinib and ketoconazole. Our previous report, along with this data shows that the DUSP proteins, especially DUSP1 and DUSP6, may serve as potential therapeutic targets when designing therapies to overcome EGFR TKI resistance.
Next, we produced a gene map showing genes that were commonly up or down regulated in more than half of the datasets but have no experimentally validated known interactions with the EGFR pathway in the IPA database. Literature searches of the upregulated genes identified a few that may play a role in EGFR TKI resistance. In one study, SEMA3C (Semaphorin 3C) was found to drive activation of multiple RTKs, including EGFR, and SEMA3C expression levels increased in castration-resistant prostate cancer, where it functioned to promote cancer cell growth and resistance to androgen receptor pathway inhibition.[72] A tandem duplication at 7q34 leading to a fusion between KIAA1549 and BRAF was found in 66% of pilocytic astrocytomas, and lead to constitutive BRAF kinase activity and was able to transform NIH3T3 cells.[73] In another study, a systems analysis of a model of EGFR-mutated NSCLC resistant to targeted therapy was performed using whole exome sequencing, global time-course discovery phospho-proteomics and computational modeling to identify resistance pathways and one of the nine phosphoproteins whose corresponding genes were directly mutated was found to be AHNAK2 (AHNAK Nucleoprotein 2).[74] Further experimentation is needed to confirm the possible roles these genes may play in the development of EGFR TKI resistance.
Intrinsic Disorder Analysis:
Lastly, we have performed intrinsic disorder analysis on each protein in our master list to investigate the possible role of intrinsic disorder in resistance to EGFR TKIs. Studies have shown the value of including protein structural information into analysis of protein networks.[75] The results of these studies point to a general involvement of intrinsically disordered proteins in numerous processes in the cell: transcriptional activation, cell-cycle regulation, membrane transport, molecular recognition and signaling.[76] Protein disorder may be involved directly in mediating interactions between highly connected proteins in signaling networks. Experimental evidence of disorder in signaling proteins and oncoproteins, such as FOS, DUSPs, and GAB2 suggest an increased amount of predicted disorder in these two protein datasets, which is proposed to relate to the signaling and regulatory functions of these proteins.[77] For example, the C-terminal activation domain of FOS is structurally disordered, giving the protein structural freedom which allows it to interact with multiple transcription factors. Another protein predicted to have a high level of intrinsic disorder is Gab2, which is involved in the amplification of signal transduction from growth factors, cytokines and antigen receptors and has been implicated in promoting tumor cell metastasis, migration and recurrence.[78] Through these interactions, the Gab2 protein triggers various downstream signal effectors, including Met-ERK1/2 and IGF1R-AKT, which has been implicated in the acquisition of EGFR TKI resistance.[79] The common occurrence of intrinsic disorder in cancer-associated and signaling proteins suggest that disorder information should be employed in the development of new strategies for the discovery of anti-cancer drugs.[77]
Study Limitations:
There are several limitations to the methods used in this study that could have impeded our ability to find a truly universal resistance mechanism. First, there were significant variations in the duration of TKI treatment between studies, variations in the number of biological replicates resulting in limited statistical power, and variations in the methods used to collect the original sequencing data. Second, we analyzed abundant public data, but most of the EGFR TKI DR datasets are from the PC9 cells, which could skew the results towards this specific cell line. Third, since the RNASEQ studies were done in vitro, we decided to validate our findings in a xenograft model of EGFR TKI DR. However, our model uses lapatinib as the EGFR TKI and all of the RNASEQ studies, but one, used gefitinib as the EFGR TKI. Due to this, our model might not replicate some of gene expression changes observed within the RNASEQ experiments. However, it should only strengthen the conclusions found within this manuscript, as the point of this study is to determine potential mechanisms of EGFR TKI DR that are consistent between resistance to multiple EGFR TKIs.
Conclusion:
One of the main reasons that EGFR TKI resistance has proven so difficult to overcome is the molecular and genetic variability that can lead to EGFR TKI resistance. Using a set of bioinformatics analyses on available RNA-Sequencing and protein array datasets, we have identified several possible EGFR TKI resistance targets. Datasets were analyzed for differential gene expression, and then pathway analysis, as well as structural disorder analysis of key proteins in these pathways, was performed. Our results suggest several key proteins, including DUSP1, DUSP6, GAB2, and FOS, that may play roles in the development and maintenance of EGFR TKI resistance in lung cancer due to the high number of predicted interactions these proteins have with DEGs in EGFR TKI DR cell lines and known markers of EGFR TKI DR. By studying the cellular effects of acquired EGFR TKI resistance, as well as the mechanism through which this resistance is maintained, we can better understand how lung cancer cells become drug resistant to EGFR TKIs and help design more efficient combination treatment strategies that will stop the development of resistance.
Materials and Methods:
1. RNASEQ Dataset Acquisition and Differential Gene Expression Analysis:
The fasta files for each dataset were downloaded from the GEO database and analyzed for differential gene expression using tools available on Galaxy, an open-source, free of charge, web-based platform.[43, 44]. The main public Galaxy server (https://usegalaxy.org), online since 2007, features a robust toolset for large-scale omics analyses, a large amount of public data for use, and analysis histories and workflows. A schematic of the workflow for RNA-SEQ data analyses is shown in Figure 1. Briefly, after uploading the data to Galaxy, the raw sequencing data was quality checked using FASTQC.[80] FastQC (Galaxy ID: toolshed.g2.bx.psu.edu/repos/devteam/fastqc/fastqc/0.73+galaxy0; URL: https://usegalaxy.org/root?tool_id=toolshed.g2.bx.psu.edu/repos/devteam/fastqc/fastqc/0.73+galaxy0) performs quality control checks on raw sequence data by using a modular set of analyses to give the researcher a short impression of whether the data has any problems that could affect downstream analysis. Then low-quality transcripts, adaptor sequences, and PCR primers sequences were removed using Trimmomatic.[81] Trimmomatic (Galaxy ID: toolshed.g2.bx.psu.edu/repos/pjbriggs/trimmomatic/trimmomatic/0.38.0; URL: https://usegalaxy.org/root?tool_id=toolshed.g2.bx.psu.edu/repos/pjbriggs/trimmomatic/trimmomatic/0.38.0) performs a selection of trimming steps designed specifically for illumina paired end and single ended data. The cleaned sequences were then aligned to the human genome using HISAT2.[82] HISAT2 (Galaxy ID: toolshed.g2.bx.psu.edu/repos/iuc/hisat2/hisat2/2.2.1+galaxy0; URL: https://usegalaxy.org/root?tool_id=toolshed.g2.bx.psu.edu/repos/iuc/hisat2/hisat2/2.2.1+galaxy0) is a spliced alignment program that allows for fast and sensitive alignment of reads to a reference genome. Count tables measuring gene expression were then created using featureCounts.[83] FeatureCounts (Galaxy ID: toolshed.g2.bx.psu.edu/repos/iuc/featurecounts/featurecounts/2.0.1+galaxy2; URL: https://usegalaxy.org/root?tool_id=toolshed.g2.bx.psu.edu/repos/iuc/featurecounts/featurecounts/2.0.1+galaxy2) is a read counting program that can be used to count both gDNA-seq and RNA-seq reads for genomic features. Differential gene expression was then calculated from the count tables using DESeq2.[84] Using a negative binomial distribution model, DESeq2 (Galaxy ID: toolshed.g2.bx.psu.edu/repos/iuc/deseq2/deseq2/2.11.40.7+galaxy1; URL: https://usegalaxy.org/root?tool_id=toolshed.g2.bx.psu.edu/repos/iuc/deseq2/deseq2/2.11.40.7+galaxy1) takes count data, estimates variance-mean dependence, and calculates differential expression. Gene fold changes represent drug resistant against parental, meaning the values correspond to up or down regulations of genes in drug resistant samples, and were considered significantly differentially expressed if the corrected p-value was <.05.
2. Cell culture
H1650 cells were purchased from the American type culture collection (ATCC) and passaged no more than 25 times. Cells were cultured in a humidified incubator at 37°C in a 5% CO2 atmosphere. Cells were cultured in tissue culture treated plates in the appropriate complete cell culture media [RPMI (GE Healthcare) containing 10% fetal bovine serum (FBS) (Atlanta Biologicals) and 1% penicillin/streptomycin (GE Healthcare)]. H1975 cells have stable expression of luciferase and green fluorescent protein (GFP), were purchased from Genecopoeia, and were grown according to the manufactures’ protocol. Drug tolerant H1975 cells were generated according to a previously published protocol.[46]
3. Phospho-Explorer Antibody Array
Total protein was extracted from tumors using RIPA buffer and protein concentration was determined using the Pierce Coomassie Protein Assay Kit (Thermo Fisher Scientific) according to the manufactures’ protocol. Tumor protein samples from each group were then equally pooled (N=3) and protein expression was determined using the Phospho-Explorer Antibody Array (Full Moon Biosystems, California USA) according to the manufactures’ protocol). Cy-3 developed array chips were scanned, and images were analyzed using ImageJ to determine relative normalized staining intensity for each antibody. β-actin was used as a house keeping gene to quantify relative protein expression. Average relative fluorescent units ±SEM (N=2) were calculated for each protein. Statistical significance for each experiment was determined using two-way analysis of variance (ANOVA) and the Bonferroni post hoc test * = p<0.05, ** = p<0.01, *** = P<0.001. Calculations were performed and graphs produced using Prism 6.0 software (GraphPad, California USA). Graphs of results show the mean and error bars depict the average plus or minus the standard error of the mean.
4. Pathway Analysis
Ingenuity Pathway Analysis (IPA) (Qiagen) was used for analysis and interpretation of the acquired differential expressed genes lists from RNA sequencing.[45] A detailed description of this software is available at ingenuity.com. IPA was then used to identify pathways that could affected by the significantly differential expressed genes. IPA uses a database of prior biological knowledge to interpret gene-expression data and create predictions of the status of regulatory molecules that could explain the observed expression changes in datasets. Available tools include Upstream Regulator Analysis, Mechanistic Networks, Causal Network Analysis, and Downstream Effects Analysis.
To begin our analysis, a core analysis was performed on each dataset, and a comparison analysis was then run to compare the parental vs drug tolerant data obtained from each dataset. We then examined the datasets for the similarly affected canonical pathways responsible for the observed gene expression changes. To determine the most significantly affected pathways, we sorted them according to their average Z scores. Z score are statistical measures of the match between expected relationship direction and observed gene expression, between datasets. Heatmapper software was used to produce heat maps from the data obtained from IPA.[85] A detailed description of this software is available at heatmapper.ca.
We then used the My Pathway/Path Designer tools in IPA to plot all known interactions between the pathway of interest and EGFR. The sum of each genes fold change compared to control from each dataset was used to color the genes. To explore these pathways further we produced a master list containing significantly changed genes that are commonly up or down regulated in 6 or more of the RNASeq datasets and added the significantly changed proteins from the PhosphoExplorer array. We then used the My Pathway/Path Designer tools in IPA to plot all known interactions between this master list and EGFR. The data from the comparison analysis was used to color the genes based on up or down regulation. Data from the PhosphoExplorer array was used to color the proteins based on up or down regulation. Known pathways in IPA for cellular signaling/ metabolic processes were then overlaid on this plot and we kept only genes or proteins that directly interacted with our pathways of interest. Finally, we produced a gene map showing genes that were commonly up or down regulated in six or more datasets but have no experimentally validated known interactions with the EGFR pathway in the IPA database. A literature search was then performed on the upregulated genes to determine any roles these genes may play in resistance to EGFR TKIs.
5. Quantitative Reverse Transcriptase PCR (qPCR)
Total cellular RNA was extracted from cell pellets using Trizol (Thermo Fisher Scientific) according to the manufactures’ protocol. RNA was quantified using the Nanodrop (Thermo Fisher Scientific). One microgram of RNA was then reverse transcribed using the Maxima cDNA Reverse Transcription Kit (Thermo Fisher Scientific) according to the manufactures’ protocol. qPCR performed on the cDNA was used to quantitate the relative expression levels of certain genes to β−2-Microglobulin or β-actin as a control. Real time analysis was performed using BlazeTaq SYBR Green qPCR Mix 2.0 (Genecoepia), according to the manufactures’ protocol, in a Bio Rad CFX-384 thermocycler using primers obtained from Integrated DNA Technologies (IDT) and RealTimerPrimers (RTP). β-Actin (IDT)- Forward: CAAACATGATCTGGGTCATCTTCT, Reverse: CAAACATGATCTGGGTCATCTTCT. β−2-Microglobulin (IDT)- Forward: TTCTGGCCTGGAGGCTATC, Reverse: TCAGGAAATTTGACTTTCCATTC. DUSP1 (RTP)- Cat# VHPS-2768. DUSP6 (RTP)- Cat# VHPS-2780. The data was analyzed using ΔCt and ΔΔCt calculations and expression of all genes was normalized to β−2-Microglobulin or β-actin expression as a housekeeping gene. Average fold change ±SEM, compared to control, was then calculated. Data analysis was performed using the CFX Maestro software (Bio-Rad).
6. Computational Characterization of Intrinsic Disorder
The master gene list was then analyzed by six per-residue predictors, such as PONDR® VLXT,[86] PONDR® FIT,[87] PONDR® VSL2,[88, 89] and PONDR® VL3,[90] available at http://www.pondr.com, and the IUPred computational platform that allows identification of either short or long regions of intrinsic disorder, IUPred-L and IUPred-S.[48, 49] The outputs of these tools are represented as real numbers between 1 (ideal prediction of disorder) and 0 (ideal prediction of order). A threshold of ≥0.5 was used to identify disordered residues and regions in query proteins. Mean disorder scores calculated by averaging the outputs of individual predictors together with the distribution of standard deviations are shown as well. For each query protein in this study, the predicted percentage of intrinsic disorder (PPID) was calculated based on the outputs of per-residue disorder predictors. Here, PPID in a query protein represents a percent of residues with disorder scores exceeding 0.5. Finally, the PPIDmean values calculated for each all proteins in this study were plotted against their PPIDPONDR-FIT values representing the PONDR® FIT-based percent of the predicted disordered residues.
Next, the outputs of two binary predictors, the charge-hydropathy (CH) plot[91, 92] and the cumulative distribution function (CDF) plot[92–94] were combined to conduct a CH-CDF analysis.[94–97] In the CH-CDF plot, the x-axis separates proteins predicted by CH-plot to be intrinsically disordered (positive y values) or compact (negative y values), whereas the y-axis separates proteins predicted by CDF to be ordered (positive x values) or intrinsically disordered (negative x values). Proteins can be classified by their position within each quadrant of the resultant CH-CDF plot. Here, the lower-right quadrant (Q1) includes proteins predicted to be ordered by both CDF and CH, the lower-left quadrant (Q2) contains proteins predicted to be disordered by CDF but compact by CH-plot, the upper-left quadrant (Q3) contains proteins predicted to be disordered by both methods, and the upper-right quadrant (Q4) contains proteins predicted to be disordered by CH-plot but ordered by CDF.
7. Animal Experiments
H1650 tumors were grown in mice and treated with vehicle or lapatinib using a previously published subcutaneous flank model.[46] Nu/Nu nude mice were purchased from Envigo. In brief, mice were injected subcutaneously on the flank with 3 million H1650 cells. Mice began treatment when tumors became palpable (2–3mm diameter). Mice were randomized and treated every day with vehicle control or 50mg/kg lapatinib until collection. Drugs were injected peritoneal. Drugs were dissolved in 0.1% Tween 20 with 5%DMSO in water. Tumors were collected when controls reached about 10mm X 10mm. Animals were housed in the University of South Florida comparative medicine facility at the Morsani College of Medicine and all protocols were reviewed and approved by the USF institutional animal care and use committee.
8. Statistics
Experiments have been repeated at least twice. The specific statistical test used for each experiment is described in the appropriate figure legend, * = p<0.05, ** = p<0.01, *** = P<0.001. Calculations were performed and graphs produced using Prism 6.0 software (GraphPad, California USA). Graphs of results show the mean and error bars depict the mean plus or minus the standard error of the mean.
Acknowledgements and Funding Details:
This work is supported by Veterans Affairs Merit Review grant (BX003413) to Dr. Subhra Mohapatra, and Research Career Scientist Awards to Dr. Subhra Mohapatra (IK6BX004212) and Dr. Shyam Mohapatra (IK6 BX003778). Though this report is based upon work supported, in part, by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, the contents of this report do not represent the views of the Department of Veterans Affairs or the United States Government. This work is also supported by National Institute of Health (NIH) grant to Dr. Subhra Mohapatra (R01CA152005).
Abbreviations
- ABL1
ABL Proto-Oncogene 1
- ADRA1B
Adrenoceptor Alpha 1B
- AKT
Serine-threonine protein kinase AKT1
- ALK
Anaplastic lymphoma kinase
- ANOVA
Analysis of variance
- AHNAK2
AHNAK Nucleoprotein 2
- ATP
Adenosine triphosphate
- ATP5F1A
ATP Synthase F1 Subunit Alpha
- AXL
AXL Receptor Tyrosine Kinase
- BIM
Bcl-2-like protein 11
- BIRC3
Baculoviral IAP Repeat Containing 3
- BMPR2
Bone Morphogenetic Protein Receptor Type 2
- BRAF
B-Raf Proto-Oncogene
- DEGs
Differentially Expressed Genes
- DMSO
Dimethyl sulfoxide
- DUSP1
Dual specificity phosphatase 1
- DUSP6
Dual specificity phosphatase 6
- EGFR
Epidermal growth factor Receptor
- EIF5A
Eukaryotic Translation Initiation Factor 5A
- EIF4EBP1
Eukaryotic Translation Initiation Factor 4E Binding Protein 1
- EMT
Epithelial-mesenchymal transition
- ERK
Extracellular signal-regulated kinases
- ERRFI1
ERBB Receptor Feedback Inhibitor 1
- F2R
Coagulation Factor II Thrombin Receptor
- FBS
Feta Bovine Serum
- FGFR
Fibroblast growth factor receptor
- FGFR1
Fibroblast growth factor receptor 1
- FGF2
Fibroblast growth factor 2
- FOS
Fos Proto-Oncogene, AP-1 Transcription Factor Subunit
- FSTL1
Follistatin Like 1
- GAB2
Growth Factor Receptor Bound Protein 2-Associated Protein 2
- GATA6
GATA Binding Protein 6
- GEO
Gene Expression Omnibus database
- HGF
Hepatocyte Growth Factor
- Hh
Hedgehog
- HER2
Human Epidermal Growth Factor Receptor 2
- HER3
Human Epidermal Growth Factor Receptor 3
- HER4
Human Epidermal Growth Factor Receptor 4
- IGF1R
Insulin Like Growth Factor 2 Receptor
- IPA
Ingenuity Pathway Analysis
- KRAS
KRAS Proto-Oncogene
- LYN
Lck/Yes-Related Novel Protein Tyrosine Kinase
- MAPK
Mitogen-activated protein kinase
- MAPK1
Mitogen-activated protein kinase 1
- Mcl-1
Induced myeloid leukemia cell differentiation protein
- MEK
Mitogen-activated protein kinase kinase
- MET
c-Met proto-oncogene protein
- mTOR
Mammalian target of rapamycin
- mTORC2
Mammalian target of rapamycin complex 2
- Myc
MYC Proto-Oncogene
- NCBI
National Center for Biotechnology Information
- NF-κB
Nuclear Factor kappa-light-chain-enhancer of activated B cells
- NSCLC
Non-small-cell lung carcinoma
- OxPhos
Oxidative phosphorylation
- PI3K
Phosphoinositide 3-kinase
- PIK3CA
Phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha
- PTEN
Phosphatase and tensin homolog
- RAF
Rapidly Accelerated Fibrosarcoma kinase
- RAS
p21/Ras family small GTPase
- RNASeq
RNA sequencing
- ROS
Reactive oxygen species
- SEMA3C
Semaphorin 3C
- SRC
Proto-oncogene tyrosine-protein kinase Src
- STAT1
Signal transducer and activator of transcription 1
- STAT3
Signal transducer and activator of transcription 3
- TKI
Tyrosine kinase inhibitor
- VEGFR2
Vascular endothelial growth factor receptor 2
- Wnt
Proto-Oncogene Wnt-1
Footnotes
Declarations:
Competing Interest: The authors declare no potential conflicts of interest.
Ethics approval and consent to participate: Not applicable.
Consent for publication: All authors have read and agreed to the published version of the manuscript.
Availability of data and material:
The raw and processed data used in this study was downloaded from the Gene Expression Omnibus (GEO) database and can be found using the indicated accession number (Table 1) for each dataset.
References
- 1.Society AC, Cancer Facts and Figures 2022. 2022. [Google Scholar]
- 2.Fedor D, Johnson WR, and Singhal S, Local recurrence following lung cancer surgery: Incidence, risk factors, and outcomes. Surgical Oncology, 2013. 22(3): p. 156–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aliperti LA, et al. , Local and systemic recurrence is the Achilles heel of cancer surgery. Ann Surg Oncol, 2011. 18(3): p. 603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Economopoulou P. and Mountzios G, The emerging treatment landscape of advanced non-small cell lung cancer. Ann Transl Med, 2018. 6(8): p. 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Housman G, et al. , Drug resistance in cancer: an overview. Cancers (Basel), 2014. 6(3): p. 1769–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doroshow DB and Herbst RS, Treatment of Advanced Non-Small Cell Lung Cancer in 2018. JAMA Oncol, 2018. 4(4): p. 569–570. [DOI] [PubMed] [Google Scholar]
- 7.Bhullar KS, et al. , Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer, 2018. 17(1): p. 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krause DS and Van Etten RA, Tyrosine kinases as targets for cancer therapy. N Engl J Med, 2005. 353(2): p. 172–87. [DOI] [PubMed] [Google Scholar]
- 9.Aisner DL, et al. , The Impact of Smoking and TP53 Mutations in Lung Adenocarcinoma Patients with Targetable Mutations-The Lung Cancer Mutation Consortium (LCMC2). Clin Cancer Res, 2018. 24(5): p. 1038–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tong CWS, et al. , Drug combination approach to overcome resistance to EGFR tyrosine kinase inhibitors in lung cancer. Cancer Lett, 2017. 405: p. 100–110. [DOI] [PubMed] [Google Scholar]
- 11.Han W. and Lo HW, Landscape of EGFR signaling network in human cancers: biology and therapeutic response in relation to receptor subcellular locations. Cancer Lett, 2012. 318(2): p. 124–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blakely CM and Bivona TG, Resiliency of lung cancers to EGFR inhibitor treatment unveiled, offering opportunities to divide and conquer EGFR inhibitor resistance. Cancer Discov, 2012. 2(10): p. 872–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohashi K, et al. , Epidermal growth factor receptor tyrosine kinase inhibitor-resistant disease. J Clin Oncol, 2013. 31(8): p. 1070–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Douillard JY, et al. , First-line gefitinib in Caucasian EGFR mutation-positive NSCLC patients: a phase-IV, open-label, single-arm study. Br J Cancer, 2014. 110(1): p. 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allison M, Turning the tide in lung cancer. Nat Biotechnol, 2010. 28(10): p. 999–1002. [DOI] [PubMed] [Google Scholar]
- 16.Parikh P. and Puri T, Personalized medicine: Lung Cancer leads the way. Indian J Cancer, 2013. 50(2): p. 77–9. [DOI] [PubMed] [Google Scholar]
- 17.Kwapiszewski R, Pawlak SD, and Adamkiewicz K, Anti-EGFR Agents: Current Status, Forecasts and Future Directions. Target Oncol, 2016. 11(6): p. 739–752. [DOI] [PubMed] [Google Scholar]
- 18.Troiani T, et al. , Therapeutic value of EGFR inhibition in CRC and NSCLC: 15 years of clinical evidence. ESMO Open, 2016. 1(5): p. e000088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blackledge G, et al. , Anti-EGF receptor therapy. Prostate Cancer Prostatic Dis, 2000. 3(4): p. 296–302. [DOI] [PubMed] [Google Scholar]
- 20.Pollack VA, et al. , Inhibition of epidermal growth factor receptor-associated tyrosine phosphorylation in human carcinomas with CP-358,774: dynamics of receptor inhibition in situ and antitumor effects in athymic mice. J Pharmacol Exp Ther, 1999. 291(2): p. 739–48. [PubMed] [Google Scholar]
- 21.Xia W, et al. , Anti-tumor activity of GW572016: a dual tyrosine kinase inhibitor blocks EGF activation of EGFR/erbB2 and downstream Erk1/2 and AKT pathways. Oncogene, 2002. 21(41): p. 6255–63. [DOI] [PubMed] [Google Scholar]
- 22.Hirsh V, Afatinib (BIBW 2992) development in non-small-cell lung cancer. Future Oncol, 2011. 7(7): p. 817–25. [DOI] [PubMed] [Google Scholar]
- 23.Zhang XY, et al. , Osimertinib (AZD9291), a Mutant-Selective EGFR Inhibitor, Reverses ABCB1-Mediated Drug Resistance in Cancer Cells. Molecules, 2016. 21(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oronsky B, et al. , Navigating the “No Man’s Land” of TKI-Failed EGFR-Mutated Non-Small Cell Lung Cancer (NSCLC): A Review. Neoplasia, 2018. 20(1): p. 92–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhong WZ, Zhou Q, and Wu YL, The resistance mechanisms and treatment strategies for EGFR-mutant advanced non-small-cell lung cancer. Oncotarget, 2017. 8(41): p. 71358–71370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Passaro A, et al. , Overcoming therapy resistance in EGFR-mutant lung cancer. Nat Cancer, 2021. 2(4): p. 377–391. [DOI] [PubMed] [Google Scholar]
- 27.He J, et al. , Mechanisms and management of 3rdgeneration EGFRTKI resistance in advanced nonsmall cell lung cancer (Review). Int J Oncol, 2021. 59(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosell R, et al. , Coregulation of pathways in lung cancer patients with EGFR mutation: therapeutic opportunities. Br J Cancer, 2021. 125(12): p. 1602–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang Z, et al. , Dual MET-EGFR combinatorial inhibition against T790M-EGFR-mediated erlotinib-resistant lung cancer. Br J Cancer, 2008. 99(6): p. 911–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bai XY, et al. , Blockade of Hedgehog Signaling Synergistically Increases Sensitivity to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Non-Small-Cell Lung Cancer Cell Lines. PLoS One, 2016. 11(3): p. e0149370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song JY, et al. , Dual inhibition of MEK1/2 and EGFR synergistically induces caspase-3-dependent apoptosis in EGFR inhibitor-resistant lung cancer cells via BIM upregulation. Invest New Drugs, 2013. 31(6): p. 1458–65. [DOI] [PubMed] [Google Scholar]
- 32.Sen M, et al. , Targeting Stat3 abrogates EGFR inhibitor resistance in cancer. Clin Cancer Res, 2012. 18(18): p. 4986–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blakely CM, et al. , NF-kappaB-activating complex engaged in response to EGFR oncogene inhibition drives tumor cell survival and residual disease in lung cancer. Cell Rep, 2015. 11(1): p. 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brady SW, et al. , Enhanced PI3K p110alpha signaling confers acquired lapatinib resistance that can be effectively reversed by a p110alpha-selective PI3K inhibitor. Mol Cancer Ther, 2014. 13(1): p. 60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhang HE, et al. , Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nat Med, 2015. 21(5): p. 440–8. [DOI] [PubMed] [Google Scholar]
- 36.Hata AN, et al. , Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med, 2016. 22(3): p. 262–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Okimoto RA, et al. , Inactivation of Capicua drives cancer metastasis. Nat Genet, 2017. 49(1): p. 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scarborough HA, et al. , AZ1366: An Inhibitor of Tankyrase and the Canonical Wnt Pathway that Limits the Persistence of Non-Small Cell Lung Cancer Cells Following EGFR Inhibition. Clin Cancer Res, 2017. 23(6): p. 1531–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song KA, et al. , Increased Synthesis of MCL-1 Protein Underlies Initial Survival of EGFR-Mutant Lung Cancer to EGFR Inhibitors and Provides a Novel Drug Target. Clin Cancer Res, 2018. 24(22): p. 5658–5672. [DOI] [PubMed] [Google Scholar]
- 40.Ware KE, et al. , A mechanism of resistance to gefitinib mediated by cellular reprogramming and the acquisition of an FGF2-FGFR1 autocrine growth loop. Oncogenesis, 2013. 2: p. e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Casas-Selves M, et al. , Tankyrase and the canonical Wnt pathway protect lung cancer cells from EGFR inhibition. Cancer Res, 2012. 72(16): p. 4154–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lim SM, et al. , Acquired resistance to EGFR targeted therapy in non-small cell lung cancer: Mechanisms and therapeutic strategies. Cancer Treat Rev, 2018. 65: p. 1–10. [DOI] [PubMed] [Google Scholar]
- 43.(NCBI), N.C.f.B.I., National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database [Google Scholar]
- 44.Afgan E, et al. , The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res, 2018. 46(W1): p. W537–W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kramer A, et al. , Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics, 2014. 30(4): p. 523–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Howell Mark C., R.G., Khalil Roukiah, Foran Elspeth, Quarni Waise, Nair Rajesh, Stevens Stanley, Grinchuk Aleksandr, Hanna Andrew, Mohapatra Shyam, and Mohapatra Subhra, Lung Cancer Cells Survive Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Exposure Through Upregulation of Cholesterol Synthesis. FASEB BioAdvances (In Press), 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chesnokova LS and Yurochko AD, Using a Phosphoproteomic Screen to Profile Early Changes During HCMV Infection of Human Monocytes. Methods Mol Biol, 2021. 2244: p. 233–246. [DOI] [PubMed] [Google Scholar]
- 48.Van Bibber NW, et al. , Intrinsic Disorder in Tetratricopeptide Repeat Proteins. Int J Mol Sci, 2020. 21(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coelho Ribeiro Mde L, et al. , Malleable ribonucleoprotein machine: protein intrinsic disorder in the Saccharomyces cerevisiae spliceosome. PeerJ, 2013. 1: p. e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Katayama Y, et al. , Heterogeneity among tumors with acquired resistance to EGFR tyrosine kinase inhibitors harboring EGFR-T790M mutation in non-small cell lung cancer cells. Cancer Med, 2022. 11(4): p. 944–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Du X, et al. , Acquired resistance to third-generation EGFR-TKIs and emerging next-generation EGFR inhibitors. Innovation (N Y), 2021. 2(2): p. 100103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chiang CT, et al. , mTORC2 contributes to the metabolic reprogramming in EGFR tyrosine-kinase inhibitor resistant cells in non-small cell lung cancer. Cancer Lett, 2018. 434: p. 152–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.de Fijter JW, Cancer and mTOR Inhibitors in Transplant Recipients. Transplantation, 2017. 101(1): p. 45–55. [DOI] [PubMed] [Google Scholar]
- 54.Okon IS, et al. , Gefitinib-mediated reactive oxygen specie (ROS) instigates mitochondrial dysfunction and drug resistance in lung cancer cells. J Biol Chem, 2015. 290(14): p. 9101–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ye M, et al. , Combined Inhibitions of Glycolysis and AKT/autophagy Can Overcome Resistance to EGFR-targeted Therapy of Lung Cancer. J Cancer, 2017. 8(18): p. 3774–3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.De Rosa V, et al. , Reversal of Warburg Effect and Reactivation of Oxidative Phosphorylation by Differential Inhibition of EGFR Signaling Pathways in Non-Small Cell Lung Cancer. Clin Cancer Res, 2015. 21(22): p. 5110–20. [DOI] [PubMed] [Google Scholar]
- 57.Sun Y, et al. , Metabolic and transcriptional profiling reveals pyruvate dehydrogenase kinase 4 as a mediator of epithelial-mesenchymal transition and drug resistance in tumor cells. Cancer Metab, 2014. 2(1): p. 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim S, et al. , Enhanced Sensitivity of Nonsmall Cell Lung Cancer with Acquired Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors to Phenformin: The Roles of a Metabolic Shift to Oxidative Phosphorylation and Redox Balance. Oxid Med Cell Longev, 2021. 2021: p. 5428364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martin MJ, et al. , Inhibition of oxidative phosphorylation suppresses the development of osimertinib resistance in a preclinical model of EGFR-driven lung adenocarcinoma. Oncotarget, 2016. 7(52): p. 86313–86325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Caiazza F, et al. , Cytosolic phospholipase A2-alpha expression in breast cancer is associated with EGFR expression and correlates with an adverse prognosis in luminal tumours. Br J Cancer, 2011. 104(2): p. 338–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cipriano R, et al. , Hyperactivation of EGFR and downstream effector phospholipase D1 by oncogenic FAM83B. Oncogene, 2014. 33(25): p. 3298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dong Z, et al. , Secretory phospholipase A2-IIa upregulates HER/HER2-elicited signaling in lung cancer cells. Int J Oncol, 2014. 45(3): p. 978–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee CS, et al. , The roles of phospholipase D in EGFR signaling. Biochim Biophys Acta, 2009. 1791(9): p. 862–8. [DOI] [PubMed] [Google Scholar]
- 64.Zhao C, et al. , Phospholipase D2-generated phosphatidic acid couples EGFR stimulation to Ras activation by Sos. Nat Cell Biol, 2007. 9(6): p. 706–12. [DOI] [PubMed] [Google Scholar]
- 65.Gringeri E, et al. , Lyn-mediated mitochondrial tyrosine phosphorylation is required to preserve mitochondrial integrity in early liver regeneration. Biochem J, 2009. 425(2): p. 401–12. [DOI] [PubMed] [Google Scholar]
- 66.Goetzman ES and Prochownik EV, The Role for Myc in Coordinating Glycolysis, Oxidative Phosphorylation, Glutaminolysis, and Fatty Acid Metabolism in Normal and Neoplastic Tissues. Front Endocrinol (Lausanne), 2018. 9: p. 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Papa S, Choy PM, and Bubici C, The ERK and JNK pathways in the regulation of metabolic reprogramming. Oncogene, 2019. 38(13): p. 2223–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cheng SC, et al. , mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science, 2014. 345(6204): p. 1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lin YC, et al. , DUSP1 expression induced by HDAC1 inhibition mediates gefitinib sensitivity in non-small cell lung cancers. Clin Cancer Res, 2015. 21(2): p. 428–38. [DOI] [PubMed] [Google Scholar]
- 70.Kesarwani M, et al. , Targeting c-FOS and DUSP1 abrogates intrinsic resistance to tyrosine-kinase inhibitor therapy in BCR-ABL-induced leukemia. Nat Med, 2017. 23(4): p. 472–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang Z, et al. , Dual specificity phosphatase 6 (DUSP6) is an ETS-regulated negative feedback mediator of oncogenic ERK signaling in lung cancer cells. Carcinogenesis, 2010. 31(4): p. 577–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Peacock JW, et al. , SEMA3C drives cancer growth by transactivating multiple receptor tyrosine kinases via Plexin B1. EMBO Mol Med, 2018. 10(2): p. 219–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jones DT, et al. , Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res, 2008. 68(21): p. 8673–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Treue D, et al. , Proteogenomic systems analysis identifies targeted therapy resistance mechanisms in EGFR-mutated lung cancer. Int J Cancer, 2019. 144(3): p. 545–557. [DOI] [PubMed] [Google Scholar]
- 75.Kim PM, et al. , The role of disorder in interaction networks: a structural analysis. Mol Syst Biol, 2008. 4: p. 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wright PE and Dyson HJ, Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol, 1999. 293(2): p. 321–31. [DOI] [PubMed] [Google Scholar]
- 77.Iakoucheva LM, et al. , Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol, 2002. 323(3): p. 573–84. [DOI] [PubMed] [Google Scholar]
- 78.Ding CB, et al. , Structure and function of Gab2 and its role in cancer (Review). Mol Med Rep, 2015. 12(3): p. 4007–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yamaoka T, et al. , Acquired Resistance Mechanisms to Combination Met-TKI/EGFR-TKI Exposure in Met-Amplified EGFR-TKI-Resistant Lung Adenocarcinoma Harboring an Activating EGFR Mutation. Mol Cancer Ther, 2016. 15(12): p. 3040–3054. [DOI] [PubMed] [Google Scholar]
- 80.Andrews S, FastQC A Quality Control tool for High Throughput Sequence Data. [Google Scholar]
- 81.Bolger AM, Lohse M, and Usadel B, Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 2014. 30(15): p. 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim D, Langmead B, and Salzberg SL, HISAT: a fast spliced aligner with low memory requirements. Nature Methods 2015. 30(7): p. 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liao Y, Smyth GK, and Shi W, featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics, 2013. 30(7): p. 923–930. [DOI] [PubMed] [Google Scholar]
- 84.Love MI, Huber W, and Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology, 2014. 15(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sasha Babicki DA, Ana Marcu, Yongjie Liang, Jason R. Grant, Adam Maciejewski, and David S. Wishart., Heatmapper: web-enabled heat mapping for all. Nucleic Acids Research, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Romero P, et al. , Sequence complexity of disordered protein. Proteins, 2001. 42(1): p. 38–48. [DOI] [PubMed] [Google Scholar]
- 87.Xue B, et al. , PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim Biophys Acta, 2010. 1804(4): p. 996–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Obradovic Z, et al. , Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins: Structure, Function, and Bioinformatics, 2005. 61(S7): p. 176–182. [DOI] [PubMed] [Google Scholar]
- 89.Peng K, et al. , Length-dependent prediction of protein intrinsic disorder. BMC Bioinformatics, 2006. 7: p. 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Peng K, et al. , Optimizing long intrinsic disorder predictors with protein evolutionary information. J Bioinform Comput Biol, 2005. 3(1): p. 35–60. [DOI] [PubMed] [Google Scholar]
- 91.Uversky VN, Gillespie JR, and Fink AL, Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins, 2000. 41(3): p. 415–27. [DOI] [PubMed] [Google Scholar]
- 92.Oldfield CJ, et al. , Comparing and combining predictors of mostly disordered proteins. Biochemistry, 2005. 44(6): p. 1989–2000. [DOI] [PubMed] [Google Scholar]
- 93.He B, et al. , Predicting intrinsic disorder in proteins: an overview. 2009. 19(8): p. 929–949. [DOI] [PubMed] [Google Scholar]
- 94.Xue B, et al. , CDF it all: consensus prediction of intrinsically disordered proteins based on various cumulative distribution functions. FEBS Lett, 2009. 583(9): p. 1469–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Huang F, et al. , Subclassifying disordered proteins by the CH-CDF plot method. Pac Symp Biocomput, 2012: p. 128–39. [PubMed] [Google Scholar]
- 96.Mohan A, et al. , Intrinsic disorder in pathogenic and non-pathogenic microbes: discovering and analyzing the unfoldomes of early-branching eukaryotes. Mol Biosyst, 2008. 4(4): p. 328–40. [DOI] [PubMed] [Google Scholar]
- 97.Huang F, et al. , Improving protein order-disorder classification using charge-hydropathy plots. BMC Bioinformatics, 2014. 15 Suppl 17: p. S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dosztányi Z, et al. , IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics, 2005. 21(16): p. 3433–3434. [DOI] [PubMed] [Google Scholar]
- 99.Dosztanyi Z, et al. , The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J Mol Biol, 2005. 347(4): p. 827–39. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw and processed data used in this study was downloaded from the Gene Expression Omnibus (GEO) database and can be found using the indicated accession number (Table 1) for each dataset.