

Abstract
Chlamydia pneumoniae is a widespread pathogen of humans causing pneumonia and bronchitis. There are many reports of an association between C.pneumoniae infection and atherosclerosis. We determined the whole genome sequence of C.pneumoniae strain J138 isolated in Japan in 1994 and compared it with the sequence of strain CWL029 isolated in the USA before 1987. The J138 circular chromosome consists of 1 226 565 nt (40.7% G+C) with 1072 likely protein-coding genes that is 3665 nt shorter than the CWL029 genome. Plasmids, phage- or transposon-like sequences were not identified. The overall genomic organization, gene order and predicted proteomes of the two strains are very similar, suggesting a high level of structural and functional conservation between the two unrelated isolates. The most conspicuous differences in the J138 genome relative to the CWL029 genome are the absence of five DNA segments, ranging in size from 89 to 1649 nt, and the presence of three DNA segments, ranging from 27 to 84 nt. The complex organization of these ‘different zones’ may be attributable to a unique system of recombination.
INTRODUCTION
Chlamydia are obligate intracellular eubacteria. Chlamydia pneumoniae, a causative agent of acute respiratory diseases, is frequently associated with complex chronic diseases such as atherosclerosis (1–4), asthma and multiple sclerosis (5). Comparison of the genome sequences of various strains will facilitate understanding of the common biological processes required for the infection and unique properties that may differentiate in virulence and diseases. We determined the whole genome sequence of C.pneumoniae strain J138 isolated in Japan and compared it with the sequence of strain CWL029 isolated in the USA (6).
MATERIALS AND METHODS
Sequencing
Chlamydial EBs were purified from infected HEp2 cells by urografin gradients as described previously (7). DNA was isolated, cloned into pUC18 and sequenced to an average coverage of 10-fold using dye-labeled primers as described previously (6). Gaps were closed either by sequencing a spanning PCR product or by using custom oligonucleotides to extend the sequence from the ends of adjacent contigs. PCR products were sequenced to improve quality in remaining low-quality regions.
Annotation
A systematic sequence similarity search of the ORFs was conducted against the annotated genome sequences of Chlamydia trachomatis serover D strain and C.pneumoniae CWL029 strain (GenBank accession numbers AE001273 and NC000922) using the BLAST2 program (8). The start codons for conserved protein-coding genes were estimated by identifying start codons adjacent to ribosome-binding motifs and on the basis of sequence conservation using the FASTA and CLUSTAL-W alignments (9). When more than one potential start codon was predicted, the first was arbitrarily chosen for annotation.
The genome sequence, annotation and gene alignments of DNA fragments will appear in GenBank under accession numbers AB036071–AB036089. Further information is available in the C.pneumoniae genome database (http://w3.grt.kyushu-u.ac.jp/J138/ ).
RESULTS AND DISCUSSION
Chlamydia pneumoniae strain J138 was isolated in Japan in 1994 from pharyngeal mucosa of a 5-year-old boy with acute bronchitis. The whole genome was sequenced and compared with the previously sequenced C.pneumoniae CWL029 genome. The identity in restriction sites in physical maps of the two strains has been interpreted to mean that C.pneumoniae strains are highly conserved in their genomic organization and gene order (Fig. 1). The sequence comparison showed that three DNA segments, ranging in size from 27 to 84 nt, are unique to the J138 genome (G2, G4 and G6), while five DNA segments, ranging in size from 89 to 1649 nt, are unique to the CWL029 genome (G1, G3, G5, G7 and G8) (Fig. 2). With the exception of these eight regions, the nucleotide sequences throughout the genomes are almost identical (identity, 99.9%), and the orders of the homologous sets of genes are the same. To our surprise, each of the sequences in region G2 of J138 and regions G1, G3, G5 and G7 of CWL029 has an almost identical counterpart in the adjacent region (Fig. 2). Thus, these elements might have a role in generating the organizational differences by deletion or duplication. In contrast to the regions described above, regions G4, G6 and G8 have no such elements. The assembled sequence of the J138 genome in these regions was further confirmed by re-sequencing the PCR products from the genome.
Figure 1.
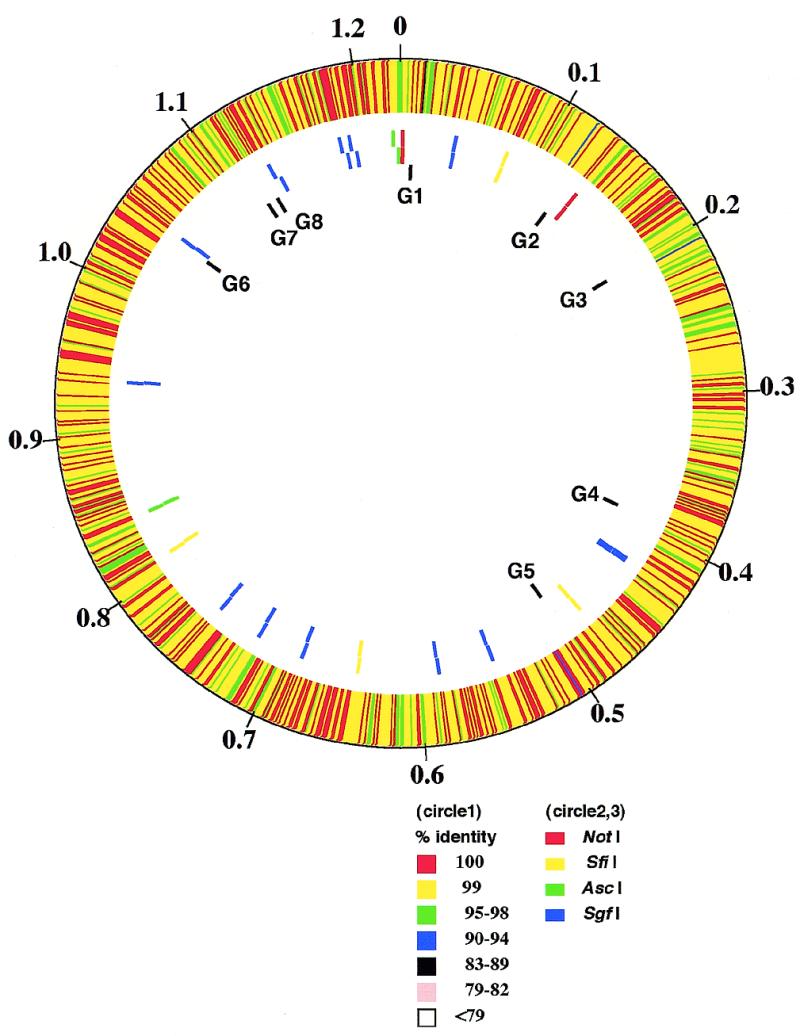
Comparison of the two C.pneumoniae genomes based on the chromosomal organization of strain CWL029. The genome-wide view is shown on the physical map. Circles are numbered starting from the outermost concentric ring. Circle 1, nucleotide similarity between J138 and CWL029 (the minimum width of color line indicates 1002 bp). Circles 2 and 3 represent the locations of the AscI, SgfI, SfiI and NotI sites in the CWL029 and J138 genomes, respectively. The relative location of each J138 or CWL029-specific sequence (G1~8) is shown immediately inside the third circle. The largest J138-specific region shown is composed of a segment of 1649 bp. To align homologous regions of the two genomes, we needed to artificially insert and/or delete eight DNA segments, ranging in size from 27 to 1649 bp, of the J138 sequence. Sizes of regions in the figure are shown in megabases (Mb).
Figure 2.


Genomic map of DNA segments unique to J138 or CWL029. The CWL029 ORFs are shown in the order and location that they are found (upper bar with nucleotide numbers and ORFs with CPn gene numbers at the top). The J138 ORFs are shown as lower bars with nucleotide numbers. ORFs with CPn gene numbers are shown at the bottom. The boxes and bars show the regions homologous between the two strains. The relative orientation of the J138 segments and genome with respect to that of the CWL029 segments is completely the same as indicated by arrows (closed arrowheads). The J138- or CWL029-specific DNA segments were delimited by lines. The organization of J138 segments that share 87.5~100% identical direct repeats (or duplications) of the regions G1, G2, G3, G5 and G7 in J138 and CWL029 are depicted with arrows (open arrowheads).
Repeat sequences may be responsible for integration of new DNA segments into the chromosome and for transfer of these segments between strains. The direct repeats in regions G1, G2, G3, G5 and G7 are not found in other regions throughout the chromosome. Clustering of DNA with a lower or higher (G+C) percentage suggestive of horizontal DNA transfer was not found in these regions, thus the strain-specific differences are not consistent with different origins for these DNA segments. The distribution of genomic reorganizations or rearrangements is not even on the chromosomes, as the region between C.pneumoniae J138 genome encoding CPn0911–CPn0979 (region G6–G8) contains substantially more reorganizations than the other areas of the genome (Fig. 1). This region does not coincide with any important regions, such as the predicted chromosome replication terminus (6).
Six coding genes in the CWL029 genome are different in J138 (Fig. 2). Thus, CPn0007 for a hypothetical protein (G1) and pmp-6 for Chlamydia-specific polymorphic outer membrane protein (G5) encode truncated proteins lacking 110 and 131 amino acids in the middle region of the homologs in CWL029, respectively, pfkA for fructose-6-phosphate phosphotransferase lacks the C-terminal 14 amino acids of the CWL029 homolog (G3), and htrB for lauroyl acyltransferase is separated into CPn0098.1 and htrB (G2) in J138. In addition, the J138 genome lacks CPn0979 for a hypothetical protein (G8) and one of the two copies of tyrP for tyrosine transporter as well as yccA for a transport permease (G7).
The low proportion of strain-specific ORFs that may be attributed to the absence of insertion sequence, phage or transposon-like homologs indicates that the C.pneumoniae genome may be limited in its flexibility. Striking similarity between the C.pneumoniae genome sequences of the two unrelated strains might be linked to the unique intracellular biology of this organism.
Recent analysis of the genomic polymorphism of chlamydial species revealed that the genome of C.pneumoniae is highly conserved, whereas that of C.trachomatis is diverse (10). Identification of the minimal genetic diversity in the whole genome will facilitate understanding of the biology of C.pneumoniae. The high genomic conservation of C.pneumoniae is in contrast to the strain-specific genetic diversity of Helicobacter pylori, the human gastric pathogen, that has been proposed to be involved in the organism’s ability to cause different diseases and to participate in the lifelong chronicity of infection (11). The genome sequence of C.pneumoniae AR39, which has just been published and found to be identical to the sequence of CWL029 except a truncated ORF (~300 bp shorter in AR39) and a bacteriophage (T.Read, personal communication), provides additional information on the genomic stability of this organism (12).
Previous genomic-sequence comparison of the two chlamydial species revealed that the C.pneumoniae CWL029 genome contains 187 711 additional nucleotides including 214 coding sequences compared with the C.trachomatis serovar D genome (6,13). In addition, there is a substantially low level of similarity for individual encoded proteins between the orthologs from the two species. These results are consistent with the recent reclassification of genus Chlamydia based on the phylogenetic analysis. A new genus, genus Chlamydophila was proposed to assimilate the current species, C.pneumoniae, Chlamydia pecorum and Chlamydia psittaci (14).
Acknowledgments
ACKNOWLEDGEMENTS
We thank N. Ogasawara and H. Yoshikawa (Nara University of Technology and Science) for helpful suggestions, C. Tanaka, H. Matsushima, J. Nishida, K. Shibata, H. Takeuchi and R. Fujinaga (Yamaguchi University) for assistance in finalizing sequence information, and K. Oshima, S. Nagao and R. Fukawa (Hitachi Instruments Service Co., Ltd) for technical assistance in sequencing. This work has been supported by the Japan Society for the Promotion of Science, Research for the Future Program (97L00101).
DDBJ/EMBL/GenBank accession nos AB036071–AB036089
REFERENCES
- 1.Grayston J.T., Kuo,C.C., Campbell,L.A. and Wang,S.P. (1989) Int. J. Syst. Bacteriol., 39, 88–90. [Google Scholar]
- 2.Kuo C.C., Shor,A., Campbell,L.A., Fukushi,H., Patton,D.L. and Grayston,J.T. (1993) J. Infect. Dis., 167, 841–849. [DOI] [PubMed] [Google Scholar]
- 3.Kuo C.C., Grayston,J.T., Campbell,L.A., Goo,Y.A., Wissler,R.W. and Benditt,E.P. (1995) Proc. Natl Acad. Sci. USA, 92, 6911–6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mlot C. (1996) Science, 272, 1422. [DOI] [PubMed] [Google Scholar]
- 5.Sriram S., Stratton,C.W., Yao,S., Tharp,A., Ding,L., Bannan,J.D. and Mitchell,W.M. (1999) Ann. Neurol., 46, 6–14. [PubMed] [Google Scholar]
- 6.Kalman S., Mitchell,W., Marathe,R., Lammel,C., Fan,J., Hyman,R.W., Olinger,L., Grimwood,J., Davis,R.W. and Stephens,R. (1999) Nature Genet., 21, 385–389. [DOI] [PubMed] [Google Scholar]
- 7.Caldwell H.D., Kromhout,J. and Schachter,J. (1981) Infect. Immun., 31, 1161–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thompson J.D., Higgins,D.G. and Gibson,T.J. (1994) Nucleic Acids Res., 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meijer A., Morre,S.A., Van Den Brule,A.J.C., Savelkoul,P.H.M. and Ossewaarde,J.M. (1999) J. Bacteriol., 181, 4469–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alm R.A., Ling,L.S., Moir,D.T., King,B.L., Brown,E.D., Doig,P.C., Smith,D.R., Noonan,B., Guild,B.C., deJonge,B.L. et al. (1999) Nature, 397, 176–180. [DOI] [PubMed] [Google Scholar]
- 12.Read T.D., Brunham,R.C., Shen,C., Gill,S.R., Heidelberg,J.F., White,O., Hickey,E.K., Peterson,T., Utterback,J., Berry,K. et al. (2000) Nucleic Acids Res., 28, 1397–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stephens R.S., Kalman,S., Lammel,C., Fan,J., Marathe,R., Aravind,L., Mitchell,W., Olinger,L., Tatusov,R.L., Zhao,Q. et al. (1998) Science, 282, 754–759. [DOI] [PubMed] [Google Scholar]
- 14.Everett K.D., Bush,R.M. and Amndersen,A.A. (1999) Int. J. Syst. Bacteriol., 49, 415–440. [DOI] [PubMed] [Google Scholar]