

Abstract
Eukaryotic cells possess several distinct mismatch repair pathways. A mismatch can be introduced in retroviral double-stranded DNA by a pre-existing mutation within the primer binding site (PBS) of the viral RNA genome. In order to evaluate mismatch repair of retroviral double-stranded DNA, Moloney leukemia virus (MLV)-based vectors with a mutation in their PBS were used to infect mismatch repair-competent as well as mismatch repair-deficient cell lines. If the target cells were capable of repairing the mismatch before an infected cell divided, the mismatch within the PBS could be repaired to the wild-type or mutant PBS. If the target cells were unable to repair the mismatch, half the cells in the colony should contain the mutant PBS while the other half should contain the wild-type PBS. To evaluate these predictions, individual colonies were isolated and analyzed by PCR. Almost all mismatch-deficient cell colonies analyzed (cell lines HCT 116 and PMS2–/–) contained both the wild-type and mutated PBS, therefore, mismatches within retroviral double-strand DNA could not be repaired by the mismatch-deficient cells. In contrast, mismatches in ~25% of the mismatch repair-competent cell clones analyzed (cell lines HeLa and PMS2+/+) were repaired, while 75% were not. Therefore, the cellular mismatch repair system is able to repair mismatches within viral double-stranded DNA, but at a low frequency.
INTRODUCTION
DNA repair systems act to maintain genome integrity in the face of replication errors, environmental insults and the cumulative effects of age. More than 70 human genes directly involved in five major pathways of DNA repair have been described (1). Mismatch repair systems in eukaryotic cells are generally characterized by a broad mismatch specificity and this is believed to be responsible for correcting DNA biosynthetic errors and for processing recombination heteroduplexes that contain mismatched base pairs (2). In humans, mismatch repair malfunctions manifest themselves as mutator phenotypes, in instabilities of microsatellite sequences and in increased levels of somatic recombination. Microsatellites are tandem iterations of simple di-, tri- or tetranucleotides. Mismatch repair deficiencies have been linked to increased mutability and tumor development, for example, hereditary non-polyposis colorectal cancer (3).
Retroviral RNA molecules are positive sense in polarity, equivalent to mRNA. After entering host cells, reverse transcriptase synthesizes minus strand DNA using the positive or sense viral RNA as template. This strand is called minus DNA because it is complementary to the positive sense viral RNA. The second strand synthesized is the plus strand DNA using the minus strand DNA as template (4). The minus strand and plus strand DNAs form a double-stranded DNA. This double-stranded DNA is a component of the pre-integration complex (PIC), which translocates to the nucleus. The viral double-stranded DNA then integrates into the host chromosomal DNA. The integrated viral DNA is called a provirus.
Like all DNA replication, reverse transcription also requires a primer, and minus strand DNA synthesis utilizes a host tRNA as primer (5,6). The host tRNA binds to an 18 base sequence termed the primer binding site (PBS), which is downstream of the 5′ long terminal repeat (LTR). All viral plus strand DNA is copied from the minus strand DNA except for the PBS. The plus strand PBS is synthesized by copying the 3′-end of the acceptor arm of the host tRNA molecule. Therefore, even if a mutation occurs within the minus strand DNA, the plus strand DNA will remain wild-type and a mismatch will be formed within the viral double-stranded DNA. Reverse transcription occurs within the viral PIC. The PIC translocates into the nucleus with the double-stranded DNA, which then integrates into the host chromosomal DNA. Cells possess pathways to repair mismatches. Some cell lines, such as HCT 116 (7,8) and PMS2–/– cells (9,10), contain a defect(s) in their repair system(s) which renders them incapable of repairing mismatches. If the cellular mismatch repair system corrects the mismatch within the viral double-stranded DNA before division of the infected cells, the mismatch will be corrected into either a mutant type or the wild-type. Consequently, all the offspring will contain either a mutant or wild-type PBS. If the cellular mismatch repair system is unable to repair this mismatch, one daughter cell of the infected cell will copy the mutant strand and the other one will copy the wild-type strand. If the offspring of the infected cell form a colony, the cells within the colony will contain both the mutant and wild-type PBS.
We previously demonstrated that a dog osteosarcoma cell line, D17 (ATCC CRL-8468), was unable to repair the majority of mismatches within Molony leukemia virus (MLV) double-stranded DNA (6). It is unknown whether the mismatch repair system in D17 cells is competent or not. It is also not clear if mismatches within a retroviral double-stranded DNA can be repaired by the normal host cellular DNA repair system.
In this report we extend our study of D17 cells to four well-characterized mismatch repair-competent and repair-defective cell lines. We demonstrate that the mismatch repair-deficient cell lines HCT 116 and PMS2–/– appeared unable to repair any mismatches within the PBS of retroviral double-stranded DNA, while the mismatch repair-competent cell lines, HeLa and PMS2+/+, could repair the same mismatches with low efficiency.
MATERIALS AND METHODS
Nomenclature
Plasmids are designated, for example, pLT5; viruses made from these plasmids are designated, for example, LT5.
Vector constructions
All recombinant techniques were carried out by conventional procedures (11). All vector sequences are available upon request.
Construction of pLT5. Construction of a retroviral vector containing one mutated base in the PBS was previously described (6). Briefly, pLT5 was derived from pLN (12), which contains a neomycin resistance (neor) gene between two MLV LTRs. The PBS was replaced with a mutated PBS by annealing two oligonucleotides, GGGTCTTTCATTTGGGGGCTCGc and gCGAGCCCCCAAATGAAAGACCC (lower case represents the mutated nucleotide). The thymidine (T) within the PBS at position 744 was changed to cytosine (C) so that the mutated PBS could be digested with SmaI (cCCGGG).
Construction of pLT10. pLT10 (Fig. 1) contained the same introduced SmaI site, but contained a different drug resistance gene, hyg. A AscI–BstEII fragment containing the mutated PBS was inserted into the AscI and BstEII sites of pJZ206. pJZ206 is an MLV vector containing a hyg gene between the two MLV LTRs (13).
Figure 1.

MLV-based retrovirus vector containing the T→C mutation within the PBS. LT5 and LT10 are derived from the MLV. LT5 encodes the neomycin resistance gene (neo) while LT10 carries the hygromycin resistance gene (hyg). The (18 base) PBS of the two vectors has been mutated. A T→C mutation within the PBS allows digestion of the mutated PBS with SmaI (cCCGGG). The lower case represents the difference from the wild-type PBS sequence.
Isolation of neor (hygr) cellular DNA and amplification of the PBS
HeLa and HCT 116 (or PMS2+/+ and PMS2–/–) cells were infected with LT5 (or LT10) and infected cells were selected for neor (or hygr). Visible colonies formed ~12 days after infection. Well separated clones were propagated in 24-well plates. The cells were harvested upon reaching confluence. Cellular DNAs were isolated from each clone using the Wizard genomic DNA purification kit (Promega, Madison, WI).
The PBS was amplified using primer U3 601 (GGCAAGCTAGCTTAAGTAACGC), which annealed within the U3 region, and primer Neo-rev (CCACCCAAGCGGCCGGAGAA), which annealed within the neo gene [or primer Hygro 1579 (AGTCCTCGGCCCAAAGCATCAG) which annealed within the hyg gene] for 25–30 cycles. Amplified DNA was purified with the Wizard PCR Preps DNA purification kit (Promega, Madison, WI) to remove the primers. The purified PCR fragment was amplified using another pair of primers: U3-2770 (CCAGATGCGGTCCAGCCC), which annealed within the U3 region (downstream of U3601), and MLV-BstEII, which anneals within the sequence of the package signal. Platinum High Fidelity Taq DNA Plus polymerase (Life Technologies, Grand Island, NY) was used to reduce the mutation frequency during amplification.
Cells, transfection and infection
The handling of PG13 (ATCC no. CRL-10686) and helper cells, DNA transfections, virus harvesting and virus infections were performed as previously described (13).
RESULTS
Construction of a retroviral vector that contained a mutation in the PBS
Like all DNA replication, reverse transcription also requires a primer, and minus strand DNA synthesis utilizes a host tRNA as its primer (5,6). The host tRNA binds to the 18 base sequence downstream the 5′ LTR of the MLV genome (Fig. 2A) (4). The 18 base sequence TGGGGGCTCGTCCGGGAT is complementary to the tRNAPro 3′-end of the acceptor arm. The nt 11 T was changed to a C using PCR mutagenesis so that nt 11–16 within the PBS could be digested with SmaI (CCCGGG). This vector is designated LT5 (6) (Fig. 1). Another MLV vector, LT10, was constructed and contains the same PBS mutation as LT5 but additionally carries the hyg resistance gene (Fig. 1).
Figure 2.

Experimental protocol to determine whether cells are able to repair mismatches within retroviral double-stranded DNA. (A) MLV virion. The retrovirus contains single-stranded genomic RNA with a host tRNAPro bound to the 18 base PBS sequence. (B) Retroviral double-stranded DNA product. The single-stranded genomic RNA is reverse transcribed into double-stranded DNA. The PBS within the minus strand DNA is synthesized using the viral RNA molecule as its template while the plus-strand PBS DNA is synthesized using the host tRNAPro as template. With the presence of a pre-existing mutation within the PBS, reverse transcription results in a mismatch within this PBS on the viral double-stranded DNA. (C) Integration. Viral double-stranded DNA integrates into the host chromosome. The zigzag lines represent the host cellular DNA. (D) Mismatch repair. If the target cell repairs this mismatch, the double-stranded DNA will be either wild-type or mutant. (E) No mismatch repair. The target cells cannot repair this mismatch. (F) Daughter cells of a mismatch repaired cell. After the cell divides, the two daughter cells remain either both wild-type or both mutant. (G) Daughter cells of a cell with a mismatch. After the cell divides, one of the daughter cells is a mutant and the other is wild-type.
Low efficiency repair of mismatches within MLV double-stranded DNA by the cellular mismatch repair system
To determine whether the cellular mismatch repair system is able to repair mismatches within the MLV double-stranded DNA, a mismatch was introduced into the viral double-stranded DNA. Both the LT5 and LT10 vectors contained a T→C mutation within the PBS so that the mutated PBS could be digested with SmaI (cCCGGG). To transcribe retroviral RNA from the MLV-based vectors, LT5 and LT10 were each transfected into the helper cell line PG13 (14). The viral RNA transcribed from LT5 (or LT10) should also contain the mutation within its PBS. Viruses were collected 10 h after transfection and were used to infect mismatch-competent (HeLa and PMS2+/+) and mismatch-defective (HCT 116 and PMS2–/–) cell lines (7–10). Since these target cells do not contain the retroviral structural genes (gag-pol and env), no virus is released from the infected target cells (13). Therefore, this infection assay represents only a single round of retroviral replication. After the viruses entered the cells, reverse transcription occurred within the viral PIC in the cytoplasm. The minus strand PBS was copied from the mutant PBS which contained the SmaI site while the plus strand PBS was copied from the host tRNAPro (4). A mismatch (T:C) formed within this PBS (Fig. 2B). The PIC translocated into the nucleus and the double-stranded DNA was integrated into the host chromosomal DNA (Fig. 2C). Infected cells were selected for neor (or hygr for LT10). LT5 (or LT10) carried a neomycin resistance gene, neo (or hygromycin resistance gene, hyg). As a consequence, every infected cell formed a colony.
If the target cells were capable of repairing the T:G mismatch before division of the infected cells, the PBS mismatch of all cells in the neor or hygr colony would be repaired to the wild-type (i.e. a T:A base pair) or to a mutant (i.e. a C:G base pair). In other words, the cells in such colonies would contain either all mutant PBS or all wild-type PBS (Fig. 2D and F). It should be remembered that the mutant PBS can be digested with SmaI while the wild-type PBS cannot. However, if the mismatch was not repaired, both the wild-type (i.e. a T:A base pair) and mutant PBS (i.e. a C:G base pair) would be passed to the two daughter cells of the infected cell and cells within such a colony should contain half wild-type and half mutant PBS (Fig. 2E and G). Cellular DNAs of well-separated colonies were isolated and the PBS sequences were amplified by PCR. Analysis of SmaI digestion of each PBS allowed the wild-type and the mutant PBS to be distinguished (Fig. 3). A colony containing both wild-type and mutant PBS represented a PBS that was not repaired in the infected cells.
Figure 3.
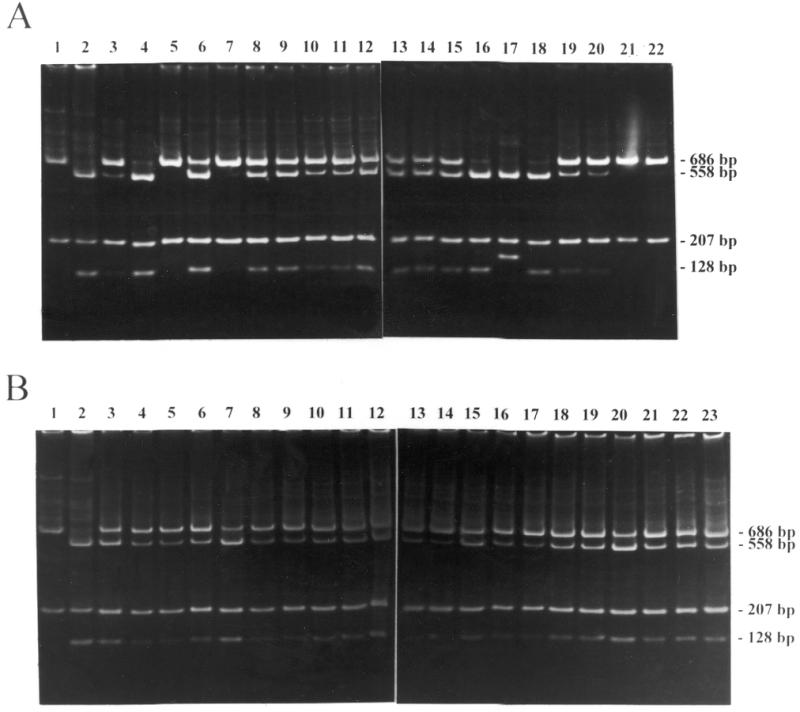
PCR and SmaI digestion analyses of the PBS within the proviral DNA isolated from HeLa (A) and HCT 116 cells (B) infected with LT5. LT5 carrying a T→C mutation within the PBS were used to infect mismatch repair-competent HeLa cells and mismatch repair-defective HCT 116 cells. Each infected cell formed a colony. The PBS from the cells within this colony was amplified by PCR and the PCR products were digested with SmaI. The amplified fragments containing wild-type PBS carry a SmaI site within the R region so that digestion with SmaI results in two fragments of 686 and 207 bp in length (lane 1). The fragments containing mutant PBS carry two SmaI sites so that digestion with SmaI results in three fragments of 558, 207 and 128 bp in length (lane 2). If a colony has a mixture of the wild-type and mutant PBS, digestion with SmaI results in four fragments of 686, 558, 207 and 128 bp in length. All lanes contain SmaI-digested PCR product of DNA isolated from individual neor colonies infected with LT5. In (A) lane 17 contains three fragments of 558, 207 and 163 bp in length. Sequence analysis indicates that a 34 bp sequence (from position 687 to 720) was duplicated at position 687 with an additional SmaI site (CCCGGG) within the PBS. Therefore, sample 17 is a mutant.
Almost all of the mismatch repair-defective colonies examined (Fig. 3B, lanes 3–23) contained both wild-type and mutant PBS sequences in the DNA isolated from a single colony. Two of 45 colonies analyzed showed a pattern corresponding to a repaired PBS (Table 1). Approximately 25% of the mismatch repair-competent cell line colonies examined contained only the wild-type PBS (Fig. 2A, lanes 5, 7, 21 and 22) or contained only the mutant type PBS (Fig. 2A, lane 17; Table 1). Therefore, although these mismatch repair-competent cell lines were able to repair mismatches within retroviral DNAs more efficiently than the mismatch repair-defective cell lines, the cellular mismatch repair system was not able to repair 75% of mismatches within the viral double-stranded DNA. The results demonstrate that the frequency of the mismatch repair in infected competent cells was low before cell division.
Table 1. Analysis of PBS from a single colony containing LT5 or LT10.
| Mismatch-competent cell line |
Mismatch-defective cell line |
|||
|---|---|---|---|---|
| HeLa | PMS2+/+ | HCT 116 | PMS2–/– | |
| Wild-type | 4 | 1 | 0 | 0 |
| Mutant | 1 | 5 | 0 | 2 |
| Mixture | 15 | 17 | 22 | 21 |
| Total | 20 | 23 | 22 | 23 |
The LT5 and LT10 vectors contained a T→C mutation in the PBS and were used to infect mismatch repair-competent and mismatch repair-defective cell lines. Infected cells were selected for neor (or hygr) and, as a consequence, each infected cell formed a colony. PBS sequences were amplified by PCR and digested with SmaI. Only the mutant PBS could be digested with SmaI, while the wild-type could not. If a portion of the PBS in a colony could be digested with SmaI and the other portion could not, this colony was designated a mixed colony.
DISCUSSION
Mismatch repair-defective cells display frequent alterations in microsatellite sequences. Nuclear extracts derived from mismatch repair-defective cells fail to correct mispairs within open circular DNA heteroduplexes in vitro (7). To date, there are no in vivo systems to introduce DNA mismatches into eukaryotic cells and quantitatively analyze repair. There is a large volume of literature concerning the frequency of mismatch repair using transfection of mismatched plasmids. We have also constructed a plasmid containing a mismatch at the start codon of the gfp gene. This mismatched plasmid was transfected into the mismatch repair-deficient cell line HCT 116. Mismatches in the HCT 116 cell line were repaired with the same efficiency as observed in the mismatch repair-competent cell lines. However, HCT 116 nuclear extracts could not repair any mismatches in vitro because its mismatch repair systems are defective (7). One possible interpretation was that the plasmids might replicate in transfected cells. Only repair of mismatches within retroviral double-stranded DNA distinguished between the mismatch repair-deficient and mismatch repair-competent cell lines.
In this report a mutation has been introduced into the PBS so that the retroviral double-stranded DNA contains a mismatch. Analysis of the proviral DNA in both the mismatch repair-competent and mismatch repair-defective cells indicates that the mismatch repair-defective cell lines cannot repair most of the mismatches within the retroviral double-strand DNA. We had expected that no mismatched PBS could be repaired in the two mismatch repair-deficient cell lines, however, two of 45 colonies analyzed were found to display a digestion pattern corresponding to a repaired PBS. Previous work demonstrated that retroviral titers determined by a color reporter gene were usually higher than the corresponding colony forming units using drug selection (15). Some infected cells are unable to survive in the selection media, probably due to the low production of the drug resistance gene product. Furthermore, Hajihosseini et al. (16) demonstrated that daughter cells did not always survive. Therefore, the two colonies with a digestion pattern corresponding to a repaired PBS probably resulted from death of the daughter cell of the infected cell with a mismatched PBS. The surviving daughter cell formed a colony so that the PBS of all cells in this colony corresponded to the repaired one.
We had also anticipated that all mismatched PBSs could be repaired in both mismatch repair-competent cell lines. However, only ~25% of the mismatched PBSs were repaired. Retroviral reverse transcription occurs in the viral PIC and the double-stranded DNA remains in the PIC until integration. When the mismatched PBS is packaged within the PIC it is possible that the cellular mismatch repair system cannot access the mismatch. Simple retroviruses, including our MLV-based vector, are unable to enter the nucleus until mitosis. Following PIC translocation into the nucleus, the double-stranded DNA is then able to integrate into the host chromosomal DNA (4,16). Therefore, the window for the cellular mismatch repair system to repair a mismatch within retroviral double-stranded DNA would be very narrow. We deduce that mismatches within a lentiretrovirus, such as human immunodeficiency virus, are more likely to be repaired by the cellular mismatch repair system since lentiviruses can enter the nucleus of growth-arrested cells (17,18) and thus the time frame for mismatch recognition and repair would be longer.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Michael Liskay for providing the PMS2+/+ and PMS2–/– cell lines. We thank William Bargmann, Kathleen Boris-Lawrie, Alan Kaplan, Ting Li, Yan Ma and Alan Simmons for helpful comments on the manuscript. This research was supported by Public Health Service research grant CA70407.
REFERENCES
- 1.Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis. ASM Press, Washington, DC.
- 2.Modrich P. and Lahue,R. (1996) Annu. Rev. Biochem., 65, 101–133. [DOI] [PubMed] [Google Scholar]
- 3.Aaltonen L.A., Peltomaki,P., Leach,F.S., Sistonen,P., Pylkkanen,L., Mecklin,J.P., Jarvinen,H., Powell,S.M., Jen,J., Hamilton,S.R. et al. (1993) Science, 260, 812–816. [DOI] [PubMed] [Google Scholar]
- 4.Coffin J.M., Hughes,S.H. and Varmus,H. (1997) Retroviruses. Cold Spring Harbor Laboratory Press, Plainview, NY. [PubMed]
- 5.Berwin B. and Barklis,E. (1993) Nucleic Acids Res., 21, 2399–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang J., Tang,L.Y., Li,T., Ma,Y. and Sapp,C.M. (2000) J. Virol., 74, 2313–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parsons R., Li,G.M., Longley,M.J., Fang,W.H., Papadopoulos,N., Jen,J., de la Chapelle,A., Kinzler,K.W., Vogelstein,B. and Modrich,P. (1993) Cell, 75, 1227–1236. [DOI] [PubMed] [Google Scholar]
- 8.Drummond J.T., Li,G.M., Longley,M.J. and Modrich,P. (1995) Science, 268, 1909–1912. [DOI] [PubMed] [Google Scholar]
- 9.Baker S.M., Bronner,C.E., Zhang,L., Plug,A.W., Robatzek,M., Warren,G., Elliott,E.A., Yu,J., Ashley,T., Arnheim,N. et al. (1995) Cell, 82, 309–319. [DOI] [PubMed] [Google Scholar]
- 10.Prolla T.A., Baker,S.M., Harris,A.C., Tsao,J.L., Yao,X., Bronner,C.E., Zheng,B., Gordon,M., Reneker,J., Arnheim,N., Shibata,D., Bradley,A. and Liskay,R.M. (1998) Nature Genet., 18, 276–279. [DOI] [PubMed] [Google Scholar]
- 11.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 12.Miller A.D. and Rosman,G.J. (1989) Biotechniques, 7, 980–982, 984,–986, 989–990. [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang J. and Temin,H.M. (1993) Science, 259, 234–238. [DOI] [PubMed] [Google Scholar]
- 14.Miller A.D., Garcia,J.V., von Suhr,N., Lynch,C.M., Wilson,C. and Eiden,M.V. (1991) J. Virol., 65, 2220–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sapp C.M., Li,T. and Zhang,J. (1999) J. Biomed. Sci., 6, 342–348. [DOI] [PubMed] [Google Scholar]
- 16.Hajihosseini M., Iavachev,L. and Price,J. (1993) EMBO J., 12, 4969–4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weinberg J.B., Matthews,T.J., Cullen,B.R. and Malim,M.H. (1991) J. Exp. Med., 174, 1477–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lewis P., Hensel,M. and Emerman,M. (1992) EMBO J., 11, 3053–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]