

Summary
Recent evidences have implicated that SENP3 is a deSUMOylase which possesses neuronal damage effects in cerebral ischemia. However, its role in microglia remains poorly understood. Here, we found that SENP3 was upregulated in the peri-infarct areas of mice following ischemic stroke. Furthermore, knockdown of SENP3 significantly inhibits the expression of proinflammatory cytokines and chemokines in microglial cells. Mechanistically, SENP3 can bind and then mediated the deSUMOylation of c-Jun, which activated its transcriptional activity, ultimately followed by the activation of MAPK/AP-1 signaling pathway. In addition, microglia-specific SENP3 knockdown alleviated ischemia-induced neuronal damage, and markedly diminished infract volume, ameliorated sensorimotor and cognitive function in animals subjected to ischemic stroke. These results indicated SENP3 functions as a novel regulator of microglia-induced neuroinflammation by activating the MAPK/AP-1 signaling pathway via mediating the deSUMOylation of c-Jun. Interventions of SENP3 expression or its interaction with c-Jun would be a new and promising therapeutic strategy for ischemic stroke.
Subject areas: Neurology, Molecular biology, Cellular neuroscience
Graphical abstract
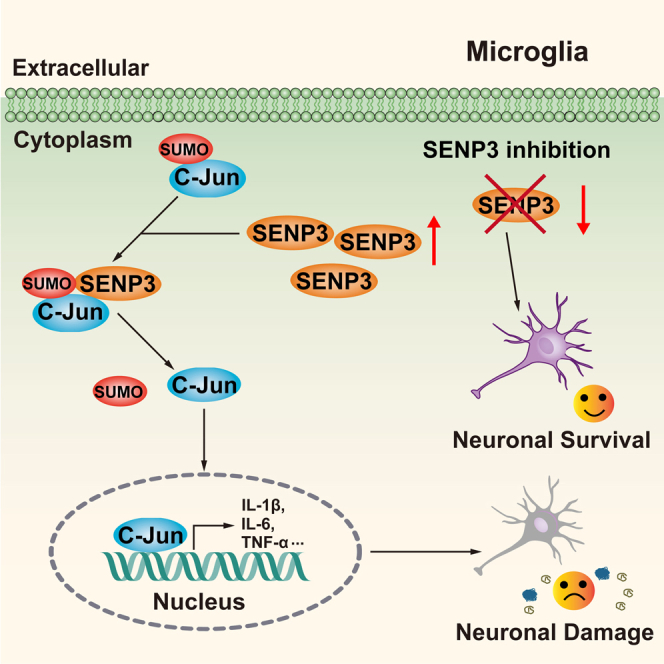
Highlights
-
•
SENP3 is increased in microglia after cerebral ischemia and reperfusion injury
-
•
SENP3 promotes the expression of proinflammatory cytokines and chemokines in microglia
-
•
SENP3 binds and mediates the deSUMOylation of c-Jun
-
•
Microglia-specific SENP3 knockdown ameliorates ischemic brain injury in mice
Neurology; Molecular biology; Cellular neuroscience
Introduction
Ischemic stroke is a major cause of disability and death around the world. The lack of specific therapeutic targets for ischemic stroke stresses the need for developing new therapeutic regimens.1 In the past decade, researchers have focused on exploring effective protection strategies that only target to neurons, which cannot produce better outcome post ischemic injury. Increasing evidence has shown that excessive neuroinflammation induced by overactivated microglia is an important factor that contributes to neuronal apoptosis and magnifies brain damage following cerebral ischemia and reperfusion (I/R) injury.2,3 Stroke-induced microglial excessive activation causes the expression and secretion of pro-inflammatory factors, such as interleukin (IL)-1β, interleukin (IL)-6, tumor necrosis factor (TNF)-α, C-X-C motif chemokine ligand 1 (CXCL1) and C-C motif chemokine ligand 2 (CCL2), which in turn exacerbate the neuroinflammation, and ultimately contributes to delayed deterioration of ischemia brain tissue.4 Thus, the development of novel agents to inhibit neuroinflammation could potentially prevent neuronal death and contribute to the treatment of ischemic brain injury.
SUMOylation is one of the reversible post-translational modifications (PTM) in which substrate proteins are deconjugated by members of the sentrin/SUMO-specific protease (SENP) family.5 SUMOylation alters inter- and/or intra-molecular interactions of substrate proteins to change their localization, interaction, stability, and activity, thus regulating cellular signaling activation and gene transcription in various tissue.6 Among the SENP family members, researchers have demonstrated that the SUMO protease SENP3, which can remove SUMO2/3 conjugation from substrates specifically, is a redox-sensitive isopeptidase.7 In experimental and clinical studies, Rawlings et al. reported that SENP3-mediated deSUMOylation promoted cardiomyocyte survival after ischemic insult.8 Meanwhile, previous studies have reported that SENP3-mediated deSUMOylation contributes to cell death during reoxygenation after ischemic insult9,10 and spinal cord injury.11 Furthermore, SENP3 promotes lipopolysaccharide (LPS)-activated inflammatory response in macrophages.12,13 Although the effects of SENP3 on central nervous system (CNS) have been introduced, whether SENP3 is a critical regulator in microglia-induced neuroinflammation after ischemic stroke remain to be further studied.
The mammalian family of mitogen-activated protein kinase (MAPK), including extracellular signal-regulated kinase (ERK), p38 MAP kinases, and c-Jun NH2-terminal kinase (JNK) have regulated a variety of important biological processes, such as cell differentiation, proliferation, migration, apoptosis, and inflammation response.14,15 In addition, it is well known that MAPK/activator protein-1 (AP-1) pathways have been implicated as critical transcriptional regulators for pro-inflammatory activation of microglia.16,17 The transcription factors c-Jun function as one of the important components of the inducible transcriptional complex AP-1, whose activity is regulated by SUMOylation modification.18,19,20,21 c-Jun has previously been described to be a target for SUMOylation on lysins 229 and 257, which entails inhibit its transactivation activity.18,21 Nevertheless, the key enzymes which control the SUMOylation modification of c-Jun and its effect on the activation of microglia after cerebral I/R injury remain poorly investigated.
In the present study, we explore the pathological role of SENP3 involved in microglial-mediated neuroinflammation after cerebral ischemia. We provide profound evidences that SENP3 induces the deSUMOylation of c-Jun, followed by the activation of MAPK/AP-1 signaling, thereby enhancing the expression of proinflammatory cytokines and chemokines, ultimately leading to excessive neuroinflammation and neuronal apoptosis after cerebral ischemia. On the contrary, SENP3 knockdown efficiently alleviated cerebral I/R injury via an anti-neuroinflammatory effect and improves neurological function following ischemic stroke. Therefore, this study suggests that interventions of SENP3 expression or its interaction with c-Jun might serve as potential therapeutic strategies for the therapy of ischemic stroke.
Results
SENP3 expression level was increased in microglia after cerebral I/R injury
To study the potential effects of SENP3 on microglia-induced neuroinflammation after ischemic stroke, we firstly determined whether SENP3 expression was altered in vivo and in vitro. Primary microglia were challenged with OGD and reoxygenation (OGD/R) at several different time points, the mRNA expression of Senp3 rapidly increased as early as 1 h following OGD/R, peaking at 24 h, as shown by RT-qPCR analysis (Figure 1A). Consistent with the mRNA level, the immunoblots assay also demonstrated that SENP3 protein expression rapidly increased in a pattern that started after 1 h and peaked after 24 h following OGD (Figures 1B and 1C). In contrast, immunoblots revealed that the global SUMO1 and SUMO2/3 conjugation in primary microglia progressively decreased with longer periods of OGD/R (Figure S1). Next, the SENP3 level was investigated in vivo. Mice undergo MCAO operation for 1 h followed by various periods of reperfusion from 1 to 72 h. RT-qPCR analysis demonstrated that Senp3 mRNA transcription increased markedly as early as 1 h following I/R, peaking at 24 h (Figure 1D). Meanwhile, similar changes in expression at the protein level were confirmed by immunoblots assay (Figures 1E and 1F). Furthermore, immunofluorescence staining of brain sections showed that SENP3 were primarily expressed in microglia and neurons and to a lesser extent in astrocytes (Figures 1G–1K). Moreover, these results were consistent with the RT-qPCR and immunoblots results, showing that SENP3 staining in microglia and neurons was more intense in the mice subjected to MCAO operation (Figures 1H–1L). Collectively, our results demonstrated that SENP3 increased dramatically in a time-dependent manner after cerebral I/R injury.
Figure 1.

Microglial SENP3 is upregulated after cerebral ischemia
(A) Primary cultured microglia were treated with OGD and reoxygenation at the indicated time points. RT-qPCR analysis for determining mRNA levels of Senp3. n = 3 per time point.
(B and C) Immunoblot analysis for determining the protein expression of SENP3 in primary cultured microglia. The representative images of immunoblot bands (B) and analysis of relative band intensity (C) are depicted. n = 3.
(D) Mouse brain homogenates were extracted after MCAO operation for 1 h and reperfusion at different time as indicated. RT-qPCR analysis for determining mRNA transcription of Senp3 in cerebral tissues. n = 4 per time point.
(E and F) Immunoblots for determining the protein level of SENP3 in cerebral tissues. The representative images of immunoblot bands (E) and analysis of relative band intensity (F) are depicted. n = 4 per time point.
(G, I, and K) Representative double immunostaining of SENP3 (red) with Iba-1 (a microglial marker, green), GFAP (an astrocyte glial marker, green) and NeuN (a neuronal marker, green) from ischemic penumbra of brain tissue after MCAO operation. Scale bars: 20 μm.
(H, J, and L) Quantification of Iba-1+/SENP3+, NeuN+/SENP3+, and GFAP+/SENP3+ fluorescence intensity was quantified using ImageJ. Data are presented as means ± SEM and quantified by one-way ANOVA followed by Dunnett’s post hoc test or unpaired Student’s t test (H, J, L) ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗∗p < 0.0001.
SENP3 facilitates microglia overactivation under OGD/R conditions
Given that microglial activation has a fundamental role in the pathophysiology of ischemic stroke,22 we then sought to examine the role of SENP3 in microglial-induced neuroinflammation after cerebral ischemia. Frist, primary microglial cells were transfected with recombinant adenovirus carrying SENP3 coding sequence (Ad-SENP3) or shRNA sequence (Ad-sh.SENP3). The overexpression and knockdown efficiency of these adenovirus are presented in Figures S2 and S3, showing successful SENP3 overexpression or knockdown in primary cultured microglial cells. Then, RT-qPCR assays were conducted to determine the mRNA expression of proinflammatory marker genes, including Il-1β, Il-6, Tnf-α, Cxcl1 and Ccl2, and anti-inflammatory marker genes, including Il-4, Il-10, Tgf-β, Arginase-1 and Cd206. The results showed that overexpressing SENP3 further promoted OGD/R-induced proinflammatory gene expression and inhibited anti-inflammatory gene expression, whereas microglia infected with Ad-sh. SENP3 showed the opposed effects (Figures 2A and S4A). To further confirm the role of SENP3 in microglial polarization, ELISA was performed to reveal the secretion of proinflammatory marker cytokines (IL-1β, IL-6, TNF-α, CXCL1 and CCL2) and anti-inflammatory marker cytokines (IL-4, IL-10, TGF-β, IL-13 and IL-1ra), and the results were consistent with the mRNA expression results (Figures 2B and S4B). Meanwhile, immunoblot was used to examine the protein levels of proinflammatory and anti-inflammatory mediators. The results revealed that SENP3 upregulation significantly increased iNOS and CD16/32 expression, and decreased Arginase-1 and CD206, while SENP3 knockdown showed the opposite effect (Figures 2C, 2D, S4C, and S4D). Last, immunofluorescence staining also confirmed these results. We found that the level of iNOS and Iba-1 were dramatically increased after SENP3 overexpression, but decreased after SENP3 knockdown in microglia after OGD/R (Figures 2E and 2F). Taken together, these data indicate that SENP3 promotes microglia-induced neuroinflammation in response to OGD/R.
Figure 2.

SENP3 promotes the expression of proinflammatory factors in microglia after cerebral I/R injury
(A) Primary cultured microglia were transduced with adenovirus expressing empty vector (Ad-vector), SENP3 coding sequence (Ad-SENP3), scramble (Ad-sh.NC) or SENP3 shRNA sequence (Ad-sh.SENP3), and then treated with OGD/R. The mRNA level of Il-β, Il-6, Tnf-α, Cxcl1 and Ccl2 in microglial cells was quantified by RT-qPCR assay.
(B) The secretion level for IL-β, IL-6, TNF-α, CXCL1 and CCL2 in primary microglia supernatants was detected by ELISA analysis.
(C and D) The protein expression of iNOS and CD16/32 was detected by immunoblots assay (C) and the analysis of relative band intensity was depicted (D). n = 3 per group.
(E) The intensity of iNOS (Green) and Iba-1 (Red) in primary cultured microglia was determined by immunofluorescence assays. Scale bars: 20 μm.
(F) The fluorescence intensity of iNOS and Iba-1 was analyzed via ImageJ software. Data are presented as means ± SEM and analyzed by one-way ANOVA followed by Tukey’s post hoc test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
SENP3 deficiency restrains the activation of MAPK/AP-1 signaling in microglia following OGD/R
To explore the potential mechanism by which SENP3 mediates microglia-induced neuroinflammation, we detected the activation of the NF-κB and MAPK/AP-1 signaling pathways, which play critical roles in microglial activation, as previously reported.14,15 First, primary cultured microglia were infected with Ad-SENP3 or Ad-sh.SENP3, and then challenged with OGD/R. The results revealed that the phosphorylation level of p65, ERK1/2, JNK1/2, p38 MAPK, and c-Jun markedly increased in response to OGD/R stimulation, whereas OGD/R-induced phosphorylation of c-Jun were further increased by SENP3 overexpression. On the contrary, SENP3 deficiency showed the oppose effects. However, the phosphorylation levels of p65, ERK1/2, JNK1/2, and p38 MAPK were both unaffected with either up-regulation or down-regulation of SENP3 (Figures 3A–3F). Next, dual-luciferase reporter assay revealed that the transcriptional activity of p65 and AP-1 was significantly increased after OGD/R. Of interest, either upregulation or downregulation of SENP3 had little impact on the transcriptional activity of p65 (Figure 3G). However, SENP3 overexpression significantly increased AP-1 transcriptional activity, and SENP3 deficiency showed the opposite effect (Figure 3H). Meanwhile, we determined the DNA binding activity of AP-1. The results revealed that SENP3 deficiency robustly reversed the OGD/R-induced increase of AP-1 DNA binding activity (Figure 3I). Finally, we attempted to examine the subcellular distribution of c-Jun. We found that SENP3 overexpression further promoted OGD/R-induced nuclear transport of c-Jun. However, SENP3 downregulation robustly decreased the nuclear accumulation of c-Jun (Figures 3J–3L). Consistently, the immunofluorescence assay demonstrated that SENP3 knockdown significantly inhibited OGD/R-induced c-Jun nuclear translocation in microglial cells (Figures S5A and S5B). Altogether, these results suggest that a selective role of SENP3 functions as a positive regulator to potentiate the activation of MAPK/AP-1 pathways, thereby enhancing the production of proinflammatory factors in OGD/R-treated microglia.
Figure 3.

SENP3 enhances the activation of MAPK/AP-1 signaling in microglia after OGD/R
(A–F) Primary microglia were transfected with Ad-vector, Ad-SENP3, Ad-sh.NC or Ad-sh.SENP3, and then treated with OGD/R, respectively. Effects of SENP3 on the activation of NF-κB and MAPK/AP-1 signaling was determined. Total protein expression and the phosphorylation levels of p65, ERK1/2, JNK1/2, p38 MAPK, and c-Jun were examined by immunoblot (A) and analysis of relative band intensity (B-F) was depicted.
(G and H) The transcriptional activity of p65 (G) and AP-1 (H) in HEK293 cells was examined by dual-luciferase reporter assay.
(I) The DNA binding activity of AP-1 in primary microglia was examined by an ELISA-based (Trans-AM) method.
(J–L) Effects of intervention of SENP3 expression on c-Jun nuclear translocation was determined by immunoblotting (J) and the analysis of relative band intensity was depicted (K, L). Data are presented as means ± SEM from at least three independent experiments. Quantitative analysis was conducted by one-way ANOVA (K and L), and all others were analyzed by two-way ANOVA followed by Tukey’s post hoc test. ns: no significance, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
SENP3 mediated deSUMOylation of c-Jun after OGD/R
It has been reported that c-Jun can be modified by SUMOylation, which inhibits its transcriptional activity.18,23 As SENP3 functions as a deSUMOylation enzyme, we investigated whether SENP3 affects the transcriptional activation of c-Jun through regulating its SUMOylation level. To this end, we firstly confirmed whether c-Jun could be modified by SUMOylation. The Ni2+-NTA agarose affinity pull-down assay demonstrated that c-Jun can be conjugated by all three SUMO proteins. Among these, SUMO2 modification was much stronger than the other two (Figure 4A). Thus, we concentrate on the SUMO2 conjugation of c-Jun in following studies. Next, co-IP assays demonstrated that the SUMOylation of c-Jun was obviously decreased in primary microglia after OGD/R (Figure 4B). Next, HEK293T cells were transduced with plasmids expressing wild-type (WT) or a catalytic mutant SENP3 (C532A, Cysteine-to Alanine mutation at residue 532).24 We found that SENP3-WT markedly decreased the SUMOylation of c-Jun, whereas the catalytic-domain-null SENP3-C532A mutant had little effect (Figure 4C). Meanwhile, we found that SENP3 knockdown by specific shRNA in HEK293T cells increased the SUMOylation level of c-Jun (Figures 4D, S6, and S7). Last, we examined whether SENP3 interacts with c-Jun. HEK293T cells were transient transduced with Myc-SENP3 and HA-c-Jun. As expected, co-IP assays revealed that ectopically expressed SENP3 could bind with c-Jun (Figures 4E and 4F). Meanwhile, co-IP assay also demonstrated that the endogenous binding of SENP3 with c-Jun was obviously enhanced in primary microglia after OGD/R (Figures 4G and 4H). Collectively, these data suggest that SENP3 bind with c-Jun and mediated deSUMOylation of c-Jun after cerebral I/R injury.
Figure 4.

SENP3 induced the deSUMOylation of c-Jun
(A) The SUMOylation level of c-Jun was determined by Ni2+-NTA pull down. HA-c-Jun, His-SUMO1/2/3, and Flag-Ubc9 were co-transduced into HEK293T cells. Cell lysates were used for Ni2+-NTA resin pull down and determined by immunoblot analysis.
(B) The SUMOylation of endogenous c-Jun in primary microglia treated with OGD/R or not was examined by co-immunoprecipitation (co-IP) assay.
(C) SENP3 downregulates the SUMOylation level of c-Jun. HA-c-Jun, His-SUMO2, and Flag-Ubc9 were co-transduced into HEK293T cells along with Myc-SENP3 or Myc-SENP3 C532A mutant. The SUMOylation level of c-Jun was examined by Ni2+-NTA pull down and analyzed by immunoblot.
(D) Representative Ni2+-NTA pull-down results demonstrating the SUMOylation level of c-Jun in HEK293T cells when SENP3 is knocked down by specific shRNA.
(E and F) The interaction of ectopically expressed c-Jun with SENP3 was determined by co-IP. Flag-SENP3 and HA-c-Jun were co-transduced into HEK293T cells, and cell lysates were subjected to co-IP.
(G and H) The binding of endogenous SENP3 with c-Jun in primary microglia subjected to OGD/R or not was detected by co-IP. All the experiments were repeated three times.
Microglial SENP3 deficiency attenuates damage of neuron co-cultured with microglia after OGD/R
In the above experimental results, we concluded that SENP3 mediated deSUMOylation of c-Jun, and activated AP-1 signaling, resulting in increased levels of proinflammatory factors in microglia after OGD/R treatment. We then sought to investigated the effect of SENP3 on neuronal damage in vitro. To this end, a microglia-neuron transwell system was applied (Figure 5A). We transfected primary cultured microglia with recombinant adenovirus carrying SENP3 coding sequence or shRNA sequence. The results showed that SENP3 upregulation in microglia greatly increased neuronal apoptosis induced by OGD/R, while SENP3 downregulation obviously decreased neuronal apoptosis (Figures 5B and 5C). Furthermore, lactate dehydrogenase (LDH) assay assays showed that SENP3 upregulation in microglia greatly increased LDH release, whereas SENP3 knockdown significantly decreased LDH release (Figure 5D). Meanwhile, we performed CCK-8 assay to detect neuronal viability, the results demonstrated that SENP3 upregulation in microglia greatly reduced neuronal viability, but SENP3 downregulation showed the oppose effect (Figure 5E). Last, immunoblot assays revealed that SENP3 upregulation in microglia greatly decreased the level of antiapoptotic gene, such as Bcl-xl, but promoted the production of proapoptotic molecules in neurons, including Bax, cleaved caspase-3, cleaved caspase-9 and cleaved PARP. However, SENP3 downregulation represented opposite regulation (Figures 5F–5K). Based on these results, we conclude that microglial SENP3 deficiency relieves the neurotoxic effects of ischemic neuronal damage induced by a microglial overactivation.
Figure 5.

SENP3 deletion alleviated the neurotoxic effects of pro-inflammatory microglia in cocultured neurons after OGD/R
(A) Primary cultured microglia were transduced with Ad-SENP3 or Ad-sh.SENP3, then co-cultured with primary neurons via transwell system, following by OGD/R treatment. Schematic representation as above.
(B and C) The apoptotic neurons were determined by TUNEL staining (B) and quantification analysis of TUNEL+ neurons (C). Scale bar: 20 μm.
(D) The cytotoxicity was examined by LDH release assay.
(E) The neuronal viability was determined by CCK-8 assay.
(F–K) (F) Immunoblot assays showed the protein expression of representative anti-apoptosis or pro-apoptosis molecules in co-cultured neurons. (G-K) Quantitative analysis of the Bcl-xl (G), Bax (H), cleaved PARP (I), cleaved caspase-9 (J), and cleaved caspase-3 (K) protein expression in (F). Data are presented as the means ± SEM. from at least three dependent experiments and analyzed by two-way ANOVA followed by Tukey’s post hoc test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
Microglial SENP3 deficiency alleviates ischemia-induced neuronal apoptosis, diminish infarct volume, and ameliorates neurological functions in mice
The above studies suggested that SENP3 deficiency decreased the expression of proinflammatory factors induced by microglia and protected against ischemia-damaged neurons, then we detected the protective effects of SENP3 inhibition against ischemic stroke in vivo. To this end, we employed a method to knockdown the expression of SENP3 in microglia of mouse brain.25,26,27 We generated an adeno-associated virus (AAV) type 2/6 construct expressing SENP3 shRNA only in cells expressing Cre recombinase driven by the Cx3cr1 promoter (Figure 6A). Each AAV vector was stereotactically injected into the hippocampus CA1 region, cerebral cortex, and striatum of Cx3cr1-Cre mice. Four weeks after virus injection, animals were conducted with MCAO operation. Next, we conducted serious of histological and behavioral experiments at different time points (Figure 6B). We evaluated the silencing efficiency of AAV-sh-SENP3 by immunoblot and the result revealed reduced SENP3 expression in isolated microglia, showing successful SENP3 knockdown (Figures 6C and 6D). Furthermore, the sh.NC and sh.SENP3–treated mice exhibited comparable regional cortical blood flow (CBF) during MCAO and reperfusion (Figures 6E and 6F). We then conducted morphometric microglial cell analysis to examine the activation state of microglia in the penumbra of mice. Labeled microglia were performed detailed quantitative morphometric analysis to determine cell body size, number and length of branches. As shown in Figure S8, microglia from sh.SENP3-treated mice had smaller cell bodies, but more and longer branch length than sh.NC-treated mice, indicating a less activated morphological profile. Next, TUNEL staining was utilized to detect the apoptotic cells, and the results demonstrated that SENP3 knockdown significantly decreased the number of apoptosis cells in the hippocampus and cerebral cortex regions under cerebral I/R injury (Figure 6G). Next, we conducted TTC staining to evaluate the cerebral infarct size. The data revealed that SENP3 knockdown obviously reduced ischemic infarct volume after cerebral ischemia (Figures 6H and 6I). Last, the neurological dysfunction of animals was scored at five time points by mNSS. The data revealed that SENP3 knockdown significantly alleviated the neurological deficits (Figure 6J). These data collectively demonstrate that microglial SENP3 knockdown protects against neuronal apoptosis, diminish infarct volume, and ameliorates neurological functions following ischemic stroke.
Figure 6.

Microglial SENP3 knockdown is neuroprotective during cerebral ischemia-reperfusion injury
(A) Experimental design for microglia-specific knockdown of SENP3 in mice. AAV2/6 vectors carrying SENP3 or scramble shRNA were injected into hippocampus CA1 region, cerebral cortex and striatum of Cx3cr1-Cre mice.
(B) Schematic representation showing the experimental process.
(C and D) The interference efficiency of SENP3 was determined by western blotting.
(E) Cerebral blood flow monitored using 2-dimensional laser speckle imaging techniques before, during MCAO, and reperfusion.
(F) Results were expressed as percent change from baseline (pre-MCAO).
(G) The neuronal apoptosis in hippocampus CA1 region, cerebral cortex was detected by TUNEL staining. Scale bar: 50 μm.
(H and I) The infarct volume was examined by TTC staining (H) and the analysis of infarct size was shown (I). n = 8–9 mice per group.
(J) The neurological deficits scores were evaluated by the mNSS. Data are presented as means ± SEM and analyzed by unpaired Student’s t test (panel D), one-way ANOVA followed by Dunnett’s post hoc test (panel I) or RM ANOVA followed by Tukey’s post hoc test (panel J). n.s. for p > 0.05, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 and ∗∗∗∗p < 0.0001.
Microglial SENP3 knockdown improves neurobehavioral function following ischemic stroke
To further investigated the neuroprotective effects of SENP3 deficiency against ischemic stroke, a battery of the indicated behavioral tests was performed on mice before and up to 28 days after cerebral ischemia. As shown in Figures 7A–7D, cerebral ischemia induced severe damage of sensorimotor functions in scramble mice as shown by increased time to touch and tear, increased asymmetric rate, and decreased latency to fall, but SENP3 knockdown treatment caused a superior neurological functional recovery. Next, the MWM test was conducted to ascertain the spatial learning and memory function. Representative tracings showing sample paths of the animals from the latency trials and probe trials are shown in Figures 7E and 7F, respectively. As expected, the mice subjected to MCAO operation represented a significant increase in the time to reach the hidden platform and decreased the time spent in the targeting quadrant, as compared to the scramble control. However, SENP3 knockdown significantly represented cognitive improvement; SENP3 deficient animals spent less time to reach the hidden platform than the scramble control animals (Figures 7G and 7H). Meanwhile, SENP3 deficient animals also spent more time in the targeting quadrant and crossed the platform location more times during the probe trials (Figures 7I and 7J). Collectively, these data revealed that microglial SENP3 knockdown offered protection against ischemic infarct.
Figure 7.

Knockdown of SENP3 in microglia alleviated cognitive and motor function after ischemic stroke in mice
(A–D) Sensorimotor deficits were assessed before (Pre) and up to 28 days after cerebral I/R injury by a series of behavioral tests. (A) Adhesive touch test. (B) Adhesive removal test. (C) Cylinder test. (D) Rotarod test.
(E–J) Latency trial and probe trial results in the Morris water maze (MWM) tests. (E) Representative path tracings on day 35 during the latency trial. (F) Representative path tracings on day 36 during the probe trial. (G) The mean escape latency during days 30–35 of testing. (H) Mean escape latency to the hidden platform on day 36. (I) The percentage of time searching for the hidden platform in the target quadrant on day 36. (J) Number of times crossing the target platform location on day 36. n = 8–10 mice per group. Data are presented as means ± SEM. Statistical difference in panel (A-D and G) was analyzed by the RM ANOVA followed by Tukey’s post hoc test. Data in panel (J) was determined by Kruskal–Wallis non-parametric test, and all others were used one-way ANOVA followed by Tukey’s post hoc test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 and ∗∗∗∗p < 0.0001.
Discussion
Excessive neuroinflammation induced by overactivated microglia plays a critical role in the brain injury following cerebral ischemia,28 but little is understood about what brain factors control neuroinflammation resolution and the potential mechanisms. The current research was performed to clarify the pro-inflammatory effects of SENP3 and the molecular mechanisms. We confirmed that SENP3 functioned as an important positive regulator of neuroinflammation after ischemia stroke. Specifically, cerebral I/R injury upregulated the level of SENP3 in microglia in vitro and in vivo. Accordingly, suppressing SENP3 significantly reduces the pro-inflammatory factors expression in OGD/R-stimulated microglia. We also found that the biological actions of SENP3 were correlated with the activation of the MAPK/AP-1 signaling via deSUMOylation of c-Jun. In the in vivo study, microglia-selective knockdown of SENP3 resulted in dramatically reduced infarct volumes and ameliorated neurological outcomes after cerebral ischemia, possibly because of the suppression of the production of pro-inflammatory mediators.
SUMOylation modification have been reported to restrained inflammation in various types of cells, exemplified by SUMOylation of Annexin-A1, MKK7, NLRP3, and so on.13,27,29 SENP3 has been found to participate in inflammatory response. Karin et al. have reported that SENP3 potentiates the activation of TLR4 signaling pathway and enhances expression of inflammatory cytokines in macrophages subjected to LPS via mediating the de-SUMOylation of MKK7.13 Similarly, SENP3 also participates in fine-tuning macrophage polarization via deSUMOylation of Akt1.30 Consistent with these reports, we found that SENP3 also play a crucial role in microglia-induced neuroinflammation response after cerebral ischemia. Intriguingly, our data suggest the SENP3 facilitates microglia overactivation and enhances the production of proinflammatory mediators, including TNF-α, IL-6, IL-1β, CCL2 and CXCL1 under OGD/R conditions. On the other hand, there are also considerable evidences showing that microglia phagocytosis playing important roles in neurological recuperation after cerebral ischemia.31 As microglial phagocytosis involves in the removal of massive damage cells and debris induced by cerebral I/R injury.32 This study only determined the impact of SENP3 on inflammatory cytokines production. It is not clear whether SENP3 participates in the regulation of microglia phagocytosis after ischemic stroke, which will be investigated in the future.
Growing evidence has demonstrated that NF-κB and MAPK/AP-1 pathway are the crucial signaling pathways that controls the production of multiple proinflammatory factors, and it is also important for microglial activation.33,34,35 Evidences from this study revealed that SENP3 knockdown dramatically restricted the activation of the MAPK/AP-1 pathway triggered by OGD/R treatment, but had little effect on the activation of the NF-κB pathway. Of interest, Lao et al. also found that SENP3 selectively increases the activation of MAPK/AP-1 signaling, whereas it confers no effect on NF-κB signaling in the LPS-induced inflammatory responses in macrophages.13 Unlike SENP6, which has been reported to negatively control TLR inflammatory signaling pathway by mediating the deSUMOylation of IκB kinase γ (IKKγ), thus inhibiting LPS activated NF-κB signaling in macrophages.36 These findings indicate that SENP family members play different roles via different substrates even in a similar inflammatory context.
Previous studies have shown that JNK activation induces direct phosphorylation of its substrate c-Jun.37,38 Following phosphorylation, c-Jun forms homo or heterodimers with other AP-1 family members to form an active transcription complex.39 In the current study, we found that SENP3 in microglia activates the AP-1 transcriptional activity in cerebral ischemia-induced inflammatory responses through selectively catalyzing deSUMOylation of c-Jun. As we known, PTMs play critical roles in regulating the functions of substrate proteins.40 SUMOylation, one of the reversible PTMs, which function in regulating protein stability, localization and interaction with other molecules.41 It has been demonstrated that c-Jun can be SUMOylated at lysine residues 229 and 257, and SUMOylation of c-Jun can attenuate the transcriptional activity of AP-1.18,21 In the present study, we found the deSUMOylating enzyme SENP3 functions as a negative regulator of c-Jun SUMOylation and enhances its transactivation capacity. Many PTM sites from the same (intra) or different (inter) substrates often cooperate with each other to serve a function, which is described as PTM cross-talk.42 Previous study showed that inhibition of SENP3-mediated deSUMOylation of Akt1 can result in the hyperphosphorylation and activation of Akt1.30 As JNK-mediated phosphorylation of c-Jun is critical for its transcriptional activity.43 Therefore, whether cross-talk exists between c-Jun phosphorylation and SUMOylation remains to be systematically characterized.
Neuronal apoptosis is a leading cause in the occurrence and development of cerebral ischemia. Neuronal apoptosis after ischemic stroke seems to be induced by several mechanisms, including inflammatory reactions, energy deficiency, intracellular calcium overload, glutamate excitotoxicity, electrogenic pump failure, and excessive generation of free radicals.44,45 In addition, correlation between SENP3 and neuronal apoptosis has been reported in several studies. Guo et al. found that SENP3-mediated deSUMOylation of Drp1 could enhance its interaction with Mff, thus promoting Drp1 mitochondrial localization and ultimately resulting in neuronal apoptosis induced by OGD/R.46,47 Furthermore, it is demonstrated that there was a positive regulation among SENP3 and cleaved caspase-3 in the cerebral cortex after subarachnoid hemorrhage.48 In the present research, the expression of cleaved caspase-3 and other pro-apoptotic molecules was dramatically elevated on SENP3 was overexpressed in microglia. Our further work claimed that microglia-specific SENP3 knockdown decreased cleaved caspase-3 expression, thereby markedly decreasing neuronal apoptosis after cerebral ischemia. This study confirmed that SENP3 aggravated neuronal apoptosis triggered by cerebral I/R injury.
We observed that microglia-specific SENP3 knockdown protected against neuronal damage, indicating that SENP3 plays a role in microglia function. However, many studies have demonstrated SENP3 is also expressed in endothelial cells, neurons and astrocytes.49,50 We cannot rule out that the effect of SENP3 on cerebral ischemia may be because of the synergistic effect of multiple cells in ischemic tissue. In this study, we demonstrated that SENP3 induces excessive neuroinflammation and neuronal apoptosis after cerebral ischemia via mediating the deSUMOylation of c-Jun. Consistently, Guo et al. also reported that SENP3-mediated deSUMOylation of Drp1 promotes neuronal cell death following cerebral ischemia.51 Nevertheless, the role of SENP3 in endothelial cells and astrocytes remains unknown after ischemia stroke, and need to be further studied. In addition, Cx3cr1-Cre mice expressed Cre recombinases under the Cx3cr1 promoter in the mononuclear phagocyte system, including monocyte and macrophage chambers and microglia.52 Thus, the AAV particles may also infect macrophages and infiltrating monocytes, and the effects of injecting AAV particles on infiltrating macrophages and infiltrating monocytes need to be further studied. Furthermore, AAV-mediated gene expression requires a certain amount of time in vivo. In this study, mice were pretreated with AAV injection four weeks before MCAO surgery. Therefore, more detailed work is needed to confirm the efficacy with a post treatment. Nevertheless, whether there are alternative approaches that rapidly reduce endogenous SENP3 activity or inhibit its binding with c-Jun, which may be a promising approach for post-stroke therapy, and these remains to be investigated in future studies.
In conclusion, this study recognized an uncovered mechanism by which SENP3 participates in microglia-induced neuroinflammation following ischemic stroke. We have provided compelling evidence demonstrating that SENP3 interacts with and mediates the deSUMOylation of c-Jun, thereby upregulating its transcriptional activity, activating MAPK/AP-1 signaling pathway and aggravating microglia-induced neuroinflammation after cerebral I/R injury, whereas specifically knockdown of SENP3 in microglia markedly decreased pro-inflammatory proinflammatory cytokines and chemokines expression, thereby exhibiting profound neuroprotective and cognitive-preserving effects against cerebral ischemia in experimental animals. Overall, our findings clarified a previously undiscovered role of SENP3 and showed that blocking the expression of SENP3 or its interaction with c-Jun would be a new and promising therapeutic approach for ischemic stroke and probably other neuroinflammatory disorders.
Limitations of the study
Limitations of this study include the absence of in vivo experiments on microglial-specific SENP3-knockout mice. This was mostly because of time constraints and resource scarcity in terms of mice breed featuring microglia-specific SENP3 deleted, and we alternatively adopted the method of using shRNA to silence SENP3 expression specific in microglia in vivo. We plan to study this area in the near future by breeding mice with specific gene types to ensure a rigorous experimental design.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-HA | Santa Cruz Biotechnology | Cat#sc-7392; RRID:AB_627809 |
| Mouse anti-Flag | Santa Cruz Biotechnology | Cat#sc-166355; RRID:AB_2017593 |
| Rabbit anti-Myc | Proteintech | Cat#16286-1-AP; RRID:AB_11182162 |
| Rabbit anti-His | Sigma-Aldrich | Cat#SAB1306085; |
| Rabbit anti-SENP3 | Proteintech | Cat#17659-1-AP; RRID:AB_2301618 |
| Mouse anti-Iba1 | Abcam | Cat#ab283319; RRID:AB_2924797 |
| Mouse anti-NeuN | Millipore | Cat#MAB377; RRID:AB_2298772 |
| Mouse anti-GFAP | Santa Cruz Biotechnology | Cat#sc-33673; RRID:AB_627673 |
| Rabbit anti-iNOS | Proteintech | Cat#18985-1-AP; RRID:AB_2782960 |
| Mouse anti-CD16/32 | R&D systems | Cat#AF1460; RRID:AB_354811 |
| Mouse anti-β-actin | Santa Cruz Biotechnology | Cat#sc-47778; RRID:AB_626632 |
| Rabbit anti-NF-κB p65 | Cell Signaling Technology | Cat#8242; RRID:AB_10859369 |
| Rabbit anti-Phospho-NF-κB p65 | Cell Signaling Technology | Cat#3033; RRID:AB_331284 |
| Rabbit anti-JNK | R&D systems | Cat#AF1387; RRID:AB_2140743 |
| Rabbit anti-Phospho-JNK | R&D systems | Cat#MAB1205; |
| Mouse anti-ERK1/2 | R&D systems | Cat#MAB1576; RRID:AB_2140121 |
| Rabbit anti-Phospho-ERK1/2 | R&D systems | Cat#AF1018; RRID:AB_354539 |
| Rabbit anti-p38 MAPK | Cell Signaling Technology | Cat#8690; RRID:AB_10999090 |
| Rabbit anti-Phospho-p38 MAPK | R&D systems | Cat#AF869; RRID:AB_2141896 |
| Mouse anti-c-Jun | Santa Cruz Biotechnology | Cat#sc-74543; RRID:AB_1121646 |
| Rabbit anti-Phospho-c-Jun | Cell Signaling Technology | Cat#3270; RRID:AB_2129575 |
| Mouse anti-α-tubulin | Santa Cruz Biotechnology | Cat#sc-8035; RRID:AB_628408 |
| Rabbit anti-Histone H3 | Cell Signaling Technology | Cat#4499; RRID:AB_10544537 |
| Mouse anti-SUMO2/3 | Santa Cruz Biotechnology | Cat#sc-393144; RRID:AB_2905545 |
| Rabbit anti-Bcl-xL | Cell Signaling Technology | Cat#2764; RRID:AB_2228008 |
| Rabbit anti-Bax | Cell Signaling Technology | Cat#41162; RRID:AB_2924730 |
| Rabbit anti-cleaved caspase-3 | Cell Signaling Technology | Cat#9664; RRID:AB_2070042 |
| Rabbit anti-cleaved caspase-9 | Cell Signaling Technology | Cat#20750; RRID:AB_2798848 |
| Rabbit anti-cleaved PARP | Cell Signaling Technology | Cat#5625; RRID:AB_10699459 |
| Mouse anti-SUMO1 | Santa Cruz Biotechnology | Cat#sc-5308; RRID:AB_628300 |
| Bacterial and virus strains | ||
| Ad-vector | Vigene Biosciences | N/A |
| Ad-wild-type (WT) SENP3 | Vigene Biosciences | N/A |
| Ad-scramble control (sh.NC) | Vigene Biosciences | N/A |
| Ad-shRNA SENP3 (sh.SENP3) | Vigene Biosciences | N/A |
| AAV2/6-U6-DIO-scramble control-EGFP | Vigene Biosciences | N/A |
| AAV2/6-U6-DIO-SENP3/shRNA-EGFP | Vigene Biosciences | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Isoflurane | Baxter | HDG9623 |
| Ethanol | Sinopharm | 64–17–5 |
| TRIzol | Invitrogen | 15596018 |
| RIPA Lysis buffer | Boster | AR0101 |
| Fetal bovine serum (FBS) | Gibco | 10099 |
| Dulbecco’s Modified Eagle’s Minimum Essential Medium (DMEM) | Gibco | 11965092 |
| Glucose-free DMEM | Gibco | 11966025 |
| Penicillin-streptomycin | Beyotime Biotechnology | C0222 |
| 0.25% trypsin–EDTA | Sigma-Aldrich | SM-2003 |
| Lipofectamine 3000 | Invitrogen | L3000015 |
| TBST | CWBIO | Cat #CW0043S |
| DAPI | Beyotime Biotechnology | C1002 |
| ProLong Gold Antifade reagent | Life Technologies | P36930 |
| N-Ethylmaleimide (NEM) | Thermo Scientific | 23030 |
| Ni2+-NTA beads | Qiagen | Cat. No./ID: 30210 |
| Protein A + G agarose beads | Beyotime Biotechnology | P2055 |
| PVDF- membranes | Millipore | Cat # IPVH00010 |
| Pierce™ ECL Western | Thermo Scientific | 32106 |
| Triphenyltetrazolium chloride (TTC) | Biosharp | BS095 |
| Critical commercial assays | ||
| SYBR Premix ExTaq Quantitative PCR Kit | TaKaRa | RR820A |
| ReverTra Ace-α-TM First Strand cDNA Synthesis Kit | Toyobo | Code No. FSK-100 |
| IL-1β ELISA kits | Dakewe Biotech | #1210122 |
| IL-6 ELISA kits | Dakewe Biotech | #121720 |
| TNF-α ELISA kits | Dakewe Biotech | #1210602 |
| CXCL1 ELISA kits | Abcam | ab100717 |
| CCL2 ELISA kits | Abcam | ab208979 |
| IL-4 ELISA kits | Dakewe Biotech | #1210402 |
| IL-10 ELISA kits | Dakewe Biotech | #1211002 |
| TGF-β ELISA kits | Dakewe Biotech | #1217102 |
| IL-13 ELISA kits | Abcam | ab219634 |
| IL-1ra ELISA kits | Abcam | ab113348 |
| Trelief SoSoo Cloning Kit | TSINGKE | TSV-S1 |
| In situ cell death detection kit | Roche | 12156792910 |
| Experimental models: Cell lines | ||
| Human Embryonic Kidney (HEK) 293T cells | ATCC | CBP60440 |
| Experimental models: Organisms/strains | ||
| C57BL/6 mice | Vital River Laboratories | https://www.vitalriver.com |
| Cx3cr1-Cre mice | Jackson Laboratory | JAX: stock 000664 |
| Oligonucleotides | ||
| Senp3F ACTCCCAGCGAACTCTAA | This paper | primer for RT-qPCR |
| Senp3R TAATACAAAGGCACCACA | This paper | primer for RT-qPCR |
| Il-1βF GAAAGACGGCACACCCAC | This paper | primer for RT-qPCR |
| Il-1βR TGTGACCCTGAGCGACCT | This paper | primer for RT-qPCR |
| Il-6 F TCTCTGGGAAATCGTGGAA | This paper | primer for RT-qPCR |
| Il-6 R GATGGTCTTGGTCCTTAGCC | This paper | primer for RT-qPCR |
| Tnf-α F ACGGCATGGATCTCAAAGAC | This paper | primer for RT-qPCR |
| Tnf-α R AGATAGCAAATCGGCTGACG | This paper | primer for RT-qPCR |
| Cxcl1 F GAGCTTGAAGGTGTTGCCCT | This paper | primer for RT-qPCR |
| Cxcl1 R CGCGACCATTCTTGAGTGTG | This paper | primer for RT-qPCR |
| Ccl2 F GCAGGTCCCTGTCATGCTTC | This paper | primer for RT-qPCR |
| Ccl2 R GTGGGGCGTTAACTGCATCT | This paper | primer for RT-qPCR |
| Arginase-1 F CAAGACAGGGCTCCTTTCAG | This paper | primer for RT-qPCR |
| Arginase-1 R TGGCTTATGGTTACCCTCCC | This paper | primer for RT-qPCR |
| Il-4 F CCCCCAGCTAGTTGTCATCC | This paper | primer for RT-qPCR |
| Il-4 R AGGACGTTTGGCACATCCAT | This paper | primer for RT-qPCR |
| Il-10 F CTGCCTGCTCTTACTGACTG | This paper | primer for RT-qPCR |
| Il-10 R AAATCACTCTTCACCTGCTC | This paper | primer for RT-qPCR |
| Tgf-β F TGCGCTTGCAGAGATTAAAA | This paper | primer for RT-qPCR |
| Tgf-β R CGTCAAAAGACAGCCACTCA | This paper | primer for RT-qPCR |
| Cd206 F TCAGCTATTGGACGCGAGGCA | This paper | primer for RT-qPCR |
| Cd206 R TCCGGGTTGCAAGTTGCCGT | This paper | primer for RT-qPCR |
| β-actin F TTCGTTGCCGGTCCACACCC | This paper | primer for RT-qPCR |
| β-actin R GCTTTGCACATGCCGGAGCC | This paper | primer for RT-qPCR |
| Cx3cr1 Cre F CAACGAGTGATGAGGTTCGCAAG | This paper | Genotyping primers |
| Cx3cr1 Cre R ACACCAGAGACGGAAATCCATCG | This paper | Genotyping primers |
| Recombinant DNA | ||
| HA-c-Jun | This paper | N/A |
| Flag-Ubc9 | This paper | N/A |
| His-SUMO1 | This paper | N/A |
| His-SUMO2 | This paper | N/A |
| His-SUMO3 | This paper | N/A |
| Myc-SENP3-WT | This paper | N/A |
| Myc-SENP3-CA | This paper | N/A |
| Software and algorithms | ||
| GraphPad Prism 8.0 | GraphPad Software | http://www.graphpad-prism.cn |
| Image-Pro Plus software 7 | Media Cybernetics | https://mediacy.com/image-pro/ |
| Adobe Illustrator CS6 | Adobe Systems | https://www.adobe.com/products/illustrator.html |
| Adobe Photoshop CS6 | Adobe Systems | www.adobe.com/cn/products/photoshop.html |
| ImageJ | National Institutes of Health | https://imagej.net/software/imagej/ |
| Other | ||
| Stepper motor-driven micro-injector | Hamilton | N/A |
| Laser Doppler flowmetry | RWD Life Science | RFLSI III |
| Fluorescence microscope | Olympus | BX53 |
| Enspire™ multilabel reader 2300 | PerkinElmer | EnSpire |
| ABI 7900 fast real-time PCR system | Applied Biosystems | Cat No.4364014 |
| Homeothermic blanket | Harvard Apparatus | N/A |
| Mouse brain matrix | RWD Life Science | Cat No.68707 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Xing Li (lixing88@hust.edu.cn).
Materials availability
This study did not generate new unique reagents.
Experimental model and study participant details
Ethics statement
All experiments were approved by the Experimental Animal Care and Use Committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, and were in agreement with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
Animals
C57BL/6J male mice obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd. were used for experiments. The mice were maintained under pathogen-free conditions in a barrier-sustained facility and provided with a normal diet and free water intake. Cx3cr1-Cre mice (C57BL/6J background) were generated from Jackson Laboratory (JAX stock 000664). A total of 46 wild-type C57BL/6J male mice and 104 Cx3cr1-Cre mice male mice were used in this study. 13 mice were excluded because of death (5 mice), cerebral hemorrhage (3 mice), disturbance of consciousness (3 mice), or failure of ischemia induction (2 mice) (as shown in Figure S9). Animals were monitored after surgery according to the IMPROVE guidelines and the experiments have been reported following the ARRIVE guidelines.53 All mice were randomized for the experiment. The operator was blinded to the experimental procedures and data analysis.
Transient focal cerebral ischemia
We induced the ischemic stroke model with left middle cerebral artery occlusion (MCAO) as previously described.54 First, the animals were anesthetized with 2.5% isoflurane, and the entire procedure was conducted at 37.0 ± 0.5 °C by maintaining the mice on a homeothermic blanket (Harvard Apparatus, Holliston, MA, USA). The left common carotid artery (CCA), external carotid artery (ECA) and internal carotid artery (ICA) was exposed through an incision in front of the midline of the neck. Then, CCA and ECA were ligated. Subsequently, blocked ICA with artery clip, and sheared at the place where ECA 5 mm from bifurcation with a small 0.2 mm wide opening. Then, inserted the suture to the ICA through ECA until there was resistance. After 1 h, the suture was withdrawn lightly. The cerebral blood flow was monitored using a Laser Speckle Imaging System (RFLSI III, RWD Life Science, Shenzhen, China). For sham surgery, surgical procedure was conducted without suture inserting treatment.
Cell culture and oxygen-glucose deprivation/reperfusion (OGD/R) procedure
Primary neurons were dissected from embryonic (E16–E18) mice with the procedure as we have previously reported.55 Briefly, after dissecting and cutting of the cerebral cortex under anatomic microscope, we digested the neurons with 0.25% trypsin–EDTA (Sigma-Aldrich, St. Louis, MO, USA) solution for dissociation. Next, centrifugated and gathered the cell suspensions. After counting the cells, seeded the cells in 6-well culture plates with normal Dulbecco’s Modified Eagle’s Minimum Essential Medium (DMEM, Thermo Fisher Scientific, Waltham, MA, USA) and 10% fetal bovine serum (FBS, Gibco, Gaithersburg, MD, USA).
Primary microglia were cultured as we have previously described.56 Briefly, mixed glia cells were prepared from the whole brain of neonatal mice at postnatal P1–P2 and were cultured in high-glucose DMEM (Gibco) medium supplemented with 20% FBS (Gibco) at 37 °C in a 95% O2 and 5% CO2 incubator for 8–10 days, and the medium was changed every 3 days. Microglia were isolated from the mixed glial cultures by mild agitation at 200 rpm for 6 h in a rotary shaker at 37 °C based on the distinct adhesive features of microglia and astrocytes. The obtained microglial cells were seeded into 6-well plates in high-glucose DMEM medium supplemented with 20% FBS at a density of 1 × 106/well for 24 h before ready for further treatment. The purity of the adherent cells was verified by immunocytochemical staining, which indicated more than 95% of the cells in the cultures were positive for the microglia-specific marker Iba1. A highly enriched population of microglia/macrophages was isolated from adult mice by Percoll density centrifugation using a protocol described previously.57
HEK293T cells were cultivated in high-glucose DMEM supplemented with 10% FBS and 1% penicillin-streptomycin (Beyotime Biotechnology, Shanghai, China) at 37 °C in a humidified 5% CO2-containing atmosphere. Transfections were conducted using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) when the cells were 80–90% confluent. OGD/R was conducted as previously.58 In brief, replaced the culture supernatant with glucose-free DMEM (Gibco), then incubated in 1% O2, 5% CO2, and 94% N2 at 37 °C. After 1 h of cultivation, replaced the medium to high-glucose DMEM, then returned to the incubator in 95% O2 and 5% CO2 at 37 °C, and began reperfusion.
Co-culture of neuron and microglia
In brief, microglia and neurons were isolated by transwell (Corning, Tewksbury, MA, USA) co-culture system. The cells were cultured in two chambers with a semi-permeable membrane of 0.4 μm between the two chambers, in which cytokines could be diffused. Primary cultured neurons were cultured in the lower cavity, while microglia were in the upper cavity 0.8 mm away from the lower cavity. After adenovirus transfected into microglia cells for 48 h infection, the supernatants were replaced with free fresh medium and the transwell insert was moved to the neurons. Afterward, cells were treated with OGD/R.
Viral vectors transduction in mice
Adenoviruses encompassing vector, wild-type (WT) SENP3, scramble control and short hairpin RNA (shRNA) against SENP3 were employed to infect primary cultured microglia. The sequences of shRNA target SENP3 are designed and verified as follows: mouse SENP3 5′- GCT TCC GGG TAT CCT ATA AGC-3′, human SENP3: 5′-GGA TGC TGC TCT ACT CAA A-3′, and a scramble shRNA used as a negative control. Primary microglia were infected using diluted recombinant adenovirus at an optimal multiplicity of infection (MOI) about 50:1 to 100:1 referring our pre-tests. After 48h infection, cells were subjected to further experiments. For in vivo viral infection studies, Cx3cr1-Cre male mice aged 8 weeks were infected with AAV2/6 viruses (2-3 × 1012 vg/ml) encoding CMV-DIO-Vector, CMV-DIO-His-SENP3, U6-DIO-shcontrol-EGFP, U6-DIO-SENP3/shRNA-EGFP by stereotactic brain injection, which induced SENP3 upregulation or downregulation in microglia specifically. Briefly, the animals were anesthetized with 2.5% isoflurane and fixed in a stereotaxic apparatus (RWD Life Science, Shenzhen, China). 500 nL of virus (50 nL/min) solution were injected into the hippocampal CA1 area, cerebral cortex, and striatum of the left hemisphere with a stepper motor-driven micro-injector (Hamilton, Reno, NV, USA).
Method details
TdT-mediated dUTP-X nick end labeling (TUNEL) staining
Neuronal apoptosis was determined via the in situ cell death detection kit (Roche, Rotkreuz, Switzerland) following manufacturer’s instructions. In brief, slides were washed in PBS for three times, and exposed to immersion fixation for 20 min in 4% paraformaldehyde (PFA). After washing 3 × 5 min in PBS, slides were incubated with TUNEL reagent at 37°C for 1 h in a humidified dark chamber. Then slides were then incubated with DAPI (Beyotime Biotechnology) for 8 min. Last, mount the cells with ProLong Gold Antifade reagent (Life Technologies, Carlsbad, CA, USA). Fluorescent images were visualized by a fluorescence microscope (BX53, Olympus, Tokyo, Japan) with equal exposure times.
Western blotting
Briefly, brain tissues and cultured cells were lysed with RIPA buffer at 4°C for 15 min, and then, the extract was mixed with sample buffer and boiled for 10 min, followed by centrifuged at 14,000 × g for another 15 min. The supernatant was collected and used for immunoblot. Equal amounts of protein extracts were separated by 10% or 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis, and then transferred to polyvinylidene fluoride membranes. After blocked with 5% BSA, the membranes were incubated with primary antibodies at 4°C overnight. And then, a secondary antibody (1:20,000; Jackson ImmunoResearch, West Grove, USA) was added to the membranes and incubated for 1 h. Finally, immunodetection was performed with a chemiluminescence substrate kit (Thermo Pierce, Rockford, USA).
Immunofluorescence
Fixed brain slices were permeabilized with PBS containing 0.5% Triton X-100 for 30 min at room temperature, followed by incubated in blocking buffer for another 30 min at 37 °C. After incubated with primary antibodies at 4 °C for 24 h, the slices were added with the secondary antibodies and incubated for 1 h at 37 °C. Finally, DAPI was added to the slices for 10 min at room temperature in the dark. Images were acquired using a fluorescence microscope (BX53, Olympus).
SUMOylation assay
SUMOylation of c-Jun was determined by Ni2+-NTA pull down as our previous publication described.27 In Brief, HEK293T cells were transduced with HA-c-Jun, Flag-Ubc9 and His-SUMO for 48 h. After washing 2 × 5 min in cold PBS, cells were lysed in 800 μL of Ni2+-NTA denaturing buffer, concluding 20 mM N-Ethylmaleimide (NEM). Then, cells were sonicated until the lysate became fluid. The lysates were collected by centrifugation at 15 000g for 10 min and supplemented with 50 μL of Ni2+-NTA beads (Qiagen, Dusseldorf, Germany). Then, the beads were washed continuously in 1 mL Ni2+-NTA washing buffer. Finally, the beads were eluted in 40 μL of 2× loading buffer containing 200 mM imidazole and boiled at 95°C for 10 min, and followed by western blotting analysis.
Co-immunoprecipitation (Co-IP)
For immunoprecipitation analysis, the clarified lysate was immunoprecipitated overnight with the indicated primary antibodies, and then mixed with 40 μL Protein A + G agarose beads (Beyotime Biotechnology) to catch the immune complex at 4°C for 4 h. Rabbit or mouse IgG antibody were utilized as the negative control. After washing 3 × 5 min in cold lysis buffer, the immune complexes on beads were resuspended with 2× loading buffer and boiled at 95°C for 10 min, and then analyzed by immunoblots assay.
Plasmids construction
The cDNA of SENP3 was obtained by PCR and cloned into indicated vectors including pMyc-CMV2, the c-Jun or Ubc9 coding sequence was cloned into pHA-CMV2 or pFlag-CMV2 using recombinase connection method. SUMO1/2/3 coding sequence was cloned into pHis-CMV2 using recombinase connection method. For site-directed mutagenesis, SENP3 site-directed mutant (SENP3-C532A) were produced by recombination with Trelief SoSoo Cloning Kit (TSINGKE, Beijing, China) per the manufacturer′s protocol.
Real-time quantitative PCR (RT-qPCR)
The PCR experiments were performed with an SYBR Premix ExTaq Quantitative PCR Kit (TaKaRa, Tokyo, Japan) on an ABI 7900 fast real-time PCR system (Applied Biosystems, Carlsbad, CA, USA). Total RNA was isolated using the TRIzol reagent (Invitrogen) following the manufacturer’s instructions. To determine the mRNA expression of Senp3, Il-1β, Il-6 and Tnf-α, Cxcl1 and Ccl2, Il-4, Il-10, Tgf-β, Arginase-1, Cd206, 0.5 to 2 μg of total RNA was converted to cDNA by a ReverTra Ace-α-TM First Strand cDNA Synthesis Kit (Toyobo, Osaka, Japan). The 2−ΔΔCt method was performed to analysis of mRNA level. The specific primers employed in this study are present in key resources table.
Enzyme-linked immunosorbent assay (ELISA)
To evaluate the cytokines and chemokines secretion in the culture medium of treated and untreated microglia, the culture medium was centrifuged at 15 000 g for 15 min at 4 °C. Then the supernatants were gathered, and the cytokines and chemokines concentration were determined with ELISA kits according to the manufacturers’ protocol. The absorbance was measured at 450 nm via an EnspireTM multilabel reader 2300 (PerkinElmer, Waltham, MA, USA).
TTC (2,3,5-triphenyl-tetrazolium chloride) staining
To detected infarct areas after MCAO surgery, mouse brain was frozen at −80 °C for 8 min, and then six 2-mm thick slices were sliced using a series of histological knives assembled as a unit and a mouse brain matrix (RWD Life Science). The slices were incubated in 1% solution of TTC (Biosharp, Hefei, China) at 37 °C in dark for 20 min. Subsequently, the samples were fixed in 4% PFA. The brain infarction volume and a total volume of brain slice were analyzed for each slide through Image-Pro Plus (version 7.0; Media Cybernetics, Rockville, MD, USA). The infarct volume was measured and indicated as: Infarct volume (%) = (contralateral volume − ipsilateral non-infarct volume)/contralateral volume × 100%.
Neurological deficit evaluation
Neurological deficiency of the animals was determined by the independent blind researchers. After 24 h of MCAO and reperfusion, modified neurological severity score (mNSS) was selected to assess the neurological dysfunction which includes beam balance tests (score 0 to 6), reflexes absent & abnormal movements (score 0 to 2) and motor tests (scored 0 to 6). Accumulative scores of f 1 to 4, 5 to 9, or 10 to 14 respectively indicated slight, moderate, or serious brain injury.
Morris water maze (MWM) test
The spatial learning and memory of mice were examined by MWM as reported previously.59 Briefly, the MWM test was performed in a circular tank with a circular platform (6 cm in diameter). The pool was filled with opaque water at 22 ± 2°C, and the platform was submerged 1 cm beneath the surface. During the incubation period, six consecutive days of subsea platform training were conducted, with each phase consisting of four tests. Animals that cannot find the platform within 60 s were guided to the platform and maintained on the platform for 15 s. During the probe trial, the animals were allowed to search for the platform for 60 s and record the animal’s performance by a digital tracking device.
Rotarod test
The rotarod test was conducted as described previously.60 Briefly, animals were acclimatized to the rotarod apparatus for one day at 0 rpm and one day at a constant 2 rpm speed. Before the test, animals were placed in the center of a rotating pole from 5 to 10 rpm per minute, and then trained for 30 min. The speed of the rotating rod then accelerated from 5 to 40 rpm in 5 min. Latency to fall was recorded automatically.
Adhesive test
The adhesive test was conducted to evaluate animals’ sensorimotor function.61 In brief, adhesive-backed paper dots (a round 6.35 mm sticker) were gently placed on the paralyzed front paw and used as bilateral tactile stimuli on the dorsal paw. Next, the mice were returned to the cage. The latency to touch the sticker was recorded as the touch time, and the latency to successful removal of the sticker by the mouse was recorded as the removal time. Animals received 3 trials per day for each forepaw with an inter-trial interval of 5 min.
Cylinder test
The cylinder test was conducted to assess forepaw use and rotation asymmetry.62 In brief, the animals were placed in a transparent Plexiglas cylinder (15 cm in high, 9 cm in diameter) surrounded by mirrored panels to allow for evaluation of all movements and videotaped for 10 min. The total number of contacts by forepaws (left, L; right, R; both, B) was totaled. For analysis, the asymmetric rate was recorded as (L − R)/(L + R + B) × 100 (%).
Quantification and statistical analysis
All data are presented as the means ± SEM from at least three independent experiments. The statistical analyses were performed by GraphPad Prism 8.0 (GraphPad Software, La Jolla, CA, USA). Student’s t test was performed to examine the significance of differences between two groups of data. Multiple group (>2 groups) comparisons were conducted by one- or two-way ANOVA followed by Dunnett’s or Tukey’s post-hoc test. The nonparametric Kruskal–Wallis rank-sum test was conducted to analyze nonnormal distributions. p values were considered significant for less than 0.05.
Acknowledgments
This study was supported by funding from the National Natural Science Foundation of China (No.32271037, No.82201472, No.31800896), China Postdoctoral Science Foundation (No. 2018T110752 and No. 2017M620310) and Tongji Hospital (HUST) Foundation for Excellent Young Scientist (No. 2020YQ17).
Author contributions
X.L. and Q.X. conceived the study, obtained financial support, and performed the laboratory experiments. M.M. and G.F.Z. conducted the data processing and statistical analysis. Z.Z.L. and Z.Y. provided helpful discussions. The manuscript was written by Q.X. with approval from all of the authors.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: May 25, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106953.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon reasonable request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Campbell B.C.V., De Silva D.A., Macleod M.R., Coutts S.B., Schwamm L.H., Davis S.M., Donnan G.A. Ischaemic stroke. Nat. Rev. Dis. Prim. 2019;5:70. doi: 10.1038/s41572-019-0118-8. [DOI] [PubMed] [Google Scholar]
- 2.Wang Y., Tian M., Tan J., Pei X., Lu C., Xin Y., Deng S., Zhao F., Gao Y., Gong Y. Irisin ameliorates neuroinflammation and neuronal apoptosis through integrin αVβ5/AMPK signaling pathway after intracerebral hemorrhage in mice. J. Neuroinflammation. 2022;19:82. doi: 10.1186/s12974-022-02438-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lambertsen K.L., Finsen B., Clausen B.H. Post-stroke inflammation-target or tool for therapy? Acta Neuropathol. 2019;137:693–714. doi: 10.1007/s00401-018-1930-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lambertsen K.L., Biber K., Finsen B. Inflammatory cytokines in experimental and human stroke. J. Cerebr. Blood Flow Metabol. 2012;32:1677–1698. doi: 10.1038/jcbfm.2012.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Q., Wang J.C.Y., Xie S.Z. SUMO wrestling cancer stem cells. Cell Chem. Biol. 2021;28:1390–1392. doi: 10.1016/j.chembiol.2021.10.001. [DOI] [PubMed] [Google Scholar]
- 6.Chang H.M., Yeh E.T.H. SUMO: from bench to bedside. Physiol. Rev. 2020;100:1599–1619. doi: 10.1152/physrev.00025.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cai Z., Wang Z., Yuan R., Cui M., Lao Y., Wang Y., Nie P., Shen L., Yi J., He B. Redox-sensitive enzyme SENP3 mediates vascular remodeling via de-SUMOylation of β-catenin and regulation of its stability. EBioMedicine. 2021;67:103386. doi: 10.1016/j.ebiom.2021.103386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rawlings N., Lee L., Nakamura Y., Wilkinson K.A., Henley J.M. Protective role of the deSUMOylating enzyme SENP3 in myocardial ischemia-reperfusion injury. PLoS One. 2019;14:e0213331. doi: 10.1371/journal.pone.0213331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson C.A., Blackstone C. SUMO wrestling with Drp1 at mitochondria. EMBO J. 2013;32:1496–1498. doi: 10.1038/emboj.2013.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun G., Qin W., Wang Q., Sun X., Chen H., Li J., Sun L., Shi F., Zhang G., Wang M. Selective-cerebral-hypothermia-induced neuroprotection against-focal cerebral ischemia/reperfusion injury is associated with an increase in SUMO2/3 conjugation. Brain Res. 2021;1756:147311. doi: 10.1016/j.brainres.2021.147311. [DOI] [PubMed] [Google Scholar]
- 11.Wei H., Teng H., Huan W., Zhang S., Fu H., Chen F., Wang J., Wu C., Zhao J. An upregulation of SENP3 after spinal cord injury: implications for neuronal apoptosis. Neurochem. Res. 2012;37:2758–2766. doi: 10.1007/s11064-012-0869-z. [DOI] [PubMed] [Google Scholar]
- 12.Chen X., Lao Y., Yi J., Yang J., He S., Chen Y. SENP3 in monocytes/macrophages up-regulates tissue factor and mediates lipopolysaccharide-induced acute lung injury by enhancing JNK phosphorylation. J. Cell Mol. Med. 2020;24:5454–5462. doi: 10.1111/jcmm.15199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lao Y., Yang K., Wang Z., Sun X., Zou Q., Yu X., Cheng J., Tong X., Yeh E.T.H., Yang J., Yi J. DeSUMOylation of MKK7 kinase by the SUMO2/3 protease SENP3 potentiates lipopolysaccharide-induced inflammatory signaling in macrophages. J. Biol. Chem. 2018;293:3965–3980. doi: 10.1074/jbc.M117.816769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ronkina N., Gaestel M. MAPK-activated protein kinases: servant or partner? Annu. Rev. Biochem. 2022;91:505–540. doi: 10.1146/annurev-biochem-081720-114505. [DOI] [PubMed] [Google Scholar]
- 15.Jiang M., Zhang Y., Li P., Jian J., Zhao C., Wen G. Mitogen-activated protein kinase and substrate identification in plant growth and development. Int. J. Mol. Sci. 2022;23:2744. doi: 10.3390/ijms23052744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao D., Kwon S.H., Chun Y.S., Gu M.Y., Yang H.O. Anti-neuroinflammatory effects of fucoxanthin via inhibition of Akt/NF-κB and MAPKs/AP-1 pathways and activation of PKA/CREB pathway in lipopolysaccharide-activated BV-2 microglial cells. Neurochem. Res. 2017;42:667–677. doi: 10.1007/s11064-016-2123-6. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y.P., Wu Y., Li L.Y., Zheng J., Liu R.G., Zhou J.P., Yuan S.Y., Shang Y., Yao S.L. Aspirin-triggered lipoxin A4 attenuates LPS-induced pro-inflammatory responses by inhibiting activation of NF-kappaB and MAPKs in BV-2 microglial cells. J. Neuroinflammation. 2011;8:95. doi: 10.1186/1742-2094-8-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bossis G., Malnou C.E., Farras R., Andermarcher E., Hipskind R., Rodriguez M., Schmidt D., Muller S., Jariel-Encontre I., Piechaczyk M. Down-regulation of c-Fos/c-Jun AP-1 dimer activity by sumoylation. Mol. Cell Biol. 2005;25:6964–6979. doi: 10.1128/MCB.25.16.6964-6979.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng J., Perkins N.D., Yeh E.T.H. Differential regulation of c-Jun-dependent transcription by SUMO-specific proteases. J. Biol. Chem. 2005;280:14492–14498. doi: 10.1074/jbc.M412185200. [DOI] [PubMed] [Google Scholar]
- 20.Schmidt D., Müller S. Members of the PIAS family act as SUMO ligases for c-Jun and p53 and repress p53 activity. Proc. Natl. Acad. Sci. USA. 2002;99:2872–2877. doi: 10.1073/pnas.052559499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tempé D., Vives E., Brockly F., Brooks H., De Rossi S., Piechaczyk M., Bossis G. SUMOylation of the inducible (c-Fos:c-Jun)/AP-1 transcription complex occurs on target promoters to limit transcriptional activation. Oncogene. 2014;33:921–927. doi: 10.1038/onc.2013.4. [DOI] [PubMed] [Google Scholar]
- 22.Subedi L., Gaire B.P. Phytochemicals as regulators of microglia/macrophages activation in cerebral ischemia. Pharmacol. Res. 2021;165:105419. doi: 10.1016/j.phrs.2021.105419. [DOI] [PubMed] [Google Scholar]
- 23.Muller S., Berger M., Lehembre F., Seeler J.S., Haupt Y., Dejean A. c-Jun and p53 activity is modulated by SUMO-1 modification. J. Biol. Chem. 2000;275:13321–13329. doi: 10.1074/jbc.275.18.13321. [DOI] [PubMed] [Google Scholar]
- 24.Liu K., Guo C., Lao Y., Yang J., Chen F., Zhao Y., Yang Y., Yang J., Yi J. A fine-tuning mechanism underlying self-control for autophagy: deSUMOylation of BECN1 by SENP3. Autophagy. 2020;16:975–990. doi: 10.1080/15548627.2019.1647944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nie X., Kitaoka S., Tanaka K., Segi-Nishida E., Imoto Y., Ogawa A., Nakano F., Tomohiro A., Nakayama K., Taniguchi M., et al. The innate immune receptors TLR2/4 mediate repeated social defeat stress-induced social avoidance through prefrontal microglial activation. Neuron. 2018;99:464–479.e7. doi: 10.1016/j.neuron.2018.06.035. [DOI] [PubMed] [Google Scholar]
- 26.Lee C.H., Kim H.J., Lee Y.S., Kang G.M., Lim H.S., Lee S.H., Song D.K., Kwon O., Hwang I., Son M., et al. Hypothalamic macrophage inducible nitric oxide synthase mediates obesity-associated hypothalamic inflammation. Cell Rep. 2018;25:934–946.e5. doi: 10.1016/j.celrep.2018.09.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li X., Xia Q., Mao M., Zhou H., Zheng L., Wang Y., Zeng Z., Yan L., Zhao Y., Shi J. Annexin-A1 SUMOylation regulates microglial polarization after cerebral ischemia by modulating IKKalpha stability via selective autophagy. Sci. Adv. 2021;7:eabc5539. doi: 10.1126/sciadv.abc5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao S.C., Ma L.S., Chu Z.H., Xu H., Wu W.Q., Liu F. Regulation of microglial activation in stroke. Acta Pharmacol. Sin. 2017;38:445–458. doi: 10.1038/aps.2016.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barry R., John S.W., Liccardi G., Tenev T., Jaco I., Chen C.H., Choi J., Kasperkiewicz P., Fernandes-Alnemri T., Alnemri E., et al. SUMO-mediated regulation of NLRP3 modulates inflammasome activity. Nat. Commun. 2018;9:3001. doi: 10.1038/s41467-018-05321-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiao M., Bian Q., Lao Y., Yi J., Sun X., Sun X., Yang J. SENP3 loss promotes M2 macrophage polarization and breast cancer progression. Mol. Oncol. 2022;16:1026–1044. doi: 10.1002/1878-0261.12967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berglund R., Guerreiro-Cacais A.O., Adzemovic M.Z., Zeitelhofer M., Lund H., Ewing E., Ruhrmann S., Nutma E., Parsa R., Thessen-Hedreul M., et al. Microglial autophagy-associated phagocytosis is essential for recovery from neuroinflammation. Sci. Immunol. 2020;5:eabb5077. doi: 10.1126/sciimmunol.abb5077. [DOI] [PubMed] [Google Scholar]
- 32.Jia J., Yang L., Chen Y., Zheng L., Chen Y., Xu Y., Zhang M. The role of microglial phagocytosis in ischemic stroke. Front. Immunol. 2021;12:790201. doi: 10.3389/fimmu.2021.790201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim B.W., More S.V., Yun Y.S., Ko H.M., Kwak J.H., Lee H., Suk K., Kim I.S., Choi D.K. A novel synthetic compound MCAP suppresses LPS-induced murine microglial activation in vitro via inhibiting NF-kB and p38 MAPK pathways. Acta Pharmacol. Sin. 2016;37:334–343. doi: 10.1038/aps.2015.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang L., Yin C., Liu T., Abdul M., Zhou Y., Cao J.L., Lu C. Pellino1 regulates neuropathic pain as well as microglial activation through the regulation of MAPK/NF-κB signaling in the spinal cord. J. Neuroinflammation. 2020;17:83. doi: 10.1186/s12974-020-01754-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou Y., Wu Z., Cao X., Ding L., Wen Z., Bian J.S. HNO suppresses LPS-induced inflammation in BV-2 microglial cells via inhibition of NF-κB and p38 MAPK pathways. Pharmacol. Res. 2016;111:885–895. doi: 10.1016/j.phrs.2016.08.007. [DOI] [PubMed] [Google Scholar]
- 36.Liu X., Chen W., Wang Q., Li L., Wang C. Negative regulation of TLR inflammatory signaling by the SUMO-deconjugating enzyme SENP6. PLoS Pathog. 2013;9:e1003480. doi: 10.1371/journal.ppat.1003480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kciuk M., Gielecińska A., Budzinska A., Mojzych M., Kontek R. Metastasis and MAPK pathways. Int. J. Mol. Sci. 2022;23:3847. doi: 10.3390/ijms23073847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao Y., Kuca K., Wu W., Wang X., Nepovimova E., Musilek K., Wu Q. Hypothesis: JNK signaling is a therapeutic target of neurodegenerative diseases. Alzheimers Dement. 2022;18:152–158. doi: 10.1002/alz.12370. [DOI] [PubMed] [Google Scholar]
- 39.Fan F., Podar K. The role of AP-1 transcription factors in plasma cell biology and multiple myeloma pathophysiology. Cancers. 2021;13:2326. doi: 10.3390/cancers13102326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang S., Osgood A.O., Chatterjee A. Uncovering post-translational modification-associated protein-protein interactions. Curr. Opin. Struct. Biol. 2022;74:102352. doi: 10.1016/j.sbi.2022.102352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang Y., He Y., Wang X., Liang Z., He G., Zhang P., Zhu H., Xu N., Liang S. Protein SUMOylation modification and its associations with disease. Open Biol. 2017;7:170167. doi: 10.1098/rsob.170167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vu L.D., Gevaert K., De Smet I. Protein Language: post-translational modifications talking to each other. Trends Plant Sci. 2018;23:1068–1080. doi: 10.1016/j.tplants.2018.09.004. [DOI] [PubMed] [Google Scholar]
- 43.Kallunki T., Deng T., Hibi M., Karin M. c-Jun can recruit JNK to phosphorylate dimerization partners via specific docking interactions. Cell. 1996;87:929–939. doi: 10.1016/s0092-8674(00)81999-6. [DOI] [PubMed] [Google Scholar]
- 44.Yang J.L., Mukda S., Chen S.D. Diverse roles of mitochondria in ischemic stroke. Redox Biol. 2018;16:263–275. doi: 10.1016/j.redox.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mao R., Zong N., Hu Y., Chen Y., Xu Y. Neuronal death mechanisms and therapeutic strategy in ischemic stroke. Neurosci. Bull. 2022;38:1229–1247. doi: 10.1007/s12264-022-00859-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guo C., Hildick K.L., Jiang J., Zhao A., Guo W., Henley J.M., Wilkinson K.A. SENP3 promotes an mff-primed bcl-x L -Drp1 interaction involved in cell death following ischemia. Front. Cell Dev. Biol. 2021;9:752260. doi: 10.3389/fcell.2021.752260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo C., Wilkinson K.A., Evans A.J., Rubin P.P., Henley J.M. SENP3-mediated deSUMOylation of Drp1 facilitates interaction with Mff to promote cell death. Sci. Rep. 2017;7:43811. doi: 10.1038/srep43811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang Y.Q., Li H., Zhang X., Wang C.X., Sun Q., Li S., Li W., Li W., Ding K., Liu M., et al. Expression and cell distribution of SENP3 in the cerebral cortex after experimental subarachnoid hemorrhage in rats: a pilot study. Cell. Mol. Neurobiol. 2015;35:407–416. doi: 10.1007/s10571-014-0136-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang D.P., Liu K.J., Kasper G., Lin Q., Hai J. Inhibition of SENP3 by URB597 ameliorates neurovascular unit dysfunction in rats with chronic cerebral hypoperfusion. Biomed. Pharmacother. 2017;91:872–879. doi: 10.1016/j.biopha.2017.05.021. [DOI] [PubMed] [Google Scholar]
- 50.Yu Z., Li H., Yan H.Y., Yang Y.Q., Zhang D.D., Huang L.T., Xie G.B., Liu M., Tohti M., Hang C.H. Expression and cell distribution of SENP3 in brain tissue after traumatic brain injury in mice: a pilot study. Cell. Mol. Neurobiol. 2015;35:733–740. doi: 10.1007/s10571-015-0169-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo C., Hildick K.L., Luo J., Dearden L., Wilkinson K.A., Henley J.M. SENP3-mediated deSUMOylation of dynamin-related protein 1 promotes cell death following ischaemia. EMBO J. 2013;32:1514–1528. doi: 10.1038/emboj.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yona S., Kim K.W., Wolf Y., Mildner A., Varol D., Breker M., Strauss-Ayali D., Viukov S., Guilliams M., Misharin A., et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38:79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Percie du Sert N., Alfieri A., Allan S.M., Carswell H.V., Deuchar G.A., Farr T.D., Flecknell P., Gallagher L., Gibson C.L., Haley M.J., et al. The IMPROVE guidelines (ischaemia models: procedural refinements of in vivo experiments) J. Cerebr. Blood Flow Metabol. 2017;37:3488–3517. doi: 10.1177/0271678X17709185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xia Q., Zhan G., Mao M., Zhao Y., Li X. TRIM45 causes neuronal damage by aggravating microglia-mediated neuroinflammation upon cerebral ischemia and reperfusion injury. Exp. Mol. Med. 2022;54:180–193. doi: 10.1038/s12276-022-00734-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li X., Zhao Y., Xia Q., Zheng L., Liu L., Zhao B., Shi J. Nuclear translocation of annexin 1 following oxygen-glucose deprivation-reperfusion induces apoptosis by regulating Bid expression via p53 binding. Cell Death Dis. 2016;7:e2356. doi: 10.1038/cddis.2016.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mao M., Xia Q., Zhan G.F., Chu Q.J., Li X., Lian H.K. SENP6 induces microglial polarization and neuroinflammation through de-SUMOylation of Annexin-A1 after cerebral ischaemia-reperfusion injury. Cell Biosci. 2022;12:113. doi: 10.1186/s13578-022-00850-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee J.K., Tansey M.G. Microglia isolation from adult mouse brain. Methods Mol. Biol. 2013;1041:17–23. doi: 10.1007/978-1-62703-520-0_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li X., Zheng L., Xia Q., Liu L., Mao M., Zhou H., Zhao Y., Shi J. A novel cell-penetrating peptide protects against neuron apoptosis after cerebral ischemia by inhibiting the nuclear translocation of annexin A1. Cell Death Differ. 2019;26:260–275. doi: 10.1038/s41418-018-0116-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xia Q., Mao M., Zeng Z., Luo Z., Zhao Y., Shi J., Li X. Inhibition of SENP6 restrains cerebral ischemia-reperfusion injury by regulating Annexin-A1 nuclear translocation-associated neuronal apoptosis. Theranostics. 2021;11:7450–7470. doi: 10.7150/thno.60277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xia Q., Li X., Zhou H., Zheng L., Shi J. S100A11 protects against neuronal cell apoptosis induced by cerebral ischemia via inhibiting the nuclear translocation of annexin A1. Cell Death Dis. 2018;9:657. doi: 10.1038/s41419-018-0686-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xia Y., Hu G., Chen Y., Yuan J., Zhang J., Wang S., Li Q., Wang Y., Deng Z. Embryonic stem cell derived small extracellular vesicles modulate regulatory T cells to protect against ischemic stroke. ACS Nano. 2021;15:7370–7385. doi: 10.1021/acsnano.1c00672. [DOI] [PubMed] [Google Scholar]
- 62.Shi Y., Jiang X., Zhang L., Pu H., Hu X., Zhang W., Cai W., Gao Y., Leak R.K., Keep R.F., et al. Endothelium-targeted overexpression of heat shock protein 27 ameliorates blood-brain barrier disruption after ischemic brain injury. Proc. Natl. Acad. Sci. USA. 2017;114:E1243–E1252. doi: 10.1073/pnas.1621174114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon reasonable request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.